 Afonso R. M. Almeida1†
Afonso R. M. Almeida1† Inês F. Amado2,3,4† Joseph Reynolds5
Inês F. Amado2,3,4† Joseph Reynolds5 Julien Berges2,3,6 Grant Lythe5
Julien Berges2,3,6 Grant Lythe5 Carmen Molina-París5
Carmen Molina-París5 Antonio A. Freitas2,3*
Antonio A. Freitas2,3*- 1 Immunobiology Unit, Institute for Molecular Medicine, Lisboa, Portugal
- 2 Institut Pasteur, Département d’Immunologie, Unité de Biologie des Populations Lymphocytaires, Paris, France
- 3 CNRS, URA1961, Paris, France
- 4 GABBA, ICBAS, Universidade do Porto, Porto, Portugal
- 5 Department of Applied Mathematics, School of Mathematics, University of Leeds, Leeds, UK
- 6 Université Pierre et Marie Curie, Cellule Pasteur UPMC, Paris, France
Homeostasis of lymphocyte numbers is believed to be due to competition between cellular populations for a common niche of restricted size, defined by the combination of interactions and trophic factors required for cell survival. Here we propose a new mechanism: homeostasis of lymphocyte numbers could also be achieved by the ability of lymphocytes to perceive the density of their own populations. Such a mechanism would be reminiscent of the primordial quorum-sensing systems used by bacteria, in which some bacteria sense the accumulation of bacterial metabolites secreted by other elements of the population, allowing them to “count” the number of cells present and adapt their growth accordingly. We propose that homeostasis of CD4+ T cell numbers may occur via a quorum-sensing-like mechanism, where IL-2 is produced by activated CD4+ T cells and sensed by a population of CD4+ Treg cells that expresses the high-affinity IL-2Rα-chain and can regulate the number of activated IL-2-producing CD4+ T cells and the total CD4+ T cell population. In other words, CD4+ T cell populations can restrain their growth by monitoring the number of activated cells, thus preventing uncontrolled lymphocyte proliferation during immune responses. We hypothesize that malfunction of this quorum-sensing mechanism may lead to uncontrolled T cell activation and autoimmunity. Finally, we present a mathematical model that describes the key role of IL-2 and quorum-sensing mechanisms in CD4+ T cell homeostasis during an immune response.
Introduction to CD4+ T Cell Homeostasis
As in other organs and tissues of the body, the number of cells of the immune system is typically maintained throughout the adult life of an individual. This type of control is akin to the mechanisms that act to maintain nutrient levels or temperature, which were first identified by Bernard (1865) and later termed homeostasis (Cannon, 1932). T cell numbers are essentially maintained at a stable level throughout adult life, despite their daily production and export from the thymus and their peripheral division. The contribution of the thymus to the establishment and maintenance of T cell numbers in peripheral pools has been quantified (Metcalf, 1965; Miller, 1965; Leuchars et al., 1978; Berzins et al., 1998; Almeida et al., 2001). The degree of thymus restoration is determined by the availability of competent precursors, but the number of peripheral T cells is largely independent of the number of thymus precursor cells, for example, mice with reduced thymus production showed replete peripheral compartments (Almeida et al., 2001). These experimental findings have indicated that T cell numbers are controlled by peripheral mechanisms, and lead to the suggestion that competition is an important factor in maintaining T cell homeostasis. Indeed, in normal mice with full thymus production, if the generation of new T cells exceeds the minimal requirements to fill the peripheral pools and their overall peripheral number is kept constant, it follows that each newly generated cell must compete for survival with other newly produced or resident cells (Freitas et al., 1996; Freitas and Rocha, 2000). Therefore, lymphocyte survival in peripheral pools is not a passive phenomenon, but rather a continuous active process driven by highly selective environmental cues (Freitas et al., 1996; Freitas and Rocha, 2000). However, all T cells are not equal; the peripheral T cell pool is composed of a diverse set of subpopulations that play different essential roles in establishing immune responses and immune-competence. Thus, the mechanisms responsible for guiding the survival and homeostasis of peripheral T cell numbers must include a qualitative dimension, allowing for the coexistence of different T cell populations (Almeida et al., 2005). How is this achieved?
The peripheral αβ TCR+ T cell pool can be divided into CD4+ “helper” and CD8+ “cytotoxic” T cells, and each of these two major subpopulations consists of naïve (not having encountered antigen) and activated/memory (antigen-experienced) pools. These populations represent the potential to react to newly encountered antigens, and to provide fast and efficient responses to recurrent antigens, respectively. For naïve T cells, the trophic survival signals are contingent on TCR-mediated signals upon interactions with MHC Class-I or MHC Class-II molecules, and on γc-dependent cytokines, namely IL-7 (Surh and Sprent, 2008). The roles of TCR signals and TCR-MHC interactions were demonstrated by transfer of T cells to MHC-devoid hosts (Takeda et al., 1996; Brocker, 1997; Kirberg et al., 1997; Tanchot et al., 1997) and by ablation of TCRs (Labrecque et al., 2001); both approaches resulted in the loss of T cells. IL-7-dependency was shown upon either transferring T cells into IL-7-deficient animals (Schluns et al., 2000; Tan et al., 2001) or treatment of host animals with anti-IL-7 antibodies (Vivien et al., 2001); again, both approaches resulted in T cell death. IL-7 also modulates MHC Class-II expression in dendritic cells, establishing a relationship between cytokines and TCR-mediated signals in naïve CD4+ T cell homeostasis (Guimond et al., 2009). IL-7 is present in the secondary lymph nodes (LN), where it is produced by fibroblastic reticular cells (FRCs; Link et al., 2007). Expression of the CD62L and CCR7 homing receptors allow T cells to follow gradients of CCL19 and CCL21 chemokines made by the LN stromal cells, and thus access IL-7. Interestingly, the transcription factor Foxo1 regulates expressions of IL-7R, CD62L, and CCR7 in naïve T cells (Freitas and Rocha, 2009; Kerdiles et al., 2009; Ouyang et al., 2009), establishing a close link between lymphocyte migration and survival. Migration ensures the distribution of lymphocytes between different environments and allows the cells to find the appropriate niche for survival.
Memory T cells have less stringent requirements for survival. CD8+ memory T cells can survive in absence of the restricting MHC Class-I element (Tanchot et al., 1997), while the survival of CD4+ memory T cells is MHC Class-II independent (Swain et al., 1999; Polic et al., 2001). Thus, memory T cell survival seems mostly dependent on IL-7 and IL-15 (Surh and Sprent, 2008). While there is some degree of overlap in survival signals, naïve, and memory T cells do not seem to compete, allowing for the coexistence of these populations. This was shown in studies where animals manipulated to have only naïve CD8+ T cells showed halved total CD8+ T cell numbers, as well as in studies where animals with only memory T cells were unable to fill up the entire T cell pool (Tanchot and Rocha, 1995; Freitas and Rocha, 2000). Coexistence of these populations is ensured through a mechanism of niche segregation; separated control of these populations ensures that both are maintained, and avoids competition between them (Tanchot et al., 1997; Freitas and Rocha, 2000; Almeida et al., 2005).
The above-described division, however, fails to fully account for the complexity of peripheral T cell pools. Several other major subpopulations should be considered, including populations of CD4+FOXP3+ regulatory T cells (Sakaguchi, 2004), CD4+ and CD8+ effector T cells, and the division of memory pools into central-memory and effector-memory, which relies on differences in migratory behavior and T cell differentiation status (Sallusto et al., 2004).
