

 Johanna K. Morales
Johanna K. Morales
- Department of Biology, The Asthma and Allergic Disease Cooperative Research Center, Virginia Commonwealth University, Richmond, VA, USA
Mast cells are central players in immune surveillance and activation, positioned at the host–environment interface. Understanding the signaling events controlling mast cell function, especially those that maintain host homeostasis, is an important and still less understood area of mast cell-mediated disease. With respect to allergic disease, it is well established that IgE and its high affinity receptor FcεRI are major mediators of mast cell activation. However, IgG-mediated signals can also modulate mast cell activities. Signals elicited by IgG binding to its cognate receptors (FcγR) are the basis for autoimmune disorders such as lupus and rheumatoid arthritis. Using knowledge of IgE-mediated mast cell signaling, recent work has begun to illuminate potential overlap between FcεRI and FcγR signal transduction. Herein we review the importance of Src family kinases in FcεRI and FcγR signaling, the role of the transcription factor STAT5, and impingement of the regulatory cytokines IL-4, IL-10, and TGFβ1 upon this network.
Introduction
Mast cells are key agents in the direction of inflammatory processes throughout most tissues, especially at the mucosal interface. Immediately identifiable consequences of mast cell activity are the symptoms of atopic disease: itching, sneezing, and allergic asthma. Mast cells enact these processes through degranulation of stored histamine, various proteases, and the secretion of prostaglandins and leukotrienes. Mast cells further affect inflammatory responses by the later (hours) de novo production and secretion of a plethora of cytokines, chemokines, and growth factors, including TNF-α, GM-CSF, IL-4, -6, -13, MIP-1α, MCP-1, and VEGF. Thus mast cells also act as effectors of other cellular responses, e.g., maintenance of IgE production, and the activities of regulatory T cells (Treg) and myeloid derived suppressor cells (MDSC; Gauchat et al., 1993; Frandji et al., 1996; Pawankar et al., 1997; Lu et al., 2006; Forward et al., 2009; Kambayashi et al., 2009; Piconese et al., 2009; Yang et al., 2010b). Notwithstanding their very important and deeply studied roles in atopy and parasite infections, mast cells are now implicated in the inflammatory responses associated with autoimmune disease, atherosclerosis, and resistance to bacterial infection (Levi-Schaffer et al., 1991; Malaviya et al., 1996; Secor et al., 2000; Hara et al., 2002; Lee et al., 2002; McLachlan et al., 2003). This multifunctional role in the immune response makes mast cell biology a critical area of research that could greatly change the treatment of inflammatory diseases.
Investigation of signals proximal to the high affinity IgE receptor, FcεRI, has been a topic of great interest due to the importance of IgE in allergic pathologies. Central points from these observations are that Src family kinases Lyn and Fyn are critical to IgE-mediated mast cell activation, and that the signal transducer and activator of transcription (STAT) 5 transcription factor mediates mast cell vitality and function, particularly in the late phase of cytokine production. Knowledge gained from FcεRI signaling could prove valuable in the setting of IgG-mediated inflammation, which is the foundation for many autoimmune diseases, including lupus and rheumatoid arthritis. Once deposited either as anti-host antibodies or as an immune complex, IgG activates innate immune cells via FcγR. These are expressed as pro-inflammatory (FcγRI, FcγRIIA/C, FcγRIIIA/B, FcγRIV) or anti-inflammatory (FcγRIIB) receptors, some in a species-restricted manner, e.g., FcγRIV in mice only, as discussed later. Macrophages, basophils, neutrophils, and mast cells are activated by FcγR signals, often via FcγRIII, with counter-regulation by FcγRIIB (Malbec and Daeron, 2007; Nimmerjahn and Ravetch, 2008). Understanding signals emanating from these receptors will offer new treatments for IgG-mediated diseases. Here we review highlights of IgE signaling such as Lyn, Fyn, and STAT5. We will also describe interplay with regulatory cytokines IL-4, IL-10, and TGFβ1, and the potential signaling overlap between IgE and IgG receptors in mast cells.
Src Family Kinases are Integral to IgE Signaling
Mast cell activation and IgE are almost synonymous topics that have been studied in-depth. An early phase of degranulation and eicosanoid secretion, and a late phase inflammatory cytokine production, are both potently stimulated by multivalent interactions between FcεRI-bound IgE molecules and antigen at the mast cell surface. As a result, the crosslinking of these receptors into lipid raft domains brings into proximity protein tyrosine kinases (PTK) that may or may not be pre-associated with FcεRI. This makes for a ripe setting of immunoreceptor tyrosine-based activation motif (ITAM) trans-phosphorylation (Pribluda et al., 1994) and further recruitment of other PTK, via Src homology 2 (SH2) domains, that propagate the signal.
Crosslinking of FcεRI activates a variety of Src family kinases (Eiseman and Bolen, 1992; Gilfillan and Rivera, 2009), this review focuses on two wings of signaling that emerge from such an event. First, the stimulation of tyrosine kinase Syk through activity of Lyn (a Src family kinase); Syk phosphorylates linker of activated T cells (LAT) which in turn discharges its duty as a SH2-based scaffold, eventually stimulating extracellular signal-regulated kinases (ERK) and de novo transcription (Rivera and Gilfillan, 2006; Gilfillan and Rivera, 2009). The second pathway involves the activity of another Src family kinase, Fyn. Through the Grb2 associated binding protein (Gab) 2, Fyn elicits signal transduction down the canonical PI3K-Akt axis (Kim et al., 2008). Fyn is also essential for early phase degranulation of mast cells through the transient receptor potential channel 1 (TRPC1), a non-selective cation channel that is required for Ca2+ influx, F-actin depolymerization, and resultant release of packed granules (Suzuki et al., 2010).
Both Lyn and Fyn are positive mediators of FcεRI activity (Parravicini et al., 2002; Gomez et al., 2005a; Rivera and Gilfillan, 2006). However, a notable distinction exists in that Lyn also contributes to the abatement of IgE-mediated signaling (Hernandez-Hansen et al., 2004; Odom et al., 2004; Kalesnikoff and Galli, 2008). Beyond its role in Syk recruitment, Lyn influences several inhibitory proteins, including SHIP-1, SHP-2, DOK-1, and Cbp, leading to decreased signaling in the PI-3 kinase and the Ras-MAPK cascades (Satterthwaite et al., 1998; Takeshita et al., 1998; Hibbs et al., 2002; Xu et al., 2005).
Lyn induces the activity of C-terminal Src kinase (Csk) by directly phosphorylating the membrane-anchored Csk-binding protein (Cbp; Odom et al., 2004; Kovarova et al., 2006), also known as phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG). This event places Csk in close proximity to the FcεRI-associated Lyn and Fyn. Csk phosphorylates in such a way to sterically hinder Src family kinase protein–protein interactions (Gilfillan and Rivera, 2009). Two groups independently uncovered Cbp/PAG as the essential factor potentiating Csk localization at the plasmalemma (Brdicka et al., 2000; Kawabuchi et al., 2000). Cbp is ubiquitously expressed and constitutively tyrosine phosphorylated in some cell types; with a number of SH2-mediated binding interactions occurring as a result of this (Brdicka et al., 2000). Most notable is a positive interaction with Csk upon phosphorylation of Cbp Y314 in rat (Takeuchi et al., 2000) and Y317 in human (Brdicka et al., 2000). The functional relationship between Src family kinases and Cbp was first defined in T cells and, Raji and Jurkat cell lines. Csk and Cbp are pre-associated in resting T-cells, however during T cell receptor (TCR) aggregation Cbp is swiftly dephosphorylated, releasing Csk, resulting in the delocalization of the latter from the membrane (Brdicka et al., 2000); thus the inhibitory phosphorylation on Lyn and Fyn is lost. Interestingly the same study demonstrated an association between Fyn and Cbp independent of an SH2 interaction (Brdicka et al., 2000). Indeed in the setting of T cell activation, Fyn eventually rephosphorylates Cbp, reinstating the Csk elicited brake on Src family kinase (Lindquist et al., 2003). However, in the context of mast cells and FcεRI, Csk recruitment/activity through Cbp seems to be more a function of post-aggregation Lyn activity (Honda et al., 1997; Ohtake et al., 2002), with Lyn knockouts possessing un-phosphorylated Cbp, therefore non-functional Csk (Odom et al., 2004). Moreover, unlike resting T cells, Cbp is not constitutively active in mast cells (Ohtake et al., 2002; Lindquist et al., 2003).
