

- 1 Immunotherapy Center, Georgia Health Sciences University, Augusta, GA, USA
- 2 Department of Radiology, Georgia Health Sciences University, Augusta, GA, USA
- 3 NewLink Genetics Corporation, Ames, IA, USA
- 4 Department of Medicine, Georgia Health Sciences University, Augusta, GA, USA
The term “immune privilege” was coined to describe weak immunogenicity (hypo-immunity) that manifests in some transplant settings. We extended this concept to encompass hypo-immunity that manifests at local sites of inflammation relevant to clinical diseases. Here, we focus on emerging evidence that enhanced tryptophan catabolism is a key metabolic process that promotes and sustains induced immune privilege, and discuss the implications for exploiting this knowledge to improve treatments for hypo-immune and hyper-immune syndromes using strategies to manipulate tryptophan metabolism.
Introduction
Immune privilege refers to the phenomenon of low immunogenicity associated with certain organs such as the anterior chamber of the eye, the brain, the testes, and the placenta (Munn and Mellor, 2006). Mechanistically, immune privilege probably arises from the combined effects of physical barriers to immunity and active immune regulatory processes that are particularly potent in some organs and tissues. The concept of immune privilege can also be extended to some chronic inflammatory syndromes where enhanced immune regulation (hypo-immunity) manifests as a factor in disease progression such as persistent infections and cancer (Mellor and Munn, 2008). In this sense, immune privilege is not a unique feature of certain specialized tissues; rather, it is a functional state induced by certain conditions in most – perhaps all – tissues as one consequence of chronic inflammation. In this review we focus on a specific molecular pathway that creates induced immune privilege in a range of disease settings involving expression of the inducible enzyme indoleamine 2,3 dioxygenase (IDO). When biochemically active the IDO enzyme catabolizes tryptophan to produce metabolites known collectively as kynurenines. Local metabolic effects of IDO-expressing cells exert profound effects on immune and tissue cells that suppress pro-inflammatory and immune stimulatory responses to a variety of insults. IDO is not the only molecular pathway able to create immune privilege in inflamed tissues, and several other metabolic pathways have been linked to regulatory outcomes recently. By focusing exclusively on the IDO pathway our aim is to use this particular metabolic pathway to illustrate “good” and “bad” aspects of immune privilege in relation to clinical disease syndromes.
A Conceptual Model of Induced Immune Privilege
A diverse range of primary insults stimulate local inflammation, including wounding, infections, pre-malignancies, radiation, vaccines, and chemical or biological toxins. However, the mucosal surfaces of the respiratory, gastro-intestinal-tracts, and reproductive tracts (especially the uterus during pregnancy) exhibit many features of inflamed tissues during homeostasis. The ability of insults to elicit potent innate and adaptive immunity varies considerably depending on which tissue experiences the primary insult. In this review we focus on natural and experimentally induced insults that stimulate inflammatory responses with potent immune regulatory components as examples of where induced immune privilege manifests. Previously, we proposed a model to explain how IDO enzyme activity induced in local tissues as a response to inflammatory cues may create immune privilege (Mellor and Munn, 2008). We proposed four key checkpoints that promote and maintain immune privilege in local inflamed tissues. In this section, we further develop this model in light of recent novel insights (Figure 1).
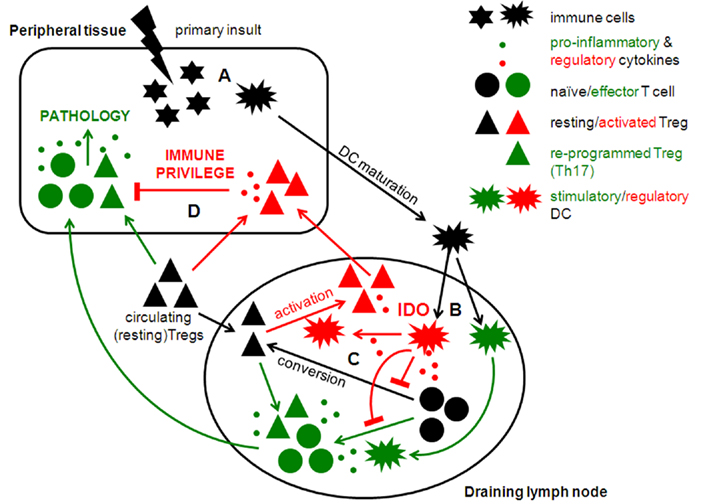
Figure 1. Potential regulatory checkpoints involved in induced immune privilege in local tissue microenvironments.
Checkpoint A
Innate immune responses to inflammatory insults
Typically, microbial infections induce rapid release of pro-inflammatory cytokines such as interferons (IFNαβ, IFNγ), tumor necrosis factor (TNFα), interleukins (IL-1β, IL-6, etc.), and chemokines by stromal cells and innate immune cells to provoke effective adaptive immunity. However some inflammatory stimuli – including some microbes – elicit innate immune responses that promote production of regulatory cytokines such as IL-10, and transforming growth factor (TGFβ) that suppress local release of pro-inflammatory cytokines and activate (or attract) immune regulatory cells. Thus initial local responses to primary insults exert profound influences on the subsequent course of immune responses, and elucidating how cells resident in primary tissue lesions respond to inflammatory cues is pivotal to understand the source of immune privilege. For example, pre-malignancies arise frequently in many tissues, but their ability to progress to form tumors is acquired over long periods, and overt tumors develop only when immune surveillance mechanisms are evaded successfully. Similar considerations may apply to pathogens that cause some persistent infections, though pathogens have probably evolved numerous mechanisms to evade – and even actively suppress – innate immune surveillance mechanisms to induce immune privilege.
In light of the considerations above it is probably no coincidence that tumors and chronic infections are often associated with induced local IDO expression because IDO has potent regulatory effects on immune and non-immune cell types (Uyttenhove et al., 2003; Boasso, 2011;Makala et al., 2011). Ablating the IDO1 gene in mice also enhanced their resistance to tumor formation in the standard of inflammation-dependent, carcinogen-induced (DMBA/TPA) cancer model (Banerjee et al., 2008). The unrelated enzyme tryptophan dioxygenase (TDO) is also expressed by some tumors and, like IDO catalyzes oxidative tryptophan catabolism to produce kynurenines (Opitz et al., 2011). Thus pre-malignant cells expressing IDO or TDO may create immune privilege via (a) tryptophan depletion to activate the general control non-derepressible-2 (GCN2) dependent integrated stress response (ISR) to amino acid withdrawal, and (b) kynurenine that binds to the aryl hydrocarbon receptor (AhR). Early induction of these (and perhaps other) metabolic processes may be critical steps in disease progression leading to the hypo-immune syndromes that characterize tumor growth and chronic infections. In this regard it may be significant that UV-irradiation (UVB) creates skin edema and associated inflammatory responses that include local IDO induction (unpublished data). Interestingly, skin tanning in response to UVB is mediated by the AhR (Jux et al., 2011), suggesting that exposure to UVB irradiation has both desirable and detrimental effects due to altered tryptophan metabolism. If these notions are correct early treatments to block immune regulatory processes may be effective in reducing the risk of contracting cancer and chronic infections. However, it is important to stress that IDO may serve dual functions as (a) an immune regulatory pathway that benefits developing tumors and pathogens, or (b) a host defense mechanism that impedes tumor growth and microbial infections to the benefit of hosts. This is a key consideration as the dominant effects of IDO at sites of inflammation may not be clear from simple observation, and must be determined by appropriate manipulation of experimental systems. For example, IDO is a frequent component of chronic inflammation associated with autoimmune destruction of healthy tissues, a correlation that could be interpreted as evidence that IDO promotes autoimmune pathology. However, exposure to IDO inhibitors accelerates autoimmune progression and potentiates disease severity in many autoimmune disease models, revealing that IDO regulates destructive autoimmunity in these syndromes. A helpful analogy to bear in mind is that firefighters are always in attendance at fires, but their presence does not guarantee that fires will eventually be brought under control to prevent total destruction. In summary, pre-malignancies and pathogens may exploit innate “host” regulatory mechanisms such as IDO to create local conditions that permit these agents of disease to evade immune surveillance during the early phases of tumor development and infections. This paradigm implies that IDO-mediated regulation that creates immune privilege may be a key factor in disease progression, and not simply a target for therapy to break immune privilege in patients with established tumors and chronic infections. If correct, treatments that target the IDO pathway may reduce the risk that pre-malignancies and persistent pathogens establish immune privilege before they can create pathologic disease. However, such interventions may also interfere with homeostatic control of immunity to self-antigens and innocuous antigens such as allergens, and it remains to be determined if appropriate balances between hyper-immunity and prevention of diseases due to hypo-immunity can be engineered through early therapeutic interventions in individuals with high risk profiles.
Checkpoint B
Antigen presentation and lymphocyte activation in local draining lymph nodes
Presenting antigens to lymphocytes is necessary, but may not be sufficient to provoke adaptive immunity. Antigens in the local inflammatory lesion, usually skin or a mucosal surface, are captured by resident tissue macrophages (MΦs) and dendritic cells (DCs) via phagocytosis or pinocytosis. DCs are specialized to undergo rapid maturation in response to innate inflammatory cues (generated at Checkpoint A), and migrate to local draining lymph nodes (dLNs) where they present processed antigens, in the form of peptides bound to surface MHC molecules, to T cells. Hence mature DCs are “professional antigen presenting cells” (APCs) that present antigens from (a) external sources such as pathogens or innocuous substances (food, allergens commensal microbes), and (b) tissue (self) antigens to T cells. T cells that recognize MHC/peptides in dLNs then undergo rapid activation and differentiation to generate helper T cells that promote optimal cellular and humoral responses, and effector (cytolytic) T cells. The processes of acquiring, transporting, and presenting antigens to T cells in dLNs may be subject to regulation in some settings of inflammation. Inhibiting DC maturation or migration would impede the process of generating helper/effector T cells. Moreover, immature DCs that enter dLNs may present antigens (signal 1) in the context of sub-optimal B7-CD28 co-stimulation (signal 2) leading to weak and ineffective T cell responses (Hackstein and Thomson, 2004). Even fully mature DCs may suppress effector responses if they acquire regulatory attributes that promote anergy or apoptosis in T cells that respond to antigens they present, and regulatory DCs may also induce naïve T cells to convert into regulatory T cells (Tregs; Reis e Sousa, 2006). Negative co-stimulatory pathways (e.g., PD-1/PD-L, ICO/ICOS-L) and certain metabolic processes (see below) may also attenuate effector responses and promote tolerance in dLNs. For example, DCs expressing IDO possess mature phenotypes but block effector T cell responses, promote CD4 T cell conversion into Foxp3-lineage Tregs, and activate pre-formed Tregs (Mellor et al., 2004, 2005; Munn et al., 2004;Baban et al., 2005, 2009, 2011;Sharma et al., 2007; Chen et al., 2008; Brenk et al., 2009;Chung et al., 2009). Thus DCs competent to express IDO or other regulatory pathways in response to appropriate cues may be pivotal in shaping adaptive immune responses to primary insults contingent on whether they are – or are not – induced to acquire regulatory phenotypes in particular settings of inflammation.