Regulatory CD4+CD25+FOXP3+ T Cells: An Essential Population of the Peripheral T Cell Pools
The physiologic function of regulatory CD4+ T (Treg) cells is central in establishing self-tolerance and in controlling autoimmune diseases. These cells were initially identified in models of autoimmune disease or inflammatory bowel disease (IBD) following the adoptive transfer of naïve T cells into lymphopenic animals, due to their ability to prevent the provoked autoimmune manifestations (Powrie et al., 1993; Sakaguchi et al., 1995; Asano et al., 1996; Sakaguchi, 2000; Maloy and Powrie, 2001). Treg cells specifically express the transcription factor FOXP3, which is strictly required for their development (Fontenot et al., 2003; Hori et al., 2003). Indeed, carriers of spontaneous FOXP3 mutations – e.g., the IPEX (immunodysregulation, polyendocrinopathy, enteropathy, x-linked) syndrome in humans (Wildin et al., 2001) and scurfy mice (Blair et al., 1994; Bennett et al., 2001) – develop rapid and lethal autoimmune syndromes that can be rescued upon transfer of Treg cell populations (Fontenot et al., 2003; Hori et al., 2003). Similar results were obtained in FOXP3-deficient animals generated by homologous recombination (Fontenot et al., 2003) or upon the selective ablation of Treg cells in adult mice (Kim et al., 2007a; Lahl et al., 2007). However, CD4+ Treg cell actions can also counter-productively suppress immune responses against tumors and viral infections (Antony et al., 2005; Belkaid and Rouse, 2005), indicating that regulatory T cells are capable of influencing all T cell-mediated responses (Sakaguchi, 2004).
Others and we have shown that CD4+ Treg cells can prevent IBD and autoimmune diseases caused by transfer of naïve CD4+ T cells into lymphopenic animals, as well as control the expansion and regulate the total number of these T cells (Annacker et al., 2000; Almeida et al., 2002). The critical role of CD4+ Treg cells on peripheral T cell homeostasis was further demonstrated in experiments with mouse chimeras reconstituted with bone marrow cells from CD25−/− (Almeida et al., 2002) or FOXP3−/− (Fontenot et al., 2003) donors; the co-transfer of small numbers of CD4+CD25+ Treg cells rescued these mice from massive lymphoproliferation, autoimmunity, and death. Importantly, the rescued chimeras exhibited normal absolute numbers of recovered T cells, and relative proportions of naïve, memory, and effector CD4+ and CD8+ T cells were restored (Almeida et al., 2002; Fontenot et al., 2003). These observations demonstrated that the lymphoid hyperplasia observed in CD25−/− or FOXP3−/− mice was not cell autonomous, and that restoring the CD4+ Treg population sufficed to restore the equilibrium of the total peripheral T cell pool. Furthermore, mice transgenic for FOXP3 overexpress the “scurfin” protein and show an increased fraction of Treg cells within reduced numbers of peripheral T cells (Khattri et al., 2001). We conclude from these studies that the presence of CD4+ Treg cells is essential to the homeostasis of naïve, memory, and effector T cells. Two important questions arise: (1) what mechanisms do regulatory T cells use to control CD4+ T cell numbers, and (2) how is homeostasis of these regulatory T cells achieved?
Function of CD4+ Treg Cells: The Possible Role of IL-2
Previous studies have identified a vast number of putative mechanisms of suppression used by CD4+ Treg cells (Shevach, 2009). Notably, these include both cell-contact dependent and independent mechanisms, and while initial studies pointed to a segregation into those categories according to the experimental system used, with in vitro studies falling into the first category and in vivo studies in the second, later studies have shown that both direct and indirect mechanisms may occur in vivo (Shevach, 2000). Indeed, strong experimental evidence indicates that in vivo the suppressive activity of Treg cells may be cell-contact dependent and APC-mediated. Treg cells from mice deficient in LAG-3 (a CD4-related molecule that binds MHC Class-II) show reduced regulatory activity (Huang et al., 2004). More importantly, recent findings using Treg cells with a specific CTLA-4 deficiency indicate that CTLA-4 is required for Treg cells to suppress immune responses by disrupting the ability of antigen-presenting cells to activate other T cells (Wing et al., 2008). A hallmark feature of CD4+FOXP3+ Treg cells is the expression of CD25, the α-chain of the high-affinity IL-2 receptor (Nelson and Willerford, 1998); therefore, it has also been proposed that the suppressive function of Treg cells could be due to their higher ability to use available IL-2, which could prevent IL-2 utilization by other naïve or effector cells that lack expression of the high-affinity IL-2 receptor (Barthlott et al., 2003; de la Rosa et al., 2004).
This concept of suppression via IL-2 consumption was based on the belief that IL-2 is a critical essential factor for T cell responses and expansion, as supported by in vitro evidence (de la Rosa et al., 2004). The dissociation kinetics, the directionality of IL-2 production within the immunological synapse, and the TCR-dependency of both IL-2 production and IL-2R expression all contributed to the notion of a requirement for proximity between producers and users of IL-2, suggesting a role of IL-2 consumption in the mechanism of suppression (Malek and Castro, 2010). However, the view that IL-2 acts as a major growth factor for in vivo T cell proliferation has since been challenged. Indeed, IL-2−/−, IL-2Rα−/−, and IL-2Rβ−/− mice develop lymphoproliferative syndromes associated with both autoimmunity and immunodeficiency (Kundig et al., 1993; Suzuki et al., 1995; Willerford et al., 1995; Almeida et al., 2006b). In these mice the strong activation state of T cells renders them refractory to further in vitro stimulation and is responsible for an “immunodeficiency” state. In IL-2R-deficient mice disease can be prevented by the transfer of relatively small numbers of Treg cells (Almeida et al., 2002; Malek et al., 2002). The increased lymphocyte proliferation and the progressive accumulation of considerable numbers of T cells observed argue against the strict requirement of IL-2 for T cell expansion in vivo. It also suggests that other γc-chain dependent cytokines, i.e., IL-15 and IL-7 or IL-7 alone in the case of the IL-2Rβ−/− mice, could induce extensive T cell proliferation in vivo. Nevertheless, it has also been shown that CD4+ T cell expansion in response to antigen can occur in the absence of IL-2Rγ-chain (γc) expression (Lantz et al., 2000). Moreover, mice deficient in STAT5, the transcription factor downstream of the IL-2R signaling that has been reported to be critical for lymphocyte cell cycle progression in vitro (Moriggl et al., 1999), also develop extensive lymphoproliferation (Snow et al., 2003; Burchill et al., 2007; Yao et al., 2007), suggesting that TCR signals alone or associated with other γc-independent cytokines suffice to elicit T cell expansion in vivo. Overall these observations make apparent the discrepancies between in vitro and in vivo studies and clearly demonstrate that in vivo, TCR signals can bypass IL-2 requirements for CD4+ T cell proliferation and expansion. It should be pointed out that although IL-2 does not seem to be strictly required for T cell proliferation and primary responses in vivo, as T cell responses have been shown to resolve infection in the absence of IL-2 (Malek, 2008), it appears to be critical in secondary responses, in particular for the differentiation and development of CD8 T cell memory cells (Williams et al., 2006) hinting to an impact of IL-2 in the quality of the immune response rather than to an absolute requirement for IL-2 in T cell responses. Thus the precise quantitative contribution of IL-2 for CD4 T cell proliferation in vivo remains yet to be established.