Importantly, genetic background has been shown to be key to the negative responses associated with Lyn. It has been postulated that this could be due to a Lyn:Fyn balance, e.g., C57BL/6 mice express significantly less Fyn than the 129/Sv strain. Lyn null bone marrow-derived mast cells (BMMC) from the C57BL/6 strain have an impaired degranulation response to antigen, while a similar knock-out (KO) in 129/Sv mast cells results in hyperdegranulation. In contrast, Lyn deletion in either strain enhances IgE-mediated cytokine production, revealing Lyn as a negative regulator (Yamashita et al., 2007). As no similar negative roles for Fyn have been observed in mast cells, one could hypothesize that increased Fyn kinase, e.g., on the 129/Sv background, predisposes to FcεRI hyperactivity when the negative effects of Lyn are absent. Yet other work has demonstrated that the function of Lyn changes with the intensity of IgE-Antigen stimulus on mast cells of similar background. Low intensity receptor activation induces a positive signaling role for Lyn, leading to eventual activation of Akt and p38, while high intensity activation of FcεRI elicits negative responses (Xiao et al., 2005). This bears resemblance to the dual roles of Fyn in the context of TCR signaling.
STAT5 Activity is Modulated by FcεRI in a FyN-Dependent Manner
The STAT family of transcription factors is best known as containing the intermediaries of cytokine and growth factor signals. Typically, aggregating cytokine receptor subunits with their associated Janus (JAK) tyrosine kinases induce JAK auto- or trans-phosphorylation and subsequent receptor phosphorylation, yielding SH2 binding sites to which STATs dock. Generally, STATs are then targeted by the JAK kinases, eliciting phosphotyrosine (pY)-induced STAT dimers that shuttle into the nucleus and stimulate target gene transcription (Schindler et al., 2007). In mast cells one such process is the induction of STAT5 phosphorylation by stem cell factor (SCF)/c-Kit, or IL-3, via JAK2 activation (Mui et al., 1995; Ryan et al., 1997; Brizzi et al., 1999). One or both of these cytokines are essential for the development and survival of mast cells in vitro and in vivo. A series of work by our group and others has established the critical part fulfilled by STAT5 in mast cell development and survival. In particular, STAT5A appears to be more important for survival as demonstrated by dominant negative and gene KO approaches (Shelburne et al., 2003; Ikeda et al., 2005). Bone marrow devoid of STAT5A has markedly higher rates of apoptosis and lesser proliferation compared to wild type when cultured in IL-3 or SCF alone. It should be noted, however, in vitro mast cell development can be achieved by dual stimulation with IL-3 + SCF (Shelburne et al., 2003). This could be due to the overlap of STAT signaling, particularly from STAT3 (Sonnenblick et al., 2005) or STAT6 (Malaviya and Uckun, 2002). In vivo, mast cells are present at birth, but cannot be detected in total STAT5 KO mice by 12 weeks of age (Shelburne et al., 2003; Barnstein et al., 2006; and our unpublished observations). Later work demonstrated that STAT5 tyrosine phosphorylation is induced by crosslinking FcεRI. STAT5 KO BMMC have an attenuated yet functional early activation response, measured by histamine and leukotriene B4 release, but a near complete inability to induce cytokine production – the hallmark of the late phase (Barnstein et al., 2006). Surprisingly, this defect is not the result of poor cytokine mRNA induction. Instead, STAT5 appears to inhibit mRNA-destabilizing factors as one mechanism to regulate mast cell cytokines. The expression of one such protein, tristetrapolin (TTP), is enhanced in STAT5 KO BMMC during FcεRI crosslinkage; inhibiting TTP partially restored cytokine production (Barnstein et al., 2006). The potential effects of STAT5 on other RNA binding factors such as HuR (human antigen R), remains as yet unknown.
Until recently, the mechanism by which FcεRI stimulates phosphotyrosine (pY)-STAT5 also remained unknown. Through EM co-localization studies (similar to those in Wilson et al., 2007; Xue et al., 2007; Andrews et al., 2008) clustering of STAT5 with FcεRI upon antigen exposure has been observed. Our group has found that Fyn is the indispensible kinase eliciting pY-STAT5 proximal to FcεRI in an IgE-dependent, IL-3/SCF-independent manner (Pullen et al., 2012). STAT5 and Fyn were found to co-immunoprecipitate in resting mast cells, an association that dissipated with IgE-antigen stimulation. Surprisingly, knock out of Gab2 – the scaffolding protein linking Fyn to PI3K signaling – allowed a slight enhancement in IgE-mediated pY-STAT5. Deficiency of SH2 tyrosine phosphatase (SHP) -1, a known Gab2 associate, replicates the Gab2 KO phenotype to a lesser extent (Pullen et al., 2012). These data with other accumulating evidence in non-mast cell studies (Yu et al., 2002; Kim et al., 2009), which can also include SHP-2, paint a more complicated picture of the role of Gab2. It is interesting to note that the latter work (Kim et al., 2009) reports an interaction between Lyn and Gab2 that inhibits osteoclastogenesis. Furthermore, a direct association between STAT3 and Gab2 has been reported (Ni et al., 2007), warranting its examination in mast cells in consideration of signal overlap with STAT5. In our hands, Lyn suppression enhances pY-STAT5 in a manner similar to Gab2 and SHP-1 loss. This is in keeping with the known negative effects of Lyn, where its deficiency on a C57BL/6 background enhances STAT5 activation.
Cytokine Regulation of FcεRI Signaling
While Lyn and Fyn offer a means of fine-tuning IgE signaling, external regulation of the IgE receptor can be achieved through cytokines, primarily TGFβ1, IL-10, and IL-4. TGFβ1 is a multipotent cytokine regulating tissue repair, inflammation, proliferation, migration, and angiogenesis (Kehrl et al., 1986; Yang et al., 2010a; Yoshimura et al., 2010). It is linked to autoimmune diseases and cancer (Letterio and Roberts, 1998; Blobe et al., 2000), and recently shown to have a critical role in Treg and Th17 differentiation (Bettelli et al., 2006; Lee et al., 2009; Ziegler and Buckner, 2009). Mast cells evoke TGFβ1-mediated paracrine signaling, by secreting TGFβ1 as well as proteases that activate its latent form (Pennington et al., 1992; Kanbe et al., 1999; Lindstedt et al., 2001). IL-10 belongs to a family of related cytokines including IL-19, IL-20, IL-22, IL-24, and IL-26 (Kotenko, 2002). Cells involved in the allergic response, including Th2 cells and mast cells, can produce IL-10 (Moore et al., 1993, 2001). The well-documented ability of IL-10 to inhibit macrophage activation and antigen presenting function suggests an important role in regulating the immune response. This is clearly demonstrated in IL-10-deficient mice, which develop a severe autoimmune phenotype (Kuhn et al., 1993), as IL-10-deficient dendritic cells are highly potent activators of Th1 responses (He et al., 2005). The presence of IL-10 has been shown to regulate inflammatory responses to pathogen infection (Gazzinelli et al., 1996; Hunter et al., 1997), protect from endotoxic shock (Howard et al., 1993; Berg et al., 1995), and control both acute and chronic inflammatory responses (Roers et al., 2004). Clinically, IL-10 inhibits progression of rheumatoid arthritis when delivered directly to the site of inflammation (Trachsel et al., 2007). As such, IL-10 is being further studied as an inflammatory therapeutic (Murray, 2006).
Our group and others have found that TGFβ1 and IL-10 suppress mast cell function and survival, offering homeostatic feedback regulation (Bissonnette et al., 1997; Miller et al., 1999; Gomez et al., 2005b; Norozian et al., 2006; Pemberton et al., 2006; Wiener et al., 2007; Woodman et al., 2008). TGFβ1 inhibited IL-3-dependent proliferation of mouse BMMC (Broide et al., 1989; Toyota et al., 1995) and blocked their rescue from apoptosis by SCF (Mekori and Metcalfe, 1994). Inhibitory effects of TGFβ1 on mast cell function have also been shown. Bissonnette et al. (1997) reported that antigen-induced histamine and TNF production from rat peritoneal mast cells were inhibited by TGFβ1, whereas Meade et al. (1992) showed that in vivo treatment with TGFβ1 blunted IgE-mediated, mast cell-dependent, immediate hypersensitivity responses in mice. These effects could be partly due to TGFβ1-mediated suppression of FcεRI expression (Gomez et al., 2005b). Despite these studies supporting the theory that TGFβ1 inhibits mast cell function, contradicting evidence exists. This includes a report by Kim and Lee (1999) that TGFβ1 potentiates IgE-dependent cutaneous anaphylaxis in rats. Like TGFβ1, IL-10 can inhibit FcεRI expression and IgE-mediated cytokine production in mast cells (Marshall et al., 1996; Lin and Befus, 1997; Royer et al., 2001a,b; Gillespie et al., 2004). Our recent work showed that IL-10 is produced endogenously during mast cell development, and suppresses mast cell numbers and FcεRI expression (Speiran et al., 2009). Exogenous IL-10 can inhibit the differentiation and survival of immature mouse mast cells, eliciting apoptosis via reduction of c-Kit signaling, caspase activation, and damage to the mitochondria (Bailey et al., 2006; Speiran et al., 2009).
