 Elina A. Kiss1,2,3 Andreas Diefenbach1,2,3,4*
Elina A. Kiss1,2,3 Andreas Diefenbach1,2,3,4*- 1 Institute of Medical Microbiology and Hygiene, University of Freiburg, Freiburg, Germany
- 2 Spemann Graduate School of Biology and Medicine, Freiburg, Germany
- 3 Research Training Group of Organogenesis (GRK1104), Freiburg, Germany
- 4 BIOSS, Centre for Biological Signaling Studies, Freiburg, Germany
Mucosal retinoic receptor-related orphan receptor (ROR)γt-expressing innate lymphoid cells (ILC) play an important role in the defense against intestinal pathogens and in promoting epithelial homeostasis and adaptation, thereby effectively protecting the vertebrate host against intestinal inflammatory disorders. The functional activity of RORγt+ ILC is under the control of environmental cues. However, the molecular sensors for such environmental signals are largely unknown. Recently, the aryl hydrocarbon receptor (AhR) has emerged as a master regulator for the postnatal maintenance of intestinal RORγt+ ILC and intraepithelial lymphocytes. AhR is a highly conserved transcription factor whose activity is regulated by environmental and dietary small molecule ligands. Here, we review the role of AhR signaling for the maintenance of intestinal immune cells and its impact on the immunological protection against intestinal infections and debilitating chronic inflammatory disorders.
Introduction
The mucosal immune system is constantly exposed to environmental antigens such as bacteria, viruses, and nutrients. In recent years, it has become clear that the intestinal ecosystem is best viewed as a network of mutualistic interactions. In contrast to previous assumptions, the design principle of the mucosal immune system is not one that aims for strict separation between environmental antigens and immune cells by an impenetrable epithelial or mucus barrier. In contrast, specialized routes of antigen access to immune cells [e.g., microfold (M) cells, transepithelial dendrites of dendritic cells (DC)] allow for the controlled induction of immune signals at specifically designated entry ports that generate homeostatic rather than inflammatory signals (Round and Mazmanian, 2009; Cerf-Bensussan and Gaboriau-Routhiau, 2010; Hill and Artis, 2010; Hooper and Macpherson, 2010; Sanos et al., 2011). A recently identified group of innate lymphocytes now widely referred to as innate lymphoid cells (ILC) has emerged as an important player in generating homeostatic signals to preserve mutualism at mucosal surfaces (Spits and Di Santo, 2011). In contrast to other innate lymphocyte subsets namely natural killer (NK) cells, ILC co-express markers of lymphoid progenitors such as the receptor tyrosine kinase Kit (CD117) and the IL-7 receptor (CD127; Moro et al., 2010; Satoh-Takayama et al., 2010). Two ILC subsets have now been recognized, retinoic acid receptor-related orphan receptor (ROR)γt-expressing ILC and natural helper cells (also referred to as nuocytes, type 2 ILC or ILC2; Sanos et al., 2011; Spits and Di Santo, 2011). RORγt+ ILC are a group of innate lymphocytes that express and developmentally depend on RORγt (Kurebayashi et al., 2000; Sun et al., 2000; Eberl et al., 2004; Sawa et al., 2010; Vonarbourg et al., 2010). RORγt+ ILC have lymphoid tissue-inducing (LTi) function and are absolutely required for the prenatal development of secondary lymphoid organs such as lymph nodes (LN) and Peyer’s patches (Kurebayashi et al., 2000; Sun et al., 2000; Eberl et al., 2004). In addition, RORγt+ ILC are also required for postnatally developing intestinal lymphoid organs such as cryptopatches (CP) and isolated lymphoid follicles (ILF; Eberl and Littman, 2004). CP are lymphoid clusters located directly underneath the crypts of Lieberkühn containing mainly RORγt+ ILC which are surrounded by a wall of DC (Kanamori et al., 1996; Pabst et al., 2005). B cells are recruited to some CP transforming them into ILF (Hamada et al., 2002) that are important locations for the production of T cell-independent IgA (Bouskra et al., 2008; Tsuji et al., 2008). While the formation of CP is independent of the intestinal microbiota, formation of ILF requires signals from intestinal bacteria (Pabst et al., 2006; Bouskra et al., 2008).
Several subsets of RORγt+ ILC have been recognized (Satoh-Takayama et al., 2008; Luci et al., 2009; Sanos et al., 2009; Takatori et al., 2009; Cella et al., 2010). Currently, CD4+ and CD4− LTi cells (LTi4 cells, LTi0 cells) and NK cell receptor (NKR)-expressing LTi cells (NKR-LTi cells also known as NK-22 cells) are discernable (Sawa et al., 2010; Vonarbourg et al., 2010). Current evidence supports the view that NKR+RORγt+ ILC are the progeny of NKR-RORγt+ ILC (Sawa et al., 2010; Vonarbourg et al., 2010). No experimental data is available assigning a specific function to these various subsets of RORγt+ ILC.
In addition to ILC, external surfaces of the body such as skin and intestinal epithelium contain specialized T cell populations. Among these are intestinal intraepithelial lymphocytes (IEL) and dendritic epidermal T cells (DETC) found in the skin of mice. Both IEL and DETC directly interact with epithelial cells and are an important part of the mucosal host defense. In addition, both IEL and DETC have been involved in epithelial homeostasis and repair (Jameson et al., 2002; Jameson and Havran, 2007). Skin-resident DETC express an invariant γδ TCR composed of the γ3 and δ1 chains (according to the nomenclature by Garman) but its cognate antigen remains unknown (Hayday, 2009). IEL are a heterogeneous lymphocyte population of unconventional T cells with antigen-experienced phenotype. IEL are composed of γδ and αβ T cell receptor-expressing T cells. Among the αβ T cells are conventional CD8αβ heterodimer-bearing conventional cytotoxic T cells and unconventional CD8αα homodimer-expressing T cells (Cheroutre, 2004). Both DETC and IEL seed skin or intestine already before birth preceding the colonization with the microflora. In fact, later in life microbiota has an essential role in shaping the development and function of the IEL (Hayday and Tigelaar, 2003; Cheroutre et al., 2011).
In addition to being cells with LTi function, all RORγt+ ILC subsets are an important source of the cytokines IL-22 and IL-17, albeit with different efficiency (Sawa et al., 2010; Vonarbourg et al., 2010). While subsets of NKR−RORγt+ ILC produce high levels of IL-22 and IL-17, NKR+RORγt+ ILC produce less IL-22 and do not express IL-17 (Satoh-Takayama et al., 2008; Sanos et al., 2009; Vonarbourg et al., 2010; Kiss et al., 2011; Sonnenberg et al., 2011b). The extent of IL-22 production by RORγt+ ILC is under the control of signals from the commensal microbiota (Satoh-Takayama et al., 2008; Sanos et al., 2009; Reynders et al., 2011; Sawa et al., 2011). The IL-22 receptor is exclusively expressed by non-hematopoietic cells such as epithelial cells and stromal cells (Wolk et al., 2004). Although the overall epithelial gene expression program controlled by constitutive IL-22 signaling has not yet been analyzed in detail, inflammation-induced IL-22 clearly regulates expression of antimicrobial lectins of the regenerating islet-derived (Reg)3 family and of antimicrobial calcium-binding proteins such as S100A8 (calgranulin A) or S100A9 (calgranulin B; Zheng et al., 2008). Given the tissue-protective and antimicrobial functions of these genes, it has been proposed that RORγt+ ILC may contribute to epithelial adaptation during epithelial stress (Sanos et al., 2011; Sonnenberg et al., 2011a). Indeed, mice genetically deficient for IL-22 or those lacking RORγt+ ILC are more susceptible to bacterial infections (Satoh-Takayama et al., 2008; Zheng et al., 2008; Cella et al., 2009; Sonnenberg et al., 2011b) and to experimental forms of colitis (Sugimoto et al., 2008; Zenewicz et al., 2008). However, the sensors of environmental factors and the transcriptional regulators adapting gene expression of mucosal ILC in response to environmental cues are not well defined.