Checkpoint C
Prevailing regulatory conditions in draining LNs
Lymphoid tissues exhibit differential capacities to promote and support adaptive immunity. Thus skin dLNs typically support robust adaptive immune responses to a range of topical insults (though not all, see below), while LNs draining mucosal surfaces generally support weak adaptive immune responses due to prevailing physiologic conditions at these sites (Kraal et al., 2006). Thus, even if tissue DCs mature efficiently, and migrate in large numbers from primary lesions to dLNs their ability to promote effective adaptive immune responses to antigens they present is critically dependent on the prevailing physiologic status of dLNs. Some factors and pathways discussed in Checkpoint B may also help establish and sustain regulatory conditions that prevail in some lymphoid tissues, and regulatory cytokines such as TGFβ may be critical to maintain such conditions during homeostasis. The constitutive presence of certain cytokines may dictate the local “immunologic tone” in homeostatic tissues, but they may also reflect local cellular states pre-determined during homeostasis. For example, immunologic tone may be pre-determined by the relative proportions of pre-formed Tregs in a given lymphoid tissue, which in turn affects the ability of APCs to present antigens in a regulatory or stimulatory context through differential “licensing” of APCs by Tregs or helper T cells. As discussed below, the presence or absence of DCs expressing IDO may be a pivotal determinant of prevailing immunologic tone in lymphoid tissues.
Checkpoint D
Induced immune regulation in inflamed target tissues
Responses to primary insults elaborated via local dLNs may include regulatory components that impact what happens in target tissues affected by the primary insult. Over time, certain cell types with immune regulatory attributes may accumulate in primary lesions to dampen down initial pro-inflammatory responses. Indeed, delayed suppression of overt immunity is considered essential to attenuate induced immune responses as local infections are brought under control and pathogens are cleared from primary lesions. Several cell types possessing a variety of regulatory pathways may help down-regulate immune responses, including (but probably not limited to) lymphocytes, myeloid cells (MΦs, DCs), natural killer (NK) cells, mast cells, and enigmatic myeloid-derived suppressor cells (MDSCs) and mesenchymal stem cells found in many settings of inflammation where hypo-immunity manifests (Ding et al., 2010; De Vries et al., 2011; Mantovani et al., 2011; Murphy et al., 2011;Ostrand-Rosenberg et al., 2012). In transplant settings tolerogenic factors that protect allografted tissues are complex, involving immunologic and metabolic processes required on a continuous basis, as disrupting these mechanisms usually leads to graft rejection even after prolonged periods of allograft acceptance (Cobbold, 2010). Thus regulatory cells and factors such as cytokines and IDO that promote immune regulation may be essential to maintain hypo-immunity following adaptive immune responses as well as mediating early responses that promote hypo-immunity (Checkpoint A). It remains to be seen if similar processes promote and maintain immune privilege in inflamed tissues.
IDO Biochemistry and Metabolic Effects on Immune Cells
IDO is a highly conserved, heme-containing intracellular enzyme that catabolizes compounds containing indole rings such as the essential amino acid tryptophan. Two closely linked, homologous IDO genes (IDO1, IDO2) are located in syntenic regions of chromosome 8 in humans and mice. IDO1 and IDO2 may respond to distinct signals since their patterns of gene expression are not identical. IDO1 encodes functional IDO protein that regulates T cell responses when expressed in some DCs while IDO2 is expressed in other cell types. Unlike IDO genes, TDO gene expression is controlled by stress factors such as glucocorticoids, not inflammatory IFNs. Here we focus on the role of IDO1 gene expression immune regulation. Though IDO is expressed by few cell types during homeostasis a number of cell types mediate IDO enzyme activity in response to inflammatory insults including stromal, hematopoietic, and tumor cells. Interferons type I (IFNαβ) and II (IFNγ) are potent IDO inducers due to the presence of interferon stimulated response element (ISRE) and interferon-gamma activated sequence (GAS) elements in the proximal gene promoters of mammalian IDO genes (Dai and Gupta, 1990; Chon et al., 1995). Other molecular signals from cytokines (TGFβ, IL-10), Toll-like receptor (TLRs), B7, CD200, GITR, OX40, PD-1, and AhR ligands may also promote IDO expression, though in some cases IDO induction may be an indirect response to IFNs elicited by primary signals (see below). MΦs expressing IDO in response to certain infections such as Toxoplasma also mediate innate host defense. Though IDO does not affect sterile clearance of Toxoplasma infections in mice, IDO ablation led to 100% mortality showing that IDO plays a crucial regulatory role in host–pathogen interactions in this chronic infection (Divanovic et al., 2012). IDO was first shown to regulate T cell immunity to fetal tissues during pregnancy (essentially a tolerated allograft) since treatment with the IDO inhibitor 1-methyl-tryptophan (1MT) caused pregnancy failure in mice due to maternal T cell mediated destruction of allogeneic but not syngeneic fetal tissues (Munn et al., 1998; Mellor et al., 2001). Since then IDO activity has been linked to tumor-induced tolerance, persistence of chronic infections, and attenuation of autoimmune, and allergic disease syndromes.
As mentioned above DCs expressing IDO suppress antigen-specific T cell responses. IDO activity in DCs does not attenuate antigen presentation and T cell activation but causes activated T cells to undergo cell cycle arrest and apoptosis, or induce anergy and promote Tregs conversion. IDO activity may suppress T cell responses in three ways: (a) tryptophan withdrawal, (b) production of kynurenines, and (c) altered redox potentials due to consumption of superoxide radicals (Figure 2). Reduced access to tryptophan during T cell activation triggers cell cycle arrest by activating GCN2 kinase, which senses reduced amino acid levels when uncharged tRNA molecules bind to ribosomes. GCN2 kinase phosphorylates eIF2α at serine 51 to inhibit general protein synthesis, and activate downstream stress response genes such as CCAAT/enhancer-binding protein homologous protein (CHOP) which has pro-apoptotic functions. T cells from GCN2-deficient mice were resistant to the regulation by DCs expressing IDO (Munn et al., 2005), and resting Tregs from GCN2-deficient mice did not acquire regulatory phenotypes in response to DCs expressing IDO (Sharma et al., 2007; Baban et al., 2009). Arginase activity in MΦs had similar effects on T cells and Tregs indicating that GCN2 kinase activation is a common downstream pathway by which amino acid catabolism promotes immune regulation (Bronte and Zanovello, 2005; Rodriguez et al., 2007). Some tryptophan catabolites made by IDO-expressing DCs also possess regulatory attributes by inhibiting T cell proliferation, inducing T cell apoptosis, and blocking differentiation of Th1 helper cells (Frumento et al., 2002; Terness et al., 2002;Fallarino et al., 2003; Chen et al., 2008). Consistent with this notion 3-hydroxyanthranilic acid (3-HAA) blocked T cell responses and promoted T cell apoptosis by depletion of intracellular glutathione (GSH) and by inhibition of PDK1, which is an essential mediator of CD28-induced NFκB activation (Lee et al., 2010). Recent studies showing that kynurenine binds to AhR and that AhR signaling regulates T cell responses and activates Tregs also provide mechanistic explanations for IDO-mediated T cell regulation (Quintana et al., 2008; Vogel et al., 2008;Bankoti et al., 2010; Mezrich et al., 2010; Opitz et al., 2011). T cells may also be sensitive to changes in redox potential mediated by IDO activity in DCs. Reactive oxygen species (ROS) produced by phagocytes and other activated immune cells such as neutrophils have potent effects on cell metabolism and biological functions. Thus, mice with a genetic defect (pHox49-deficient) in NADPH oxidase-dependent ROS production exhibited defective IDO activity that contributed to T cell hyperactivity in a model of pulmonary aspergillosis, analogous to patients susceptible to chronic granulomatous disease (CGD; Romani et al., 2008). In vitro studies also showed that treatment of human NK cell line with IDO metabolite, L-kynurenine, significantly reduced cell viability through generation of ROS (Song et al., 2011).
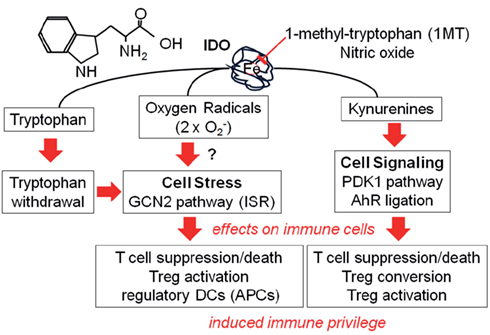
Figure 2. Metabolic effects of IDO activity on immune cells.
IDO activity also blocks functional re-programming of pre-formed, resting Tregs into polyfunctional helper/effector (Th17) T cells in certain settings of inflammation. Thus, IDO activity in DCs activated GCN2 kinase in Tregs, and inhibited IL-6 production in tumor dLNs and in mice treated with TLR9 ligands to induce IDO (Baban et al., 2009; Sharma et al., 2009). Moreover, IDO-mediated Treg activation in response to systemic TLR9 ligands was dependent on regulatory cytokines (TGFβ, IL-10) and negative co-stimulation via CTLA4/B7 and PD-1/PD-L interactions between Tregs and DCs that maintained competence of DCs to express IDO in response to TLR9-induced IFN type I signaling (Baban et al., 2011). These data suggest that multiple pathways maintain default responsiveness via IDO to create regulatory outcomes during homeostasis, and that inflammatory signals must overcome these default regulatory pathways to promote pro-inflammatory, immune stimulatory outcomes. For example, vaccination with antigen plus (low dose) TLR9 ligands promoted Treg conversion into effector T cells, which were essential for initial priming of effector CD8 T cells by cross-presented antigens (Sharma et al., 2010). However IDO expression induced by established tumors prevented Treg re-programming, indicating that IDO was critical in regulating early effector T cell responses during inflammation and that vaccines were rendered ineffective, unless IDO activity was ablated. In human immunodeficiency virus (HIV) infected patients, elevated IDO activity by myeloid DCs was associated with loss of Th17 cells and increase of Tregs (Favre et al., 2010). Thus IDO has a critical role in maintaining the balance of Tregs and helper/effector T cells, and may be critical for pathogens to persist in immunocompetent hosts despite host defense functions of IDO in some infections.
IDO-Competent DCs
Dendritic cells are the only APC type capable of activating naïve T cells, underscoring their critical role in initiating T cell immunity. Few DCs in humans and mice are competent to express functional IDO. Though murine CD8− and CD8+ DC subsets expressed IDO protein in response to IFNγ treatment, only CD8+ DCs produced kynurenine, indicating IDO enzyme activity (Fallarino et al., 2002). Systemic treatments with soluble CTLA4 (CTLA4-Ig) and relatively high doses of TLR9 ligands (CpGs) were also able to induce CD8+ DCs to express functional IDO by ligating B7 or TLR9 molecules and inducing IDO via IFNα signaling in a rare, but distinctive subset of splenic CD19+ DCs co-expressing CD8, the plasmacytoid DC (pDC) marker B220, as well as CD19 and other B cell markers (Grohmann et al., 2002; Mellor et al., 2003, 2005;Baban et al., 2005;Johnson et al., 2010). In humans, monocyte-derived DCs competent to express IDO in response to IFNγ were a discrete subset of DCs expressing the chemokine receptor CCR6 and the pDC marker CD123 (Munn et al., 2002), and human DCs expressing IDO in response to CpGs also exhibited pDC attributes (Chen et al., 2008).
