 Wei Guang1
Wei Guang1
- 1 Department of Pediatrics, University of Maryland School of Medicine, Baltimore, MD, USA
- 2 Department of Pathology, University of Maryland School of Medicine, Baltimore, MD, USA
Interleukin (IL)-8-driven neutrophil infiltration of the gastric mucosa is pathognomonic of persistent Helicobacter pylori infection. Our prior study showed that ectopic over-expression of MUC1 in human AGS gastric epithelial cells reduced H. pylori-stimulated IL-8 production compared with cells expressing MUC1 endogenously. Conversely, Muc1 knockout (Muc1−/−) mice displayed an increased level of transcripts encoding the keratinocyte chemoattractant (KC), the murine equivalent of human IL-8, in gastric mucosa compared with Muc1+/+ mice during experimental H. pylori infection. The current study tested the hypothesis that a decreased IL-8 level observed following MUC1 over-expression is mediated through the ability of MUC1 to associate with β-catenin, thereby inhibiting H. pylori-induced β-catenin nuclear translocation. Increased neutrophil infiltration of the gastric mucosa of H. pylori-infected Muc1−/− mice was observed compared with Muc1+/+ wild type littermates, thus defining the functional consequences of increased KC expression in the Muc1-null animals. Protein co-immunoprecipitation (co-IP) studies using lysates of untreated or H. pylori-treated AGS cells demonstrated that (a) MUC1 formed a co-IP complex with β-catenin and CagA, (b) MUC1 over-expression reduced CagA/β-catenin co-IP, and (c) in the absence of MUC1 over-expression, H. pylori infection increased the nuclear level of β-catenin, (d) whereas MUC1 over-expression decreased bacteria-driven β-catenin nuclear localization. These results suggest that manipulation of MUC1 expression in gastric epithelia may be an effective therapeutic strategy to inhibit H. pylori-dependent IL-8 production, neutrophil infiltration, and stomach inflammation.
Introduction
Stomach cancer is the fourth most common cancer worldwide and the second most common cause of cancer death1 (∼800,000 per year). In the U.S., 21,520 new cases of gastric cancer with 10,340 deaths are estimated to occur in 20112. Colonization of the gastric mucosa by Helicobacter pylori is the strongest identified risk factor for malignancies that arise within the stomach, with infection by the bacteria estimated to cause ∼70% of all gastric cancers (Peek et al., 2010). However, the vast majority of H. pylori-infected individuals are asymptomatic beyond histologic inflammation. Within the subset of bacteria-infected individuals who do display clinical symptoms, epidemiologic, and pathologic studies have confirmed a progressive increase in disease symptomatology and severity over several decades starting with chronic inflammation (gastritis), followed by the development of non-cancerous or precancerous lesions, and finally gastric adenocarcinoma (Correa, 1995). The current paradigm of H. pylori-induced gastric cancer proposes that chronic and persistent gastritis during a lifetime of infection generates an environment awash with inflammatory mediators and signaling pathways that ultimately lead to tumorigenesis (Correa and Piazuelo, 2011). Indeed, the host inflammatory response to H. pylori infection is a primary factor involved in the development of stomach cancer. In support of this hypothesis, chronic stomach infection with H. pylori is associated with epithelial cell gene mutations, inhibition of apoptosis, stimulation of angiogenesis, and increased cell proliferation (MacFarlane and Stover, 2007). Therefore, better understanding of host proinflammatory mechanisms, as well as the counter-regulatory pathways that normally attenuate inflammation, will provide important new information to treat H. pylori-infected patients with gastritis who are at increased risk for developing gastric cancer.
Epithelial cells of the mucosa, including the gastric epithelium, express multiple surface receptors that signal the presence of mucosal pathogens and activate host inflammation (Brown et al., 2010). While much is known concerning the inflammatory pathways that are activated following microbial colonization, relatively few studies have characterized the counterbalancing anti-inflammatory responses. Using a mouse model of H. pylori stomach infection, we recently reported that expression of the Mucin-1 (Muc1) protein counter-regulated H. pylori-induced gastric inflammation (Guang et al., 2010). MUC1/Muc1 (by convention, MUC1 in humans, Muc1 in animals) is a membrane bound protein with a highly glycosylated ectodomain and a tyrosine phosphorylated intracellular domain that is expressed by all secretory epithelia and most hematopoietic cells. The MUC1 cytoplasmic region interacts with β-catenin through a -S-X-X-X-X-X-S-S-L- motif (where X represents any amino acid) that is conserved with other β-catenin-binding partners, including the cadherins (Yamamoto et al., 1997; Li et al., 2001a; Schroeder et al., 2003; Molock and Lillehoj, 2006). β-Catenin was originally identified in the Wnt signaling pathway, a major oncogenic pathway associated with gastric tumorigenesis (Oshima and Oshima, 2010). Subsequently, β-catenin was demonstrated to form a multiprotein complex that anchors the cytoplasmic domain of E-cadherin to the actin cytoskeleton, thereby regulating epithelial barrier formation, the paracellular pathway, and polarity in normal cells (Baum and Georgiou, 2011). However, the role of MUC1/β-catenin interaction in regulating H. pylori gastritis is unknown.