We have investigated the means by which IL-10 and TGFβ1 suppress mast cell activation. In recent work, we found that adding exogenous IL-10 or TGFβ1 to mast cells leads to a gradual decline in Fyn and STAT5 protein levels, reaching its nadir in 3–4 days (Kennedy Norton et al., 2008 and data not yet published). This slow onset could be argued to allow protective mast cell functions prior to reinstating immune homeostasis. By targeting the Fyn-STAT5 pathway, these cytokines effectively disrupt mast cell cytokine production and survival. Our most recent findings show that a set of microRNAs (miRs) is regulated by IL-10 and TGFβ1; miRs that could target Fyn and STAT5, based on in silico analyses. We look forward to unraveling this area, in hopes of providing clues to molecular therapies targeting mast cells.
IL-4 is a pleiotropic cytokine that participates in driving the polarization of T cells toward a Th2 phenotype by inhibiting IFN-γ production and inducing further production of IL-4 by T cells (Foster et al., 2002; Galli et al., 2008). IL-4 signals by binding to the IL-4Rα chain, present as a heterodimer, paired with either the common gamma chain or the IL-13Rα1, creating the type I and type II receptors, respectively. IL-4 plays an important role in the development of asthma by supporting the early development of Th2 cells and promoting B cell isotype switching to IgE. Along with IL-13, IL-4 contributes to the pathophysiology of asthma by inducing bronchial hyperresponsiveness and goblet cell mucus production (Wills-Karp and Finkelman, 2008). Like IL-10, IL-4 can be secreted by mast cells (Brown and Hanifin, 1989), and is produced constitutively in cultures of developing mast cells (Speiran et al., 2009). The combination of exogenous IL-4 and IL-10 can inhibit mast cell differentiation and survival (Yeatman et al., 2000; Bailey et al., 2004; Bouton et al., 2004) through mechanisms not used independently by either cytokine. Moreover, deletion of IL-4 [or IL-10] enhanced BMMC growth and FcεRI expression (Speiran et al., 2009). Also similar to IL-10 and TGFβ1, IL-4 acts on mature mouse mast cells to decrease FcεRI expression and signaling through a STAT6 dependent pathway (Ryan et al., 1998). However, IL-4 has been repeatedly shown to have an enhancing effect on human FcεRI expression (Toru et al., 1996; Bischoff et al., 1999; Lorentz and Bischoff, 2001; Macey et al., 2010). For this reason, IL-4 and its receptor have been attractive candidates for the treatment of asthma. Unfortunately, therapies targeting IL-4 have not yielded promising results due to possible redundancy from IL-13 through the type II receptor. Currently in clinical trials is a monoclonal antibody targeting IL-4Rα, an antisense oligonucleotide that prevents translation of the IL-4Rα chain, and an IL-4 mutein that interferes with the binding of endogenous IL-4 (Nguyen and Casale, 2011).
We recently found interplay between IL-4 and TGFβ1 that could contribute to immune homeostasis. TGFβ1 antagonizes IL-4 signaling in mast cells, by down-regulating IL-4 receptor expression and suppressing STAT6 activation (Macey et al., 2010). TGFβ1 has also been shown to prevent IL-4 expression, by inducing the adaptor protein Nedd4 family-interacting protein 1 (Ndfip1) in Treg. Ndfip1 participates in the ubiquitination of the Jun family transcription factors needed for IL-4 transcription (Beal et al., 2011). Interestingly, IL-4 demonstrates similar antagonism of TGFβ1. We found that IL-4 suppresses TGFβ receptor expression and signaling in mast cells, showing these two cytokines are able to fine tune mast cell responses (Macey et al., 2010).
IgG-Mediated Mast Cell Activity
As discussed earlier, mast cell activation can be achieved through a variety of stimuli, ranging from cytokines to allergen-specific immunoglobulins. In allergic and inflammatory pathologies, allergen-specific IgE crosslinks mast cell FcεRI and triggers the activation of intracellular signaling molecules, ultimately resulting in mediator release and chemotaxis. However in addition to FcεRI, mast cells express the IgG receptor family (FcγR). Of the high-affinity IgG receptors, both humans and mice possess FcγRI, while FcγRIV is restricted to mice. The low affinity FcγRII and FcγRIII, are found in humans and mice. However, there are three FcγRII (A, B, and C) and two FcγRIII (A and B) in humans, whereas mice have one receptor of each type (FcγRIIB and FcγRIIIA; Nimmerjahn and Ravetch, 2008). Further complicating receptor distribution among species is evidence that receptor type expression is also variable according to mast cell phenotype (Benhamou et al., 1990), e.g., peritoneal vs. BMMC, along with the profile of mediator release (Katz et al., 1992), e.g., leukotriene C4 vs. prostaglandin D2.
Activating FcγR on mouse cells consist of a ligand-binding alpha-chain (α) and a signal-transducing gamma-chain (γ) bearing ITAMs, which once phosphorylated upon receptor crosslinking, act as docking sites for signaling molecules such as Syk kinase; this results in the activation of signaling pathways. In contrast, ligand binding can also result in downregulation of antigen-dependent mediator release due to the existence of the immunoreceptor tyrosine-based inhibitory motifs (ITIMs) on the cytosolic portion of the FcγRIIB α-chain. Phosphorylation of ITIMs upon FcγRIIB ligation induces the activation of Lyn kinase, which can recruit and activate phosphatases like SHIP in addition to directly acting upon ITIMs, leading to decreased mediator release (Fong et al., 1996; Ono et al., 1996). FcγRIIB inhibits several aspects of mast cell and basophil functions such as IgE-dependent degranulation and IL-4 production (Daëron et al., 1993, 1995). In addition to mast cells, FcγRIIB is expressed by other immune cells including dendritic cells, macrophages, activated neutrophils, basophils, and B cells. When expressed by these cells, FcγRIIB inhibits the functions of activating FcγRs, e.g., phagocytosis and pro-inflammatory cytokine release (Daëron et al., 1993, 1995; Nimmerjahn and Ravetch, 2008).
Fcγ receptors share the common gamma subunit with FcεRI, and not surprisingly, similar signaling pathways (Kurosaki et al., 1992; Dombrowicz et al., 1997). Although the importance of Syk kinase in IgG signaling has been demonstrated (Crowley et al., 1997; Lach-Trifilieff et al., 2000) little is known about Src family kinase functions in FcγR-mediated mast cell activation. Our latest data demonstrates FcγR-induced mast cell activation of Fyn and Lyn kinases, and downstream initiation of the PI3K-Akt, Erk, p38, and JNK pathways (Falanga et al., 2012). In the same report, it was revealed that Fyn and Lyn kinases have opposing roles upon FcγR crosslinking, since Fyn-deficient (KO) mast cells displayed decreased degranulation and mediator release, whereas an exacerbated response was observed with Lyn KO mast cells. This demonstrates that FcγR-induced mast cell mediator release is regulated in a Fyn- and Lyn-dependent manner.
In pathologic and extremely rare conditions, mast cell activation with antigen-specific IgE can lead to anaphylaxis, a rapid, and life-threatening hypersensitivity reaction. Historically, the onset and regulation of anaphylaxis was exclusively attributed to the generally known classical pathway of anaphylaxis involving IgE-mediated FcεRI mast cell activation and subsequent histamine release (Miyajima et al., 1997; Galli et al., 2005). However, elegantly conducted research on murine models demonstrated that an anaphylactic reaction can be induced through an IgG-FcγR pathway (Strait et al., 2002; Obata et al., 2007; Tsujimura et al., 2008; Jonsson et al., 2011a). In this alternative pathway, activation of basophils and macrophages (FcγRI and FcγRIII; Strait et al., 2002; Obata et al., 2007; Tsujimura et al., 2008), and recently FcγRIV on neutrophils (Jonsson et al., 2011a) elicits anaphylactic symptoms, including a drop in core body temperature. Although mast cell contribution in IgG-mediated anaphylaxis was insignificant – as mast cell-deficient mice still developed IgG-induced anaphylaxis via platelet activating factor (PAF) – recent data demonstrate that mast cells account for the majority of circulating histamine during FcγR-induced passive systemic anaphylaxis (PSA), which is regulated by Fyn and Lyn (Falanga et al., 2012). Additionally, mast cells have recently been reported to be responsible for FcγRIIA-dependent cutaneous anaphylaxis whereas monocytes/macrophages and neutrophils were found to be involved in FcγRIIA-dependent systemic anaphylaxis (Jonsson et al., 2011b). Taken together, these findings bring new insights to the function of Fyn and Lyn kinases downstream of FcγR stimulation and the role of mast cells in IgG-mediated pathologies.