The observations that in vitro FOXP3+ Treg cells lacking CD25 retain intact suppressive capacities (Fontenot et al., 2005a) and, more importantly, that a FOXP3 transgene restored Treg cell function and protected against the onset of autoimmunity in IL-2Rβ−/− mice (Soper et al., 2007), indicate that the suppressive activity of Treg cells can occur in the complete absence of IL-2 signals. Thus, it is likely that the suppressive activity of Treg cells can occur both by inhibiting naïve T cell activation and IL-2 production and possibly by influencing the response via IL-2 consumption. Indeed, recent reports have suggested suppression via IL-2 consumption by CD4+CD25+ Treg cells as a mechanism of controlling immune responses in vivo (Chen et al., 2011; Pandiyan et al., 2011). These studies may also be interpreted as suggesting that Treg cells modify environmental factors and immune responses to favor specific TH17 mimicking suppression of other T cell fates. Nevertheless, there exists a vast body of evidence demonstrating other non-IL-2 related mechanisms of Treg-mediated suppression, including IL-10-mediated regulation of gut IL-17 responses (Chaudhry et al., 2011; Huber et al., 2011), implying that not all Treg function is translated into TH17 responses by the suppressed effectors. It remains to be established if these effects can occur simultaneously and what cues determine the triggering of these diverse mechanisms.
Homeostasis of Regulatory CD4+ T Cells: The Role of IL-2
Our investigation of CD4+ Treg cell homeostasis revealed that the absolute number of Treg cells and the ratio of non-Treg/Treg CD4+ T cells are tightly regulated under steady-state in vivo conditions (Almeida et al., 2006b). Adoptive transfer of a limited number of CD25+ T cells rescues CD25−/− bone marrow (BM) chimeras (Almeida et al., 2002), neonatal FOXP3sf mice (Fontenot et al., 2003), and IL-2Rβ−/− mice (Malek et al., 2002) by reconstituting a normal CD4+CD25+ Treg pool that persists indefinitely without new thymus output. Our strategy involved mixed BM chimeras that were reconstituted with precursor cells from WT donors (that were competent to generate CD4+CD25+ Treg cells) diluted at different ratios with precursor cells from CD25−/− donors (that were incompetent to generate CD4+CD25+ Treg cells). We found that similar numbers and proportions of peripheral CD4+CD25+ Treg cells were recovered regardless of the fraction of precursor BM cells that were competent to generate CD4+CD25+ Treg cells (Almeida et al., 2006b). These findings and the constancy of Treg cell numbers and proportions demonstrated that these cells occupy a limited and specialized niche in the peripheral T cell pool. The expression of CD25 (IL-2Rα) by the vast majority of CD4+ Treg cells suggests a major role of IL-2 in CD4+ Treg biology. Indeed, mice that are genetically deficient in IL-2 (Schorle et al., 1991; Sadlack et al., 1995; Wolf et al., 2001), IL-2Rα (Willerford et al., 1995), IL-2Rβ (Suzuki et al., 1995; Malek et al., 2000), or STAT5 (the transcription factor downstream of the IL-2R signaling; Snow et al., 2003; Burchill et al., 2007; Yao et al., 2007) each lack sizeable populations of CD4+ Treg cells and, consequently, develop lethal lymphoid hyperplasia and autoimmune diseases. Moreover, IL-2-dependent signaling is involved in the maintenance of FOXP3 expression and in the maintenance of the FOXP3-dependent gene signature, even at low levels of IL-2Rβ-dependent signaling (Gavin et al., 2002; Hill et al., 2007; Yu et al., 2009). Others and we have shown that IL-2 is essential for the peripheral survival and expansion of CD4+CD25+ Treg cells. Thus, Treg cells from IL-2-deficient donors fail to survive in IL-2−/− hosts (Almeida et al., 2006b) or to accumulate in the periphery in the absence of IL-2R signals (Almeida et al., 2002, 2006b; Fontenot et al., 2005b), and blocking IL-2R or neutralizing IL-2 reduces Treg cell numbers (Bayer et al., 2005; Setoguchi et al., 2005).
The observation that CD4+CD25+ T cell numbers and proportions are remarkably stable does not explain how CD4+CD25+ Treg homeostasis is achieved or the mode of action of IL-2. Parsimony suggests that the expression of high-affinity IL-2Rα and the dependence on IL-2 for survival are critical and connected clues to understanding the homeostasis of the Treg cell subpopulation. We showed that expression of high levels of high-affinity IL-2Rα specializes the CD4+CD25+ Treg cells for exploiting the IL-2 resource in vivo. In fact, in the CD4+ T cell pool of mouse chimeras reconstituted with a mixture of precursors differing only in their potential to exploit IL-2, the WT CD4+ T cells that are able to express CD25 have a noticeable seeding advantage over the CD25−/−CD4+ cells (Almeida et al., 2006b). Thus, CD25 expression by Treg cells permits them to occupy a niche defined by the IL-2 resource, and allows them to avoid direct competition with other CD4+ T cells that do not express the high-affinity IL-2Rα. This explains how the immune system ensures the presence of this subpopulation of cells, but it fails to explain their constancy or their relative proportion.
We have shown that complementation chimeras reconstituted with a mix of donor BM from CD25−/− and IL-2−/− animals present a normal peripheral T cell pool, including a normal proportion of CD4+CD25+ Treg cells (Almeida et al., 2002). This indicates that paracrine IL-2 is sufficient for the peripheral survival of CD4+CD25+ Treg cells. We also reported that, in steady-state conditions, establishment of a fully sized and functional Treg cell population that is capable of preventing development of autoimmune syndromes only occurs in the presence of IL-2-competent αβ T cells (Almeida et al., 2006b). This suggests that αβ T cells (particularly activated CD4+ T cells) represent the major source of the IL-2 required for the survival of a functional population of Treg cells in the peripheral pools (Curotto de Lafaille et al., 2004; Setoguchi et al., 2005; Almeida et al., 2006b). We have also used transgenic mice overexpressing IL-7 to greatly increase peripheral T cell numbers recovered at equilibrium time-points, and we found that the numbers of CD4+CD25+ Treg cells increased proportionally to the absolute number of CD4+ T cells (Almeida et al., 2006b). Since CD4+ Treg cells are unable to produce IL-2 (Shevach, 2000) due to active FOXP3-dependent repression of the il2 gene (Wu et al., 2006; Ono et al., 2007), the corollary conclusion from these observations is that Treg cells are reliant on other IL-2-producing T cells for survival. To demonstrate this point, we reconstituted irradiated Rag2−/−IL-2−/− hosts with mixes of IL-2-sufficient and IL-2-deficient BM cells, and we found a direct correlation between the numbers of IL-2-competent cells and CD4+ Treg cells (Almeida et al., 2006b). We concluded that the size of the Treg niche corresponds to the available quantity of IL-2, meaning that the number of CD4+CD25+ Treg cells is tied to the number of IL-2-producing CD4+ T cells. This explains why the relative proportion of the two populations is stably maintained. Importantly, the size of the IL-2 niche is not fixed by factors external to the overall T cell pool; instead, it is mainly dependent on IL-2 produced by other T cells (Almeida et al., 2006b).
This mechanism of homeostasis implies that the survival of a given Treg and the capacity for expansion will be dependent on the amounts of IL-2 produced by other T cells and, thus, Treg cells are capable of extended expansion and renewal when faced with an empty Treg niche. Indeed, when very low numbers of Treg cells are co-transferred with IL-2-producing CD4+ T cells into lymphopenic animals, they expand considerably and tend to occupy the vacant CD4+ Treg niche (Almeida et al., 2006b; Komatsu et al., 2009). A similar situation occurs in the first days of life, as the first wave of CD4+ Treg cells expand considerably between days 3 and 7 (Bayer et al., 2005). Interestingly, the capacity of expansion of “converted” or CD25low-expressing FOXP3+ contaminants in transfers of CD4+CD25− T cells is greatly affected by the presence of CD4+CD25+ co-transferred cells, suggesting strong competition for the occupancy of the Treg niche and further demonstrating that initial CD25 expression is a major competitive advantage in IL-2 usage (Almeida et al., 2006a).