The aryl hydrocarbon receptor (AhR) is an excellent candidate gene because it is a transcription factor whose activity is controlled by environmental ligands. The AhR belongs to the basic helix-loop-helix/Per-Arnt-Sim (bHLH/PAS) family of proteins that play important roles in adapting multicellular organisms to the environment (e.g., hypoxia response, circadian rhythm etc.; Gu et al., 2000). AhR is a ligand-activated transcription factor located in the cytoplasm that upon ligand binding translocates to the nucleus, dimerizes with another bHLH/PAS member [AhR nuclear translocator (ARNT) or HIF-1β] and binds to xenobiotic response elements (XRE) in the promoter regions of AhR response genes (Gu et al., 2000; McIntosh et al., 2010). AhR is known to induce transcription of genes encoding xenobiotic metabolizing enzymes of the cytochrome P450 monooxidase family (e.g., Cyp1a1 and Cyp1b1; Gu et al., 2000; Kewley et al., 2004). AhR was originally described as a receptor for dioxin, an environmental toxin formed as a by-product of numerous industrial processes (Fernandez-Salguero et al., 1995). However, several other small molecular ligands for the AhR have been identified (Nguyen and Bradfield, 2008; McIntosh et al., 2010). Among them are naturally occurring AhR ligands predominantly found in plants which include flavonoids such as quercetin in apples (Ciolino et al., 1999; Ross and Kasum, 2002), resveratrol in red wine (Ciolino et al., 1998), and indole-3-carbinol (I3C), a hydrolytic product of the glucosinolate glucobrassicin contained in cruciferous plants such as broccoli and Brussel’s sprouts (Bjeldanes et al., 1991; Dixon, 2004). AhR is an evolutionary conserved protein that is present in invertebrate organisms. For example in Drosophila melanogaster, AhR signaling is required for the proper development of the antennae and leg. The AhR in invertebrates lacks the ability to interact with xenobiotics, suggesting that the recognition of environmental toxins is not the original function of AhR (Barouki et al., 2007; Stevens et al., 2009). Given the developmental role of the AhR system in invertebrates, it appears likely that AhR-mediated signals in vertebrates have significance beyond the recognition of recently emerging industrial toxins. However, experimental data linking AhR signals to important developmental functions have been largely lacking (Gu et al., 2000).
Although the role of AhR in regulating gene expression in response to low-molecular-weight chemicals acting as AhR ligands is extensively studied, its role in the immune system has only recently attracted wider attention (Esser et al., 2009; Stevens et al., 2009). Among the first observations was the finding that T and B lymphocytes of Ahr-deficient mice were slower to seed secondary lymphoid organs (Fernandez-Salguero et al., 1995). This phenotype was found in Ahr-deficient mice generated by Gonzalez and colleagues (Fernandez-Salguero et al., 1995), whereas two independently generated targeted Ahr alleles did not show any robust perturbations of immune system development (Schmidt et al., 1996; Shimizu et al., 2000). At that time, Ahr−/− mice had mixed genetic backgrounds, which may have affected the results observed in different mouse lines. Today, Ahr−/− mice on a pure C57BL/6 background are available, but the initial studies regarding the immune system phenotype were never repeated (Lahvis and Bradfield, 1998; Esser, 2009). Due to the discrepancies in the immune phenotype of the various Ahr-deficient mice, the role of the AhR in the immune system remained unclear. Recently, it was demonstrated that AhR signals were required for T helper cell fate decisions in particular for the commitment of Th17 cells (Quintana et al., 2008; Veldhoen et al., 2008). These data linked environmental toxins to the development of Th17-mediated inflammatory disorders such as experimental autoimmune encephalitis (EAE), a mouse model of multiple sclerosis. Th17 cells share a number of similarities with RORγt+ ILC, such as the expression of the transcription factor RORγt and a similar profile of cytokine production. Recently, a number of independent reports investigated the role of the AhR for development and function of RORγt+ ILC that we will review here.
AHR is Required for the Formation of Postnatally Developing Intestinal Lymphoid Organs
An essential role of AhR for the formation of postnatally developing intestinal lymphoid clusters such as CP and ILF was observed because mice genetically deficient for the Ahr gene lacked CP and ILF (Kiss et al., 2011; Lee et al., 2011). Although AhR is constitutively expressed by several cell types, such as hepatocytes, intestinal epithelial cells (IEC), subsets of T cells, and DC (Esser et al., 2009; Chmill et al., 2010), tissue-specific deletion of AhR in RORγt+ ILC was sufficient to impair the formation of lymphoid clusters in the small intestine (Kiss et al., 2011). In contrast, mice with deletion of Ahr in all CD11c+ cells or IEC showed normal development of CP and ILF ruling out an important role of AhR signaling in CD11c+ DC or IEC for the development of intestinal lymphoid organs (Kiss et al., 2011). Thus, AhR-controlled transcriptional programs in RORγt+ ILC with LTi function are required for the postnatal formation of intestinal lymphoid clusters (Figure 1).
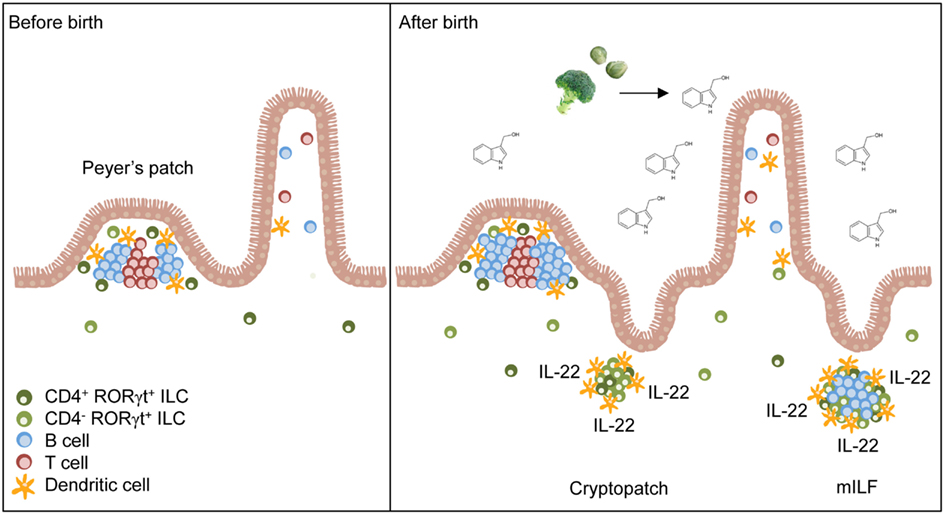
Figure 1. Natural AhR ligands control the pool size of RORγt+ ILC. Secondary lymphoid organs (e.g., Peyer’s patches) of the small intestine form before birth and their development is independent of AhR signals. After birth, the amount of dietary AhR ligands such as glucosinolate-derived indole-3-carbinol increases in the intestine due to the beginning resorptive capacity of enterocytes. AhR signaling induces the expansion of CD4− RORγt+ ILC leading to the formation of cryptopatches and ILF. Furthermore, AhR increases IL-22 production by RORγt+ ILC and consequently reinforces epithelial protection against bacteria.
Interestingly, the failure to form lymphoid organs in the absence of AhR did not extend to prenatally developing intestinal lymphoid organs such as Peyer’s patches, cecal patches, and mesenteric LN or peripheral, non-mucosal LN (Kiss et al., 2011; Lee et al., 2011). RORγt+ ILC were present in normal numbers in the LN and in the spleen of Ahr-deficient mice, indicating that AhR signaling is not needed for the lineage commitment or differentiation of RORγt+ ILC at non-mucosal sites (Kiss et al., 2011). In addition, RORγt+ ILC were normally represented in the small intestine of newborn Ahr-deficient mice (Kiss et al., 2011; Qiu et al., 2011). These results have two important implications. First, AhR is not required for the prenatal development or differentiation of intestinal RORγt+ ILC but AhR signals are likely required for their postnatal functional program or their maintenance. Second, AhR signaling required for the development of CP and ILF may be specifically induced at mucosal sites.
How does AhR signaling in RORγt+ ILC control the development of CP and ILF? The development of intestinal lymphoid clusters requires signals integrated by the lymphotoxin β receptor (LTβR) expressed by mesenchymal stroma (Futterer et al., 1998; Taylor et al., 2004). LTβR interacts with two TNF superfamily ligands expressed by RORγt+ ILC, LTα1β2, or LIGHT (TNFSF14; Ware, 2005). In the light of these data (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2011), it was conceivable that AhR controls expression of these genes. However, expression of TNF superfamily genes including Lta, Ltb, and Tnfsf14 was normal in Ahr−/−mice (Kiss et al., 2011). Thus, AhR does not control the expression of genes that confer LTi function to RORγt+ ILC.