Closing Remarks
Exploration of the biological chemistry behind IgG signaling will be an important activity contributing to efforts in understanding IgG-mediated hypersensitivity disorders. Lessons from FcεRI studies serve as a valuable model offering clues in this pursuit. We posit that the Fyn/STAT5 pathway will be an important regulator in IgG-mediated mast cell pathology, and here we have reviewed key areas of IgE-mediated signaling where IgG cues are likely to intersect, summarized by Figure 1. The similarities also offer attractive lines of study with respect to the interplay of IgG and other exogenous signals such as IL-4, IL-10, and TGFβ1, which can have their own effects on Fyn and STAT5. Notwithstanding this potential, there are still many avenues of investigation that need attention.
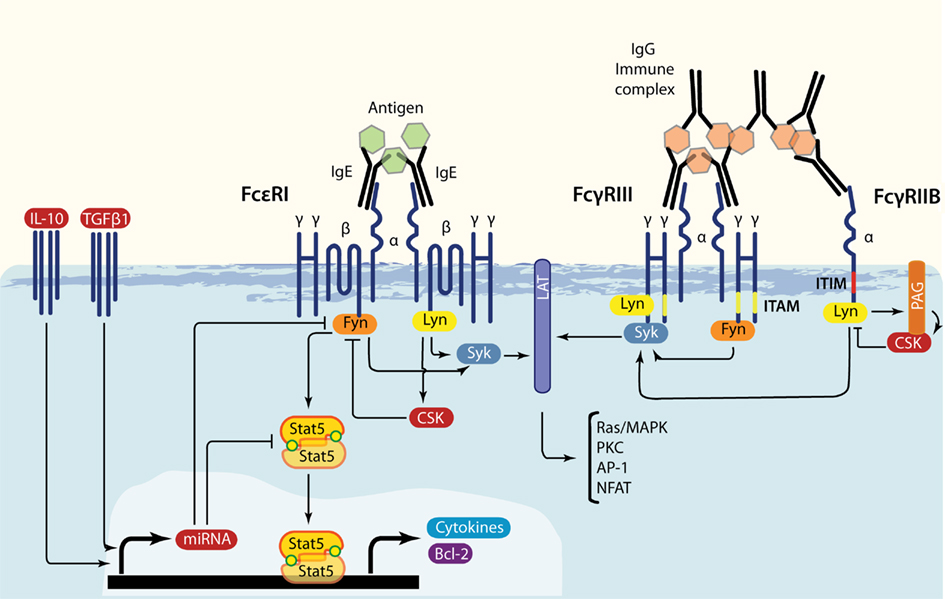
Figure 1. Overlap in FcεRI, FcγRIII, and FcγRIIB signaling. IgE-bound multivalent antigen or IgG immune complexes induce FcεRI or FcγRIII/FcγRIIB crosslinking, respectively. These events lead to the swift activation of Src family kinases Lyn and Fyn. Both positively influence downstream signaling through Syk and LAT, but Lyn also produces negative feedback by phosphorylating ITIMs and by inducing the Csk recruitment via SH2 interactions with phosphorylated Cbp/PAG. Additionally, Fyn stimulates the PI3-K axis. Under FcεRI-driven signaling, Fyn activates STAT5, transcription factors that are required for mast cell survival (STAT5A) and cytokine production (STAT5B). Regulatory cytokines such as IL-10 and TGFβ1 can inhibit Fyn and STAT5 production, possibly through the induction of specific miRs.
First is the need for a more precise dissection of STAT5 activation and mobilization as an effect of IgE and IgG receptor crosslinking. STAT5 is actually represented by two similar but distinct proteins: STAT5A and STAT5B (Grimley et al., 1999). As mentioned earlier STAT5A is important for the development and proliferation of mast cells (Shelburne et al., 2003), whereas STAT5B has been identified as the regulator of cytokine production (Pullen et al., 2012). Thus far these roles do not appear interchangeable, e.g., STAT5A depletion does not significantly affect IgE-mediated cytokine production. While the role of STAT5B in mast cell development has not been directly addressed, its impact is likely redundant since total STAT5KO bone marrow can be rescued by reconstituting STAT5A (Shelburne et al., 2003). One question needing pursuit asks if there is a specificity for which STAT5 isoform associates with Fyn and FcεRI. Furthermore, which STAT5 mobilizes to the nucleus when, and what are their targets? Complicating this matter is the fact that in addition to tyrosine at least two serines are phosphorylated, and the function of each phosphorylated residue could possibly diverge between each STAT5. For example, phosphorylation of STAT5A serines 725 and 779 has been identified as a requirement for leukemogenic transformation (Friedbichler et al., 2010), and there are slight primary sequence variations in the location of the known target serines and tyrosine between STAT5A and STAT5B.
Second is the need to further dissect the roles of other Fc receptor signal mediators. For example, double KO of the Tec kinases Itk and Btk was found to inhibit degranulation/histamine release and impaired mast cell granularity (Iyer et al., 2011). Interestingly, in the same work c-Kit expression and mast cell development were not affected, however the double KOs showed enhanced proliferation. Furthermore, FcεRI and the FcγR share common γ-chains containing ITAMs that, when phosphorylated, can recruit Syk (Crowley et al., 1997; Lach-Trifilieff et al., 2000; Nimmerjahn and Ravetch, 2008). In FcεRI signaling, the activation of Syk through Lyn is a well-known effector of mast cell function; if this happens in the context of FcγR crosslinking is open for investigation. The association of Lyn [and Fyn] with FcεRI is an enigmatic matter, as it is known to be dynamic and possibly compartmentalized (Gilfillan and Rivera, 2009). This in combination with the diversity of IgG receptors on mast cells suggests that, while the high affinity IgE receptor serves as a good model, there is much more to these signaling stories than is currently understood. For example, additional exploration of FcγR expression profiles in mast cells of different sources, phenotypes, and species will be important for interpreting data.
Finally, unraveling these discrete biochemical questions should be matched with in vivo studies of STAT5 functions in allergic and autoimmune diseases, especially those in which mast cells are a key component. Therapies targeting mast cell activation and secreted mediators have proven effective in allergic disease. If the Fyn-Stat5 pathway operates as a key signaling pathway as we hypothesize, selective targeting of this axis could prove therapeutic to many disorders. These clinical goals are critical, and should come as a natural product of efforts to understand the fundamentals of immune homeostasis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Andrews, N. L., Lidke, K. A., Pfeiffer, J. R., Burns, A. R., Wilson, B. S., Oliver, J. M., and Lidke, D. S. (2008). Actin restricts FcepsilonRI diffusion and facilitates antigen-induced receptor immobilization. Nat. Cell Biol. 10, 955–963.
Bailey, D. P., Kashyap, M., Bouton, L. A., Murray, P. J., and Ryan, J. J. (2006). Interleukin-10 induces apoptosis in developing mast cells and macrophages. J. Leukoc. Biol. 80, 581–589.
Bailey, D. P., Kashyap, M., Mirmonsef, P., Bouton, L. A., Domen, J., Zhu, J., Dessypris, E. N., and Ryan, J. J. (2004). Interleukin-4 elicits apoptosis of developing mast cells via a Stat6-dependent mitochondrial pathway. Exp. Hematol. 32, 52–59.
Barnstein, B. O., Li, G., Wang, Z., Kennedy, S., Chalfant, C., Nakajima, H., Bunting, K. D., and Ryan, J. J. (2006). Stat5 expression is required for IgE-mediated mast cell function. J. Immunol. 177, 3421–3426.
Beal, A. M., Ramos-Hernandez, N., Riling, C. R., Nowelsky, E. A., and Oliver, P. M. (2011). TGF-beta induces the expression of the adaptor Ndfip1 to silence IL-4 production during iT(reg) cell differentiation. Nat. Immunol. 13, 77–85.