Helicobacter pylori express a number of virulence factors that promote infection, including the cytotoxin-associated gene A (CagA). Individuals infected with bacterial strains carrying the Cag pathogenicity island (PAI) have a stronger gastric inflammatory response and are at a greater risk of developing stomach cancer compared with individuals infected with strains lacking the Cag PAI (Kusters et al., 2006). The Cag PAI encodes a type IV secretion apparatus that delivers the CagA protein into gastric epithelial cells where it disrupts the cytoskeleton, modifies intercellular adherence, and alters cell polarity (Backert and Selbach, 2008). Recent mechanistic studies have begun to elucidate how CagA manifests its numerous cellular effects through the modification of specific signal transduction pathways (Hatakeyama, 2004). Among the intracellular signaling cascades that are affected by CagA is the β-catenin pathway (Franco et al., 2005; Bebb et al., 2006; Murata-Kamiya et al., 2007). For example, Murata-Kamiya et al. (2007) reported that CagA formed a co-immunoprecipitation complex with E-cadherin, thereby destabilizing the E-cadherin/β-catenin complex and cytoplasmic/nuclear accumulation of β-catenin. Because MUC1 interacts with β-catenin (Li et al., 2001a), and because nuclear β-catenin transactivates the IL-8 gene promoter (Lévy et al., 2002), we hypothesized that one of the mechanisms by which up-regulation of MUC1 expression reduces H. pylori-stimulated IL-8 production involves disruption of CagA-dependent β-catenin nuclear localization. As an initial step toward testing this hypothesis, the current study investigated protein–protein interactions between MUC1, β-catenin, and CagA, and the cytoplasm/nuclear distribution of these proteins, in H. pylori-treated AGS gastric epithelial cells.
Materials and Methods
Bacteria
Helicobacter pylori strains SS1 (Ding et al., 2009) and 26695 (Talarico et al., 2009) were maintained on Columbia blood agar containing 7% defibrinated horse blood (Cleveland Scientific, Bath, OH, USA), 20 μg/ml bacitracin, 20 μg/ml trimethoprim, 16 μg/ml cefsulodin, 6.0 μg/ml vancomycin, and 2.5 μg/ml fungizone (Sigma, St. Louis, MO, USA) under microaerophilic conditions as described (Ding et al., 2009). For in vitro infection of AGS cells, bacteria were grown in static liquid cultures of Brucella broth (Difco, Detroit, MI, USA) containing 10% heat inactivated fetal bovine serum (FBS) and antibiotics at 37°C with 10% CO2.
Muc1 Knockout Mice
Muc1−/− mice on a FVB genetic background were generated by homologous recombination (Guang et al., 2010). Age- (8–12 week) and gender-matched FVB Muc1+/+ littermates were from The Jackson Labs (Bar Harbor, ME, USA). Muc1 genotypes and phenotypes were confirmed by polymerase chain reaction and Western blot analysis, respectively, using gastric epithelial tissues. All mice were caged in Static Micro-Isolator LPTM cages (Lab Products, Seaford, DE, USA), bedded in combination size corncob bedding, fed solid chow, and housed under specific pathogen-free conditions.
H. pylori Stomach Infection and Histologic Evaluation
Mice were infected by gastric intubation on two consecutive days with 0.5 ml of log-phase cultures of H. pylori strain SS1 at 1.0 × 107 colony forming units (CFU)/animal as described (Guang et al., 2010). Mice were sacrificed by intraperitoneal pentobarbital injection at 4 weeks post-infection and stomach strips were fixed in K-2 fixative (2% paraformaldehyde, 2.5% glutaraldehyde, 0.1 M sodium cacodylate buffer, 0.025% CaCl2) as described (Ding et al., 2009). The fixative was replaced with 0.1 M sodium cacodylate buffer (4°C) containing sodium azide (pH 7.35–7.40). Tissue was paraffin embedded and sectioned for analysis by Giemsa staining. Gastric inflammation was assessed as described (Stuller et al., 2008). Briefly, the area of the tissue section displaying the most severe inflammation was evaluated blindly and assigned a global score from 0 to 5 based on the following parameters: 0, absence of lesions; 1, mild infiltrate of inflammatory cells, usually along the base of the glands; 2, larger area of inflammation extending between glands and/or in submucosa; 3, inflammatory infiltrate extending between glands toward the lumen and in the underlying submucosa; 4, intense transmucosal inflammation extending across the field, distorting glandular architecture, with epithelial hyperplasia and extensive mucous cell metaplasia often present; 5, extensive mucosal and submucosal inflammation with complete disruption of glandular architecture and ulceration. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Maryland.
AGS Cells
AGS gastric adenocarcinoma cells (CRL-1739, ATCC, Manassas, VA, USA) were cultured in DMEM/F12 medium containing 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA, USA). The cells were washed with PBS and treated for 24 h with H. pylori strain 26695 at a multiplicity of infection (MOI) of 100, or with PBS as a vehicle control.
Over-Expression of MUC1
AGS cells in 60-mm dishes were transfected with the pcDNA empty vector (Invitrogen) or a MUC1 expression plasmid (pMUC1) using Lipofectamine as described (Lillehoj et al., 2003). The cells were cultured for 24 h with PBS or H. pylori, and processed for co-immunoprecipitation.
Co-Immunoprecipitation Assay
Helicobacter pylori-treated AGS cells were washed with PBS and lysed at 4°C with PBS, pH 7.2 containing 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM NaF, 1.0 mM NaVO4, and 1.0% protease inhibitor cocktail (Sigma) as described (Guang et al., 2010). Equal protein aliquots (1.0 mg) were immunoprecipitated with 2.0 μg of antibodies to MUC1, β-catenin, or H. pylori CagA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) plus protein G-agarose (Invitrogen) as described (Lillehoj et al., 2003). Immunoprecipitated proteins were resolved on 4–20% SDS-polyacrylamide gels and transferred to PVDF membranes (BioRad, Richmond, CA, USA). The membranes were blocked for 1 h with PBS containing 0.1% Tween 20 (PBS-T) and 5% non-fat dry milk (Sigma) and reacted with antibodies against MUC1, β-catenin, or CagA, each diluted 1:1,000. The membranes were washed with PBS-T, incubated for 1 h at room temperature with horseradish peroxidase-conjugated anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) diluted 1:5,000 and developed with enhanced chemiluminescence substrate (Thermo Scientific, Rockford, IL, USA). To confirm equivalent protein loading and transfer, the blots were stripped with 62.5 mM Tris–HCl, pH 6.7, 100 mM 2-mercaptoethanol, and 2.0% SDS, blocked with PBS-T, 5% milk, and probed with the same antibody used for immunoprecipitation. Immunoreactive bands were identified by co-migration of prestained protein size markers (Fermentas, Glen Burnie, MD, USA). In some experiments, AGS whole cell lysates, or cytoplasmic and nuclear fractions of the cells (Guang et al., 2010), were directly probed with antibodies against MUC1, β-catenin, or CagA followed by secondary antibodies and chemiluminescence substrate. To control for protein loading and transfer, the blots were stripped and reprobed for β-actin (Santa Cruz Biotechnology).