Quorum-Sensing and CD4+ T Cell Homeostasis
It is believed that control of lymphocyte numbers may be determined by cellular competition for a limited amount of resources, e.g., trophic factors required for survival (Freitas and Rocha, 2000). However, it is not clear what mechanisms are used to control expanding lymphocyte numbers in situations where resources are not limiting, e.g., during immune responses or an excess of self-antigens or cytokines. It remains unanswered how lymphocyte populations “count” the number of their individuals and how do they “know” when to stop growing?
Many species of bacteria use quorum-sensing mechanisms to coordinate their gene expression according to their population density (Miller and Bassler, 2001; Diggle et al., 2007). A similar quorum-sensing-like mechanism may play a critical role in lymphocyte homeostasis, if lymphocytes can assess the number of molecules with which they interact and respond accordingly once a threshold number of molecules is detected.
We propose that control of lymphocyte numbers could be achieved by the ability of lymphocytes to perceive their own population density (de Freitas, 2009). The correlation between the numbers of CD4+CD25+FOXP3+ Treg cells and of activated IL-2-producing T cells indeed suggests the presence of a quorum-sensing-like mechanism. In this case CD4+ T cell populations use the Treg cell subset expressing the high-affinity IL-2Rα-chain to monitor the number of activated CD4+ T cells by detecting (sensing) the IL-2 produced, and they adapt their collective behavior according to the quantities of IL-2 sensed (Figure 1A). In this manner, CD4+ T cells may be able to control not only the number of activated IL-2-producing CD4+ T cells, but also the size of the total CD4+ T cell pool. This mechanism prevents uncontrolled lymphocyte proliferation and maintains a constant pool size. Thus, according to the quorum-sensing hypothesis, IL-2, rather than being a mere growth factor required for the proliferation of some T cells, represents a key molecule playing a central role in CD4+ T cell homeostasis.
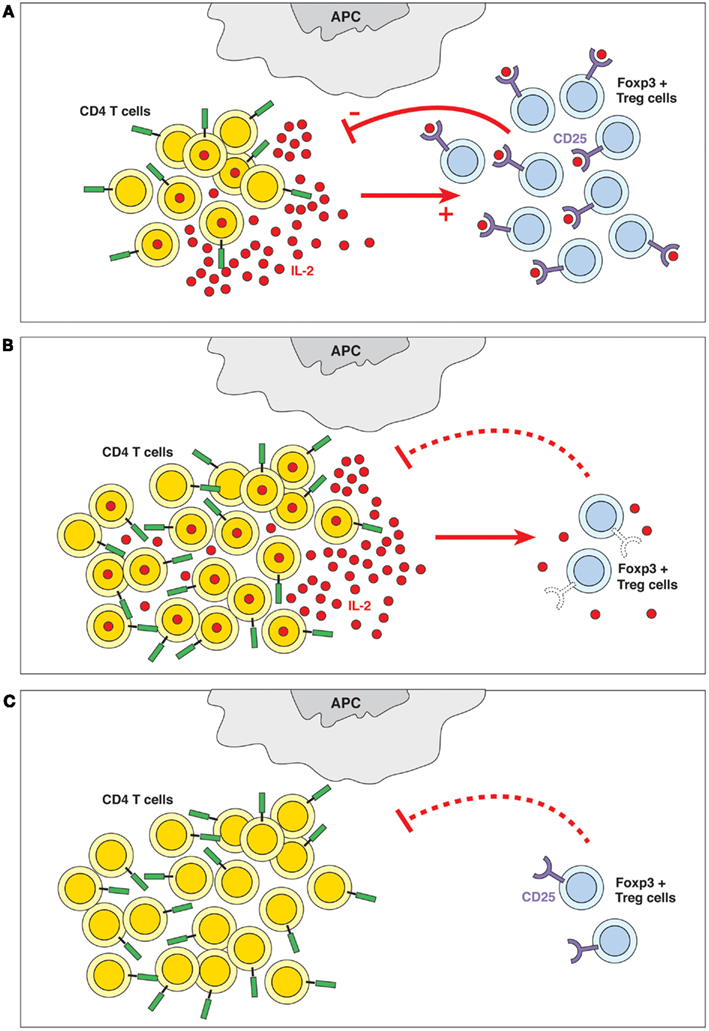
Figure 1. (A) Quorum-sensing. The presence of IL-2 and the ability of the Treg cells to detect its levels are crucial to the homeostasis of the immune system. Quorum-sensing in this case is defined as an indirect feedback loop where the IL-2 produced by a subpopulation of activated T cells (among others) is detected (sensed) by a subpopulation of CD4+ Treg cells expressing the high-affinity IL-2Rα-chain; these cells contribute to controlling the number of CD4+ T cells. In other words, the overall CD4+ T cell populations sense the produced quantities of IL-2 and adapt their behavior accordingly. (B) Failure of quorum-sensing by defective sensor molecule. The inability to detect IL-2 because of defects in IL-2R expression (in IL-2Rα−/− or IL-2Rβ−/− mice) or signaling (in STAT5−/− mice) leads to lymphoid hyperplasia and autoimmune disease. (C) Failure of quorum-sensing due to absence of the sensed molecule. In the absence of IL-2, Treg cells do not survive, which causes lymphoid hyperplasia and autoimmune pathology.
We hypothesize that failure of this quorum-sensing mechanism results in uncontrolled CD4+ T cell activation and autoimmune disease. The inability of CD4+ T cells to detect IL-2 – due to defects of IL-2R expression (Willerford et al., 1995; Malek et al., 2000) or IL-2 signaling (Burchill et al., 2007), or due to failure to produce IL-2 (Schorle et al., 1991) – leads to lymphoid hyperplasia and lethal autoimmune disease (Figures 1B,C). Other autoimmune diseases are also linked to defects in the IL-2/IL-2R signal pathways. Autoimmunity in NOD mice seems to be dependent on lower IL-2 production resulting from genetic defects that map to the il-2 region (Yamanouchi et al., 2007). Polymorphisms linked to IL-2 receptors, IL-2Rα and IL-2Rβ, are associated with several autoimmune diseases, such as type 1 diabetes, multiple sclerosis, celiac diseases, and rheumatoid arthritis (Gregersen and Olsson, 2009; Todd, 2010). It has also been reported that Dicer, the enzyme responsible for generating functional miRNAs, enhances Treg cell development (Cobb et al., 2006). FOXP3 regulates miR155 expression and optimizes Treg cell responses to limiting quantities of IL-2 by targeting suppressor of cytokine signaling 1 (SOCS1) expression (Lu et al., 2009; Stahl et al., 2009). Consequently, the selective disruption of miRNAs in the Treg cell lineage results in lethal autoimmunity (Liston et al., 2008; Zhou et al., 2008). However, it should be noted that genetic risk for autoimmunity might be more complex than the IL-2 regulation of Treg cell function, as it may also involve increased responses of the self-reactive clones to IL-2.