Benhamou, M., Bonnerot, C., Fridman, W. H., and Daëron, M. (1990). Molecular heterogeneity of murine mast cell Fc gamma receptors. J. Immunol. 144, 3071–3077.
Berg, D. J., Kuhn, R., Rajewsky, K., Muller, W., Menon, S., Davidson, N., Grunig, G., and Rennick, D. (1995). Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J. Clin. Invest. 96, 2339–2347.
Bettelli, E., Carrier, Y., Gao, W., Korn, T., Strom, T. B., Oukka, M., Weiner, H. L., and Kuchroo, V. K. (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238.
Bischoff, S. C., Sellge, G., Lorentz, A., Sebald, W., Raab, R., and Manns, M. P. (1999). IL-4 enhances proliferation and mediator release in mature human mast cells. Proc. Natl. Acad. Sci. U.S.A. 96, 8080–8085.
Bissonnette, E. Y., Enciso, J. A., and Befus, A. D. (1997). TGF-beta1 inhibits the release of histamine and tumor necrosis factor-alpha from mast cells through an autocrine pathway. Am. J. Respir. Cell Mol. Biol. 16, 275–282.
Blobe, G. C., Schiemann, W. P., and Lodish, H. F. (2000). Role of transforming growth factor beta in human disease. N. Engl. J. Med. 342, 1350–1358.
Bouton, L. A., Ramirez, C. D., Bailey, D. P., Yeatman, C. F., Yue, J., Wright, H. V., Domen, J., Rosatao, R. R., Grant, S., Fischer-Stanger, K., and Ryan, J. J. (2004). Costimulation with interleukin-4 and interleukin-10 induces mast cell apoptosis and cell-cycle arrest: the role of p53 and the mitochondrion. Exp. Hematol. 32, 1137–1145.
Brdicka, T., Pavlistová, D., Leo, A., Bruyns, E., Korinek, V., Angelisová, P., Scherer, J., Shevchenko, A., Hilgert, I., Cerný, J., Drbal, K., Kuramitsu, Y., Kornacker, B., Horejsí, V., and Schraven, B. (2000). Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J. Exp. Med. 191, 1591–1604.
Brizzi, M. F., Dentelli, P., Rosso, A., Yarden, Y., and Pegoraro, L. (1999). STAT protein recruitment and activation in c-Kit deletion mutants. J. Biol. Chem. 274, 16965–16972.
Broide, D. H., Wasserman, S. I., Alvaro-Gracia, J., Zvaifler, N. J., and Firestein, G. S. (1989). Transforming growth factor-beta 1 selectively inhibits IL-3-dependent mast cell proliferation without affecting mast cell function or differentiation. J. Immunol. 143, 1591–1597.
Crowley, M. T., Costello, P. S., Fitzer-Attas, C. J., Turner, M., Meng, F., Lowell, C., Tybulewicz, V. L., and DeFranco, A. L. (1997). A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J. Exp. Med. 186, 1027–1039.
Daëron, M., Malbec, O., Latour, S., Arock, M., and Fridman, W. H. (1995). Regulation of high-affinity IgE receptor-mediated mast cell activation by murine low-affinity IgG receptors. J. Clin. Invest. 95, 577–585.
Daëron, M., Malbec, O., Latour, S., Bonnerot, C., Segal, D. M., and Fridman, W. H. (1993). Distinct intracytoplasmic sequences are required for endocytosis and phagocytosis via murine Fc gamma RII in mast cells. Int. Immunol. 5, 1393–1401.
Dombrowicz, D., Flamand, V., Miyajima, I., Ravetch, J. V., Galli, S. J., and Kinet, J. P. (1997). Absence of Fc epsilonRI alpha chain results in upregulation of Fc gammaRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between Fc epsilonRI and Fc gammaRIII for limiting amounts of FcR beta and gamma chains. J. Clin. Invest. 99, 915–925.
Eiseman, E., and Bolen, J. B. (1992). Engagement of the high-affinity IgE receptor activates src protein-related tyrosine kinases. Nature 355, 78–80.
Falanga, Y. T., Chaimowitz, N. S., Charles, N., Finkelman, F. D., Pullen, N. A., Barbour, S., Dholaria, K., Faber, T., Kolawole, M., Huang, B., Odom, S., Rivera, J., Carlyon, J., Conrad, D. H., Spiegel, S., Oskeritzian, C. A., and Ryan, J. J. (2012). Lyn but not Fyn kinase controls IgG-mediated systemic anaphylaxis. J. Immunol. 188, 4360–4368.
Fong, D. C., Malbec, O., Arock, M., Cambier, J. C., Fridman, W. H., and Daeron, M. (1996). Selective in vivo recruitment of the phosphatidylinositol phosphatase SHIP by phosphorylated Fc gammaRIIB during negative regulation of IgE-dependent mouse mast cell activation. Immunol. Lett. 54, 83–91.
Forward, N. A., Furlong, S. J., Yang, Y., Lin, T. J., and Hoskin, D. W. (2009). Mast cells down-regulate CD4+CD25+ T regulatory cell suppressor function via histamine H1 receptor interaction. J. Immunol. 183, 3014–3022.
Foster, P. S., Martinez-Moczygemba, M., Huston, D. P., and Corry, D. B. (2002). Interleukins-4, -5, and -13: emerging therapeutic targets in allergic disease. Pharmacol. Ther. 94, 253–264.
Frandji, P., Tkaczyk, C., Oskeritizian, C., David, B., Desaymard, C., and Mecheri, S. (1996). Exogenous and endogenous antigens are differentially presented by mast cells to CD4T lymphocytes. Eur. J. Immunol. 26, 2517–2528.
Friedbichler, K., Kerenyi, M. A., Kovacic, B., Li, G., Hoelbl, A., Yahiaoui, S., Sexl, V., Müllner, E. W., Fajmann, S., Cerny-Reiterer, S., Valent, P., Beug, H., Gouilleux, F., Bunting, K. D., and Moriggl, R. (2010). Stat5a serine 725 and 779 phosphorylation is a prerequisite for hematopoietic transformation. Blood 116, 1548–1558.
Galli, S. J., Nakae, S., and Tsai, M. (2005). Mast cells in the development of adaptive immune responses. Nat. Immunol. 6, 135–142.
Galli, S. J., Tsai, M., and Piliponsky, A. M. (2008). The development of allergic inflammation. Nature 454, 445–454
Gauchat, J.-F., Henchoz, S., Mazzei, G., Aubry, J.-P., Brunner, T., Blasey, H., Life, P., Talabot, D., Flores-Romo, L., Thompson, J., Kishi, K., Butterfield, J., Dahinden, C., and Bonnefoy, J.-Y. (1993). Induction of human IgE synthesis in B cells by mast cells and basophils. Nature 365, 340–343.
Gazzinelli, R. T., Wysocka, M., Hieny, S., Scharton-Kersten, T., Cheever, A., Kuhn, R., Muller, W., Trinchieri, G., and Sher, A. (1996). In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 157, 798–805.
Gilfillan, A. M., and Rivera, J. (2009). The tyrosine kinase network regulating mast cell activation. Immunol. Rev. 228, 149–169.
Gillespie, S. R., DeMartino, R. R., Zhu, J., Chong, H. J., Ramirez, C., Shelburne, C. P., Bouton, L. A., Bailey, D. P., Gharse, A., Mirmonsef, P., Odom, S., Gomez, G., Rivera, J., Fischer-Stenger, K., and Ryan, J. J. (2004). IL-10 inhibits Fc epsilon RI expression in mouse mast cells. J. Immunol. 172, 3181–3188.
Gomez, G., Gonzalez-Espinosa, C., Odom, S., Baez, G., Cid, M. E., Ryan, J. J., and Rivera, J. (2005a). Impaired FcepsilonRI-dependent gene expression and defective eicosanoid and cytokine production as a consequence of Fyn deficiency in mast cells. J. Immunol. 175, 7602–7610.
Gomez, G., Ramirez, C. D., Rivera, J., Patel, M., Norozian, F., Wright, H. V., Kashyap, M. V., Barnstein, B. O., Fischer-Stenger, K., Schwartz, L. B., Kepley, C. L., and Ryan, J. J. (2005b). TGF-beta 1 inhibits mast cell Fc epsilon RI expression. J. Immunol. 174, 5987–5993.
Grimley, P. M., Dong, F., and Rui, H. (1999). Stat5a and Stat5b: fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 10, 131–157.