The relative paucity of IDO-competent DCs in mice (∼10% of total splenic DCs) and humans is a technical problem. To help counter this problem, we developed robust (MLR-based) assays to detect T cell suppression mediated by IDO+ DCs from mice bearing tumors or treated with IDO inducers (Table 1). Using these DC suppression assays, and analogous assays to detect suppression mediated by small numbers of Tregs from lymphoid tissues, we determined that IDO+ DCs mediate T cell suppression by blocking clonal expansion of activated T cell, and by activating pre-formed, functionally quiescent (resting) Tregs to mediate bystander suppression. A distinctive feature of IDO-activated Tregs from tumor-bearing mice and mice treated with systemic TLR9 ligands was that suppression depended on interactions between PD-1 and its ligands PD-L1 and PD-L2. Intact GCN2 genes (in Tregs) were essential for Tregs to activate in response to IDO activity in DCs, emphasizing the critical role of tryptophan depletion in driving regulatory outcomes (Sharma et al., 2007; Baban et al., 2009). Increasing evidence also points to roles for tryptophan catabolites in promoting regulatory outcomes since kynurenine is an AhR ligand that drives Treg conversion from naïve CD4 T cells (Mezrich et al., 2010; Nguyen et al., 2010). Moreover, requirements for signals from CTLA4, PD-1, TGFβ, and IL-10 for CD19+ DCs to up-regulate IDO in response to TLR9 ligands suggest that IDO-competence is maintained by constitutive interactions between CD19+ DCs and resting Tregs during homeostasis (Baban et al., 2011). Loss of any one of these molecular pathways caused Foxp3-lineage Tregs to undergo rapid functional re-programming into helper/effector T cells expressing TNFα, IL-17 IL-2 and IFNγ, probably due to IL-6 production which is suppressed by IDO activity in DCs. Thus, the relative numbers of CD19+ DCs and Tregs and several non-redundant molecular pathways may be key factors that determine “immunologic tone” in particular lymphoid tissues (Checkpoint C) by regulating the ability of Tregs to become potent suppressor cells or polyfunctional helper/effector T cells in response to inflammatory cues.
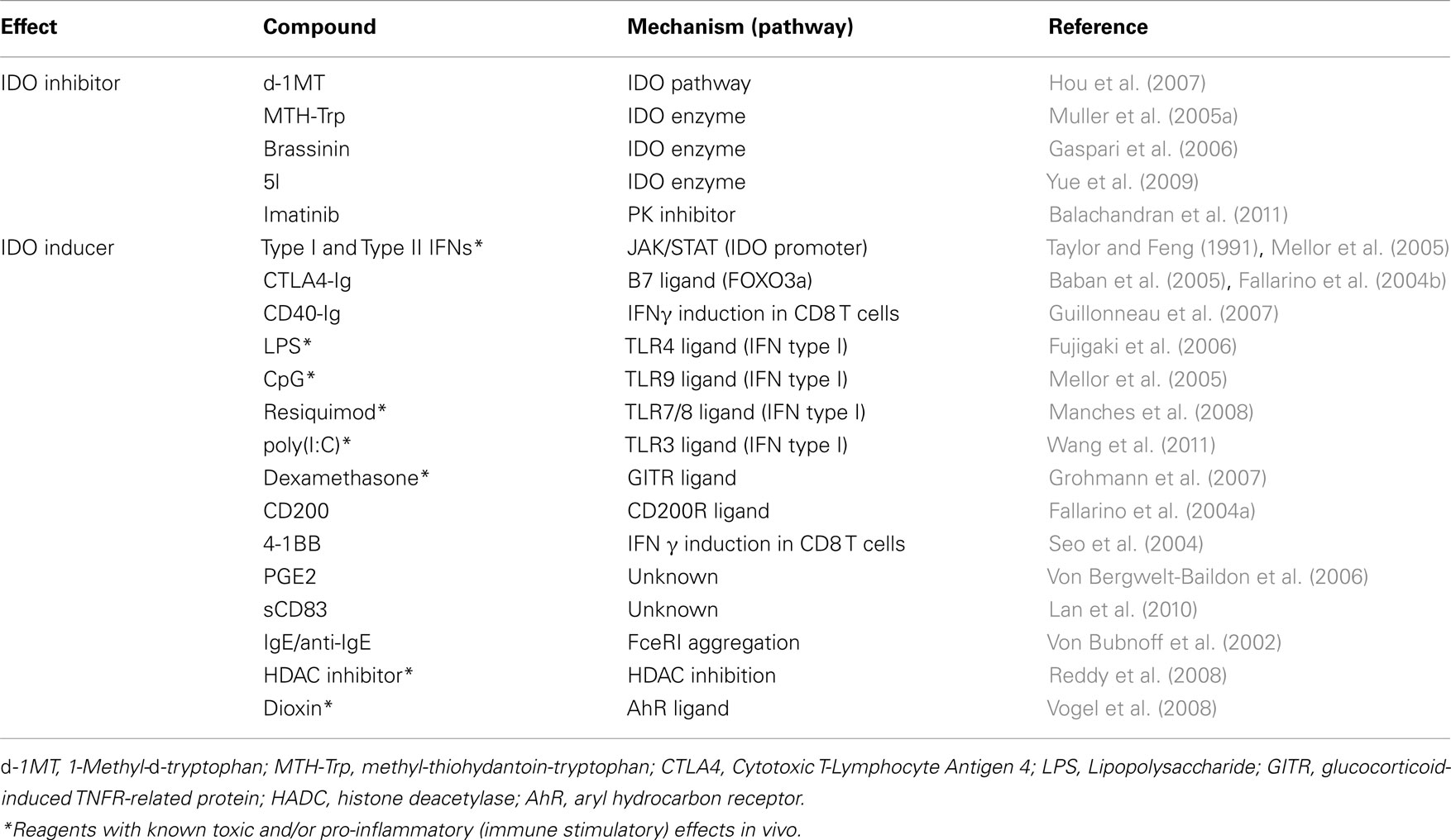
Table 1. IDO pathway inhibitors and inducers.
Manipulating IDO to Create or Destroy Immune Privilege
Immune privilege helps protect tissues from damage mediated by excess immune responses generated when inflammation lowers thresholds that keep innate autoimmunity in check during homeostasis. In transplant settings artificial creation of immune privilege to mimic local tolerance induced by tumors and some pathogens that cause chronic infections is a key strategy to enhance allograft survival. Hence, manipulating the IDO pathway to create or destroy immune privilege offers potential therapeutic strategies to treat a range of chronic inflammatory disease syndromes.
Using IDO Pathway Inhibitors to Attenuate Established Immune Privilege
Inhibiting the IDO pathway is a potential strategy to treat patients with hypo-immune syndromes (cancer and chronic infections) caused by induced immune privilege that permits persistence and progression of tumors and pathogen infections in immunocompetent individuals. In this section we discuss recent progress in developing new methods to inhibit IDO that may be effective in treating patients with cancer and chronic infections. We focus on the use of pharmacologic IDO pathway inhibitors to achieve this goal, though strategies using si/shRNA to knockdown IDO gene expression in specific cell or tissue types may be an alternative approach to achieve similar goals (Zheng et al., 2006; Flatekval and Sioud, 2009;Yen et al., 2009; Huang et al., 2011).
Cancer
At sites of tumor growth tryptophan catabolism mediated by IDO in host APCs, or IDO, or TDO in tumor cells can promote and sustain immune privilege. In humans, IDO1 expression in numerous tumor cell types is a significant predictor of poor prognosis (Sakurai et al., 2004; Astigiano et al., 2005;Brandacher et al., 2006; Ino et al., 2006;Riesenberg et al., 2007; Takao et al., 2007;Pan et al., 2008). In murine models, transfection of immunogenic tumor cell lines with recombinant IDO1 or TDO rendered them immunosuppressive and lethally progressive in vivo (Uyttenhove et al., 2003; Pilotte et al., 2012). The role of IDO in tumor immunosuppression was initially investigated using the racemic mixture of the IDO inhibitor 1MT (D,L-1MT). Thus D,L-1MT administration to mice bearing tumors that over-expressed IDO due to transfection or genetic deficiency of the tumor-suppressor Bin1 (leading to IDO1 over-expression) enhanced anti-tumor immunity and slowed tumor growth (Friberg et al., 2002; Uyttenhove et al., 2003;Muller et al., 2005b). Moreover D,L-1MT treatment synergized with several chemo-immunotherapy regimens to enhance anti-tumor effects in mice, as well as in regimens involving combination with radiotherapy (Muller et al., 2005b; Hou et al., 2007). A key question regarding D,L-1MT was whether both D and L stereoisomers of 1MT were biologically active. Based on in vitro assays using purified rabbit intestinal IDO L-1MT it was assumed that L-1MT would be more effective than D-1MT as an IDO inhibitor to promote T cell mediated anti-tumor immunity in vivo (Peterson et al., 1994). Unexpectedly, D-1MT was as effective, or more potent than L-1MT than the racemic mixture in restoring proliferation of T cells suppressed by physiologic IDO+ pDCs (Munn et al., 2002). In addition, D-1MT was more effective than L-1MT in reversing suppression of T cell proliferation created by IDO+ human monocyte-derived DCs and murine DCs from tumor-dLNs, and restored proliferation of primary CD4+ and CD8+ T cells blocked by IDO+ fibroblast cells (Hou et al., 2007; Forouzandeh et al., 2008). D-1MT was also more effective than L-1MT as an anti-cancer agent in chemo-immunotherapy regimens in mouse models of transplantable melanoma and transplantable and autochthonous breast cancer (Hou et al., 2007). Despite these indications of superior D-1MT efficacy as an anti-tumor drug the molecular target of D-1MT has not been defined as D-1MT did not inhibit enzyme activity of recombinant human and murine IDO1 protein in cell-free assays, and IDO enzyme activity in human tumor cell lines and cell lines transfected with IDO1 genes (Lob et al., 2009). Nevertheless the anti-tumor and T cell enhancing effects of D-1MT were abrogated in the absence of functional IDO1 genes, and IDO1-deficient mice developed tumors that were not sensitized to chemo-immunotherapy involving D-1MT (Hou et al., 2007). Thus, the pharmacologic effects of D-1MT manifested only when the IDO1 pathway was intact, and D-1MT targets DCs that attenuate T cell responses. Several mechanisms could explain how D-1MT inhibits the IDO pathway in DCs while not inhibiting IDO enzyme activity directly as follows; (a) inhibition of high affinity tryptophan transporters in DCs to block substrate supply to IDO; (b) inhibition of uncharacterized IDO1 enzyme isoforms produced by RNA editing, post-translational modifications or association with other proteins into supramolecular complexes in DCs; (c) D-1MT conversion into the active enzymatic inhibitor L-1MT; (d) mimicking signals of tryptophan sufficiency, or blocking signals of tryptophan deficiency or downstream signals elicited by tryptophan metabolites in DCs. Regarding the last mechanism, D-1MT may block downstream GCN2 activation or AhR signaling mediated by tryptophan withdrawal and kynurenine production. Thus D-1MT may target the “IDO pathway” but not IDO enzyme. Interestingly, the PK inhibitor Imatinib mesylate (Gleevec), an effective treatment for patients with gastrointestinal stromal tumor (GIST), blocked PK-mediated IDO induction in a mouse model of GIST (Balachandran et al., 2011). In addition halofuginone (Hf), a derivative of the bioactive compound Febrifugine found in a traditional Chinese herbal medicine, bypasses IDO by activating GCN2 kinase to block effector Th17 responses (Sundrud et al., 2009). Recently, the molecular target of Hf was identified as glutamyl-prolyl-tRNA synthetase (EPRS), and Hf was shown to inhibit prolyl-tRNA synthetase activity (Keller et al., 2012). Thus Imatinib and Hf act, at least in part, by targeting components of the IDO pathway but not IDO directly to phenocopy IDO-mediated effects, and D-1MT may have a similar mode of action. It remains to be seen if D-1MT will be effective in cancer patients. Early indications from Phase I clinical trials hinted at efficacy that manifested as hypophysitis (in which the immune system attacks the pituitary gland) in some patients who had received prior antibody immunotherapy treatment (Garber, 2012). Anti-tumor efficacy is more likely to manifest when D-1MT is used in combination with other anti-cancer treatments because blocking IDO does not incite anti-tumor immunity per se. The anti-tumor and immune enhancing effects of D-1MT treatment were also replicated by administering other compounds that inhibit IDO1-encoded IDO enzyme activity in vitro and in IDO1-transfected cells such as 5-Br-brassinin, menadione, methyl-thiohydantoin-tryptophan, and analogs of phenylimidazole (Muller and Scherle, 2006; Banerjee et al., 2008;Kumar et al., 2008). Recently, hydroxyamidine inhibitors were also reported to suppress tryptophan catabolism and possess anti-tumor activity (Yue et al., 2009; Koblish et al., 2010;Liu et al., 2010), and TDO inhibitors may also be effective anti-tumor drugs based on a recent report that Kyn from tumor cells expressing TDO promoted tumor-induced immune privilege via AhR signaling (Opitz et al., 2011).