Co-Localization Immunofluorescence Microscopy
AGS cells were cultured overnight in 4-chamber Lab-Tek slides (Nunc, Naperville, IL, USA), washed with PBS, treated for 5 h with H. pylori, washed, and fixed for 15 min with 4% paraformaldehyde. The cells were permeabilized and blocked for 15 min at 22°C with PBS containing 0.1% Triton X-100 and 5% normal goat serum (NGS), followed by 15 min at 22°C with Image-iT™ FX signal enhancer (Invitrogen). The slides were washed and incubated overnight at 4°C with rabbit or mouse primary antibodies to MUC1, β-catenin, or H. pylori CagA, all diluted 1/100 in PBS containing 2% BSA, 2% NGS, and 0.05% Tween 20. The slides were washed and incubated for 1 h at 22°C with Alexa Fluor 488-conjugated goat anti-rabbit IgG or Alexa Fluor 594-conjugated goat anti-mouse IgG secondary antibodies (1/1,000; Invitrogen). DAPI was used to counterstain nuclei. The cells were mounted with Vectashield (Vector Laboratories, Burlingame, CA, USA) and visualized using an Axio Observer.Z1 microscope (Carl Zeiss, Oberkochen, Germany) fitted with a 63× objective and standard excitation/emission filters for detection of Alexa Fluor 488, Alexa Fluor 594, and DAPI. Images were cropped and assembled into panels with Adobe Photoshop 4.0 (Adobe Systems, San Jose, CA, USA).
Results
H. pylori-Infected Muc1−/− Mice have Increased Neutrophil Infiltration in the Gastric Mucosa Compared with Muc1+/+ Mice
Our prior study demonstrated that silencing of MUC1 expression in human gastric AGS epithelial cells using a MUC1-targeting small interfering (si)RNA, or through defective Muc1 expression in Muc1 knockout mice, increased H. pylori-stimulated IL-8 or keratinocyte chemoattractant (KC) production compared with the respective MUC1/Muc1-expressing cells (Guang et al., 2010). Because IL-8 and KC are chemotactic for neutrophils, we asked whether abrogation of Muc1 expression in a mouse model of H. pylori infection similarly increased neutrophil infiltration of the gastric mucosa. Muc1+/+ and Muc1−/− mice were challenged by gastric intubation with 1.0 × 107 CFU/animal of H. pylori, and the gastric mucosa was visualized at 4 weeks post-infection by Giemsa staining as described (Ding et al., 2009). Figure 1Ai–v illustrates representative stomach sections from infected and uninfected Muc1+/+ and Muc1−/− mice. Quantitative analysis revealed no differences in neutrophilic inflammation between uninfected Muc1+/+ and uninfected Muc1−/− mice (Figure 1B). By contrast, significantly greater neutrophil infiltration of the gastric mucosa was seen in H. pylori-infected Muc1−/− mice compared with infected Muc1+/+ mice.
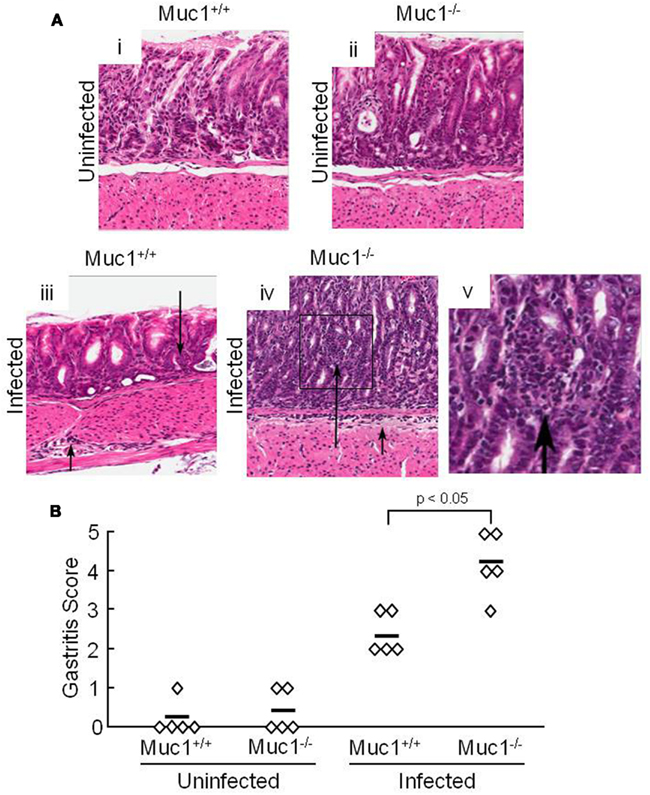
Figure 1. H. pylori-infected Muc1−/− mice have increased neutrophils in the gastric mucosa compared with Muc1+/+ mice. (A) Muc1+/+ (i,iii) and Muc1−/− (ii,iv) mice were uninfected (i,ii) or were infected with H. pylori (iii,iv), and gastric mucosa were assessed at 4 weeks post-infection by Giemsa staining. Arrows indicate inflammatory cell infiltrates. Original magnification, 200×. (v) Illustrates a higher magnification of the indicated area of (iv) to demonstrate the infiltrating cells. (B) Quantitative analysis of gastric inflammation on a scale of 0–5 based on the following parameters: 0, no significant lesions; 1, mild infiltrate of inflammatory cells; 2, larger focus of inflammation extending between glands and/or in submucosa; 3, patches of inflammation extending between glands toward the lumen and in the underlying submucosa; 4, intense transmucosal inflammatory infiltrate extending across the field distorting glandular architecture; 5, extensive mucosal and submucosal inflammation with disruption of glandular architecture and ulceration (Stuller et al., 2008). Each symbol represents an individual tissue section. Horizontal bars represent median values. Differences between median values were assessed by the Student’s t test. The results are representative of two experiments.