Intestinal RORγt+ ILC can be divided into a CD4− and a CD4+ subset (Sawa et al., 2010; Vonarbourg et al., 2010). Fetal RORγt+ ILC were described to be CD4+ cells (Mebius et al., 1997) but 80–90% of the RORγt+ ILC population found in the small intestine of adult mice is CD4-negative (Sawa et al., 2010; Vonarbourg et al., 2010). While the absolute numbers of CD4+ RORγt+ ILC remained largely steady after birth, the CD4− subset dramatically expanded in numbers during the first 3 weeks of life coinciding with the development of CP and ILF (Sawa et al., 2010; Kiss et al., 2011). Interestingly in Ahr-deficient mice, the CD4− RORγt+ ILC population failed to expand (Kiss et al., 2011). Thus, AhR signaling is required for the postnatal expansion of the pool of CD4− RORγt+ ILC (Figure 1). Although CD4+ RORγt+ ILC also express AhR, they are only mildly affected by the absence of AhR (Kiss et al., 2011; Li et al., 2011). AhR expression by CD4+ RORγt+ ILC is lower compared to CD4− RORγt+ ILC which may explain that their survival or maintenance is not affected by weaker AhR signals (Kiss et al., 2011). However, another study reported that CD4+ CD127+ cells, which mainly consist of CD4+ RORγt+ ILC, were decreased in the absence of AhR (Lee et al., 2011). Future studies will need to dissect why maintenance of the CD4− RORγt+ ILC pool is so dependent on AhR signals whereas CD4+ RORγt+ ILC are only to a lesser extent. Perhaps, negative regulators of AhR signaling such as the AhR repressor, which inhibits the transcriptional activity of AhR, should be considered (Mimura et al., 1999).
Aryl hydrocarbon receptor-mediated signals could be involved in the expansion of CD4− RORγt+ ILC by controlling their proliferation and/or survival. A substantial fraction of CD4− RORγt+ ILC proliferated after birth, for which AhR signaling was a prerequisite (Sawa et al., 2010; Kiss et al., 2011). In contrast, CD4+ RORγt+ ILC had a much lower proliferative capacity and did not substantially expand. Thus, AhR signals are required for the proliferation and expansion of CD4− RORγt+ ILC. Furthermore, AhR signals may also be involved in their survival as RORγt+ ILC from adult Ahr−/− mice had decreased expression of antiapoptotic genes such as Bcl2 and Bcl2l1 (Qiu et al., 2011). Collectively, the data demonstrate that AhR signals control the pool size of intestinal RORγt+ ILC with LTi function by regulating their expansion and/or survival during the first weeks after birth. Thus, the failure of Ahr−/− mice to form CP and ILF reflects the impairment in postnatal expansion of RORγt+ ILC.
Interestingly, AhR signals were also required for the maintenance of IEL, which consist of γδ T cells and CD8αα+ αβ T cells (Li et al., 2011). Also for IEL, AhR was shown to act in a T cell intrinsic fashion (Li et al., 2011). In addition, maintenance of invariant Vγ3+ skin DETC was also dependent on AhR signals (Kadow et al., 2011; Li et al., 2011). Before birth, IEL were found to normally develop and migrate to the intestine in Ahr-deficient mice, however similar to RORγt+ ILC their postnatal maintenance was impaired.
How is AHR Regulating the Pool Size of RORγt+ ILC?
There are several models that could explain how the AhR could regulate the pool size of RORγt+ ILC in the small intestine. First, AhR could either directly or indirectly control the expression of genes required for the development or differentiation of RORγt+ ILC such as RORγt. Second, AhR could regulate RORγt+ ILC-expressed genes that affect their gut homing properties. Third, AhR could regulate the expression of genes required for the expansion and/or maintenance of RORγt+ ILC. Previous studies with T cells have shown that the AhR does not regulate the expression of RORγt (Veldhoen et al., 2008, 2009). Moreover, it is unlikely that the AhR is required to induce RORγt expression in RORγt-negative precursor cells because the specific deletion of AhR in lineage-specified RORγt+ cells was sufficient to impair their development/maintenance (Kiss et al., 2011). Although AhR is not required for the induction of RORγt expression, it may still be a factor stabilizing RORγt in RORγt+ ILC, which needs to be addressed in future lineage tracing studies. In addition, it is unlikely that AhR affects the homing properties of RORγt+ ILC because Peyer’s patches were formed normally in the absence of AhR and the numbers of RORγt+ ILC in the newborn gut was not affected. These data indicate that the initial homing of RORγt+ ILC from the fetal liver to the gut is not impaired in the absence of AhR signals. At current, it cannot be excluded that RORγt+ ILC have a second, postnatal homing wave regulated by AhR. However, in Ahr−/−mice the RORγt+ ILC were not found to accumulate in organs where precursors of RORγt+ ILC are located, such as in the bone marrow or liver (Elina A. Kiss and Andreas Diefenbach, unpublished data).
AHR Controls Expression of Genes Required for the Homeostasis of RORγt+ ILC
The available data provide strong evidence that AhR controls the expression of genes required for the homeostasis of RORγt+ ILC. Recently, AhR-controlled candidate genes directly involved in maintaining the RORγt+ ILC pool have been identified.
Kit
In the absence of AhR signals, RORγt+ ILC-expressed lower levels of the receptor tyrosine kinase Kit (Figure 2A; Kiss et al., 2011). Kit is the receptor for stem cell factor (SCF) known to be important for the maintenance of RORγt+ ILC especially at mucosal sites (Chappaz et al., 2011). Incubation of RORγt+ ILC in the presence of SCF led to their expansion supporting the view that Kit signaling may contribute to the postnatal expansion of RORγt+ ILC (Kiss et al., 2011). This is further supported by the finding that mice with a signaling-deficient Kit receptor (KitWv/Wv) had reduced numbers of mucosal RORγt+ ILC and reduced development of CP and ILF. However, this data should be interpreted with caution because in KitWv/Wv mice, Kit signaling is deficient in all cells not only in RORγt+ ILC. Future studies enabling for genetic deletion of Kit specifically in RORγt+ ILC will allow to more precisely investigate how Kit signaling controls the homeostasis of RORγt+ ILC.
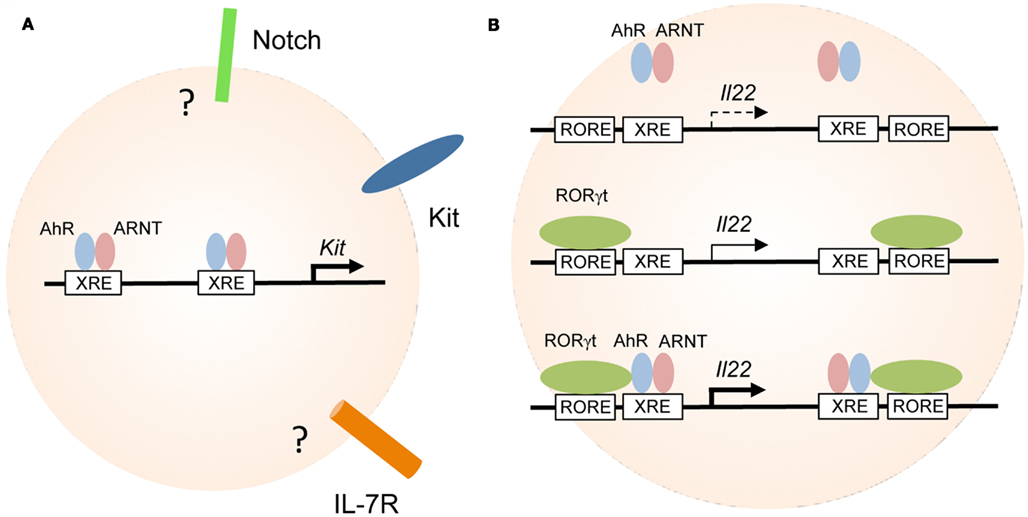
Figure 2. Aryl hydrocarbon receptor transcriptionally controls the expression of Kit and IL-22. (A) Regulation of genes involved in the maintenance of RORγt+ ILC. AhR-deficiency leads to reduced expression of Kit, IL-7R, Notch, and antiapoptotic molecules all of which have been implicated in the maintenance and/or survival of RORγt+ ILC. AhR directly binds to the two XRE elements of the Kit promoter and induces its transcription. It is not known whether the genes encoding Notch or IL-7R are direct targets of AhR in RORγt+ ILC. (B) Cooperativity between AhR and RORγt in the regulation of IL-22 expression. AhR alone does not bind to or only weakly interacts with the XRE elements in the Il22 promoter (upper panel). RORγt expression alone may induce transcription of the Il22 gene (middle panel). RORγt and AhR synergistically promote IL-22 expression (lower panel).
Several lines of evidence demonstrate that Kit is under the direct transcriptional control of the AhR. Activation of AhR by incubation of purified RORγt+ ILC in the presence of AhR ligands led to the upregulation of Kit expression in vitro (Kiss et al., 2011). Two canonical XRE elements are located in the human and mouse Kit promoters (Sun et al., 2004; Jux et al., 2011; Kadow et al., 2011; Kiss et al., 2011). Indeed, chromatin immunoprecipitation (ChIP) assays showed that AhR was bound to the XRE elements of the Kit promoter and promoter occupancy could be enhanced by treating RORγt+ ILC with AhR ligands (Figure 2A; Kiss et al., 2011). Furthermore, gene expression reporter assays revealed that binding of AhR to the XRE elements induced transcription of both human and mouse Kit and mutation of the XRE elements blocked transcription (Jux et al., 2011; Kadow et al., 2011; Kiss et al., 2011).