Chronic infections
The use of IDO inhibitors to enhance host immunity to infectious pathogens may seem paradoxical based on findings that MΦs expressing IDO can mediate innate host defense in some infectious disease settings such as Toxoplasma, Trypanosoma, Chlamydia, and (Vincendeau et al., 1999; Njau et al., 2009;Knubel et al., 2010). Moreover IDO inhibition failed to restrict herpes simplex virus type 1 (HSV-1) replication or latency, and reactivated latent toxoplasma infections in mice leading to mortality (Divanovic et al., 2012). Nevertheless from the perspective of some pathogens, the disadvantages of needing to survive against IDO-mediated innate host defense may be outweighed by the potential advantages of IDO-mediated regulation of host adaptive immunity. For example HIV-1, which causes AIDS, is a potent IDO inducer leading to altered Th17/Treg balance that may favor HIV-1 persistence (Boasso et al., 2007, 2009;Favre et al., 2010) In effect, HIV-1 may be an example of a pathogen that has evolved to exploit the IDO pathway as a means to prevent host-mediated viral clearance. If correct, this notion suggests that IDO inhibitors may destroy HIV-1-induced immune privilege that suppresses natural and vaccine-induced anti-HIV-1 immunity. Consistent with this idea, Boasso et al. (2009) reported that combined D-1MT, and anti-retroviral therapy (ART) lowered virus titers in simian immunodeficiency virus (SIV)-infected rhesus macaques. Similar effects of D-1MT on lowering parasite burdens were observed in mice infected with Leishmania major (Makala et al., 2011; Divanovic et al., 2012). In vitro data showed 1MT treatment restored M. tuberculosis-specific CD4 T cell effector functions by increasing IFNγ production (Li et al., 2011). These observations support the use of IDO pathway inhibitors as a possible method to destroy pathogen-induced immune privilege; however, opposing effects of IDO pathway inhibitors on host defense mechanisms that maintain stable host–pathogen relationships developed over evolutionary time may complicate efforts to apply this basic approach in clinical settings.
Vaccines
Regulatory pathways that create and maintain immune privilege may block or attenuate responses to vaccines designed to induce protective and therapeutic immunity in settings of cancer and chronic infections. Established tumors and infections are notoriously resistant to vaccine-induced immunity due to the intensity of local suppressive mechanisms. However, a common assumption is that regulatory pathways do not impede responses to prophylactic vaccines. This assumption may not be entirely correct in some cases. For example, the vaccine adjuvant effects of the NK cell inducer and CD1d ligand alpha-galactosylceramide (αgalcer) was masked by rapid induction of IDO in mice vaccinated with inactivated influenza A virus, and co-treatment with IDO inhibitor (1MT) augmented primary T cell responses to vaccination, though did not enhance memory T cell generation (Guillonneau et al., 2009). It is unclear why IDO was induced rapidly in this model; one potential explanation is that IFNγ from activated NK cells stimulated local IDO expression, though a subset of DCs expressing CD1d may respond directly to αgalcer by expressing IDO. In a recent report SIV infected macaques treated with a SIV-based vaccine and ART simultaneously succumbed to lethal acute pancreatitis and hyperglycemic coma when vaccination and ART were combined with D-1MT and CTLA4 mAb blockade to inhibit immune regulatory pathways (Vaccari et al., 2012). This study revealed that IDO and/or CTLA4 regulatory pathways impede catastrophic host responses to ART-related toxicities, and sounds a precautionary note that interfering with regulatory pathways during vaccination can provoke undesirable toxicities. Nevertheless, optimal vaccine design should take into account the effects of host regulatory pathways, especially in therapeutic settings when established immune privilege may completely eliminate any beneficial effects of vaccine administration where it matters, namely at local sites of tumor growth and infection.
Using IDO Inducers and Tryptophan Metabolites to Create Immune Privilege
Autoimmunity, allergy, and transplantation are all hyper-immune syndromes in which excessive immunity damages healthy tissues. Regulatory pathways are often chronically activated in such settings. Though enhanced regulation may delay disease progression and onset and reduce the severity of disease pathology enhanced regulation may not prevent disease progression completely; a pertinent analogy is driving a car with the parking brake on. Thus, IDO enzyme activity is often elevated in hyper-immune syndromes (increased Kyn in blood and tissues) and IDO slows but does not prevent disease progression. Nevertheless, enhancing IDO expression and activity may be a potential strategy to ameliorate hyper-immune syndromes and protect healthy tissues in the same way that IDO-mediated dominant regulation protects allogeneic fetal tissues from destruction by maternal T cell cells. Three broad strategies may help achieve this goal; (a) IDO gene transfer into vulnerable tissues, (b) treatment with reagents that induce IDO, and (c) treatment with reagents that mediate downstream regulatory effects of IDO. In this section we discuss progress in developing these complementary strategies to treat hyper-immune syndromes.
IDO gene transfer
IDO gene transfer to drive increased IDO expression in local tissues is the most direct strategy to attenuate hyper-immunity. IDO gene transfer into donor tissues prolonged allograft survival in corneal (Beutelspacher et al., 2006), cardiac (Li et al., 2007), and lung transplantation (Swanson et al., 2004; Liu et al., 2006a,b, 2007, 2009). The mechanism of enhanced allograft survival is not fully understood, but correlated with reduced clonal expansion of donor-specific effector T cells and attenuated effector functions of CD8 T cells that were generated and migrated to donor tissues expressing increased IDO (Liu et al., 2009). Reduced cytokine levels, T cell proliferative capacity, and increased Tregs were associated with protection of cardiac allografts transfected with IDO (Yu et al., 2008). Regardless of the exact mechanisms of transplant protection in these models, these findings support the use of IDO gene transfer as a potential means to protect healthy tissues in clinical settings. However, the creation of more robust immune privilege by over-expressing IDO may have unintended and undesirable consequences that may preclude clinical application such as increased risks of infection and tumor formation in tissues where IDO activity is artificially and constitutively elevated.
IDO inducing reagents
Table 1 lists some biologic and pharmacologic reagents that have been reported to induce IDO activity in mice or cell lines. A key consideration from a clinical perspective is that reagents to induce IDO must have minimal toxicities, as it is likely that extended (chronic) treatments will be needed to attenuate established hyper-immune disease syndromes. This consideration precludes the use of many reagents listed in Table 1 due to known toxicities. For example, type I and II IFNs that induce IDO via receptors distributed on many cell types via JAK–STAT signaling pathways have documented toxicities. However, IFN type I is the current standard-of-care for some hyper-immune syndromes, despite the undesirable side-effects of chronic exposure to exogenous IFN type I. Thus, IFNβ is used to treat patients with multiple sclerosis (MS), and pegylated-IFNα is used to treat patients with hepatitis C virus (HCV) infections in combination with Ribovirin, a broad range anti-viral drug (Masarone and Persico, 2011). The mode of action of type I IFNs in these settings is not fully understood. However signaling via IFN type I receptors on myeloid cells attenuated experimental autoimmune encephalitis (EAE) disease progression and severity in mice, a widely accepted model of MS (Prinz et al., 2008). Type I IFNs may be pivotal regulators of host immunity in a range of autoimmune and chronic infections diseases such as HIV and HCV. For this reason pDCs may be critical mediators of hyper-immune syndromes since they are major (but not exclusive) sources of type I IFNs, especially in response to pathogen-associated molecular patterns (PAMPs) such as TLRs. It may be pertinent that IDO-competent DCs in humans and mice exhibit attributes that overlap with those of conventional pDCs, as well as other lymphoid and myeloid subsets (Munn et al., 2002; Chen et al., 2008;Johnson et al., 2010). Nevertheless, the role of IFNs, and the cells that produce IFNs, in hyper-immune syndromes is far from clear as IFNs mediate multiple downstream effects that encompass immune stimulatory and immune regulatory pathways.
The B7 (CD80/86) ligand CTLA4-Ig was designed as reagents to block co-stimulation required to generate effector T cells. CTLA4-Ig induces some DC subsets to express IDO and acquire tolerogenic phenotypes due to “reverse signaling” via B7 molecules. Thus B7 ligands may promote immune privilege by co-stimulatory blockade and by inducing DCs to express IDO. B7 ligands approved for clinical treatment of hyper-immune syndromes may not induce IDO in humans as poorly defined structural features may be critical for reverse signaling via B7 to induce IDO. Hence, it is not clear to what extent the potential IDO inducing attributes of B7 ligands may account for their clinical efficacy as immune modulators. A key issue is that reagents for clinical use are based on human proteins that may not interact with homologous human and mouse receptors in identical ways. In our experience CTLA4-Ig isoforms engineered with human sequences and with modified immunoglobulin constant (Fc) domains do not induce IDO in mice. Hence the co-stimulatory blockade attributes of such reagents may manifest in mice but IDO-mediated effects will not. It remains to be seen if B7 ligands engineered specifically to induce IDO in humans are effective immune modulators in clinical settings of hyper-immune syndromes. These points notwithstanding, a recent study in mice revealed that combined CTLA4-Ig (abatacept) and donor-specific transfusion (DST) therapy promoted heart allograft acceptance in an IDO and Treg dependent manner (Sucher et al., 2012). Thus IDO induction, particularly in DCs, is a pivotal checkpoint that can promote immune privilege, though factors that control IDO induction in DCs are not fully understood.