Hara, M., Ono, K., Hwang, M. W., Iwasaki, A., Okada, M., Nakatani, K., Sasayama, S., and Matsumori, A. (2002). Evidence for a role of mast cells in the evolution to congestive heart failure. J. Exp. Med. 195, 375–381.
He, Q., Moore, T. T., Eko, F. O., Lyn, D., Ananaba, G. A., Martin, A., Singh, S., James, L., Stiles, J., Black, C. M., and Igietseme, J. U. (2005). Molecular basis for the potency of IL-10-deficient dendritic cells as a highly efficient APC system for activating Th1 response. J. Immunol. 174, 4860–4869.
Hernandez-Hansen, V., Smith, A. J., Surviladze, Z., Chigaev, A., Mazel, T., Kalesnikoff, J., Lowell, C. A., Krystal, G., Sklar, L. A., Wilson, B. S., and Oliver, J. M. (2004). Dysregulated FcepsilonRI signaling and altered Fyn and SHIP activities in Lyn-deficient mast cells. J. Immunol. 173, 100–112.
Hibbs, M. L., Harder, K. W., Armes, J., Kountouri, N., Quilici, C., Casagranda, F., Dunn, A. R., and Tarlinton, D. M. (2002). Sustained activation of Lyn tyrosine kinase in vivo leads to autoimmunity. J. Exp. Med. 196, 1593–1604.
Honda, Z., Suzuki, T., Hirose, N., Aihara, M., Shimizu, T., Nada, S., Okada, M., Ra, C., Morita, Y., and Ito, K. (1997). Roles of C-terminal Src kinase in the initiation and the termination of the high affinity IgE receptor-mediated signaling. J. Biol. Chem. 272, 25753–25760.
Howard, M., Muchamuel, T., Andrade, S., and Menon, S. (1993). Interleukin 10 protects mice from lethal endotoxemia. J. Exp. Med. 177, 1205–1208.
Hunter, C. A., Ellis-Neyes, L. A., Slifer, T., Kanaly, S., Grunig, G., Fort, M., Rennick, D., and Araujo, F. G. (1997). IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J. Immunol. 158, 3311–3316.
Ikeda, K., Nakajima, H., Suzuki, K., Watanabe, N., Kagami, S., and Iwamoto, I. (2005). Stat5a is essential for the proliferation and survival of murine mast cells. Int. Arch. Allergy Immunol. 137(Suppl. 1), 45–50.
Iyer, A. S., Morales, J. L., Huang, W., Ojo, F., Ning, G., Wills, E., Baines, J. D., and August, A. (2011). Absence of Tec family kinases interleukin-2 inducible T cell kinase (Itk) and Bruton’s tyrosine kinase (Btk) severely impairs Fc epsilonRI-dependent mast cell responses. J. Biol. Chem. 286, 9503–9513.
Jonsson, F., Mancardi, D. A., Kita, Y., Karasuyama, H., Iannascoli, B., Van Rooijen, N., Shimizu, T., Daeron, M., and Bruhns, P. (2011a). Mouse and human neutrophils induce anaphylaxis. J. Clin. Invest. 121, 1484–1496.
Jonsson, F., Mancardi, D. A., Zhao, W., Kita, Y., Iannascoli, B., Khun, H., van Rooijen, N., Shimizu, T., Schwartz, L. B., Daeron, M., and Bruhns, P. (2011b). Human FcgammaRIIA induces anaphylactic and allergic reactions. Blood 119, 2533–2544.
Kalesnikoff, J., and Galli, S. J. (2008). New developments in mast cell biology. Nat. Immunol. 9, 1215–1223.
Kambayashi, T., Allenspach, E. J., Chang, J. T., Zou, T., Shoag, J. E., Reiner, S. L., Caton, A. J., and Koretzky, G. A. (2009). Inducible MHC class II expression by mast cells supports effector and regulatory T cell activation. J. Immunol. 182, 4686–4695.
Kanbe, N., Kurosawa, M., Nagata, H., Saitoh, H., and Miyachi, Y. (1999). Cord blood-derived human cultured mast cells produce transforming growth factor beta1. Clin. Exp. Allergy 29, 105–113.
Katz, H. R., Raizman, M. B., Gartner, C. S., Scott, H. C., Benson, A. C., and Austen, K. F. (1992). Secretory granule mediator release and generation of oxidative metabolites of arachidonic acid via Fc-IgG receptor bridging in mouse mast cells. J. Immunol. 148, 868–871.
Kawabuchi, M., Satomi, Y., Takao, T., Shimonishi, Y., Nada, S., Nagai, K., Tarakhovsky, A., and Okada, M. (2000). Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature 404, 999–1003.
Kehrl, J. H., Wakefield, L. M., Roberts, A. B., Jakowlew, S., Alvarez-Mon, M., Derynck, R., Sporn, M. B., and Fauci, A. S. (1986). Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 163, 1037–1050.
Kennedy Norton, S., Barnstein, B., Brenzovich, J., Bailey, D. P., Kashyap, M., Speiran, K., Ford, J., Conrad, D., Watowich, S., Moralle, M. R., Kepley, C. L., Murray, P. J., and Ryan, J. J. (2008). IL-10 suppresses mast cell IgE receptor expression and signaling in vitro and in vivo. J. Immunol. 180, 2848–2854.
Kim, H. J., Zhang, K., Zhang, L., Ross, F. P., Teitelbaum, S. L., and Faccio, R. (2009). The Src family kinase, Lyn, suppresses osteoclastogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 2325–2330.
Kim, H. M., and Lee, Y. M. (1999). Role of TGF-beta 1 on the IgE-dependent anaphylaxis reaction. J. Immunol. 162, 4960–4965.
Kim, M. S., Radinger, M., and Gilfillan, A. M. (2008). The multiple roles of phosphoinositide 3-kinase in mast cell biology. Trends Immunol. 29, 493–501.
Kotenko, S. V. (2002). The family of IL-10-related cytokines and their receptors: related, but to what extent? Cytokine Growth Factor Rev. 13, 223–240.
Kovarova, M., Wassif, C. A., Odom, S., Liao, K., Porter, F. D., and Rivera, J. (2006). Cholesterol deficiency in a mouse model of Smith-Lemli-Opitz syndrome reveals increased mast cell responsiveness. J. Exp. Med. 203, 1161–1171.
Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K., and Muller, W. (1993). Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274.
Kurosaki, T., Gander, I., Wirthmueller, U., and Ravetch, J. V. (1992). The beta subunit of the Fc epsilon RI is associated with the Fc gamma RIII on mast cells. J. Exp. Med. 175, 447–451.
Lach-Trifilieff, E., Menear, K., Schweighoffer, E., Tybulewicz, V. L., and Walker, C. (2000). Syk-deficient eosinophils show normal interleukin-5-mediated differentiation, maturation, and survival but no longer respond to FcgammaR activation. Blood 96, 2506–2510.
Lee, D. M., Friend, D. S., Gurish, M. F., Benoist, C., Mathis, D., and Brenner, M. B. (2002). Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 297, 1689–1692.
Lee, Y. K., Mukasa, R., Hatton, R. D., and Weaver, C. T. (2009). Developmental plasticity of Th17 and Treg cells. Curr. Opin. Immunol. 21, 274–280.
Letterio, J. J., and Roberts, A. B. (1998). Regulation of immune responses by TGF-beta. Annu. Rev. Immunol. 16, 137–161.
Levi-Schaffer, F., Riesel, N., Soffer, D., Abramsky, O., and Brenner, T. (1991). Mast cell activity in experimental allergic encephalomyelitis. Mol. Chem. Neuropathol. 15, 173–184.
Lin, T. J., and Befus, A. D. (1997). Differential regulation of mast cell function by IL-10 and stem cell factor. J. Immunol. 159, 4015–4023.
Lindquist, J. A., Simeoni, L., and Schraven, B. (2003). Transmembrane adapters: attractants for cytoplasmic effectors. Immunol. Rev. 191, 165–182.
Lindstedt, K. A., Wang, Y., Shiota, N., Saarinen, J., Hyytiainen, M., Kokkonen, J. O., Keski-Oja, J., and Kovanen, P. T. (2001). Activation of paracrine TGF-beta1 signaling upon stimulation and degranulation of rat serosal mast cells: a novel function for chymase. FASEB J. 15, 1377–1388.
Lorentz, A., and Bischoff, S. C. (2001). Regulation of human intestinal mast cells by stem cell factor and IL-4. Immunol. Rev. 179, 57–60.