By modifying IL-2 levels it should be possible to increase Treg cell numbers and control auto-reactive immune responses. However, IL-2 also contributes to the effector arm of immune responses by driving CD8+ T cell proliferation and differentiation into effector functions (Kalia et al., 2010; Pipkin et al., 2010; Zaragoza et al., 2011) and development of CD8+ T cell memory (Williams et al., 2006). Treg cells have a major competitive advantage for IL-2 usage (Almeida et al., 2006b); this is due to the constitutive expression of the high densities of the IL-2Rα chain that confers a up to 1000-fold increase of affinity of binding to the IL-2Rα, β, γ trimer (Feinerman et al., 2010), the fact that even a low level of IL-2-dependent signaling sufficiently ensures maintenance of FOXP3 expression (Gavin et al., 2002; Hill et al., 2007; Yu et al., 2009), and because FOXP3 expression itself optimizes Treg cell responses to limiting quantities of IL-2 (Lu et al., 2009; Stahl et al., 2009). Therefore, Treg cells should be particularly susceptible to small increases in IL-2 availability. Indeed, administration of low doses of IL-2 (Tang et al., 2008; Grinberg-Bleyer et al., 2010) or inducible adenovirus bearing IL-2 genes (Goudy et al., 2011) can reverse disease or prevent diabetes in NOD1 mice by increasing Treg cell numbers in the target organs. Some anti-IL-2/IL-2 immune complexes have been used in vivo to prolong the half-life of the administered cytokine (Boyman et al., 2006), and can selectively favor Treg cell expansion by blocking the IL-2 binding site to the IL-2Rβ (Boyman et al., 2006; Webster et al., 2009). Differences in the IL-2R signaling pathways between Treg and T effector cells may also allow manipulation of responses to either favor or antagonize Treg function (reviewed by Malek and Castro, 2010).
Feedback loops between interacting effector and regulatory cells have been proposed to occur in the course of immune responses (Knoechel et al., 2005; Fehervari et al., 2006; Carneiro et al., 2007). However, these previous models describing regulatory feedback loops between Teff/Treg cells did not take into consideration the protagonist part of IL-2 and Treg cells in overall lymphocyte homeostasis. The quorum-sensing hypothesis extends beyond the scope of specific immune responses and predicts the behavior of the global T cell pool, including maintenance of the naïve pool and control of the size of the activated T cell pool. Indeed, the effects of the absence of IL-2 and/or of the Treg cell subset extend to all subpopulations on the CD4+ T cell pool suggesting that this homeostatic mechanism acts to maintain total T cell numbers rather than the size of effector and regulatory T cells only. The quorum-sensing hypothesis, besides its proposed overall role on T cell homeostasis, also embodies a feedback mechanism to control T cell activation while allowing immune responses to occur; this likely plays a part in the contraction phase of the CD4+ T cell responses. In the initial stages of an immune response, the presence of antigen perturbs the immune system equilibrium and incites proliferation of antigen-specific cells. Upon activation, the number of IL-2-producing cells (T and non-T) and the IL-2 concentrations increase; Treg cells respond with proliferation, and eventually re-establish a steady-state by suppressing further T cell activation and proliferation, thus decreasing the number of IL-2-producing cells. Such a mechanism might have evolved to control peripheral cross-reactive or auto-reactive T cell clones, while allowing controlled increases in overall peripheral T cell number. This mechanism definitively enables CD4+ T cells to limit their expansion when in presence of an excess of resources.
In conclusion our quorum-sensing model provides new mechanistic insights and has implications for understanding autoimmune disease and will create a new way of thinking about the homeostasis of lymphocyte populations.
Proposed Mathematical Model for the Role of “Quorum-Sensing” in CD4+ T Cell Homeostasis
Based on the suggested quorum-sensing hypothesis we here propose a mathematical model that is intended to describe in a quantitative way the role of IL-2 and quorum-sensing mechanisms in CD4+ T cell homeostasis. This model describes CD4+ T cell dynamics, that is the time evolution of four different CD4+ T cell subsets: naïve (n1), IL-2-producing (n2), activated/memory non-IL-2-producing (n3), and regulatory CD4+ T cells (n4). The model assumes that cells can divide, differentiate, or die. The assumptions of the model are as follows.
Thymus Output
We model thymus output as occurring at a constant rate, independent of the number of cells in the peripheral CD4+ T cell population. We only consider thymus output for naïve and regulatory T cells. These terms take the form
where i = 1, 4.
Natural Death
We assume that CD4+ T cells possess a natural death rate and that this constant death rate depends on the T cell subset. Thus, the death terms are proportional to the population size of each subset, namely
where i = 1, 2, 3. We have already discussed the requirement for IL-2 for the survival of regulatory T cells. This can be encoded as an IL-2-dependent death term for the subset of regulatory T cells. We do not directly include a dynamical equation for IL-2 levels, but we assume that the IL-2 levels are proportional to the number of IL-2-producing cells. The death term for the regulatory T cells takes the form
where κ2 is the number of IL-2-producing cells at which the death rate for regulatory cells is half its value in the absence of any IL-2.
TCR/IL-7-Induced Proliferation
We assume that all T cell subsets proliferate in response to TCR- and/or IL-7-mediated signals; thus, we include them as logistic growth terms with carrying capacity κ. In this way, we impose a limit on the population size that these signals can sustain, which is consistent with the idea that T cells must compete for such signals in the periphery. The proliferation due to TCR and IL-7 takes the form
where i = 1, 2, 3.
IL-2-Induced Proliferation
In addition to growth terms due to TCR and/or IL-7 stimulus we also include proliferation terms to model T cell responses to IL-2. For a given T cell subset (except for naïve T cells), the growth rate due to IL-2 signals is assumed to be proportional to the number of IL-2-producing cells. We model IL-2-induced proliferation with quadratic terms of the form
where i = 2, 3, 4.
Differentiation of naïve T Cells
Naïve T cells can be activated in response to their specific antigen. We model this differentiation by assuming naïve T cells become IL-2-producing cells at a constant rate α12. We also assume naïve T cells differentiate into memory cells at a constant rate α13. These assumptions are modeled by terms of the form
where i = 2, 3.
Differentiation of IL-2-Producing Cells
We assume IL-2-producing cells stop producing IL-2 at a constant rate α23 and thereafter differentiate to the activated/memory pool. This term is written as
Reactivation of Memory
We assume that memory T cells can become IL-2-producing cells in response to further stimulation by their specific antigen. This assumption is modeled by a term of the form
Suppression
We encode regulatory T cell suppression as follows: regulatory T cells give a signal to activated IL-2-producing T cells that causes them to stop producing IL-2. We model this as a contact term proportional to the number of regulatory T cells, with the assumption that IL-2-producing cells may become memory cells after having received this signal. The assumption is modeled by a term of the form
System of ODEs
Based on the assumptions introduced above, we can write a system of ordinary differential equations (ODEs) that fully describes the model. The ODEs are given by
We note that a detailed mathematical analysis of this system of ODEs (existence and stability of steady-states, for example) is beyond the scope of this paper and will be explored in a separate publication.