Several TLR ligands stimulate IDO expression and enzyme activity in mice. The TLR4 ligand LPS induced IDO in human peripheral blood mononuclear cells, THP-1, and U937 cell lines, and mouse bone marrow derived DCs (Fujigaki et al., 2006; Jung et al., 2007). LPS also induced human monocyte-derived DCs to express IDO, which promoted DC maturation and Treg differentiation (Hill et al., 2007). TLR9 ligands (CpGs) also stimulated mouse and human DCs to express IDO and acquire tolerogenic phenotypes by autocrine/paracrine type I IFN signaling in mice (Baban et al., 2005; Mellor et al., 2005; Chen et al., 2008). As a consequence, TLR9 ligands promoted naïve CD4 T cells to differentiate (convert) into Tregs, and caused pre-formed (resting) Tregs to acquire potent regulatory phenotypes (activate) via IDO (Baban et al., 2009, 2011). Despite these examples of TLR-mediated immune regulation, TLR ligands have well-documented pro-inflammatory and immune stimulatory attributes that preclude their use as immune modulators in clinical settings of hyper-immunity.
Other IDO inducing reagents include histone deacetylase (HDAC) inhibitors and dexamethasone that ligates glucocorticoid-induced TNF receptor (GITR; Grohmann et al., 2007; Reddy et al., 2008). Polyinosinic–polycytidylic acid [poly(I:C)], a synthetic double stranded RNA analog and TLR3 ligand, induced human trophoblast cells to express IDO (Wang et al., 2011). Surface ligand/receptors including CD200, 4-1BB, and the eicosanoid prostaglandin E2 (PGE2) were also reported to induce IDO (Fallarino et al., 2004a; Seo et al., 2004;Von Bergwelt-Baildon et al., 2006). Recently, the immunomodulatory reagent soluble CD83 (sCD83) was reported to be an IDO inducer, and the tolerizing effects of sCD83 in a murine renal transplant model were blocked by IDO inhibitor (Ge et al., 2010; Lan et al., 2010). Recently, we discovered that systemic treatment of mice with nanoparticles containing the cationic polymer polyethylenimine (PEI) complexed with DNA stimulated IDO expression in a range of mouse tissues including lymphoid tissues (manuscript submitted). This response was unexpected as DNA/PEI nanoparticles elicit well-documented pro-inflammatory and immune stimulatory responses that promote anti-tumor immunity (Rodrigo-Garzon et al., 2010). IDO activity was induced in splenic DCs, which activated resting Tregs via IDO and blocked antigen-specific T cell responses to vaccines within 24 h of treatment with DNA/PEI nanoparticles. These responses were dependent on type I, but not type II IFN signaling. Moreover, the presence of TLR9 ligands (CpG motifs) in bacterial (plasmid) DNA was not required to induce IDO or type I IFN. Removing TLR9 ligands abrogated rapid and uniform activation of NK cells, which released large amounts of IFNγ into serum in response to DNA/PEI nanoparticles containing immune stimulatory bacterial plasmid DNA containing TLR9 ligands. Treating mice with DNA/PEI nanoparticles also reduced the severity of antigen-induced arthritis, and prevented Type 1 Diabetes (T1D) progression and onset in non-obese diabetic (NOD) female mice prone to T1D onset. In ongoing studies, we are addressing the mechanism of IDO induction following DNA/PEI nanoparticle treatment, and engineering biodegradable isoforms of DNA/PEI nanoparticles as potential reagents to treat clinical hyper-immune syndromes that promote immune privilege by inducing IDO and activating Tregs.
In summary, IDO inducing reagents offer promising new approaches to slow, prevent, or reverse hyper-immune syndromes. Mechanisms of IDO induction and the cell types induced to express IDO by these reagents have are not well-documented for all IDO inducing reagents, and further studies will be necessary to evaluate if these reagents can create robust tolerogenic phenotypes in immune cells to create and sustain immune privilege that protects healthy tissue at risk from hyper-immunity.
Tryptophan metabolites
Some tryptophan catabolites generated and released by cells expressing IDO or TDO possess immunomodulatory attributes that may promote regulation in inflamed tissues and associated dLNs to create and maintain local immune privilege. Combined treatment with Kyn and 3-hydroxy-anthranilic acid (3-HAA) prolonged allograft survival in a rat allogeneic skin transplant model (Bauer et al., 2005), and 3-HAA treatment inhibited PDK1 activation and promoted T cell apoptosis to suppress T cell-mediated lung pathology in a murine model of asthma (Hayashi et al., 2007). IDO-mediated elevation of Treg:Th17 ratios characteristic of the progressive chronic inflammatory state that manifests during active HIV disease may also be mediated in part by 3-HAA, based on the finding that 3-HAA replicated this phenotype in vitro (Favre et al., 2010). Metabolic interplay between IDO and AhR signaling is mediated by tryptophan metabolites as Kyn is an AhR ligand, and AhR ligands such as Kyn, 2-(1’H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) a natural AhR ligand found in lungs, and the artificial AhR ligand iVAG539 induce IDO expression in DCs; in addition, Kyn promotes Treg conversion, possibly by direct AhR-mediated effects on Tregs or indirectly by inducing DCs to express IDO (Song et al., 2002; Hauben et al., 2008;Vogel et al., 2008; Mezrich et al., 2010;Nguyen et al., 2010; Opitz et al., 2011). These observations suggest that the IDO and AhR pathways reinforce regulatory responses via positive feedback loops involving tryptophan catabolites that are AhR ligands and other AhR ligands that induce IDO. The metabolic effects of AhR ligands on inflammatory responses by DCs were the subject of a recent review (Bankoti et al., 2010).
In summary, cells expressing functional IDO or TDO may modulate local immune effector functions by stimulating AhR in innate and adaptive immune cells, such as T cells, Th17, Tregs, and DCs, which reinforces IDO-mediated regulatory phenotypes in these cells. AhR expressed on non-immune cells including lung epithelial cells, liver hepatocytes, and endothelial cells may also mediate cell responses in tissue development, physiological function, and immune responses (Walisser et al., 2005; Chiba et al., 2011, 2012), indicating ubiquitous influences of tryptophan metabolites on both hematopoietic and non-hematopoietic cells. The relative importance of the immune modulatory effects of IDO-mediated tryptophan withdrawal to trigger the ISR in immune cells and production of tryptophan catabolites is not known. However these novel insights identify several metabolic pathways that create and sustain immune privilege, thereby providing new therapeutic targets to disrupt (or reinforce) immune privilege according to clinical need. As described above, Imatinib and Hf are already examples of compounds that target the IDO pathway indirectly, though with diametric effects on T cell immunity. These findings reinforce the hypothesis that altered amino acid metabolism at sites of local inflammation is a pivotal immune control mechanism that helps create and sustain immune privilege.
Summary and Prospects
In this review we summarize key recent developments relating to the concept that induced immune privilege explains differential immunogenicities observed at local sites of inflammation. Clearly interactions between small numbers of innate and adaptive immune cells in microenvironmental niches exert profound influences on immune outcomes and disease progression that have not been fully appreciated. This knowledge deficiency arose because key initiating events are far removed in time, and perhaps in space too from their measurable consequences in terms of disease progression and onset. The application of new imaging tools to examine interactions between small numbers of cells that lead to measurable metabolic changes has potential to improve understanding of the fundamental biochemical and metabolic changes that explain immunologic outcomes relevant to chronic inflammatory diseases. It is also clear that we are only just beginning to understand how initial responses to inflammatory insults in tissues, and the prevailing immunologic tone of lymphoid tissues draining such tissues integrate information about the primary insult that shapes subsequent responses in the innate and adaptive immune systems. Some immune modulatory drugs that interfere with processes that create local immune privilege are already approved for clinical use, and many others are in the pipeline, with more potential targets still to be considered. It will be interesting to see how these new immunotherapies fare in clinical settings, and how this field develops based on data from clinical trials and basic research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding sources were the NIH (AI083005) and the Carlos and Marguerite Mason Trust (to Andrew L. Mellor) and NewLink Genetics Corporation (to Mario Mautino).
References
Astigiano, S., Morandi, B., Costa, R., Mastracci, L., D’Agostino, A., Ratto, G. B., Melioli, G., and Frumento, G. (2005). Eosinophil granulocytes account for indoleamine 2,3-dioxygenase-mediated immune escape in human non-small cell lung cancer. Neoplasia 7, 390–396.
Baban, B., Chandler, P. R., Johnson, B. A. III, Huang, L., Li, M., Sharpe, M. L., Francisco, L. M., Sharpe, A. H., Blazar, B. R., Munn, D. H., and Mellor, A. L. (2011). Physiologic control of IDO competence in splenic dendritic cells. J. Immunol. 187, 2329–2335.
Baban, B., Chandler, P. R., Sharma, M. D., Pihkala, J., Koni, P. A., Munn, D. H., and Mellor, A. L. (2009). IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J. Immunol. 183, 2475–2483.
Baban, B., Hansen, A. M., Chandler, P. R., Manlapat, A., Bingaman, A., Kahler, D. J., Munn, D. H., and Mellor, A. L. (2005). A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell suppression via type I IFN signaling following B7 ligation. Int. Immunol. 17, 909–919.
Balachandran, V. P., Cavnar, M. J., Zeng, S., Bamboat, Z. M., Ocuin, L. M., Obaid, H., Sorenson, E. C., Popow, R., Ariyan, C., Rossi, F., Besmer, P., Guo, T., Antonescu, C. R., Taguchi, T., Yuan, J., Wolchok, J. D., Allison, J. P., and Dematteo, R. P. (2011). Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 17, 1094–1100.
Banerjee, T., Duhadaway, J. B., Gaspari, P., Sutanto-Ward, E., Munn, D. H., Mellor, A. L., Malachowski, W. P., Prendergast, G. C., and Muller, A. J. (2008). A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene 27, 2851–2857.
Bankoti, J., Rase, B., Simones, T., and Shepherd, D. M. (2010). Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicol. Appl. Pharmacol. 246, 18–28.
Bauer, T. M., Jiga, L. P., Chuang, J. J., Randazzo, M., Opelz, G., and Terness, P. (2005). Studying the immunosuppressive role of indoleamine 2,3-dioxygenase: tryptophan metabolites suppress rat allogeneic T-cell responses in vitro and in vivo. Transpl. Int. 18, 95–100.