MUC1 forms a Co-Immunoprecipitation Complex with β-Catenin and CagA
We and others have previously shown that MUC1 interacts with several members of the Armadillo protein family, including β-catenin and p120-catenin (Li et al., 2001a; Schroeder et al., 2003; Molock and Lillehoj, 2006). In addition, H. pylori CagA is known to destabilize the E-cadherin/β-catenin complex (Murata-Kamiya et al., 2007), and CagA directly interacts with p120-catenin (Oliveira et al., 2009). On the basis of these combined studies, we hypothesized that the increased IL-8 and KC levels observed with MUC1/Muc1 deficiency was mediated through the ability of MUC1 mucin to associate with β-catenin and/or CagA. To test this hypothesis, reciprocal co-immunoprecipitation (co-IP) experiments were performed between the three denoted proteins in lysates of AGS cells. MUC1 formed a co-IP complex with β-catenin in untreated and H. pylori-treated AGS cells (Figure 2A). Correspondingly, CagA formed a co-IP complex with MUC1 (Figure 2B) and with β-catenin (Figure 2C) in the infected cells. As controls, total MUC1 and β-catenin protein levels were equal in untreated and bacteria-treated cells (Figure 2D). Moreover, it appeared that MUC1/β-catenin co-IP was increased in H. pylori-treated cells compared with untreated cells (Figure 2A), suggesting that CagA may release β-catenin from E-cadherin, thus allowing it to bind to MUC1. By immunofluorescence microscopy, co-staining of H. pylori-infected AGS cells with antibodies to MUC1 and β-catenin, and with antibodies to MUC1 and CagA, demonstrated that these proteins are both found within the same cellular compartment (Figures 3i–viii). By confocal microscopy, co-localization of MUC1 with β-catenin (Figure 3ix), and MUC1 with CagA (Figure 3x), was also apparent.
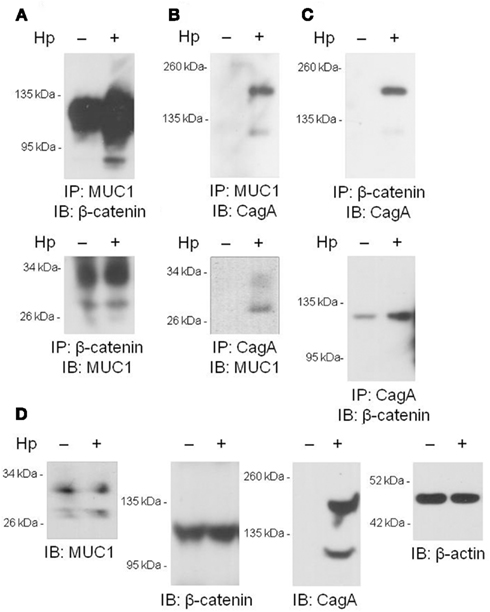
Figure 2. Co-immunoprecipitation of MUC1, β-catenin, and H. pylori CagA. (A–C) AGS cells were untreated or were treated with H. pylori (Hp) at an MOI of 100. At 24 h post-infection, the cells were washed and cell lysates were immunoprecipitated with the indicated antibodies. Immunoprecipitated proteins were resolved by SDS-PAGE, transferred to PVDF membranes, and processed for immunoblotting with the indicated antibodies. (D) Lysates of untreated or HP-treated AGS cells were resolved by SDS-PAGE, transferred to PVDF membranes, and processed for immunoblotting with antibodies to MUC1, β-catenin, CagA, or β-actin. Molecular weight markers are indicated on the left in kilodaltons (kDa). The results are representative of two experiments.
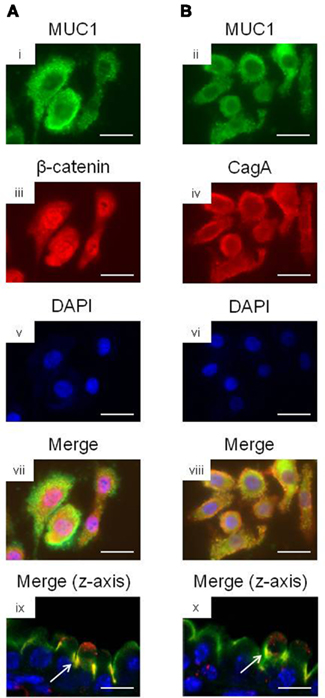
Figure 3. Co-localization microscopy of MUC1, β-catenin, and H. pylori CagA. AGS cells were treated with H. pylori and stained with (A) MUC1 (i) and β-catenin (ii) antibodies, or with (B) MUC1 (iii) and CagA (iv) antibodies. The cells were counterstained with DAPI (v,vi) and examined by immunofluorescence microscopy. Co-localization of MUC1 with β-catenin (vii), and MUC1 with CagA (viii), was revealed by yellow immunostaining of merged images. Co-localization of MUC1 with β-catenin (ix), and MUC1 with CagA (x) was also revealed by confocal microscopy along the z-axis of the cell monolayers, with the apical region of the cells facing up. Arrows in (ix,x) indicate areas of co-localization. Scale bar, 25 μm. The results are representative of two experiments.
Over-Expression of MUC1 Reduces CagA/β-Catenin co-IP
Because MUC1 interacted with both β-catenin and CagA, we asked whether over-expression of MUC1 would modulate the interaction between the latter two proteins. AGS cells transfected with a MUC1 expression plasmid, or with the empty vector control, were processed for reciprocal β-catenin/CagA and CagA/β-catenin co-IPs. As illustrated in Figure 4, reduced co-immunoprecipitation between β-catenin and CagA was observed in MUC1-transfected cells compared with the empty vector controls, suggesting that MUC1 competes with CagA for binding of β-catenin. Stripping and reprobing these blots with the immunoprecipitating antibody confirmed equal gel loading and membrane transfer of the proteins in MUC1 and vector only transfected cells.