Requirement of AhR signals for Kit expression has also been observed for skin γδ T cells and melanocytes indicating a general role of AhR signals at mucosal surfaces for stable Kit expression (Jux et al., 2011; Kadow et al., 2011). A recent report has indicated that in addition to RORγt+ ILC also the homeostasis of intestinal intraepithelial γδ T cells requires AhR signals (Li et al., 2011). In this report, the AhR-regulated genes were not investigated but previous data demonstrated that mice with a mutant Kit receptor, closely resembling the phenotype observed in Ahr−/− mice, had a significantly reduced pool size of intraepithelial γδ T cells (Puddington et al., 1994).
IL-7 and IL-7 Receptor
IL-7 and TSLP signals play an important and partially redundant role for the development of RORγt+ ILC (Satoh-Takayama et al., 2010; Vonarbourg et al., 2010). Mice lacking IL-7 (Il7−/−) or TSLP-signaling (Tslpr−/−) had reduced albeit still substantial numbers of intestinal RORγt+ ILC. Mice deficient for the IL-7R (Il7ra−/−) which is required for both IL-7 and TSLP-signaling had virtually no RORγt+ ILC demonstrating that TSLP and IL-7 play redundant roles for the differentiation and/or survival of RORγt+ ILC (Vonarbourg et al., 2010). One report has found reduced IL-7R expression in adult (6–10 weeks old) Ahr−/− mice (Qiu et al., 2011) whereas two other reports did not observe substantial differences in IL-7R expression by RORγt+ ILC (Kiss et al., 2011; Lee et al., 2011). In the same report it was shown that IL-7 expression in the colon of Ahr−/− mice is reduced (Qiu et al., 2011). The major source of IL-7 in the intestine are epithelial cells (Shalapour et al., 2010). However, it is unlikely that IL-7 expression by epithelial cells is under the direct control of AhR because normal numbers of RORγt+ ILC were found in mice lacking AhR in epithelial cells (Kiss et al., 2011). In addition, no difference in the numbers of RORγt+ ILC could be observed after transfer of Ahr-proficient RORγt+ ILC into Ahr−/− (low IL-7 environment) or Ahr+/+ mice (Qiu et al., 2011) basically ruling out an important role of reduced IL-7 in Ahr−/−mice for the maintenance of RORγt+ ILC.
Notch
Notch genes have been identified as AhR target genes (Boverhof et al., 2006; Stevens et al., 2009; Dere et al., 2011) and the population of lineage marker (Lin)-negative Kit+ cells which includes RORγt+ ILC express Notch1, 2 and 4 (Lugering et al., 2010). A recent report provided evidence that Notch signaling in CD4+ T cells may promote IL-22 production by inducing the availability of endogenous AhR ligands (Alam et al., 2010). Furthermore, Notch2 signaling has recently been identified to be important for the generation of RORγt+ ILC from adult bone marrow precursors in vitro (Possot et al., 2011). Human NKR+ RORγt+ ILC residing in Peyer’s patches, maintenance of which is independent of AhR signaling, express high levels of Notch1 and HES, a bHLH protein induced by Notch signaling and repressing Notch signals (Lee et al., 2011). In contrast, NK cells express very low levels of Notch genes. Notch1 and Notch2 expression in the lamina propria of the colon was upregulated in an AhR-dependent manner if mice were fed with dioxin (Lee et al., 2011). Mice that lack Notch signaling in the hematopoietic compartment (Vav-Cre; RBP-Jκ mice) had a reduced fraction of NKR+ CD127+ cells, a population that contains among other innate lymphocyte subsets NKR+ RORγt+ ILC. However and in contrast to Ahr−/− mice, the number of NKR− RORγt+ ILC that are the immediate precursors of NKR+ RORγt+ ILC (Sawa et al., 2010; Vonarbourg et al., 2010) and the development of CP or ILF were normal in mice lacking Notch signaling (Lee et al., 2011). Thus, Notch signals may be required for the differentiation of NKR+ RORγt+ ILC but it remains to be determined if this requires Notch signaling in RORγt+ ILC and if Notch expression is under the direct transcriptional control of AhR.
Bcl2 and Bcl2l1
RORγt+ ILC from adult mice were shown to be more prone to apoptosis in the absence of AhR (Qiu et al., 2011). Indeed, the expression of antiapoptotic genes Bcl2 and Bcl2l1 was decreased in Ahr-deficient RORγt+ ILC. Yet, it is not clear whether the AhR directly regulates the expression of Bcl2 and Bcl2l1 or whether this is rather an indirect effect caused by deprivation of Kit, Notch or IL-7R signaling. However, the combination of increased apoptosis in adult mice and decreased proliferation during the first 2 weeks after birth may both affect the RORγt+ ILC pool size in the adult small intestine.
Dietary AHR Ligands Regulate the Homeostasis of RORγt+ ILC
Transcriptional activity of AhR is regulated by a plethora of small molecule ligands. The important question arose which AhR ligands are involved in controlling the pool size of RORγt+ ILC. Microbiota-derived ligands are unlikely to be of importance because germ-free mice have normal numbers of CP (Kanamori et al., 1996; Pabst et al., 2006; Bouskra et al., 2008). In addition, germ-free mice had normal fractions of NKp46+NK1.1− or NKp46+NK1.1int cells, populations that contain NKR+RORγt+ ILC (Lee et al., 2011). This is in line with previously published data showing that microbiota is required for the stabilization of RORγt within the population of NKR+RORγt+ ILC so that no differences in the overall population size of NKR+ cells is observed but rather in the fraction of NKR+ cells co-expressing RORγt (Vonarbourg et al., 2010).
Another significant source of AhR ligands are the plant compounds of diets. Indeed, dietary AhR ligands were observed to be important for the postnatal expansion of RORγt+ ILC and for the timely development of intestinal lymphoid follicles (Kiss et al., 2011). Mice fed with synthetic diets, lacking all plant-derived components had lower levels of Kit expression by RORγt+ ILC, impaired postnatal expansion of RORγt+ ILC and delayed formation of intestinal lymphoid follicles (Figure 1). Addition of I3C, which can be converted into the high affinity AhR ligands indolo [3,2-b]carbazole (ICZ) and 3,3-diindolylmethane (DIM) in the acidic environment of the upper gastrointestinal tract (Bjeldanes et al., 1991), to the synthetic diets could rescue Kit expression, the expansion of RORγt+ ILC and the timely formation of intestinal lymphoid clusters. However, the lack of dietary AhR ligands could only delay the formation of postnatally forming intestinal lymphoid organs indicating that at later points in time, other sources of AhR ligands can compensate for the lack of dietary AhR ligands. Intriguingly, dietary AhR ligands were also shown to regulate the maintenance of IEL (Li et al., 2011). In this study it was further shown, that the absence of IEL in Ahr−/− mice resulted in increased susceptibility to dextran sodium sulfate (DSS)-induced epithelial damage (Li et al., 2011). These studies were the first to link diets to the homeostasis of intestinal immune cells and to susceptibility to intestinal diseases.
Colonna and colleagues have contested the notion that dietary AhR ligands are required for the maintenance of NKp46+NK1.1− cells, some of them are NKR+ RORγt+ ILC (Lee et al., 2011). The experiments were performed by comparing a conventional rodent diet from one vendor with a “defined diet” from another vendor. While conventional diets are mostly grain-based diets supplemented with vitamins and some micronutrients, defined diets are composed of purified compounds. The comparison of a defined diet with a conventional diet is not appropriate because these diets differ in many aspects so that the results of such a comparison are difficult to interpret. In addition, defined diets are not per se free of AhR ligands. For example, soybean oil was used as the fat compound of the defined diet in the above mentioned study. Soybeans and soybean oil are known to contain flavonoids and isoflavonoids which are potent AhR ligands and can act as AhR agonists or antagonists (Dixon, 2004; Nguyen and Bradfield, 2008; Morimoto et al., 2009; Messina, 2010). Although the effect of flavonoids on RORγt+ ILC and their effects on the immune system have not yet been thoroughly addressed, the results obtained with diets containing potential AhR ligands should be interpreted with caution.