Lu, L. F., Lind, E. F., Gondek, D. C., Bennett, K. A., Gleeson, M. W., Pino-Lagos, K., Scott, Z. A., Coyle, A. J., Reed, J. L., Van Snick, J., Strom, T. B., Zheng, X. X., and Noelle, R. J. (2006). Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 442, 997–1002.
Macey, M. R., Sturgill, J. L., Morales, J. K., Falanga, Y. T., Morales, J., Norton, S. K., Yerram, N., Shim, H., Fernando, J., Gifillan, A. M., Gomez, G., Schwartz, L., Oskeritzian, C., Spiegel, S., Conrad, D., and Ryan, J. J. (2010). IL-4 and TGF-beta 1 counterbalance one another while regulating mast cell homeostasis. J. Immunol. 184, 4688–4695.
Malaviya, R., Ikeda, T., Ross, E., and Abraham, S. N. (1996). Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature 381, 77–80.
Malaviya, R., and Uckun, F. M. (2002). Role of STAT6 in IgE receptor/FcepsilonRI-mediated late phase allergic responses of mast cells. J. Immunol. 168, 421–426.
Malbec, O., and Daeron, M. (2007). The mast cell IgG receptors and their roles in tissue inflammation. Immunol. Rev. 217, 206–221.
Marshall, J. S., Leal-Berumen, I., Nielsen, L., Glibetic, M., and Jordana, M. (1996). Interleukin (IL)-10 inhibits long-term IL-6 production but not preformed mediator release from rat peritoneal mast cells. J. Clin. Invest. 97, 1122–1128.
McLachlan, J. B., Hart, J. P., Pizzo, S. V., Shelburne, C. P., Staats, H. F., Gunn, M. D., and Abraham, S. N. (2003). Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat. Immunol. 4, 1199–1205.
Meade, R., Askenase, P. W., Geba, G. P., Neddermann, K., Jacoby, R. O., and Pasternak, R. D. (1992). Transforming growth factor-beta 1 inhibits murine immediate and delayed type hypersensitivity. J. Immunol. 149, 521–528.
Mekori, Y. A., and Metcalfe, D. D. (1994). Transforming growth factor-beta prevents stem cell factor-mediated rescue of mast cells from apoptosis after IL-3 deprivation. J. Immunol. 153, 2194–2203.
Miller, H. R., Wright, S. H., Knight, P. A., and Thornton, E. M. (1999). A novel function for transforming growth factor-beta1: upregulation of the expression and the IgE-independent extracellular release of a mucosal mast cell granule-specific beta-chymase, mouse mast cell protease-1. Blood 93, 3473–3486.
Miyajima, I., Dombrowicz, D., Martin, T. R., Ravetch, J. V., Kinet, J. P., and Galli, S. J. (1997). Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and Fc gammaRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J. Clin. Invest. 99, 901–914.
Moore, K. W., de Waal Malefyt, R., Coffman, R. L., and O’Garra, A. (2001). Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19, 683–765.
Moore, K. W., O’Garra, A., de Waal Malefyt, R., Vieira, P., and Mosmann, T. R. (1993). Interleukin-10. Annu. Rev. Immunol. 11, 165–190.
Mui, A. L., Wakao, H., O’Farrell, A. M., Harada, N., and Miyajima, A. (1995). Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J. 14, 1166–1175.
Murray, P. J. (2006). Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 6, 379–386.
Nguyen, T. H., and Casale, T. B. (2011). Immune modulation for treatment of allergic disease. Immunol. Rev. 242, 258–271.
Ni, S., Zhao, C., Feng, G. S., Paulson, R. F., and Correll, P. H. (2007). A novel Stat3 binding motif in Gab2 mediates transformation of primary hematopoietic cells by the Stk/Ron receptor tyrosine kinase in response to Friend virus infection. Mol. Cell. Biol. 27, 3708–3715.
Nimmerjahn, F., and Ravetch, J. V. (2008). Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47.
Norozian, F., Kashyap, M., Ramirez, C. D., Patel, N., Kepley, C. L., Barnstein, B. O., and Ryan, J. J. (2006). TGFbeta1 induces mast cell apoptosis. Exp. Hematol. 34, 579–587.
Obata, K., Mukai, K., Tsujimura, Y., Ishiwata, K., Kawano, Y., Minegishi, Y., Watanabe, N., and Karasuyama, H. (2007). Basophils are essential initiators of a novel type of chronic allergic inflammation. Blood 110, 913–920.
Odom, S., Gomez, G., Kovarova, M., Furumoto, Y., Ryan, J. J., Wright, H. V., Gonzalez-Espinosa, C., Hibbs, M. L., Harder, K. W., and Rivera, J. (2004). Negative regulation of immunoglobulin E-dependent allergic responses by Lyn kinase. J. Exp. Med. 199, 1491–1502.
Ohtake, H., Ichikawa, N., Okada, M., and Yamashita, T. (2002). Cutting edge: transmembrane phosphoprotein Csk-binding protein/phosphoprotein associated with glycosphingolipid-enriched microdomains as a negative feedback regulator of mast cell signaling through the FcepsilonRI. J. Immunol. 168, 2087–2090.
Ono, M., Bolland, S., Tempst, P., and Ravetch, J. V. (1996). Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature 383, 263–266.
Parravicini, V., Gadina, M., Kovarova, M., Odom, S., Gonzalez-Espinosa, C., Furumoto, Y., Saitoh, S., Samelson, L. E., O’Shea, J. J., and Rivera, J. (2002). Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat. Immunol. 3, 741–748.
Pawankar, R., Okuda, M., Yssel, H., Okumura, K., and Ra, C. (1997). Nasal mast cells in perennial allergic rhinitis exhibit increased expression of the Fc epsilonRI, CD40L, IL-4, and IL-13, and can induce IgE synthesis in B cells. J. Clin. Invest. 99, 1492–1499.
Pemberton, A. D., Brown, J. K., Wright, S. H., Knight, P. A., and Miller, H. R. (2006). The proteome of mouse mucosal mast cell homologues: the role of transforming growth factor beta1. Proteomics 6, 623–631.
Pennington, D. W., Lopez, A. R., Thomas, P. S., Peck, C., and Gold, W. M. (1992). Dog mastocytoma cells produce transforming growth factor beta 1. J. Clin. Invest. 90, 35–41.
Piconese, S., Gri, G., Tripodo, C., Musio, S., Gorzanelli, A., Frossi, B., Pedotti, R., Pucillo, C. E., and Colombo, M. P. (2009). Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17-cell differentiation. Blood 114, 2639–2648.
Pribluda, V. S., Pribluda, C., and Metzger, H. (1994). Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc. Natl. Acad. Sci. U.S.A. 91, 11246–11250.
Pullen, N. A., Barnstein, B. O., Falanga, Y. T., Wang, Z., Suzuki, R., Lama Tamang, T. D., Khurana, M. C., Harry, E. A., Draber, P., Bunting, K. D., Mizuno, K., Wilson, B. S., and Ryan, J. J. (2012). Novel mechanism for FcεRI-mediated signal transducer and activator of transcription 5 (STAT5) tyrosine phosphorylation and the selective influence of STAT5B over mast cell cytokine production. J. Biol. Chem. 287, 2045–2054.
Rivera, J., and Gilfillan, A. M. (2006). Molecular regulation of mast cell activation. J. Allergy Clin. Immunol. 117, 1214–1225; quiz 1226.
Roers, A., Siewe, L., Strittmatter, E., Deckert, M., Schlüter, D., Stenzel, W., Gruber, A. D., Krieg, T., Rajewsky, K., and Müller, W. (2004). T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J. Exp. Med. 200, 1289–1297.
Royer, B., Varadaradjalou, S., Saas, P., Gabiot, A. C., Kantelip, B., Feger, F., Guillosson, J. J., Kantelip, J. P., and Arock, M. (2001a). Autocrine regulation of cord blood-derived human mast cell activation by IL-10. J. Allergy Clin. Immunol. 108, 80–86.
Royer, B., Varadaradjalou, S., Saas, P., Guillosson, J. J., Kantelip, J. P., and Arock, M. (2001b). Inhibition of IgE-induced activation of human mast cells by IL-10. Clin. Exp. Allergy 31, 694–704.
Ryan, J. J., DeSimone, S., Klisch, G., Shelburne, C., McReynolds, L. J., Han, K., Kovacs, R., Mirmonsef, P., and Huff, T. F. (1998). IL-4 inhibits mouse mast cell Fc epsilonRI expression through a STAT6-dependent mechanism. J. Immunol. 161, 6915–6923.