Beutelspacher, S. C., Pillai, R., Watson, M. P., Tan, P. H., Tsang, J., Mcclure, M. O., George, A. J., and Larkin, D. F. (2006). Function of indoleamine 2,3-dioxygenase in corneal allograft rejection and prolongation of allograft survival by over-expression. Eur. J. Immunol. 36, 690–700.
Boasso, A. (2011). Wounding the immune system with its own blade: HIV-induced tryptophan catabolism and pathogenesis. Curr. Med. Chem. 18, 2247–2256.
Boasso, A., Herbeuval, J. P., Hardy, A. W., Anderson, S. A., Dolan, M. J., Fuchs, D., and Shearer, G. M. (2007). HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood 109, 3351–3359.
Boasso, A., Vaccari, M., Fuchs, D., Hardy, A. W., Tsai, W. P., Tryniszewska, E., Shearer, G. M., and Franchini, G. (2009). Combined effect of antiretroviral therapy and blockade of IDO in SIV-infected rhesus macaques. J. Immunol. 182, 4313–4320.
Brandacher, G., Perathoner, A., Ladurner, R., Schneeberger, S., Obrist, P., Winkler, C., Werner, E. R., Werner-Felmayer, G., Weiss, H. G., Gobel, G., Margreiter, R., Konigsrainer, A., Fuchs, D., and Amberger, A. (2006). Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin. Cancer Res. 12, 1144–1151.
Brenk, M., Scheler, M., Koch, S., Neumann, J., Takikawa, O., Hacker, G., Bieber, T., and Von Bubnoff, D. (2009). Tryptophan deprivation induces inhibitory receptors ILT3 and ILT4 on dendritic cells favoring the induction of human CD4+CD25+Foxp3+ T regulatory cells. J. Immunol. 183, 145–154.
Bronte, V., and Zanovello, P. (2005). Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 5, 641–654.
Chen, W., Liang, X., Peterson, A. J., Munn, D. H., and Blazar, B. R. (2008). The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J. Immunol. 181, 5396–5404.
Chiba, T., Chihara, J., and Furue, M. (2012). Role of the arylhydrocarbon receptor (AhR) in the pathology of asthma and COPD. J. Allergy 2012, 8.
Chiba, T., Uchi, H., Tsuji, G., Gondo, H., Moroi, Y., and Furue, M. (2011). Arylhydrocarbon receptor (AhR) activation in airway epithelial cells induces MUC5AC via reactive oxygen species (ROS) production. Pulm. Pharmacol. Ther. 24, 133–140.
Chon, S. Y., Hassanain, H. H., Pine, R., and Gupta, S. L. (1995). Involvement of two regulatory elements in interferon-gamma-regulated expression of human indoleamine 2,3-dioxygenase gene. J. Interferon Cytokine Res. 15, 517–526.
Chung, D. J., Rossi, M., Romano, E., Ghith, J., Yuan, J., Munn, D. H., and Young, J. W. (2009). Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood 114, 555–563.
Cobbold, S. P. (2010). Combining regulation with suppression and aiming for tolerance. Transplantation 89, 909–910.
Dai, W., and Gupta, S. L. (1990). Regulation of indoleamine 2,3-dioxygenase gene expression in human fibroblasts by interferon-gamma. Upstream control region discriminates between interferon-gamma and interferon-alpha. J. Biol. Chem. 265, 19871–19877.
De Vries, V. C., Pino-Lagos, K., Nowak, E. C., Bennett, K. A., Oliva, C., and Noelle, R. J. (2011). Mast cells condition dendritic cells to mediate allograft tolerance. Immunity 35, 550–561.
Ding, Y., Bushell, A., and Wood, K. J. (2010). Mesenchymal stem-cell immunosuppressive capabilities: therapeutic implications in islet transplantation. Transplantation 89, 270–273.
Divanovic, S., Sawtell, N. M., Trompette, A., Warning, J. I., Dias, A., Cooper, A. M., Yap, G. S., Arditi, M., Shimada, K., Duhadaway, J. B., Prendergast, G. C., Basaraba, R. J., Mellor, A. L., Munn, D. H., Aliberti, J., and Karp, C. L. (2012). Opposing biological functions of tryptophan catabolizing enzymes during intracellular infection. J. Infect. Dis. 205, 152–161.
Fallarino, F., Asselin-Paturel, C., Vacca, C., Bianchi, R., Gizzi, S., Fioretti, M. C., Trinchieri, G., Grohmann, U., and Puccetti, P. (2004a). Murine plasmacytoid dendritic cells initiate the immunosuppressive pathway of tryptophan catabolism in response to CD200 receptor engagement. J. Immunol. 173, 3748–3754.
Fallarino, F., Bianchi, R., Orabona, C., Vacca, C., Belladonna, M. L., Fioretti, M. C., Serreze, D. V., Grohmann, U., and Puccetti, P. (2004b). CTLA-4-Ig activates forkhead transcription factors and protects dendritic cells from oxidative stress in nonobese diabetic mice. J. Exp. Med. 200, 1051–1062.
Fallarino, F., Grohmann, U., Vacca, C., Orabona, C., Spreca, A., Fioretti, M. C., and Puccetti, P. (2003). T cell apoptosis by kynurenines. Adv. Exp. Med. Biol. 527, 183–190.
Fallarino, F., Vacca, C., Orabona, C., Belladonna, M. L., Bianchi, R., Marshall, B., Keskin, D. B., Mellor, A. L., Fioretti, M. C., Grohmann, U., and Puccetti, P. (2002). Functional expression of indoleamine 2,3-dioxygenase by murine CD8 alpha(+) dendritic cells. Int. Immunol. 14, 65–68.
Favre, D., Mold, J., Hunt, P. W., Kanwar, B., Loke, P., Seu, L., Barbour, J. D., Lowe, M. M., Jayawardene, A., Aweeka, F., Huang, Y., Douek, D. C., Brenchley, J. M., Martin, J. N., Hecht, F. M., Deeks, S. G., and Mccune, J. M. (2010). Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci. Transl. Med. 2, 32ra36.
Flatekval, G. F., and Sioud, M. (2009). Modulation of dendritic cell maturation and function with mono- and bifunctional small interfering RNAs targeting indoleamine 2,3-dioxygenase. Immunology 128, e837–e848.
Forouzandeh, F., Jalili, R. B., Germain, M., Duronio, V., and Ghahary, A. (2008). Differential immunosuppressive effect of indoleamine 2,3-dioxygenase (IDO) on primary human CD4+ and CD8+ T cells. Mol. Cell. Biochem. 309, 1–7.
Friberg, M., Jennings, R., Alsarraj, M., Dessureault, S., Cantor, A., Extermann, M., Mellor, A. L., Munn, D. H., and Antonia, S. J. (2002). Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int. J. Cancer 101, 151–155.
Frumento, G., Rotondo, R., Tonetti, M., Damonte, G., Benatti, U., and Ferrara, G. B. (2002). Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 196, 459–468.
Fujigaki, H., Saito, K., Fujigaki, S., Takemura, M., Sudo, K., Ishiguro, H., and Seishima, M. (2006). The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J. Biochem. 139, 655–662.
Garber, K. (2012). Evading immunity: new enzyme implicated in cancer. J. Natl. Cancer Inst. 104, 349–352.
Gaspari, P., Banerjee, T., Malachowski, W. P., Muller, A. J., Prendergast, G. C., Duhadaway, J., Bennett, S., and Donovan, A. M. (2006). Structure-activity study of brassinin derivatives as indoleamine 2,3-dioxygenase inhibitors. J. Med. Chem. 49, 684–692.
Ge, W., Arp, J., Lian, D., Liu, W., Baroja, M. L., Jiang, J., Ramcharran, S., Eldeen, F. Z., Zinser, E., Steinkasserer, A., Chou, P., Brand, S., Nicolette, C., Garcia, B., and Wang, H. (2010). Immunosuppression involving soluble CD83 induces tolerogenic dendritic cells that prevent cardiac allograft rejection. Transplantation 90, 1145–1156.
Grohmann, U., Orabona, C., Fallarino, F., Vacca, C., Calcinaro, F., Falorni, A., Candeloro, P., Belladonna, M. L., Bianchi, R., Fioretti, M. C., and Puccetti, P. (2002). CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 3, 1097–1101.
Grohmann, U., Volpi, C., Fallarino, F., Bozza, S., Bianchi, R., Vacca, C., Orabona, C., Belladonna, M. L., Ayroldi, E., Nocentini, G., Boon, L., Bistoni, F., Fioretti, M. C., Romani, L., Riccardi, C., and Puccetti, P. (2007). Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat. Med. 13, 579–586.
Guillonneau, C., Hill, M., Hubert, F. X., Chiffoleau, E., Herve, C., Li, X. L., Heslan, M., Usal, C., Tesson, L., Menoret, S., Saoudi, A., Le Mauff, B., Josien, R., Cuturi, M. C., and Anegon, I. (2007). CD40Ig treatment results in allograft acceptance mediated by CD8CD45RC T cells, IFN-gamma, and indoleamine 2,3-dioxygenase. J. Clin. Invest. 117, 1096–1106.
Guillonneau, C., Mintern, J. D., Hubert, F. X., Hurt, A. C., Besra, G. S., Porcelli, S., Barr, I. G., Doherty, P. C., Godfrey, D. I., and Turner, S. J. (2009). Combined NKT cell activation and influenza virus vaccination boosts memory CTL generation and protective immunity. Proc. Natl. Acad. Sci. U.S.A. 106, 3330–3335.
Hackstein, H., and Thomson, A. W. (2004). Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nat. Rev. Immunol. 4, 24–34.
Hauben, E., Gregori, S., Draghici, E., Migliavacca, B., Olivieri, S., Woisetschlager, M., and Roncarolo, M. G. (2008). Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell-mediated effects on regulatory T cells. Blood 112, 1214–1222.
Hayashi, T., Mo, J. H., Gong, X., Rossetto, C., Jang, A., Beck, L., Elliott, G. I., Kufareva, I., Abagyan, R., Broide, D. H., Lee, J., and Raz, E. (2007). 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc. Natl. Acad. Sci. U.S.A. 104, 18619–18624.
Hill, M., Tanguy-Royer, S., Royer, P., Chauveau, C., Asghar, K., Tesson, L., Lavainne, F., Remy, S., Brion, R., Hubert, F. X., Heslan, M., Rimbert, M., Berthelot, L., Moffett, J. R., Josien, R., Gregoire, M., and Anegon, I. (2007). IDO expands human CD4++CD25high regulatory T cells by promoting maturation of LPS-treated dendritic cells. Eur. J. Immunol. 37, 3054–3062.
Hou, D. Y., Muller, A. J., Sharma, M. D., Duhadaway, J., Banerjee, T., Johnson, M., Mellor, A. L., Prendergast, G. C., and Munn, D. H. (2007). Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 67, 792–801.
Huang, T. T., Yen, M. C., Lin, C. C., Weng, T. Y., Chen, Y. L., Lin, C. M., and Lai, M. D. (2011). Skin delivery of short hairpin RNA of indoleamine 2,3 dioxygenase induces antitumor immunity against orthotopic and metastatic liver cancer. Cancer Sci. 102, 2214–2220.