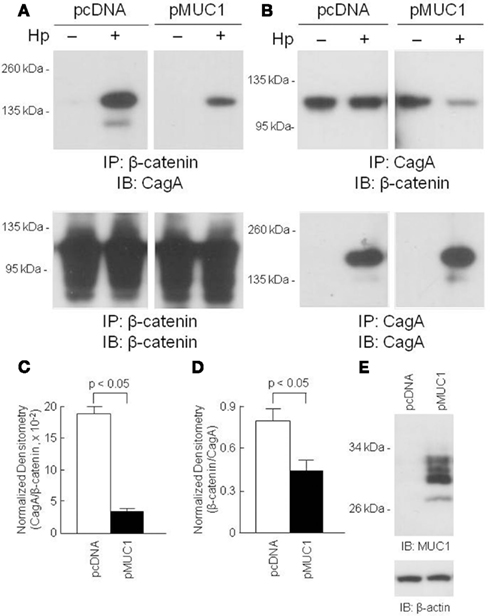
Figure 4. Over-expression of MUC1 reduces CagA/β-catenin co-immunoprecipitation. AGS cells were transfected with the pcDNA empty vector or the pMUC1 expression plasmid. At 24 h post-transfection, the cells were untreated or were treated with H. pylori (Hp) at an MOI of 100. At 24 h post-infection, the cells were washed and cell lysates were immunoprecipitated with antibodies to β-catenin (A) or CagA (B). Immunoprecipitated proteins were resolved by SDS-PAGE, transferred to PVDF membranes, and processed for immunoblotting with antibodies to CagA (A) or β-catenin (B). To control for to control for protein loading and transfer, blots were stripped and reprobed with the same antibody used for immunoprecipitation. Molecular weight markers are indicated on the left in kilodaltons (kDa). (C) Densitometry of the blots in (A). (D) Densitometry of the blots in (B). Each bar represents the mean ± SEM value of the density of the CagA band normalized to the β-catenin band [from (A)], or the β-catenin band normalized to the CagA band [from (B)], in Hp-treated cells (n = 3). (E) Increased MUC1 expression in AGS cells transfected with the pMUC1 plasmid. The results are representative of two experiments.
Over-Expression of MUC1 Inhibits H. pylori-Dependent Increased Nuclear β-Catenin Levels
Published studies have demonstrated that H. pylori infection of gastric epithelia increases nuclear translocation of β-catenin (Murata-Kamiya et al., 2007). Given that MUC1 formed a co-IP complex with β-catenin in the cytoplasm of AGS cells (Figure 2A), we hypothesized that over-expression of MUC1 would inhibit the ability of H. pylori to induce β-catenin nuclear translocation. In untreated cells transfected with the pcDNA empty vector, or with the pMUC1 expression plasmid, β-catenin levels in the nucleus were generally less than or relatively equal to cytoplasmic β-catenin levels (Figure 5, lanes 1 vs. 2 and 3 vs. 4). In H. pylori-treated cells transfected with the empty vector, the level of nuclear β-catenin was increased compared with the cytoplasmic β-catenin level (lanes 6 vs. 5), thus corroborating prior reports (Murata-Kamiya et al., 2007). However, in bacteria-treated cells that were over-expressing MUC1, the level of nuclear β-catenin was dramatically reduced compared with cytoplasmic β-catenin (lanes 8 vs. 7). In contrast, while MUC1 also translocated to the nucleus to a small extent compared with its cytoplasmic level, cytoplasmic and nuclear MUC1 levels were relatively equal in untreated and H. pylori-treated cells (lanes 11 vs. 15 and 12 vs. 16). The distribution of CagA between the cytoplasmic and nuclear fractions was unaffected by manipulation of MUC1 expression (lanes 21 vs. 23 and 22 vs. 24). Indeed, the relative distribution of β-catenin in the cytoplasmic and nuclear fractions of H. pylori-treated cells that were over-expressing MUC1 (lanes 7, 8) was similar to that seen with MUC1 (lanes 15, 16) and quite distinct from that observed with CagA (lanes 23, 24). In summary, these data are consistent with the hypothesis that over-expression of MUC1 blocks H. pylori-dependent β-catenin nuclear translocation, likely through sequestration of the MUC1/β-catenin protein complex in the cytoplasm.
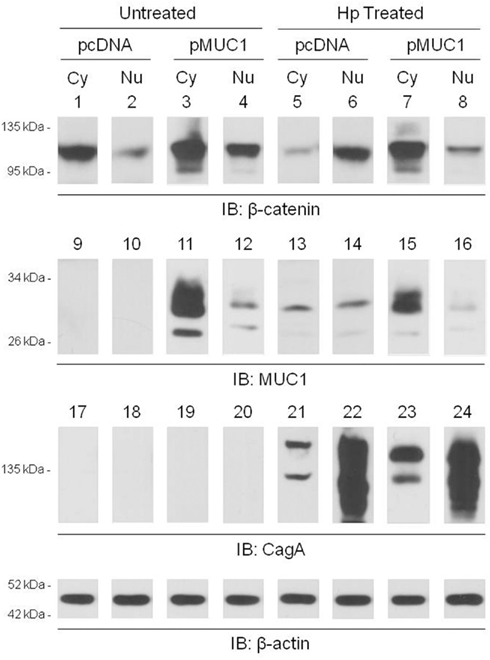
Figure 5. MUC1 over-expression inhibits H. pylori-dependent increased nuclear β-catenin level. AGS cells were transfected with the pcDNA empty vector or the pMUC1 expression plasmid. At 24 h post-transfection, the cells were untreated or were treated with H. pylori (Hp) at an MOI of 100. At 24 h post-infection, the cells were washed, cytoplasmic (Cy) and nuclear (Nu) lysate fractions were prepared, and immunoprecipitated with antibodies to β-catenin (lanes 1–8), MUC1 (lanes 9–16), or CagA (lanes 17–24). To control for to control for protein loading and transfer, blots were stripped and reprobed with antibody to β-actin. Molecular weight markers are indicated on the left in kilodaltons (kDa). The results are representative of two experiments.