Ryan, J. J., Huang, H., McReynolds, L. J., Shelburne, C., Hu-Li, J., Huff, T. F., and Paul, W. E. (1997). Stem cell factor activates STAT-5 DNA binding in IL-3-derived bone marrow mast cells. Exp. Hematol. 25, 357–362.
Satterthwaite, A. B., Lowell, C. A., Khan, W. N., Sideras, P., Alt, F. W., and Witte, O. N. (1998). Independent and opposing roles for Btk and lyn in B and myeloid signaling pathways. J. Exp. Med. 188, 833–844.
Schindler, C., Levy, D. E., and Decker, T. (2007). JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282, 20059–20063.
Secor, V. H., Secor, W. E., Gutekunst, C. A., and Brown, M. A. (2000). Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J. Exp. Med. 191, 813–822.
Shelburne, C. P., McCoy, M. E., Piekorz, R., Sexl, V., Roh, K. H., Jacobs-Helber, S. M., Gillespie, S. R., Bailey, D. P., Mirmonsef, P., Mann, M. N., Kashyap, M., Wright, H. V., Chong, H. J., Bouton, L. A., Barnstein, B., Ramirez, C. D., Bunting, K. D., Sawyer, S., Lantz, C. S., and Ryan, J. J. (2003). Stat5 expression is critical for mast cell development and survival. Blood 102, 1290–1297.
Sonnenblick, A., Levy, C., and Razin, E. (2005). Immunological trigger of mast cells by monomeric IgE: effect on microphthalmia transcription factor, STAT3 network of interactions. J. Immunol. 175, 1450–1455.
Speiran, K., Bailey, D. P., Fernando, J., Macey, M., Barnstein, B., Kolawole, M., Curley, D., Watowich, S. S., Murray, P. J., Oskeritzian, C., and Ryan, J. J. (2009). Endogenous suppression of mast cell development and survival by IL-4 and IL-10. J. Leukoc. Biol. 85, 826–836.
Strait, R. T., Morris, S. C., Yang, M., Qu, X. W., and Finkelman, F. D. (2002). Pathways of anaphylaxis in the mouse. J. Allergy Clin. Immunol. 109, 658–668.
Suzuki, R., Liu, X., Olivera, A., Aguiniga, L., Yamashita, Y., Blank, U., Ambudkar, I., and Rivera, J. (2010). Loss of TRPC1-mediated Ca2+ influx contributes to impaired degranulation in Fyn-deficient mouse bone marrow-derived mast cells. J. Leukoc. Biol. 88, 863–875.
Takeshita, H., Taniuchi, I., Kato, J., and Watanabe, T. (1998). Abrogation of autoimmune disease in Lyn-deficient mice by the mutation of the Btk gene. Int. Immunol. 10, 435–444.
Takeuchi, S., Takayama, Y., Ogawa, A., Tamura, K., and Okada, M. (2000). Transmembrane phosphoprotein Cbp positively regulates the activity of the carboxyl-terminal Src kinase, Csk. J. Biol. Chem. 275, 29183–29186.
Toru, H., Ra, C., Nonoyama, S., Suzuki, K., Yata, J., and Nakahata, T. (1996). Induction of the high-affinity IgE receptor (Fc epsilon RI) on human mast cells by IL-4. Int. Immunol. 8, 1367–1373.
Toyota, N., Hashimoto, Y., Matsuo, S., and Iizuka, H. (1995). Transforming growth factor beta 1 inhibits IL-3- and IL-4-dependent mouse connective tissue-type mast cell proliferation. Arch. Dermatol. Res. 287, 198–201.
Trachsel, E., Bootz, F., Silacci, M., Kaspar, M., Kosmehl, H., and Neri, D. (2007). Antibody-mediated delivery of IL-10 inhibits the progression of established collagen-induced arthritis. Arthritis Res. Ther. 9, R9.
Tsujimura, Y., Obata, K., Mukai, K., Shindou, H., Yoshida, M., Nishikado, H., Kawano, Y., Minegishi, Y., Shimizu, T., and Karasuyama, H. (2008). Basophils play a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity 28, 581–589.
Wiener, Z., Kohalmi, B., Pocza, P., Jeager, J., Tolgyesi, G., Toth, S., Gorbe, E., Papp, Z., and Falus, A. (2007). TIM-3 is expressed in melanoma cells and is upregulated in TGF-beta stimulated mast cells. J. Invest. Dermatol. 127, 906–914.
Wills-Karp, M., and Finkelman, F. D. (2008). Untangling the complex web of IL-4- and IL-13-mediated signaling pathways. Sci. Signal. 1, pe55.
Wilson, B. S., Pfeiffer, J. R., Raymond-Stintz, M. A., Lidke, D., Andrews, N., Zhang, J., Yin, W., Steinberg, S., and Oliver, J. M. (2007). Exploring membrane domains using native membrane sheets and transmission electron microscopy. Methods Mol. Biol. 398, 245–261.
Woodman, L., Siddiqui, S., Cruse, G., Sutcliffe, A., Saunders, R., Kaur, D., Bradding, P., and Brightling, C. (2008). Mast cells promote airway smooth muscle cell differentiation via autocrine up-regulation of TGF-beta 1. J. Immunol. 181, 5001–5007.
Xiao, W., Nishimoto, H., Hong, H., Kitaura, J., Nunomura, S., Maeda-Yamamoto, M., Kawakami, Y., Lowell, C. A., Ra, C., and Kawakami, T. (2005). Positive and negative regulation of mast cell activation by Lyn via the FcepsilonRI. J. Immunol. 175, 6885–6892.
Xu, Y., Harder, K. W., Huntington, N. D., Hibbs, M. L., and Tarlinton, D. M. (2005). Lyn tyrosine kinase: accentuating the positive and the negative. Immunity 22, 9–18.
Xue, M., Hsieh, G., Raymond-Stintz, M. A., Pfeiffer, J., Roberts, D., Steinberg, S. L., Oliver, J. M., Prossnitz, E. R., Lidke, D. S., and Wilson, B. S. (2007). Activated N-formyl peptide receptor and high-affinity IgE receptor occupy common domains for signaling and internalization. Mol. Biol. Cell 18, 1410–1420.
Yamashita, Y., Charles, N., Furumoto, Y., Odom, S., Yamashita, T., Gilfillan, A. M., Constant, S., Bower, M. A., Ryan, J. J., and Rivera, J. (2007). Cutting edge: genetic variation influences Fc epsilonRI-induced mast cell activation and allergic responses. J. Immunol. 179, 740–743.
Yang, L., Pang, Y., and Moses, H. L. (2010a). TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 31, 220–227.
Yang, Z., Zhang, B., Li, D., Lv, M., Huang, C., Shen, G. X., and Huang, B. (2010b). Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS ONE 5, e8922. doi:10.1371/journal.pone.0008922
Yeatman, C. F. II, Jacobs-Helber, S. M., Mirmonsef, P., Gillespie, S. R., Bouton, L. A., Collins, H. A., Sawyer, S. T., Shelburne, C. P., and Ryan, J. J. (2000). Combined stimulation with the T helper cell type 2 cytokines interleukin (IL)-4 and IL-10 induces mouse mast cell apoptosis. J. Exp. Med. 192, 1093–1103.
Yoshimura, A., Wakabayashi, Y., and Mori, T. (2010). Cellular and molecular basis for the regulation of inflammation by TGF-beta. J. Biochem. 147, 781–792.
Yu, W. M., Hawley, T. S., Hawley, R. G., and Qu, C. K. (2002). Role of the docking protein Gab2 in beta(1)-integrin signaling pathway-mediated hematopoietic cell adhesion and migration. Blood 99, 2351–2359.
Keywords: mast cell, Stat5, Fyn, IgE, IgG, IL-4, IL-10, TGF
Citation: Pullen NA, Falanga YT, Morales JK and Ryan JJ (2012) The Fyn-STAT5 pathway: a new Frontier in IgE- and IgG-mediated mast cell signaling. Front. Immun. 3:117. doi: 10.3389/fimmu.2012.00117
Received: 04 January 2012; Accepted: 23 April 2012;
Published online: 11 May 2012.
Edited by:
Toshiaki Kawakami, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Toshiaki Kawakami, La Jolla Institute for Allergy and Immunology, USAYoshimichi Okayama, Nihon University School of Medicine, Japan
Andrew Craig, Queen’s University, Canada
Copyright: © 2012 Pullen, Falanga, Morales and Ryan. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: John J. Ryan, Department of Biology, Virginia Commonwealth University, 1000 West Cary Street, Richmond, VA 23284-2012, USA. e-mail:ampyeWFuQHZjdS5lZHU=