The mathematical model introduced in this section assumes that the limiting factors mediating the expansion of the CD4+ Treg population are not only the available levels of IL-2, but also the number of IL-2-producing cells. Implicit in this assumption is that upon the encounter of an IL-2-producing T cell and a Treg cell, the strength of the signal which can be delivered via IL-2 is non-limiting; indeed the limiting factor is the rate at which such encounters occur. The quorum-sensing mechanism described here can be understood as follows: Treg cells “count” and regulate the numbers of activated T cells through the detection of the IL-2 and the number of interactions between these two populations, of which a specified proportion (encoded within the parameters of the model) leads to cellular events such as a division, survival, or suppression. Previous mathematical models have explored the consequences of different mechanisms for the action of Tregs on IL-2-producing cells (usually called effector T cells). In the model presented here, the suppression mechanism (represented by the term proportional to β) is mathematically encoded as a non-linear density dependent term in the equation of motion for the IL-2-producing cells. Naive T cells, at a rate proportional to the number of Treg cells and to β, become activated/memory cells. Such a suppressive mechanism is in contrast to previously studied mechanisms where the action of Treg cells is to consume available IL-2, thus limiting the number of survival or proliferative signals cells within the CD4+ T cell population can receive. We believe that our approach is more robust since the action of Treg cells is assumed to limit the number of cells which are activated and produce the resource IL-2, rather than try to limit the IL-2 resource itself by consumption of IL-2 post-production. Whilst it has been extensively shown that Treg cells are critically dependent on IL-2 for their survival and proliferation, it is not so clear whether IL-2-producing cells are solely dependent on IL-2 for their own expansion. In our model, we encode two possible mechanisms of expansion for the IL-2-producing (effector) population, one of which is IL-2 independent. In such a scenario, the action of Treg cells focused around consumption of IL-2 would only serve to limit one of these two mechanisms for expansion of the IL-2-producing T cell population. By assuming that the suppressive action of Treg cells prevents overall T cell activation and proliferation, the degree of expansion through either pathway is controlled, and thus both populations, are indexed to each other.
Plots
We conclude with a series of plots of the deterministic trajectories of the above system of equations as well as with realizations of a stochastic version of the deterministic model. It must be noted that we neglected thymus output terms (v1 = 0 and v4 = 0). Initial conditions were 100 naïve CD4+ T cells and 10 Treg cells. We choose parameter values to simulate the dynamics of a clone of CD4+ T cells in response to antigenic challenge, starting at time t = 0. The parameter values are given in Table 1. In order to make the mathematical model more realistic we have also assumed that naïve T cells differentiate progressively with cell division, i.e., they differentiate after they have divided at least three times. Additionally, we assumed that IL-2-producing cells only differentiate into activated/memory cells if they have divided at least three times. Suppression still acts across every generation of IL-2-producing cells, and memory cells are free to become IL-2-producing cells without any requirements for prior division. In the simulations this is achieved by keeping track of the generation each cell belongs to (by writing an ODE for each respective generation) and keeping parameter values constant across all generations with the exception of those parameters, which are generation-dependent. The deterministic solutions were simulated over periods of 2 and 8 weeks, respectively, are shown in Figure 2. We also present realizations from a stochastic version of the same deterministic model (Figure 3), with simulations run over the same time windows as the deterministic trajectories. The stochastic realizations shown here were produced using the Gillespie algorithm. The system of ODEs was solved using a fourth-order Runge–Kutta scheme. Initially, the model shows slow growth in the number of T cells; during the first 2 days, the total cell population approximately doubles from around 100 cells to 200 cells, with the immune response reaching a peak and at around day 7. At days 4–5 there is a sharp increase in the number of IL-2-producing cells, followed by a subsequent increase in the number of antigen-experienced cells, which are not producing IL-2. Notably, this leads to a large increase in regulatory cells around day 7, which is immediately followed by a sharp contraction phase that lasts around 2 days. A slower contraction phase that lasts around 3 weeks follows before a homeostatic equilibrium is reached. We note that for this parameter set the regulatory T cell subset takes longer to reach equilibrium than the memory T cell subset.
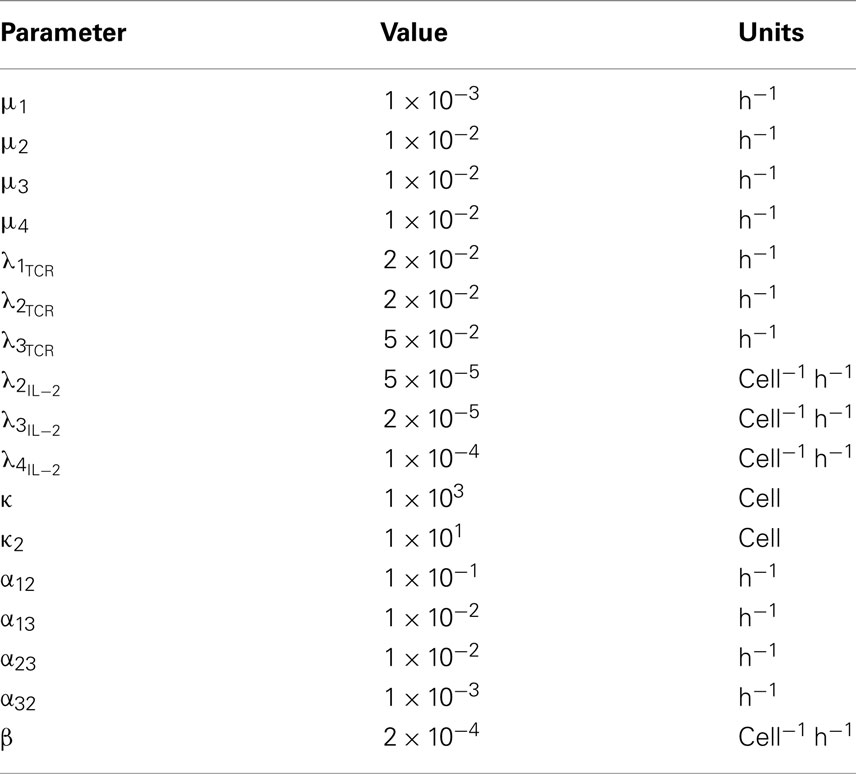
Table 1. Parameter values and dimensions for a quorum-sensing model of CD4 T cell homeostasis.
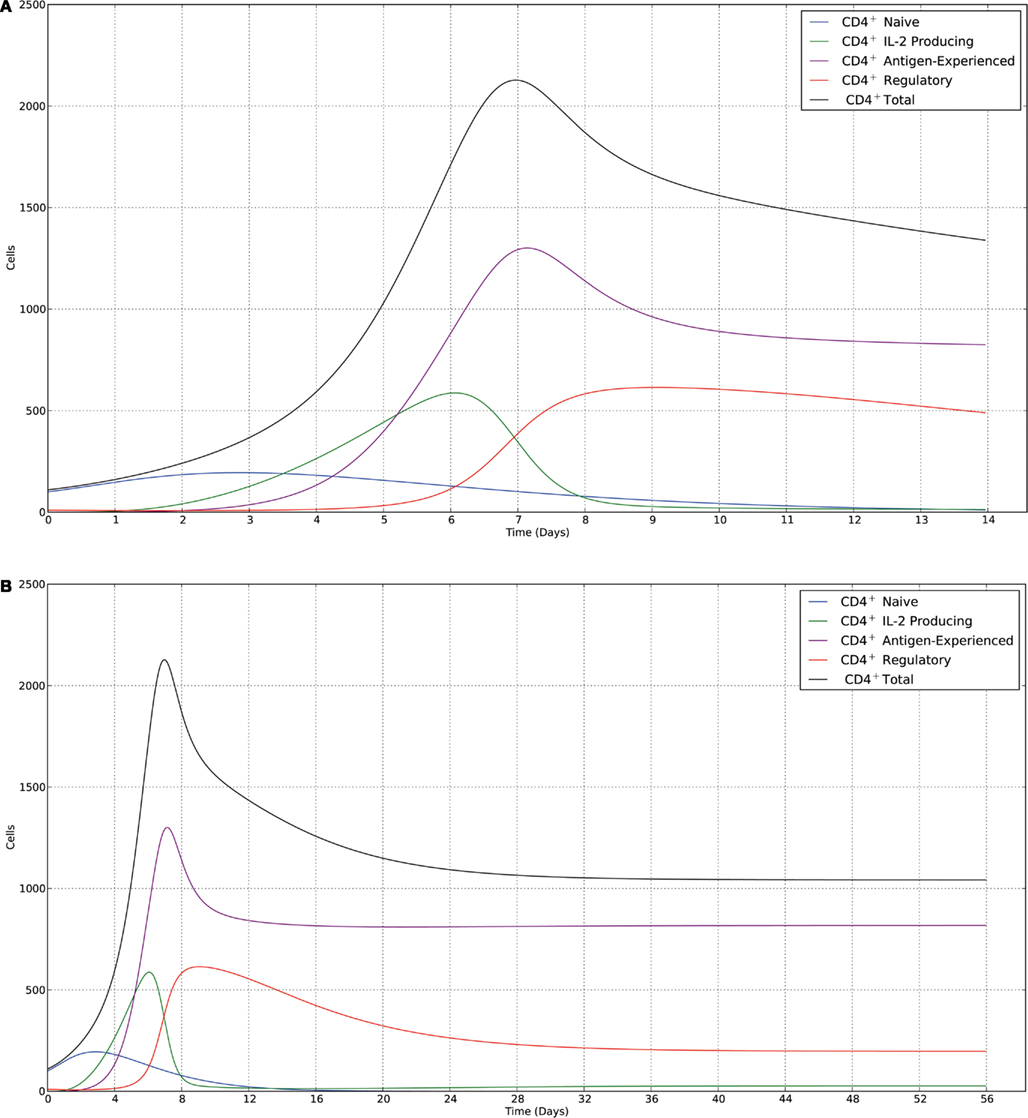
Figure 2. Trajectories of the deterministic model for the parameter set given in Table 1, with the following initial conditions: 100 naïve cells (blue) and 10 regulatory cells (red). (A) Data shown over a time course of 2 weeks. The immune response peaks at ∼7 days, then declines to a homeostatic equilibrium dominated by memory cells. (B) Data shown over a time course of 8 weeks. The contraction phase occurs over an extended time period of ∼20 days for memory cells and ∼40 days for regulatory cells.