Discussion
This study demonstrated (a) increased neutrophil infiltration of the gastric mucosa of H. pylori-infected Muc1−/− mice compared with Muc1+/+ littermates, (b) MUC1/β-catenin, MUC1/CagA, and CagA/β-catenin co-IPs complexes in lysates of AGS cells, (c) reduced level of the CagA/β-catenin co-IP complex in MUC1 over-expressing cells, (d) increased nuclear level of β-catenin in H. pylori-infected cells, and (e) inhibition of bacteria-driven nuclear β-catenin following MUC1 over-expression. These results, together with our prior data (Guang et al., 2010), can be most parsimoniously explained if over-expression of MUC1 blocks H. pylori-stimulated β-catenin nuclear translocation, thereby attenuating IL-8 production and neutrophil gastric infiltration. A schematic illustration of the putative mechanisms and pathways involving β-catenin and CagA in H. pylori-infected gastric epithelial cells with or without MUC1 over-expression with regards to the predominant protein complexes, nuclear translocation of β-catenin, and IL-8 production is shown in Figure 6.
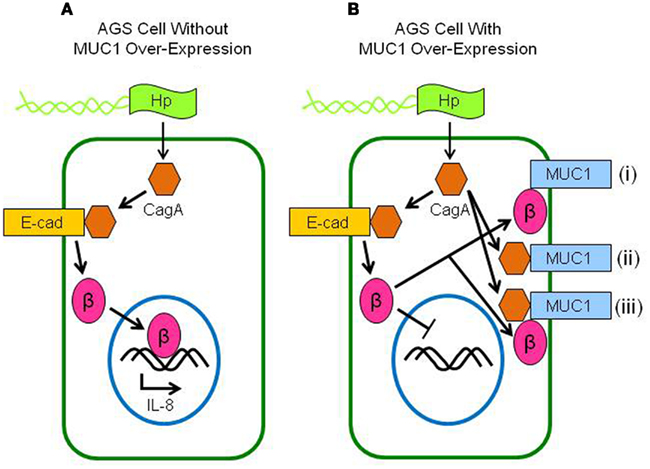
Figure 6. Schematic illustration depicting the putative interactions between MUC1, β-catenin, and CagA in H. pylori-infected gastric epithelial cells. (A) Without MUC1 over-expression, CagA delivered to the cytosol of H. pylori (Hp)-infected cells displaces β-catenin (β) from E-cadherin (E-cad), allowing β-catenin to translocate to the nucleus and activate IL-8 gene expression. (B) With MUC1 over-expression, β-catenin that is displaced by CagA binds to MUC1 (i), thus blocking its nuclear translocation and attenuating IL-8 production. CagA also binds to MUC1, possibly either without (ii) or with (iii) β-catenin.
Helicobacter pylori colonize the gastric mucosa of up to half the world’s population and play a causative role in the development of gastritis, peptic ulcers, and stomach cancer. While H. pylori infection causes a massive influx of neutrophils into the stomach epithelium, for unknown reasons, the host immune response does not clear the infection. Up to 70% of H. pylori strains in Western countries encode the Cag PAI (Peek and Crabtree, 2006). Individuals infected with Cag PAI expressing H. pylori strains have greater gastric inflammatory responses and are at an increases risk of developing peptic ulcers or stomach cancer than those infected with strains lacking the Cag PAI (Kusters et al., 2006). Following adhesion of H. pylori to gastric epithelial cells, the type IV secretion system expressed by the Cag PAI delivers several bacterial components to the cell cytosol. Among these are bacterial peptidoglycan and the CagA gene product. Peptidoglycan is an agonist for the innate immune receptor, Nod1, which stimulates expression of TNF-α and IL-8, as well as other cytokines and chemokines, that promote inflammation (Viala et al., 2004).
CagA elicits multiple cellular alterations, including disruption of the cytoskeleton, increased intercellular adhesion, altered intracellular signaling, and loss of cell polarity (Backert and Selbach, 2008). Some of these effects occur as a consequence of CagA phosphorylation at a C-terminal EPIYA-repeat motif by the c-Src and Abl cytosolic tyrosine kinases, resulting in activation of the protein tyrosine phosphatase, SHP-2 (Hatakeyama, 2004). Cag+ strains of H. pylori also have been shown to activate the receptor protein tyrosine kinase, epidermal growth factor receptor (EGFR; Keates et al., 2005), although in this case the mechanism has been reported to involve activation of toll-like receptor (TLR)4 by the H. pylori secretory protein, HP0175 (Basu et al., 2008). As with activation of SHP-2, activation of EGFR by H. pylori is associated with altered signal transduction and gene expression in host epithelial cells that may contribute to pathogenesis. In this regard, EGFR tyrosine phosphorylates the MUC1 intracellular domain, leading to binding of β-catenin (Li et al., 2001b). Interaction of MUC1 with β-catenin modulated the Wnt target gene, cyclin D1, in H. pylori-induced gastric cancer (Udhayakumar et al., 2007) and decreased nuclear translocation of β-catenin in a human embryonic kidney cell model system (Lillehoj et al., 2007).