In addition to dietary AhR ligands, several endogenous AhR ligands have been described that may contribute to AhR signals required for the maintenance of RORγt+ ILC, IEL, and/or DETC (Denison and Nagy, 2003; Nguyen and Bradfield, 2008). Arachidonic acid metabolites, such as lipoxin 4A (Schaldach et al., 1999) and prostaglandin G2 (Seidel et al., 2001) have been shown to bind to AhR. However, the concentration needed for AhR activation is high, making them unlikely to be physiologically relevant ligands. Heme metabolites, like bilirubins are known to activate AhR signaling (Nguyen and Bradfield, 2008). However, heme is usually bound to serum albumin and not freely available for intracellular signaling. Interestingly, hydrodynamic shear stress, like blood flow in vessels, is suggested to modify low density lipoprotein (LDL) to become an AhR activator (McMillan and Bradfield, 2007). More recently, tryptophan metabolites have received attention. Specifically, UV photoproducts of tryptophan generate high affinity AhR ligands (Rannug et al., 1987). For example, 6-formylindolo[3,2-b] carbazole (FICZ) has an affinity for the AhR that is comparable to dioxin, but the activation induced by FICZ is transient as it is metabolized quickly after the induction of CYP1A1 and CYP1B1 enzymes. Synthesis of FICZ can probably also occur in vivo. The exposure of human skin to UV-B induces the upregulation of CYP1A1 and CYP1B1 enzymes, indicating that sunlight generates AhR ligands in the skin (Rannug and Fritsche, 2006; Fritsche et al., 2007). Furthermore, it has been shown that FICZ can be generated by exposing tryptophan to sunlight (Diani-Moore et al., 2006). Very recently, kynurenine, a tryptophan metabolite generated by the indoleamine-2,3 dioxygenase (IDO) pathway has gained attention. IDO is upregulated in DC in response the AhR activation, which leads to the accumulation of kynurenine. Kynurenine was shown to interact with AhR in T cells promoting the formation of regulatory T cells but not Th17 cells (Mezrich et al., 2010; Nguyen et al., 2010). There is evidence that kynurenine is also formed in the small intestine. Gut CD103+ DC were shown to express IDO and promote intestinal tolerance through instruction of regulatory T cells (Matteoli et al., 2010). Interestingly, inhibition of IDO in T cell transfer colitis or DSS-induced colitis exacerbated the disease (Matteoli et al., 2010). While the role of kynurenine and AhR signaling was not investigated, it is intriguing to propose that the induction of regulatory T cells requires AhR signals. Recently, kynurenine was shown to be produced by tumor cells through another tryptophan degrading enzyme, tryptophan-2,3 dioxygenase (TDO). Kynurenine promoted tumor growth and survival and suppressed antitumor effects by the immune system (Opitz et al., 2011). Based on the available data, kynurenine likely induced regulatory T cells in the tumor, creating a tolerogenic tumor environment. Future studies will need to address whether kynurenine or FICZ can promote the expansion/maintenance of RORγt+ ILC, DETC or IEL in vivo.
Regulation of IL-22 Expression by AHR and Immunity to Citrobacter Rodentium Infection
The role of RORγt+ ILC for Citrobacter (C.) rodentium infection has been extensively studied. C. rodentium belongs to a group of enterobacteria that cause attaching and effacing (A/E) lesions (Schauer and Falkow, 1993a,b). Among the enterobacteria causing A/E lesions are human pathogens such as enteropathogenic Escherichia (E.) coli (EPEC) and enterohemorrhagic E. coli (EHEC) strains, which makes C. rodentium a very well suited mouse model for these types of infections (Eckmann, 2006). Early evidence has demonstrated that IL-22 is required for the early, T cell-independent phase of host defense against C. rodentium infection (Zheng et al., 2008). The main source of IL-22 following C. rodentium infection are innate lymphocytes, namely RORγt+ ILC (Sonnenberg et al., 2011b). During C. rodentium infection, IL-23 production by colon-resident myeloid cells increases through a process that requires LTβR signaling in myeloid cells and membrane LTα1β2 expression by RORγt+ ILC (Ota et al., 2011; Tumanov et al., 2011). Elevated IL-23 levels lead to enhanced IL-22 production by RORγt+ ILC and, consequently, to increased expression of antimicrobial proteins of the regenerating islet-derived 3 (Reg3) protein family (Zheng et al., 2008). Interestingly, the maintenance of the organization and the increase in size of colonic patches during C. rodentium infection were dependent on IL-22 expression induced by LTβR signaling. However, IL-22 was dispensable for the development and maintenance of colonic lymphoid structures during homeostatic conditions, indicating that different signals induce formation of lymphoid structures in the colon during normal development and during inflammation (Ota et al., 2011).
Aryl hydrocarbon receptor signals are known to be essential for IL-22 production by Th17 cells (Veldhoen et al., 2008). Consistent with these findings from T cells, AhR-deficient RORγt+ ILC produced less IL-22 (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2011). The fraction of IL-22-producing cells among various subsets of RORγt+ ILC was reduced. This reflects RORγt+ ILC-intrinsic AhR signaling because a similar reduction in IL-22 production was seen in mice with deletion of the AhR in RORγt+ ILC but not in mice lacking AhR expression in CD11c+ cells or IEC (Kiss et al., 2011). Indeed, IL-23R expression by RORγt+ ILC was decreased in the absence of AhR, explaining the reduced IL-22 production by these cells. Specifically, the inflammation-induced IL-22, which requires IL-23, is likely to be affected by decreased IL-23R expression in Ahr-deficient RORγt+ ILC. However, AhR signaling is not an absolute requirement for IL-22 production as maybe the case for Th17 cells because substantial residual production of IL-22 was observed (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2011). Due to the failing postnatal expansion of RORγt+ ILC in Ahr−/− mice, the absolute numbers of IL-22-producing RORγt+ ILC in small intestine and colon were dramatically decreased and could not be enhanced after C. rodentium infection (Kiss et al., 2011). As a consequence, expression of epithelium-protective Reg3 genes was critically reduced in C. rodentium-infected Ahr-deficient mice, which were highly susceptible to C. rodentium infection (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2011).
Interestingly, both the promoter and intron 1 of the Il22 gene contain one ROR response element (RORE) and one XRE (i.e., AhR response element) each. Using a T cell line (EL4) stably transfected with an epitope-tagged version of RORγt, RORγt occupancy at both RORE was demonstrated (Qiu et al., 2011). A similar approach revealed that constitutively active AhR does not bind to the two XRE elements of the AhR gene in the absence of RORγt. However, co-expression of RORγt in EL4 cells facilitated binding of the AhR to both XRE. Indeed RORγt and AhR could be co-immunoprecipitated from EL4 cells ectopically expressing these two transcription factors demonstrating that RORγt and AhR physically interact (Qiu et al., 2011). While AhR expression alone only marginally increased IL-22 expression by EL4 cells, RORγt alone induced IL-22 transcription and strong synergism was observed in cell lines transduced with both transcription factors. Collectively, the data indicate that there is cooperativity between RORγt and AhR signaling for the induction of IL-22 expression (Figure 2B; Qiu et al., 2011). However, the experiments were performed with a T cell line and it remains unknown whether the same effects could be observed with RORγt+ ILC. It has been shown that the retroviral transfection of naïve T cells with AhR and RORγt was not sufficient to induce IL-22 production, indicating that at least in T cells additional components are involved (Veldhoen et al., 2009). More research into the transcriptional circuitry underlying IL-22 production by RORγt+ ILC is required.
AHR-Deficient Mice Develop Colitis-Promoting RORγt+ ILC-Less B Cell Clusters
Although AhR-deficient mice have reduced numbers of RORγt+ ILC in small intestine and colon, we observed the tendency of Ahr−/− mice to develop RORγt+ ILC-less B cell follicles in the colon (Kiss et al., 2011). Structurally similar follicles were observed in the colon of mice lacking all RORγt+ ILC (Lochner et al., 2011). Formation of such B cell follicles was instructed by B cells and was enhanced when the intestinal epithelium was disrupted by DSS-induced damage. Interestingly, RORγt+ ILC-less B cell clusters in both Rorc(γt)−/− and Ahr−/− mice contained inappropriately activated B cells that promoted intestinal inflammation (Kiss et al., 2011; Lochner et al., 2011). Therefore, the increased inflammation following C. rodentium infection in Ahr−/− mice may reflect the lack of protective IL-22-producing RORγt+ ILC and the presence of inflammation-promoting RORγt+ ILC-less B cell clusters. Consistent with our observation that Ahr−/− mice develop inflammation-promoting RORγt+ ILC-less B cell follicles in the colon, ca. 50% of aging Ahr−/− mice (>9 months of age) were observed to develop anal prolapse, which was associated with colonic hyperplasia and inflammation (Fernandez-Salguero et al., 1997). Colonic inflammation and spontaneous anal prolapse in Ahr-deficient mice has been reported only by Gonzalez and colleagues (Fernandez-Salguero et al., 1997) but not by two other groups that generated Ahr−/−mice (Schmidt et al., 1996; Shimizu et al., 2000). Likely the differences in the susceptibility to spontaneous intestinal inflammation are due to the distinct housing environment of the mice rather than variabilities in the genetic background as we have observed spontaneous anal prolapse in Ahr−/− mice originally generated by Bradfield and colleagues (Schmidt et al., 1996). Interestingly, Ahr−/− mice presenting with anal prolapse were found to be infected with Helicobacter hepaticus which was not observed in Ahr-proficient mice suggesting that RORγt+ ILC and/or intraepithelial T cells may be involved in immunity to H. hepaticus infection (Fernandez-Salguero et al., 1997; Buonocore et al., 2010).