Ino, K., Yoshida, N., Kajiyama, H., Shibata, K., Yamamoto, E., Kidokoro, K., Takahashi, N., Terauchi, M., Nawa, A., Nomura, S., Nagasaka, T., Takikawa, O., and Kikkawa, F. (2006). Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br. J. Cancer 95, 1555–1561.
Johnson, B. A. III, Kahler, D. J., Baban, B., Chandler, P. R., Kang, B., Shimoda, M., Koni, P. A., Pihkala, J., Vilagos, B., Busslinger, M., Munn, D. H., and Mellor, A. L. (2010). B-lymphoid cells with attributes of dendritic cells regulate T cells via indoleamine 2,3-dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 107, 10644–10648.
Jung, I. D., Lee, C. M., Jeong, Y. I., Lee, J. S., Park, W. S., Han, J., and Park, Y. M. (2007). Differential regulation of indoleamine 2,3-dioxygenase by lipopolysaccharide and interferon gamma in murine bone marrow derived dendritic cells. FEBS Lett. 581, 1449–1456.
Jux, B., Kadow, S., Luecke, S., Rannug, A., Krutmann, J., and Esser, C. (2011). The aryl hydrocarbon receptor mediates UVB radiation-induced skin tanning. J. Invest. Dermatol. 131, 203–210.
Keller, T. L., Zocco, D., Sundrud, M. S., Hendrick, M., Edenius, M., Yum, J., Kim, Y. J., Lee, H. K., Cortese, J. F., Wirth, D. F., Dignam, J. D., Rao, A., Yeo, C. Y., Mazitschek, R., and Whitman, M. (2012). Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 8, 311–317.
Knubel, C. P., Martinez, F. F., Fretes, R. E., Diaz Lujan, C., Theumer, M. G., Cervi, L., and Motran, C. C. (2010). Indoleamine 2,3-dioxigenase (IDO) is critical for host resistance against Trypanosoma cruzi. FASEB J. 24, 2689–2701.
Koblish, H. K., Hansbury, M. J., Bowman, K. J., Yang, G., Neilan, C. L., Haley, P. J., Burn, T. C., Waeltz, P., Sparks, R. B., Yue, E. W., Combs, A. P., Scherle, P. A., Vaddi, K., and Fridman, J. S. (2010). Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol. Cancer Ther. 9, 489–498.
Kraal, G., Samsom, J. N., and Mebius, R. E. (2006). The importance of regional lymph nodes for mucosal tolerance. Immunol. Rev. 213, 119–130.
Kumar, S., Jaller, D., Patel, B., Lalonde, J. M., Duhadaway, J. B., Malachowski, W. P., Prendergast, G. C., and Muller, A. J. (2008). Structure based development of phenylimidazole-derived inhibitors of indoleamine 2,3-dioxygenase. J. Med. Chem. 51, 4968–4977.
Lan, Z., Ge, W., Arp, J., Jiang, J., Liu, W., Gordon, D., Healey, D., Debenedette, M., Nicolette, C., Garcia, B., and Wang, H. (2010). Induction of kidney allograft tolerance by soluble CD83 associated with prevalence of tolerogenic dendritic cells and indoleamine 2,3-dioxygenase. Transplantation 90, 1286–1293.
Lee, S. M., Lee, Y. S., Choi, J. H., Park, S. G., Choi, I. W., Joo, Y. D., Lee, W. S., Lee, J. N., Choi, I., and Seo, S. K. (2010). Tryptophan metabolite 3-hydroxyanthranilic acid selectively induces activated T cell death via intracellular GSH depletion. Immunol. Lett. 132, 53–60.
Li, J., Meinhardt, A., Roehrich, M. E., Golshayan, D., Dudler, J., Pagnotta, M., Trucco, M., and Vassalli, G. (2007). Indoleamine 2,3-dioxygenase gene transfer prolongs cardiac allograft survival. Am. J. Physiol. Heart Circ. Physiol. 293, H3415–3423.
Li, Q., Li, L., Liu, Y., Fu, X., Wang, H., Lao, S., Yang, B., and Wu, C. (2011). Biological functions of Mycobacterium tuberculosis-specific CD4+ T cells were impaired by tuberculosis pleural fluid. Immunol. Lett. 138, 113–121.
Liu, H., Liu, L., Fletcher, B. S., and Visner, G. A. (2006a). Novel action of indoleamine 2,3-dioxygenase attenuating acute lung allograft injury. Am. J. Respir. Crit. Care Med. 173, 566–572.
Liu, H., Liu, L., Fletcher, B. S., and Visner, G. A. (2006b). Sleeping Beauty-based gene therapy with indoleamine 2,3-dioxygenase inhibits lung allograft fibrosis. FASEB J. 20, 2384–2386.
Liu, H., Liu, L., Liu, K., Bizargity, P., Hancock, W. W., and Visner, G. A. (2009). Reduced cytotoxic function of effector CD8+ T cells is responsible for indoleamine 2,3-dioxygenase-dependent immune suppression. J. Immunol. 183, 1022–1031.
Liu, H., Liu, L., and Visner, G. A. (2007). Nonviral gene delivery with indoleamine 2,3-dioxygenase targeting pulmonary endothelium protects against ischemia-reperfusion injury. Am. J. Transplant. 7, 2291–2300.
Liu, X., Shin, N., Koblish, H. K., Yang, G., Wang, Q., Wang, K., Leffet, L., Hansbury, M. J., Thomas, B., Rupar, M., Waeltz, P., Bowman, K. J., Polam, P., Sparks, R. B., Yue, E. W., Li, Y., Wynn, R., Fridman, J. S., Burn, T. C., Combs, A. P., Newton, R. C., and Scherle, P. A. (2010). Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 115, 3520–3530.
Lob, S., Konigsrainer, A., Zieker, D., Brucher, B. L., Rammensee, H. G., Opelz, G., and Terness, P. (2009). IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol. Immunother. 58, 153–157.
Makala, L. H., Baban, B., Lemos, H., El-Awady, A. R., Chandler, P. R., Hou, D. Y., Munn, D. H., and Mellor, A. L. (2011). Leishmania major attenuates host immunity by stimulating local indoleamine 2,3-dioxygenase expression. J. Infect. Dis. 203, 715–725.
Manches, O., Munn, D., Fallahi, A., Lifson, J., Chaperot, L., Plumas, J., and Bhardwaj, N. (2008). HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3-dioxygenase-dependent mechanism. J. Clin. Invest. 118, 3431–3439.
Mantovani, A., Germano, G., Marchesi, F., Locatelli, M., and Biswas, S. K. (2011). Cancer-promoting tumor-associated macrophages: new vistas and open questions. Eur. J. Immunol. 41, 2522–2525.
Masarone, M., and Persico, M. (2011). Antiviral therapy: why does it fail in HCV-related chronic hepatitis? Expert Rev. Anti Infect. Ther. 9, 535–543.
Mellor, A. L., Baban, B., Chandler, P., Marshall, B., Jhaver, K., Hansen, A., Koni, P. A., Iwashima, M., and Munn, D. H. (2003). Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J. Immunol. 171, 1652–1655.
Mellor, A. L., Baban, B., Chandler, P. R., Manlapat, A., Kahler, D. J., and Munn, D. H. (2005). Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J. Immunol. 175, 5601–5605.
Mellor, A. L., Chandler, P., Baban, B., Hansen, A. M., Marshall, B., Pihkala, J., Waldmann, H., Cobbold, S., Adams, E., and Munn, D. H. (2004). Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int. Immunol. 16, 1391–1401.
Mellor, A. L., and Munn, D. H. (2008). Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat. Rev. Immunol. 8, 74–80.
Mellor, A. L., Sivakumar, J., Chandler, P., Smith, K., Molina, H., Mao, D., and Munn, D. H. (2001). Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat. Immunol. 2, 64–68.
Mezrich, J. D., Fechner, J. H., Zhang, X., Johnson, B. P., Burlingham, W. J., and Bradfield, C. A. (2010). An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198.
Muller, A. J., Duhadaway, J. B., Donover, P. S., Sutanto-Ward, E., and Prendergast, G. C. (2005a). Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 11, 312–319.
Muller, A. J., Malachowski, W. P., and Prendergast, G. C. (2005b). Indoleamine 2,3-dioxygenase in cancer: targeting pathological immune tolerance with small-molecule inhibitors. Expert Opin. Ther. Targets 9, 831–849.
Muller, A. J., and Scherle, P. A. (2006). Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat. Rev. Cancer 6, 613–625.
Munn, D. H., and Mellor, A. L. (2006). The tumor-draining lymph node as an immune-privileged site. Immunol. Rev. 213, 146–158.
Munn, D. H., Sharma, M. D., Baban, B., Harding, H. P., Zhang, Y., Ron, D., and Mellor, A. L. (2005). GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22, 633–642.
Munn, D. H., Sharma, M. D., Hou, D., Baban, B., Lee, J. R., Antonia, S. J., Messina, J. L., Chandler, P., Koni, P. A., and Mellor, A. L. (2004). Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Invest. 114, 280–290.
Munn, D. H., Sharma, M. D., Lee, J. R., Jhaver, K. G., Johnson, T. S., Keskin, D. B., Marshall, B., Chandler, P., Antonia, S. J., Burgess, R., Slingluff, C. L. Jr., and Mellor, A. L. (2002). Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science 297, 1867–1870.
Munn, D. H., Zhou, M., Attwood, J. T., Bondarev, I., Conway, S. J., Marshall, B., Brown, C., and Mellor, A. L. (1998). Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 281, 1191–1193.
Murphy, S. P., Porrett, P. M., and Turka, L. A. (2011). Innate immunity in transplant tolerance and rejection. Immunol. Rev. 241, 39–48.
Nguyen, N. T., Kimura, A., Nakahama, T., Chinen, I., Masuda, K., Nohara, K., Fujii-Kuriyama, Y., and Kishimoto, T. (2010). Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 19961–19966.
Njau, F., Geffers, R., Thalmann, J., Haller, H., and Wagner, A. D. (2009). Restriction of Chlamydia pneumoniae replication in human dendritic cell by activation of indoleamine 2,3-dioxygenase. Microbes Infect. 11, 1002–1010.
Opitz, C. A., Litzenburger, U. M., Sahm, F., Ott, M., Tritschler, I., Trump, S., Schumacher, T., Jestaedt, L., Schrenk, D., Weller, M., Jugold, M., Guillemin, G. J., Miller, C. L., Lutz, C., Radlwimmer, B., Lehmann, I., Von Deimling, A., Wick, W., and Platten, M. (2011). An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203.
Ostrand-Rosenberg, S., Sinha, P., Beury, D. W., and Clements, V. K. (2012). Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. PMID: 22313874. [Epub ahead of print]
Pan, K., Wang, H., Chen, M. S., Zhang, H. K., Weng, D. S., Zhou, J., Huang, W., Li, J. J., Song, H. F., and Xia, J. C. (2008). Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 134, 1247–1253.