Previous studies have identified H. pylori-induced CagA-dependent and CagA-independent β-catenin activation and nuclear translocation (Franco et al., 2005; Bebb et al., 2006; Murata-Kamiya et al., 2007; Weydig et al., 2007). In the nucleus, β-catenin acts as a transcription factor in association with the lymphoid enhancer-binding factor/T cell factor (LEF/TCF) proteins (Henderson and Fagotto, 2002). Among the best characterized target genes activated by β-catenin/LEF/TCF complexes are cyclin D1, c-Myc, and others that promote carcinogenesis by stimulating cell proliferation and inhibiting apoptosis. In addition, evidence exists to demonstrate that nuclear β-catenin stimulates the expression of a variety of inflammation-related genes, including IL-8. For example, Lévy et al. (2002) reported that the β-catenin/TCF4 complex transcriptionally activated IL-8 gene expression, and that ectopic expression of β-catenin in hepatoma cells increased IL-8 protein secretion. IL-8 also has been identified as a transcriptional target of β-catenin in human umbilical vein endothelial cells (Masckauchán et al., 2005). Comparison of disease and healthy tissues in a mouse model of colorectal cancer identified KC (murine IL-8) as a major up-regulated proinflammatory chemokine in colon cancer (Zhu et al., 2011). Finally, translocation of β-catenin into the nucleus of hepatocellular carcinoma cells was correlated with increased IL-8 production (Lai et al., 2011). In the current study, it may be possible that up-regulation of MUC1 expression in AGS gastric epithelial cells blocks H. pylori-dependent β-catenin activation and nuclear translocation, thereby inhibiting bacteria-induced IL-8 production. This remains a valid hypothesis for future testing. Infection of cells with suppressed β-catenin expression by CagA-positive and CagA-negative H. pylori, followed by quantification of IL-8 production would start to address this question and would greatly expand the physiological relevance of these findings.
In addition to binding to MUC1, β-catenin also formed a co-IP complex with CagA (Figure 2C). Similarly, MUC1/CagA protein interaction was observed (Figure 2B), suggesting that CagA may indirectly associate with β-catenin through their mutual binding to MUC1. Our previous study demonstrated that MUC1 interacted with β-catenin, but not with γ-catenin, in transfected HEK293 cells (Molock and Lillehoj, 2006). MUC1 contains a -S-A-G-N-G-G-S-S-L- sequence that serves as the binding site for β-catenin and that fits a canonical β-catenin-binding motif present in E-cadherin. E-cadherin also binds to γ-catenin and mapping studies have shown that β- and γ-catenin harbor overlapping binding sites on E-cadherin (Potter et al., 1999). Binding of the two catenins to E-cadherin is mutually exclusive. Because the β- and γ-catenin binding sites on E-cadherin are only partially overlapping, it is possible that additional sequences outside of the denoted motif, and not present in MUC1, may contribute to γ-catenin binding.
In summary, the results of this study raise the interesting prospect of counteracting H. pylori-induced IL-8 production, with subsequent reduction of neutrophil infiltration and gastritis, using therapeutic strategies to augment MUC1 expression in the gastric mucosa. Future investigations of the potential of augmented MUC1 expression for treatment of H. pylori gastritis may yield promising results.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by U.S. Public Health Service grants AI072291 and AI083463. The authors thank Penny Bamford for help with immunofluorescence microscopy.
Footnotes
References
Backert, S., and Selbach, M. (2008). Role of type IV secretion in Helicobacter pylori pathogenesis. Cell. Microbiol. 10, 1573–1581.
Basu, S., Pathak, S. K., Chatterjee, G., Pathak, S., Basu, J., and Kundu, M. (2008). Helicobacter pylori protein HP0175 transactivates epidermal growth factor receptor through TLR4 in gastric epithelial cells. J. Biol. Chem. 283, 32369–32376.
Baum, B., and Georgiou, M. (2011). Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J. Cell Biol. 192, 907–917.
Bebb, J. R., Leach, L., Zaitoun, A., Hand, N., Letley, D. P., Thomas, R., and Atherton, J. C. (2006). Effects of Helicobacter pylori on the cadherin-catenin complex. J. Clin. Pathol. 59, 1261–1266.
Brown, J., Wang, H., Hajishengallis, G. N., and Martin, M. (2010). TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 90, 417–427.
Correa, P. (1995). Helicobacter pylori and gastric carcinogenesis. Am. J. Surg. Pathol. 19, S37–S43.
Correa, P., and Piazuelo, M. B. (2011). Helicobacter pylori infection and gastric adenocarcinoma. US Gastroenterol. Hepatol. Rev. 7, 59–64.
Ding, H., Nedrud, J. G., Wershil, B., Redline, R. W., Blanchard, T. G., and Czinn, S. J. (2009). Partial protection against Helicobacter pylori in the absence of mast cells in mice. Infect. Immun. 77, 5543–5550.
Franco, A. T., Israel, D. A., Washington, M. K., Krishna, U., Fox, J. G., Rogers, A. B., Neish, A. S., Collier-Hyams, L., Perez-Perez, G. I., Hatakeyama, M., Whitehead, R., Gaus, K., O’Brien, D. P., Romero-Gallo, J., and Peek, R. M. Jr. (2005). Activation of β-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 102, 10646–10651.
Guang, W., Ding, H., Czinn, S. J., Kim, K. C., Blanchard, T. G., and Lillehoj, E. P. (2010). MUC1 cell surface mucin attenuates epithelial inflammation in response to a common mucosal pathogen. J. Biol. Chem. 285, 20547–20557.
Hatakeyama, M. (2004). Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 4, 688–694.
Henderson, B. R., and Fagotto, F. (2002). The ins and outs of APC and β-catenin nuclear transport. EMBO Rep. 3, 834–839.
Keates, S., Keates, A. C., Nath, S., Peek, R. M. Jr., and Kelly, C. P. (2005). Transactivation of the epidermal growth factor receptor by cag+Helicobacter pylori induces upregulation of the early growth response gene Egr-1 in gastric epithelial cells. Gut 54, 1363–1369.
Kusters, J. G., van Vliet, A. H., and Kuipers, E. J. (2006). Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490.
Lai, T. Y., Su, C. C., Kuo, W. W., Yeh, Y. L., Kuo, W. H., Tsai, F. J., Tsai, C. H., Weng, Y. J., Huang, C. Y., and Chen, L. M. (2011). β-Catenin plays a key role in metastasis of human hepatocellular carcinoma. Oncol. Rep. 26, 415–422.
Lévy, L., Neuveut, C., Renard, C. A., Charneau, P., Branchereau, S., Gauthier, F., Van Nhieu, J. T., Cherqui, D., Petit-Bertron, A. F., Mathieu, D., and Buendia, M. A. (2002). Transcriptional activation of interleukin-8 by β-catenin-Tcf4. J. Biol. Chem. 277, 42386–42393.