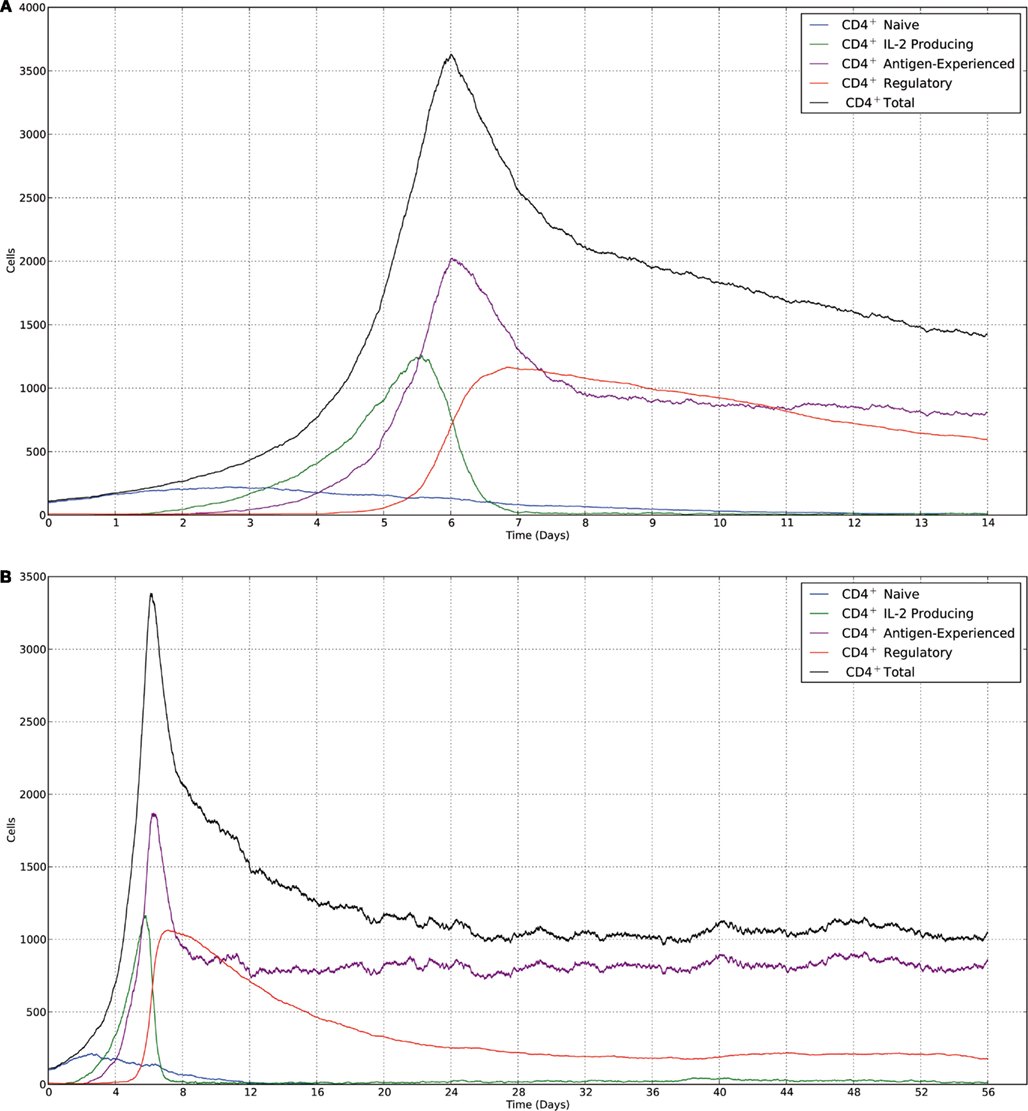
Figure 3. Realizations of the stochastic model carried out using the Gillespie algorithm. All parameters and initial conditions are the same as in the deterministic model, with (A,B), respectively, correlating to (A,B) from Figure 2. There is a higher degree of noise at equilibrium for memory cells, which is due to memory cells repeatedly receiving antigen-mediated signals to become IL-2-producing, then reverting back to a non-IL-2-producing state.
At equilibrium, the model predicts that the size of the memory T cell population is limited only by the availability of TCR/IL-7 signals, which is included in the model as the carrying capacity κ. This population acts as a source for the population of IL-2-producing cells, so that there is a small but constant supply of IL-2-producing cells at equilibrium. This source of IL-2 is then sufficient to maintain a regulatory T cell population, which in turn keeps the IL-2-producing population from expanding. The deviation away from this ratio of regulatory T cells and IL-2-producing cells allows the model to reproduce the key characteristics of the immune peak and the contraction phase before settling down to equilibrium. Indeed, it is possible to simulate a larger immune peak by specifying a larger ratio between naïve cells and regulatory cells in the initial conditions, or to dampen the immune peak by changing the ratio in the opposite manner.
In our model, we assumed that antigen is constant (encoded in the proliferation parameter ) and non-limiting; however, an effective immune response should serve to remove antigen. We could address this issue by either introducing time-dependent rates for the activation of naïve or memory cells, or by introducing within the rates a dependence on a number of cells that would be linked to the antigen-clearance rate, e.g., effector cells. We have also assumed that naïve T cells may only be activated after having proceeded through at least three divisions, and equivalently IL-2-producing cells are assumed to only revert back to a non-IL-2-producing state if they have gone through three divisions. In reality, the division/proliferation-linked differentiation program is unlikely to be so clear-cut. We could refine the model by introducing a probabilistic differentiation based on generation, where cells are increasingly likely to differentiate as they progress through more divisions. The model we introduce here has been applied only to the pool of specific T cells that can play a part in response to their cognate antigen. The model could be extended to mirror the effect this immune response would have on a bystander population of T cells that is not involved in the immune response. We note that our model can also be applied to model T cell homeostasis with a different set of parameter values. However, this is beyond the scope of what we wish to present here.