Interestingly, Ahr−/−mice or mice fed with diets devoid of dietary AhR ligands were highly susceptible to DSS-induced colitis (Li et al., 2011). In contrast, mice fed with an AhR ligand-free diet supplemented with I3C were clinically much improved. This observation was mechanistically correlated with the reduced homeostasis of intraepithelial lymphocytes in Ahr−/− mice, as reconstitution of Ahr-deficient mice with control IEL could decrease the severity of the DSS-induced colitis (Li et al., 2011). However under these experimental conditions, maintenance of both IEL (Li et al., 2011) and RORγt+ ILC (Kiss et al., 2011) were impaired and a protective role of IL-22-producing RORγt+ ILC in the context of DSS colitis has been described before (Sugimoto et al., 2008; Zenewicz et al., 2008). Future experiments will need to dissect the specific roles of AhR in IEL and RORγt+ ILC and may require the development of more discriminating genetic tools because both the Rorc(γt)-Cre and Rag1-Cre lines used in these previous studies delete the Ahr gene in most IEL and in RORγt+ ILC. This is because during their development both IEL and RORγt+ ILC transiently express RORγt (Eberl and Littman, 2004) or Rag proteins (Yang et al., 2011), respectively, which will lead to the generation of an Ahr loss allele.
Importantly, there is evidence that AhR may also play a role in human intestinal inflammation. A recent study shows that in patients with Crohn’s disease the expression of AhR is decreased compared to healthy controls (Monteleone et al., 2011). This decrease in AhR expression was prominent in lymphoid cells isolated from the intestine. AhR signaling was shown to induce IL-22 expression and downregulate the expression of IFN-γ and T-bet in CD3 and CD28-stimulated cells isolated from the intestine of Crohn’s disease patients or from healthy controls. Furthermore, low intake of fruits and vegetables in children correlates with Crohn’s disease risk indicating that diets containing high concentrations of AhR ligands have an effect on intestinal inflammation also in humans (D’Souza et al., 2008). It will be interesting to study, whether the beneficial effects of a vegetable-rich diet in humans is also partially transmitted through AhR signaling and whether RORγt+ ILC and/or IEL are involved in the protection of epithelial cells preventing the onset of chronic inflammatory disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank G. Häcker for support and the members of the Diefenbach laboratory for valuable discussions. Work in the Diefenbach laboratory is supported by the Deutsche Forschungsgemeinschaft (SGBM, GRK1104, and SFB620/A14 to Andreas Diefenbach and Elina A. Kiss), the Bundesministerium für Bildung und Forschung, Centrum für Chronische Immundefizienz (to Andreas Diefenbach).
References
Alam, M. S., Maekawa, Y., Kitamura, A., Tanigaki, K., Yoshimoto, T., Kishihara, K., and Yasutomo, K. (2010). Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U.S.A. 107, 5943–5948.
Barouki, R., Coumoul, X., and Fernandez-Salguero, P. M. (2007). The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett. 581, 3608–3615.
Bjeldanes, L. F., Kim, J. Y., Grose, K. R., Bartholomew, J. C., and Bradfield, C. A. (1991). Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc. Natl. Acad. Sci. U.S.A. 88, 9543–9547.
Bouskra, D., Brezillon, C., Berard, M., Werts, C., Varona, R., Boneca, I. G., and Eberl, G. (2008). Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456, 507–510.
Boverhof, D. R., Burgoon, L. D., Tashiro, C., Sharratt, B., Chittim, B., Harkema, J. R., Mendrick, D. L., and Zacharewski, T. R. (2006). Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in Sprague Dawley rats and C57BL/6 mice. Toxicol. Sci. 94, 398–416.
Buonocore, S., Ahern, P. P., Uhlig, H. H., Ivanov, I. I., Littman, D. R., Maloy, K. J., and Powrie, F. (2010). Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375.
Cella, M., Fuchs, A., Vermi, W., Facchetti, F., Otero, K., Lennerz, J. K., Doherty, J. M., Mills, J. C., and Colonna, M. (2009). A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457, 722–725.
Cella, M., Otero, K., and Colonna, M. (2010). Expansion of human NK-22 cells with IL-7, IL-2, and IL-1β reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. U.S.A. 107, 10961–10966.
Cerf-Bensussan, N., and Gaboriau-Routhiau, V. (2010). The immune system and the gut microbiota: friends or foes? Nat. Rev. Immunol. 10, 735–744.
Chappaz, S., Gartner, C., Rodewald, H. R., and Finke, D. (2011). Kit ligand and Il7 differentially regulate Peyer’s patch and lymph node development. J. Immunol. 185, 3514–3519.
Cheroutre, H. (2004). Starting at the beginning: new perspectives on the biology of mucosal T cells. Annu. Rev. Immunol. 22, 217–246.
Cheroutre, H., Lambolez, F., and Mucida, D. (2011). The light and dark sides of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 11, 445–456.
Chmill, S., Kadow, S., Winter, M., Weighardt, H., and Esser, C. (2010). 2,3,7,8-Tetrachlorodibenzo-p-dioxin impairs stable establishment of oral tolerance in mice. Toxicol. Sci. 118, 98–107.
Ciolino, H. P., Daschner, P. J., and Yeh, G. C. (1998). Resveratrol inhibits transcription of CYP1A1 in vitro by preventing activation of the aryl hydrocarbon receptor. Cancer Res. 58, 5707–5712.
Ciolino, H. P., Daschner, P. J., and Yeh, G. C. (1999). Dietary flavonols quercetin and kaempferol are ligands of the aryl hydrocarbon receptor that affect CYP1A1 transcription differentially. Biochem. J. 340 (Pt 3), 715–722.
Denison, M. S., and Nagy, S. R. (2003). Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43, 309–334.
Dere, E., Lo, R., Celius, T., Matthews, J., and Zacharewski, T. R. (2011). Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics 12, 365. doi:10.1186/1471-2164-12-365
Diani-Moore, S., Labitzke, E., Brown, R., Garvin, A., Wong, L., and Rifkind, A. B. (2006). Sunlight generates multiple tryptophan photoproducts eliciting high efficacy CYP1A induction in chick hepatocytes and in vivo. Toxicol. Sci. 90, 96–110.
D’Souza, S., Levy, E., Mack, D., Israel, D., Lambrette, P., Ghadirian, P., Deslandres, C., Morgan, K., Seidman, E. G., and Amre, D. K. (2008). Dietary patterns and risk for Crohn’s disease in children. Inflamm. Bowel Dis. 14, 367–373.
Eberl, G., and Littman, D. R. (2004). Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORγt+ cells. Science 305, 248–251.
Eberl, G., Marmon, S., Sunshine, M. J., Rennert, P. D., Choi, Y., and Littman, D. R. (2004). An essential function for the nuclear receptor RORγt in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 5, 64–73.
Eckmann, L. (2006). Animal models of inflammatory bowel disease: lessons from enteric infections. Ann. N. Y. Acad. Sci. 1072, 28–38.
Esser, C. (2009). The immune phenotype of AhR null mouse mutants: not a simple mirror of xenobiotic receptor over-activation. Biochem. Pharmacol. 77, 597–607.
Esser, C., Rannug, A., and Stockinger, B. (2009). The aryl hydrocarbon receptor in immunity. Trends Immunol. 30, 447–454.
Fernandez-Salguero, P., Pineau, T., Hilbert, D. M., McPhail, T., Lee, S. S., Kimura, S., Nebert, D. W., Rudikoff, S., Ward, J. M., and Gonzalez, F. J. (1995). Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726.
Fernandez-Salguero, P. M., Ward, J. M., Sundberg, J. P., and Gonzalez, F. J. (1997). Lesions of aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 34, 605–614.
Fritsche, E., Schafer, C., Calles, C., Bernsmann, T., Bernshausen, T., Wurm, M., Hubenthal, U., Cline, J. E., Hajimiragha, H., Schroeder, P., Klotz, L. O., Rannug, A., Furst, P., Hanenberg, H., Abel, J., and Krutmann, J. (2007). Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc. Natl. Acad. Sci. U.S.A. 104, 8851–8856.