Peterson, A. C., Migawa, M. T., Martin, M. J., Hamaker, L. K., Czerwinski, K. M., Zhang, W., Arend, R. A., Fisette, P. L., Ozaki, Y., Will, J. A., Brown, R. R., and Cook, J. M. (1994). Evaluation of functionalized tryptophan derivatives and related compounds as competitive inhibitors of indolamine 2,3-dioxygenase. Med. Chem. Res. 3, 531–544.
Pilotte, L., Larrieu, P., Stroobant, V., Colau, D., Dolusic, E., Frederick, R., De Plaen, E., Uyttenhove, C., Wouters, J., Masereel, B., and Van Den Eynde, B. J. (2012). Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 109, 2497–2502.
Prinz, M., Schmidt, H., Mildner, A., Knobeloch, K. P., Hanisch, U. K., Raasch, J., Merkler, D., Detje, C., Gutcher, I., Mages, J., Lang, R., Martin, R., Gold, R., Becher, B., Bruck, W., and Kalinke, U. (2008). Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 28, 675–686.
Quintana, F. J., Basso, A. S., Iglesias, A. H., Korn, T., Farez, M. F., Bettelli, E., Caccamo, M., Oukka, M., and Weiner, H. L. (2008). Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71.
Reddy, P., Sun, Y., Toubai, T., Duran-Struuck, R., Clouthier, S. G., Weisiger, E., Maeda, Y., Tawara, I., Krijanovski, O., Gatza, E., Liu, C., Malter, C., Mascagni, P., Dinarello, C. A., and Ferrara, J. L. (2008). Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J. Clin. Invest. 118, 2562–2573.
Riesenberg, R., Weiler, C., Spring, O., Eder, M., Buchner, A., Popp, T., Castro, M., Kammerer, R., Takikawa, O., Hatz, R. A., Stief, C. G., Hofstetter, A., and Zimmermann, W. (2007). Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin. Cancer Res. 13, 6993–7002.
Rodrigo-Garzon, M., Berraondo, P., Ochoa, L., Zulueta, J. J., and Gonzalez-Aseguinolaza, G. (2010). Antitumoral efficacy of DNA nanoparticles in murine models of lung cancer and pulmonary metastasis. Cancer Gene Ther. 17, 20–27.
Rodriguez, P. C., Quiceno, D. G., and Ochoa, A. C. (2007). L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 109, 1568–1573.
Romani, L., Fallarino, F., De Luca, A., Montagnoli, C., D’Angelo, C., Zelante, T., Vacca, C., Bistoni, F., Fioretti, M. C., Grohmann, U., Segal, B. H., and Puccetti, P. (2008). Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 451, 211–215.
Sakurai, K., Enomoto, K., Amano, S., Kimura, T., Sugito, K., Kimizuka, K., Ikeda, T., Shibata, M., and Negishi, N. (2004). Study of indoleamine 2,3-dioxygenase expression in patients of esophageal squamous cell carcinoma. Gan To Kagaku Ryoho 31, 1780–1782.
Seo, S. K., Choi, J. H., Kim, Y. H., Kang, W. J., Park, H. Y., Suh, J. H., Choi, B. K., Vinay, D. S., and Kwon, B. S. (2004). 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat. Med. 10, 1088–1094.
Sharma, M. D., Baban, B., Chandler, P., Hou, D. Y., Singh, N., Yagita, H., Azuma, M., Blazar, B. R., Mellor, A. L., and Munn, D. H. (2007). Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest. 117, 2570–2582.
Sharma, M. D., Hou, D. Y., Baban, B., Koni, P. A., He, Y., Chandler, P. R., Blazar, B. R., Mellor, A. L., and Munn, D. H. (2010). Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity 33, 942–954.
Sharma, M. D., Hou, D. Y., Liu, Y., Koni, P. A., Metz, R., Chandler, P., Mellor, A. L., He, Y., and Munn, D. H. (2009). Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 113, 6102–6111.
Song, H., Park, H., Kim, Y. S., Kim, K. D., Lee, H. K., Cho, D. H., Yang, J. W., and Hur, D. Y. (2011). L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int. Immunopharmacol. 11, 932–938.
Song, J., Clagett-Dame, M., Peterson, R. E., Hahn, M. E., Westler, W. M., Sicinski, R. R., and Deluca, H. F. (2002). A ligand for the aryl hydrocarbon receptor isolated from lung. Proc. Natl. Acad. Sci. U.S.A. 99, 14694–14699.
Sucher, R., Fischler, K., Oberhuber, R., Kronberger, I., Margreiter, C., Ollinger, R., Schneeberger, S., Fuchs, D., Werner, E. R., Watschinger, K., Zelger, B., Tellides, G., Pilat, N., Pratschke, J., Margreiter, R., Wekerle, T., and Brandacher, G. (2012). IDO and regulatory T cell support are critical for cytotoxic T lymphocyte-associated Ag-4 Ig-mediated long-term solid organ allograft survival. J. Immunol. 188, 37–46.
Sundrud, M. S., Koralov, S. B., Feuerer, M., Calado, D. P., Kozhaya, A. E., Rhule-Smith, A., Lefebvre, R. E., Unutmaz, D., Mazitschek, R., Waldner, H., Whitman, M., Keller, T., and Rao, A. (2009). Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science 324, 1334–1338.
Swanson, K. A., Zheng, Y., Heidler, K. M., Mizobuchi, T., and Wilkes, D. S. (2004). CDllc+ cells modulate pulmonary immune responses by production of indoleamine 2,3-dioxygenase. Am. J. Respir. Cell Mol. Biol. 30, 311–318.
Takao, M., Okamoto, A., Nikaido, T., Urashima, M., Takakura, S., Saito, M., Okamoto, S., Takikawa, O., Sasaki, H., Yasuda, M., Ochiai, K., and Tanaka, T. (2007). Increased synthesis of indoleamine-2,3-dioxygenase protein is positively associated with impaired survival in patients with serous-type, but not with other types of, ovarian cancer. Oncol. Rep. 17, 1333–1339.
Taylor, M. W., and Feng, G. S. (1991). Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 5, 2516–2522.
Terness, P., Bauer, T. M., Rose, L., Dufter, C., Watzlik, A., Simon, H., and Opelz, G. (2002). Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J. Exp. Med. 196, 447–457.
Uyttenhove, C., Pilotte, L., Theate, I., Stroobant, V., Colau, D., Parmentier, N., Boon, T., and Van Den Eynde, B. J. (2003). Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 9, 1269–1274.
Vaccari, M., Boasso, A., Fenizia, C., Fuchs, D., Hryniewicz, A., Morgan, T., Weiss, D., Doster, M. N., Heraud, J. M., Shearer, G. M., and Franchini, G. (2012). Fatal pancreatitis in simian immunodeficiency virus SIV(mac251)-infected macaques treated with 2’,3’-dideoxyinosine and stavudine following cytotoxic-T-lymphocyte-associated antigen 4 and indoleamine 2,3-dioxygenase blockade. J. Virol. 86, 108–113.
Vincendeau, P., Lesthelle, S., Bertazzo, A., Okomo-Assoumou, M. C., Allegri, G., and Costa, C. V. (1999). Importance of L-tryptophan metabolism in trypanosomiasis. Adv. Exp. Med. Biol. 467, 525–531.
Vogel, C. F., Goth, S. R., Dong, B., Pessah, I. N., and Matsumura, F. (2008). Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 375, 331–335.
Von Bergwelt-Baildon, M. S., Popov, A., Saric, T., Chemnitz, J., Classen, S., Stoffel, M. S., Fiore, F., Roth, U., Beyer, M., Debey, S., Wickenhauser, C., Hanisch, F. G., and Schultze, J. L. (2006). CD25 and indoleamine 2,3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: additional mechanisms of T-cell inhibition. Blood 108, 228–237.
Von Bubnoff, D., Matz, H., Frahnert, C., Rao, M. L., Hanau, D., De La Salle, H., and Bieber, T. (2002). FcepsilonRI induces the tryptophan degradation pathway involved in regulating T cell responses. J. Immunol. 169, 1810–1816.
Walisser, J. A., Glover, E., Pande, K., Liss, A. L., and Bradfield, C. A. (2005). Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. U.S.A. 102, 17858–17863.
Wang, B., Koga, K., Osuga, Y., Cardenas, I., Izumi, G., Takamura, M., Hirata, T., Yoshino, O., Hirota, Y., Harada, M., Mor, G., and Taketani, Y. (2011). Toll-like receptor-3 ligation-induced indoleamine 2, 3-dioxygenase expression in human trophoblasts. Endocrinology 152, 4984–4992.
Yen, M. C., Lin, C. C., Chen, Y. L., Huang, S. S., Yang, H. J., Chang, C. P., Lei, H. Y., and Lai, M. D. (2009). A novel cancer therapy by skin delivery of indoleamine 2,3-dioxygenase siRNA. Clin. Cancer Res. 15, 641–649.
Yu, G., Dai, H., Chen, J., Duan, L., Gong, M., Liu, L., Xiong, P., Wang, C. Y., Fang, M., and Gong, F. (2008). Gene delivery of indoleamine 2,3-dioxygenase prolongs cardiac allograft survival by shaping the types of T-cell responses. J. Gene Med. 10, 754–761.
Yue, E. W., Douty, B., Wayland, B., Bower, M., Liu, X., Leffet, L., Wang, Q., Bowman, K. J., Hansbury, M. J., Liu, C., Wei, M., Li, Y., Wynn, R., Burn, T. C., Koblish, H. K., Fridman, J. S., Metcalf, B., Scherle, P. A., and Combs, A. P. (2009). Discovery of potent competitive inhibitors of indoleamine 2,3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model. J. Med. Chem. 52, 7364–7367.
Zheng, X., Koropatnick, J., Li, M., Zhang, X., Ling, F., Ren, X., Hao, X., Sun, H., Vladau, C., Franek, J. A., Feng, B., Urquhart, B. L., Zhong, R., Freeman, D. J., Garcia, B., and Min, W. P. (2006). Reinstalling antitumor immunity by inhibiting tumor-derived immunosuppressive molecule IDO through RNA interference. J. Immunol. 177, 5639–5646.
Keywords: IDO, inflammation, immunity, regulation, tolerance
Citation: Li L, Huang L, Lemos HP, Mautino M and Mellor AL (2012) Altered tryptophan metabolism as a paradigm for good and bad aspects of immune privilege in chronic inflammatory diseases. Front. Immun. 3:109. doi: 10.3389/fimmu.2012.00109
Received: 15 March 2012; Accepted: 17 April 2012;
Published online: 11 May 2012.
Edited by:
Rachel R. Caspi, National Institutes of Health, USAReviewed by:
Rachel R. Caspi, National Institutes of Health, USATakashi Yamamura, National Center of Neurology and Psychiatry, Japan
Antonio Curti, Hematology Bologna, Italy
Copyright: © 2012 Li, Huang, Lemos, Mautino and Mellor. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Andrew L. Mellor, Immunotherapy Center, Georgia Health Sciences University, 1120 15th Street, Augusta, GA 30912, USA. e-mail:YW1lbGxvckBnZW9yZ2lhaGVhbHRoLmVkdQ==