Li, Y., Kuwahara, H., Ren, J., Wen, G., and Kufe, D. (2001a). The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3β and β-catenin. J. Biol. Chem. 276, 6061–6064.
Li, Y., Ren, J., Yu, W., Li, Q., Kuwahara, H., Yin, L., Carraway, K. L. III, and Kufe, D. (2001b). The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and β-catenin. J. Biol. Chem. 276, 35239–35242.
Lillehoj, E. P., Han, F., and Kim, K. C. (2003). Mutagenesis of a Gly-Ser cleavage site in MUC1 inhibits ectodomain shedding. Biochem. Biophys. Res. Commun. 307, 743–749.
Lillehoj, E. P., Lu, W., Kiser, T., Goldblum, S. E., and Kim, K. C. (2007). MUC1 inhibits cell proliferation by a β-catenin-dependent mechanism. Biochim. Biophys. Acta 1773, 1028–1038.
MacFarlane, A. J., and Stover, P. J. (2007). Convergence of genetic, nutritional and inflammatory factors in gastrointestinal cancers. Nutr. Rev. 65, S157–S166.
Masckauchán, T. N., Shawber, C. J., Funahashi, Y., Li, C. M., and Kitajewski, J. (2005). Wnt/β-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis 8, 43–51.
Molock, K. E., and Lillehoj, E. P. (2006). Biochemical interactions among intercellular adhesion molecules expressed by airway epithelial cells. Biochem. Biophys. Res. Commun. 343, 513–519.
Murata-Kamiya, N., Kurashima, Y., Teishikata, Y., Yamahashi, Y., Saito, Y., Higashi, H., Aburatani, H., Akiyama, T., Peek, R. M. Jr., Azuma, T., and Hatakeyama, M. (2007). Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 26, 4617–4626.
Oliveira, M. J., Costa, A. M., Costa, A. C., Ferreira, R. M., Sampaio, P., Machado, J. C., Seruca, R., Mareel, M., and Figueiredo, C. (2009). CagA associates with c-Met, E-cadherin, and p120-catenin in a multiprotein complex that suppresses Helicobacter pylori-induced cell-invasive phenotype. J. Infect. Dis. 200, 745–755.
Oshima, H., and Oshima, M. (2010). Mouse models of gastric tumors: Wnt activation and PGE2 induction. Pathol. Int. 60, 599–607.
Peek, R. M. Jr., Fiske, C., and Wilson, K. T. (2010). Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol. Rev. 90, 831–858.
Peek, R. M., and Crabtree, J. E. (2006). Helicobacter infection and gastric neoplasia. J. Pathol. 208, 233–248.
Potter, W., Bergwitz, C., and Brabant, G. (1999). The cadherin-catenin system: implications for growth and differentiation of endocrine tissues. Endocr. Rev. 20, 207–239.
Schroeder, J. A., Adriance, M. C., Thompson, M. C., Camenisch, T. D., and Gendler, S. J. (2003). MUC1 alters β-catenin-dependent tumor formation and promotes cellular invasion. Oncogene 22, 1324–1332.
Stuller, K. A., Ding, H., Redline, R. W., Czinn, S. J., and Blanchard, T. G. (2008). CD25+ T cells induce Helicobacter pylori-specific CD25- T-cell anergy but are not required to maintain persistent hyporesponsiveness. Eur. J. Immunol. 38, 3426–3435.
Talarico, S., Gold, B. D., Fero, J., Thompson, D. T., Guarner, J., Czinn, S., and Salama, N. R. (2009). Pediatric Helicobacter pylori isolates display distinct gene coding capacities and virulence gene marker profiles. J. Clin. Microbiol. 47, 1680–1688.
Udhayakumar, G., Jayanthi, V., Devaraj, N., and Devaraj, H. (2007). Interaction of MUC1 with β-catenin modulates the Wnt target gene cyclinD1 in H. pylori-induced gastric cancer. Mol. Carcinog. 46, 807–817.
Viala, J., Chaput, C., Boneca, I. G., Cardona, A., Girardin, S. E., Moran, A. P., Athman, R., Mémet, S., Huerre, M. R., Coyle, A. J., DiStefano, P. S., Sansonetti, P. J., Labigne, A., Bertin, J., Philpott, D. J., and Ferrero, R. L. (2004). Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 5, 1166–1174.
Weydig, C., Starzinski-Powitz, A., Carra, G., Lower, J., and Wessler, S. (2007). CagA-independent disruption of adherence junction complexes involves E-cadherin shedding and implies multiple steps in Helicobacter pylori pathogenicity. Exp. Cell Res. 313, 3459–3471.
Yamamoto, M., Bharti, A., Li, Y., and Kufe, D. (1997). Interaction of the DF3/MUC1 breast carcinoma-associated antigen and β-catenin in cell adhesion. J. Biol. Chem. 272, 12492–12494.
Keywords: Helicobacter pylori, stomach, gastric, infection
Citation: Guang W, Twaddell WS and Lillehoj EP (2012) Molecular interactions between MUC1 epithelial mucin, β-catenin, and CagA proteins. Front. Immun. 3:105. doi: 10.3389/fimmu.2012.00105
Received: 10 January 2012; Paper pending published: 20 February 2012;
Accepted: 16 April 2012; Published online: 07 May 2012.
Edited by:
Thomas Blanchard, University of Maryland School of Medicine, USAReviewed by:
Emma Slack, Universität Bern, SwitzerlandDiane Bimczok, University of Alabama at Birmingham, USA
Copyright: © 2012 Guang, Twaddell and Lillehoj. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Erik P. Lillehoj, Department of Pediatrics, University of Maryland School of Medicine, 655 West Baltimore Street, BRB 13-029, Baltimore, MD 21201, USA. e-mail:ZWxpbGxlaG9qQHBlZHMudW1hcnlsYW5kLmVkdQ==