Futterer, A., Mink, K., Luz, A., Kosco-Vilbois, M. H., and Pfeffer, K. (1998). The lymphotoxin β receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity 9, 59–70.
Gu, Y. Z., Hogenesch, J. B., and Bradfield, C. A. (2000). The PAS superfamily: sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 40, 519–561.
Hamada, H., Hiroi, T., Nishiyama, Y., Takahashi, H., Masunaga, Y., Hachimura, S., Kaminogawa, S., Takahashi-Iwanaga, H., Iwanaga, T., Kiyono, H., Yamamoto, H., and Ishikawa, H. (2002). Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J. Immunol. 168, 57–64.
Hayday, A., and Tigelaar, R. (2003). Immunoregulation in the tissues by γδ T cells. Nat. Rev. Immunol. 3, 233–242.
Hayday, A. C. (2009). γδ T cells and the lymphoid stress-surveillance response. Immunity 31, 184–196.
Hill, D. A., and Artis, D. (2010). Intestinal bacteria and the regulation of immune cell homeostasis. Annu. Rev. Immunol. 28, 623–667.
Hooper, L. V., and Macpherson, A. J. (2010). Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 10, 159–169.
Jameson, J., and Havran, W. L. (2007). Skin γδ T-cell functions in homeostasis and wound healing. Immunol. Rev. 215, 114–122.
Jameson, J., Ugarte, K., Chen, N., Yachi, P., Fuchs, E., Boismenu, R., and Havran, W. L. (2002). A role for skin γδ T cells in wound repair. Science 296, 747–749.
Jux, B., Kadow, S., Luecke, S., Rannug, A., Krutmann, J., and Esser, C. (2011). The aryl hydrocarbon receptor mediates UVB radiation-induced skin tanning. J. Invest. Dermatol. 131, 203–210.
Kadow, S., Jux, B., Zahner, S. P., Wingerath, B., Chmill, S., Clausen, B. E., Hengstler, J., and Esser, C. (2011). Aryl hydrocarbon receptor is critical for homeostasis of invariant γδ T cells in the murine epidermis. J. Immunol. 187, 3104–3110.
Kanamori, Y., Ishimaru, K., Nanno, M., Maki, K., Ikuta, K., Nariuchi, H., and Ishikawa, H. (1996). Identification of novel lymphoid tissues in murine intestinal mucosa where clusters of c-kit+ IL-7R+ Thy1+ lympho-hemopoietic progenitors develop. J. Exp. Med. 184, 1449–1459.
Kewley, R. J., Whitelaw, M. L., and Chapman-Smith, A. (2004). The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int. J. Biochem. Cell Biol. 36, 189–204.
Kiss, E. A., Vonarbourg, C., Kopfmann, S., Hobeika, E., Finke, D., Esser, C., and Diefenbach, A. (2011). Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565.
Kurebayashi, S., Ueda, E., Sakaue, M., Patel, D. D., Medvedev, A., Zhang, F., and Jetten, A. M. (2000). Retinoid-related orphan receptor gamma (RORγ) is essential for lymphoid organogenesis and controls apoptosis during thymopoiesis. Proc. Natl. Acad. Sci. U.S.A. 97, 10132–10137.
Lahvis, G. P., and Bradfield, C. A. (1998). Ahr null alleles: distinctive or different? Biochem. Pharmacol. 56, 781–787.
Lee, J. S., Cella, M., McDonald, K. G., Garlanda, C., Kennedy, G. D., Nukaya, M., Mantovani, A., Kopan, R., Bradfield, C. A., Newberry, R. D., and Colonna, M. (2011). AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 13, 144–151.
Li, Y., Innocentin, S., Withers, D. R., Roberts, N. A., Gallagher, A. R., Grigorieva, E. F., Wilhelm, C., and Veldhoen, M. (2011). Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147, 629–640.
Lochner, M., Ohnmacht, C., Presley, L., Bruhns, P., Si-Tahar, M., Sawa, S., and Eberl, G. (2011). Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORγt and LTi cells. J. Exp. Med. 208, 125–134.
Luci, C., Reynders, A., Ivanov, I. I., Cognet, C., Chiche, L., Chasson, L., Hardwigsen, J., Anguiano, E., Banchereau, J., Chaussabel, D., Dalod, M., Littman, D. R., Vivier, E., and Tomasello, E. (2009). Influence of the transcription factor RORγt on the development of NKp46+ cell populations in gut and skin. Nat. Immunol. 10, 75–82.
Lugering, A., Ross, M., Sieker, M., Heidemann, J., Williams, I. R., Domschke, W., and Kucharzik, T. (2010). CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clin. Exp. Immunol. 160, 440–449.
Matteoli, G., Mazzini, E., Iliev, I. D., Mileti, E., Fallarino, F., Puccetti, P., Chieppa, M., and Rescigno, M. (2010). Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut 59, 595–604.
McIntosh, B. E., Hogenesch, J. B., and Bradfield, C. A. (2010). Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu. Rev. Physiol. 72, 625–645.
McMillan, B. J., and Bradfield, C. A. (2007). The aryl hydrocarbon receptor is activated by modified low-density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 104, 1412–1417.
Mebius, R. E., Rennert, P., and Weissman, I. L. (1997). Developing lymph nodes collect CD4+CD3− LTβ+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 7, 493–504.
Messina, M. (2010). A brief historical overview of the past two decades of soy and isoflavone research. J. Nutr. 140, 1350S–1354S.
Mezrich, J. D., Fechner, J. H., Zhang, X., Johnson, B. P., Burlingham, W. J., and Bradfield, C. A. (2010). An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198.
Mimura, J., Ema, M., Sogawa, K., and Fujii-Kuriyama, Y. (1999). Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 13, 20–25.
Monteleone, I., Rizzo, A., Sarra, M., Sica, G., Sileri, P., Biancone, L., Macdonald, T. T., Pallone, F., and Monteleone, G. (2011). Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 141, 237–248, 248 e231.
Morimoto, M., Watanabe, T., Yamori, M., Takebe, M., and Wakatsuki, Y. (2009). Isoflavones regulate innate immunity and inhibit experimental colitis. J. Gastroenterol. Hepatol. 24, 1123–1129.
Moro, K., Yamada, T., Tanabe, M., Takeuchi, T., Ikawa, T., Kawamoto, H., Furusawa, J., Ohtani, M., Fujii, H., and Koyasu, S. (2010). Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit+ Sca-1+ lymphoid cells. Nature 463, 540–544.
Nguyen, L. P., and Bradfield, C. A. (2008). The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 21, 102–116.
Nguyen, N. T., Kimura, A., Nakahama, T., Chinen, I., Masuda, K., Nohara, K., Fujii-Kuriyama, Y., and Kishimoto, T. (2010). Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 19961–19966.
Opitz, C. A., Litzenburger, U. M., Sahm, F., Ott, M., Tritschler, I., Trump, S., Schumacher, T., Jestaedt, L., Schrenk, D., Weller, M., Jugold, M., Guillemin, G. J., Miller, C. L., Lutz, C., Radlwimmer, B., Lehmann, I., Von Deimling, A., Wick, W., and Platten, M. (2011). An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203.
Ota, N., Wong, K., Valdez, P. A., Zheng, Y., Crellin, N. K., Diehl, L., and Ouyang, W. (2011). IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat. Immunol. 12, 941–948.
Pabst, O., Herbrand, H., Friedrichsen, M., Velaga, S., Dorsch, M., Berhardt, G., Worbs, T., Macpherson, A. J., and Forster, R. (2006). Adaptation of solitary intestinal lymphoid tissue in response to microbiota and chemokine receptor CCR7 signaling. J. Immunol. 177, 6824–6832.
Pabst, O., Herbrand, H., Worbs, T., Friedrichsen, M., Yan, S., Hoffmann, M. W., Korner, H., Bernhardt, G., Pabst, R., and Forster, R. (2005). Cryptopatches and isolated lymphoid follicles: dynamic lymphoid tissues dispensable for the generation of intraepithelial lymphocytes. Eur. J. Immunol. 35, 98–107.
Possot, C., Schmutz, S., Chea, S., Boucontet, L., Louise, A., Cumano, A., and Golub, R. (2011). Notch signaling is necessary for adult, but not fetal, development of RORγt+ innate lymphoid cells. Nat. Immunol. 12, 949–958.
Puddington, L., Olson, S., and Lefrancois, L. (1994). Interactions between stem cell factor and c-Kit are required for intestinal immune system homeostasis. Immunity 1, 733–739.
Qiu, J., Heller, J. J., Guo, X., Chen, Z. M., Fish, K., Fu, Y. X., and Zhou, L. (2011). The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36, 92–104.
Quintana, F. J., Basso, A. S., Iglesias, A. H., Korn, T., Farez, M. F., Bettelli, E., Caccamo, M., Oukka, M., and Weiner, H. L. (2008). Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71.
Rannug, A., and Fritsche, E. (2006). The aryl hydrocarbon receptor and light. Biol. Chem. 387, 1149–1157.
Rannug, A., Rannug, U., Rosenkranz, H. S., Winqvist, L., Westerholm, R., Agurell, E., and Grafstrom, A. K. (1987). Certain photooxidized derivatives of tryptophan bind with very high affinity to the Ah receptor and are likely to be endogenous signal substances. J. Biol. Chem. 262, 15422–15427.
Reynders, A., Yessaad, N., Vu Manh, T. P., Dalod, M., Fenis, A., Aubry, C., Nikitas, G., Escaliere, B., Renauld, J. C., Dussurget, O., Cossart, P., Lecuit, M., Vivier, E., and Tomasello, E. (2011). Identity, regulation and in vivo function of gut NKp46+RORγt+ and NKp46+RORγt- lymphoid cells. EMBO J. 30, 2934–2947.
Ross, J. A., and Kasum, C. M. (2002). Dietary flavonoids: bioavailability, metabolic effects, and safety. Annu. Rev. Nutr. 22, 19–34.
Round, J. L., and Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323.
Sanos, S. L., Bui, V. L., Mortha, A., Oberle, K., Heners, C., Johner, C., and Diefenbach, A. (2009). RORγt and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 10, 83–91.
Sanos, S. L., Vonarbourg, C., Mortha, A., and Diefenbach, A. (2011). Control of epithelial cell function by interleukin-22-producing RORγt+ innate lymphoid cells. Immunology 132, 453–465.
Satoh-Takayama, N., Lesjean-Pottier, S., Vieira, P., Sawa, S., Eberl, G., Vosshenrich, C. A., and Di Santo, J. P. (2010). IL-7 and IL-15 independently program the differentiation of intestinal CD3−NKp46+ cell subsets from Id2-dependent precursors. J. Exp. Med. 207, 273–280.
Satoh-Takayama, N., Vosshenrich, C. A., Lesjean-Pottier, S., Sawa, S., Lochner, M., Rattis, F., Mention, J. J., Thiam, K., Cerf-Bensussan, N., Mandelboim, O., Eberl, G., and Di Santo, J. P. (2008). Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29, 958–970.
Sawa, S., Cherrier, M., Lochner, M., Satoh-Takayama, N., Fehling, H. J., Langa, F., Di Santo, J. P., and Eberl, G. (2010). Lineage relationship analysis of RORγt+ innate lymphoid cells. Science 330, 665–669.
Sawa, S., Lochner, M., Satoh-Takayama, N., Dulauroy, S., Berard, M., Kleinschek, M., Cua, D., Di Santo, J. P., and Eberl, G. (2011). RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 12, 320–326.
Schaldach, C. M., Riby, J., and Bjeldanes, L. F. (1999). Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry 38, 7594–7600.
Schauer, D. B., and Falkow, S. (1993a). Attaching and effacing locus of a Citrobacter freundii biotype that causes transmissible murine colonic hyperplasia. Infect. Immun. 61, 2486–2492.
Schauer, D. B., and Falkow, S. (1993b). The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect. Immun. 61, 4654–4661.
Schmidt, J. V., Su, G. H., Reddy, J. K., Simon, M. C., and Bradfield, C. A. (1996). Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. U.S.A. 93, 6731–6736.
Seidel, S. D., Winters, G. M., Rogers, W. J., Ziccardi, M. H., Li, V., Keser, B., and Denison, M. S. (2001). Activation of the Ah receptor signaling pathway by prostaglandins. J. Biochem. Mol. Toxicol. 15, 187–196.
Shalapour, S., Deiser, K., Sercan, O., Tuckermann, J., Minnich, K., Willimsky, G., Blankenstein, T., Hammerling, G. J., Arnold, B., and Schuler, T. (2010). Commensal microflora and interferon-γ promote steady-state interleukin-7 production in vivo. Eur. J. Immunol. 40, 2391–2400.
Shimizu, Y., Nakatsuru, Y., Ichinose, M., Takahashi, Y., Kume, H., Mimura, J., Fujii-Kuriyama, Y., and Ishikawa, T. (2000). Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 779–782.
Sonnenberg, G. F., Fouser, L. A., and Artis, D. (2011a). Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 12, 383–390.
Sonnenberg, G. F., Monticelli, L. A., Elloso, M. M., Fouser, L. A., and Artis, D. (2011b). CD4+ lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34, 122–134.
Spits, H., and Di Santo, J. P. (2011). The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 12, 21–27.
Stevens, E. A., Mezrich, J. D., and Bradfield, C. A. (2009). The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology 127, 299–311.
Sugimoto, K., Ogawa, A., Mizoguchi, E., Shimomura, Y., Andoh, A., Bhan, A. K., Blumberg, R. S., Xavier, R. J., and Mizoguchi, A. (2008). IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118, 534–544.
Sun, Y. V., Boverhof, D. R., Burgoon, L. D., Fielden, M. R., and Zacharewski, T. R. (2004). Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res. 32, 4512–4523.
Sun, Z., Unutmaz, D., Zou, Y. R., Sunshine, M. J., Pierani, A., Brenner-Morton, S., Mebius, R. E., and Littman, D. R. (2000). Requirement for RORγ in thymocyte survival and lymphoid organ development. Science 288, 2369–2373.
Takatori, H., Kanno, Y., Watford, W. T., Tato, C. M., Weiss, G., Ivanov, I. I., Littman, D. R., and O’Shea, J. J. (2009). Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206, 35–41.
Taylor, R. T., Lugering, A., Newell, K. A., and Williams, I. R. (2004). Intestinal cryptopatch formation in mice requires lymphotoxin α and the lymphotoxin β receptor. J. Immunol. 173, 7183–7189.
Tsuji, M., Suzuki, K., Kitamura, H., Maruya, M., Kinoshita, K., Ivanov, I. I., Itoh, K., Littman, D. R., and Fagarasan, S. (2008). Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29, 261–271.
Tumanov, A. V., Koroleva, E. P., Guo, X., Wang, Y., Kruglov, A., Nedospasov, S., and Fu, Y. X. (2011). Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe 10, 44–53.
Veldhoen, M., Hirota, K., Christensen, J., O’Garra, A., and Stockinger, B. (2009). Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 206, 43–49.
Veldhoen, M., Hirota, K., Westendorf, A. M., Buer, J., Dumoutier, L., Renauld, J. C., and Stockinger, B. (2008). The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109.
Vonarbourg, C., Mortha, A., Bui, V. L., Hernandez, P. P., Kiss, E. A., Hoyler, T., Flach, M., Bengsch, B., Thimme, R., Holscher, C., Honig, M., Pannicke, U., Schwarz, K., Ware, C. F., Finke, D., and Diefenbach, A. (2010). Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt+ innate lymphocytes. Immunity 33, 736–751.
Ware, C. F. (2005). Network communications: lymphotoxins, LIGHT, and TNF. Annu. Rev. Immunol. 23, 787–819.
Wolk, K., Kunz, S., Witte, E., Friedrich, M., Asadullah, K., and Sabat, R. (2004). IL-22 increases the innate immunity of tissues. Immunity 21, 241–254.
Yang, Q., Saenz, S. A., Zlotoff, D. A., Artis, D., and Bhandoola, A. (2011). Cutting edge: natural helper cells derive from lymphoid progenitors. J. Immunol. 187, 5505–5509.
Zenewicz, L. A., Yancopoulos, G. D., Valenzuela, D. M., Murphy, A. J., Stevens, S., and Flavell, R. A. (2008). Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29, 947–957.
Keywords: innate lymphoid cell, lymphoid tissue inducer cells, aryl hydrocarbon receptor, glucosinolates, RORγt, cryptopatch, isolated lymphoid follicles, interleukin 22
Citation: Kiss EA and Diefenbach A (2012) Role of the aryl hydrocarbon receptor in controlling maintenance and functional programs of RORγt+ innate lymphoid cells and intraepithelial lymphocytes. Front. Immun. 3:124. doi: 10.3389/fimmu.2012.00124
Received: 11 January 2012; Paper pending published: 08 February 2012;
Accepted: 02 May 2012; Published online: 31 May 2012.
Edited by:
Brigitta Stockinger, MRC National Institute for Medical Research, UKCopyright: © 2012 Kiss and Diefenbach. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Andreas Diefenbach, Institute of Medical Microbiology and Hygiene, University of Freiburg, Hermann-Herder-Strasse 11, D-79104 Freiburg, Germany. e-mail:YW5kcmVhcy5kaWVmZW5iYWNoQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==