Based on single-cell in vitro experiments, Feinerman et al. (2010) recently developed a quantitative model for the regulation of immune responses by IL-2 – in particular, for the competition for IL-2 between effector and regulatory CD4+ T cells during an immune response. They discuss how binding of IL-2 leads to IL-2Rα up-regulation in both effector and regulatory cells. Regulatory cells express higher basal IL-2Rα levels, which gives them a distinct advantage over effector cells in the ability to scavenge IL-2. This advantage reportedly allows regulatory cells to critically limit the amount of IL-2 available for consumption by effector cells. The authors develop a biochemical computational model of IL-2R and STAT5 signaling, which takes into account the cell-to-cell variability in IL-2 receptor expression levels, as well as a dynamical model of IL-2 competition between regulatory and effector T cells. Similarly, Busse et al. (2010) introduced a reaction-diffusion model to describe the spatio-temporal dynamics of IL-2, and regulatory and helper T cells. Their model captures the interplay between IL-2 production, binding, internalization, degradation, and recycling after antigen stimulation; it is based on the positive correlation between IL-2 binding and cell-surface number of IL-2Rα. The authors conclude that IL-2-induced up-regulation of high-affinity IL-2R establishes a positive feedback loop of IL-2 signaling. Furthermore, under conditions of limiting IL-2, their results indicate that regulatory T cells can deprive moderately stimulated helper T cells of IL-2 (Busse et al., 2010). It is important to note, however, that antigen-specific CD4+ T cells expand in the absence of the IL-2Rγ-chain (Lantz et al., 2000) and that a FOXP3 transgene restored Treg cells and protected against autoimmunity in Il-2Rβ−/− mice (Soper et al., 2007), both observations indicating that expansion of effector cells can occur in the absence of IL-2 and the suppressive activity of Treg cells can occur in the absence of IL-2 consumption and signaling.
In contrast, the model that we propose here does not include competition for IL-2 at the receptor/molecular level; however, in choosing the parameters, we imposed λ4IL-2 > λ2IL-2. Implicit in this inequality is the fact that regulatory T cells are more sensitive to IL-2 than IL-2-producing cells (Gavin et al., 2002; Almeida et al., 2006b; Hill et al., 2007; Lu et al., 2009; Stahl et al., 2009; Yu et al., 2009), but we do not assume that this limits the proliferative capacity of IL-2-producing cells. In our model, the limiting factor of proliferation for IL-2-producing cells is the number of cells themselves, for which regulatory cells play a major role through the suppressive term β.
Recent experimental work by Quiel et al. (2011) and mathematical work by Bocharov et al. (2011) provide a model based on the observation that the number of antigen-specific cells after immunization is not proportional to the number of precursors, and that the factor of expansion decreases as the number of precursors increases. The authors speculate that this phenomenon is due to crowding effects, and use a deterministic model with four CD4+ T cell populations that differ in their maturation and differentiation states (Bocharov et al., 2011). The authors assume that differentiated non-dividing T cells impose a feedback mechanism on less mature dividing T cells, which acts to increase the differentiation rates of these less mature cells, thereby limiting their proliferative capacity. This differs from our model, where proliferation will continue “unchecked” in the absence of regulatory cells.
There is already a large body of work devoted to the development of mathematical and computational models to study the dynamic interactions between the populations of Treg cells and effector CD4+ T cells. Early work focused on investigating different potential mechanisms of interaction between Treg cells and effector cells (Leon et al., 2003; Carneiro et al., 2007), occurring at conjugation sites present on the surface of antigen-presenting cells (APCs; Tang et al., 2006). Thus, Treg cells are assumed to be reliant on paracrine cytokines for their clonal expansion, and it is thought that IL-2 is the critical cytokine required for this expansion (Almeida et al., 2002, 2006b; Malek et al., 2002; Curotto de Lafaille et al., 2004; de la Rosa et al., 2004; Setoguchi et al., 2005). The “crossregulation model” explores the mathematical consequences of several of these assumptions, particularly the mechanisms by which Treg cells limit the proliferative capacity of effector cells (Carneiro et al., 2007).
Finally, we should mention other mathematical modeling efforts that have included bystander T cell activation (Kim et al., 2007b; Burroughs et al., 2011). Burroughs et al. (2011) make use of a dynamical systems approach to model a regulatory T cell population, a population of effector T cells that have received APC-mediated antigenic stimulus, and a bystander T cell population that is unresponsive to the antigen. The authors conclude that autoimmune conditions can arise following a primary immune response due to a bystander T cell population and that the equilibrium of the immune system depends on a balance between effector and regulatory T cell populations.
In conclusion, we have introduced a quantitative mathematical model of quorum-sensing for CD4+ T cell homeostasis. The model includes thymus output, cellular proliferation (TCR/IL-7 and IL-2-induced), differentiation and suppression, as well as death, linked to the levels of IL-2-producing cells in the case of regulatory T cells. The model has been used to explore the dynamics of CD4+ T cells during an immune response. Our results (Figures 2 and 3) show that there is a sharp increase in the number of IL-2-producing cells at days 4–5, followed by an increase in the number of activated/memory non-IL-2-producing cells. This leads to a large increase, around day 7 (which corresponds to the peak of the response) in the number of regulatory T cells, which is then followed by a sharp contraction phase that lasts around 2 days. Steady-state conditions or homeostasis are reached after a slower contraction phase that lasts around 3 weeks. Key features of the model are: (i) IL-2-producing cells, which correlate with IL-2 levels, dictate the proliferation rate of regulatory T cells and (ii) regulatory T cells control IL-2-producing cell differentiation to non-IL-2-producing cells. These two terms guarantee that CD4+ T cells are able to restrain their own proliferation by “sensing” the number of IL-2-producing cells, and thus preventing unbounded lymphocyte division as well as controlling the number of activated T cells and the total CD4+ T cell population.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the European Research Council (ERC), the Agence pour la Recherche sur le Cancer (ARC), the Institut Pasteur, and the Centre National pour la Recherche Scientifique (CNRS) to Antonio A. Freitas and through FP7 PIRSES-GA-2008-230665 (Joseph Reynolds, Grant Lythe, and Carmen Molina-París), BBSRC BB/F003811/1 (Grant Lythe and Carmen Molina-París), BBSRC BB/G023395/1 (Carmen Molina-París). Inês F. Amado was supported by the Fundacao para a Ciencia e Tecnologia (FCT), Portugal, Julien Berges by the Direction Recherche Enseignement et Technologie (DRET), and Joseph Reynolds by a White Rose studentship.
References
Almeida, A. R., Borghans, J. A., and Freitas, A. A. (2001). T cell homeostasis: thymus regeneration and peripheral T cell restoration in mice with a reduced fraction of competent precursors. J. Exp. Med. 194, 591–599.
Almeida, A. R., Legrand, N., Papiernik, M., and Freitas, A. A. (2002). Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J. Immunol. 169, 4850–4860.
Almeida, A. R., Rocha, B., Freitas, A. A., and Tanchot, C. (2005). Homeostasis of T cell numbers: from thymus production to peripheral compartmentalization and the indexation of regulatory T cells. Semin. Immunol. 17, 239–249.
Almeida, A. R., Zaragoza, B., and Freitas, A. A. (2006a). Competition controls the rate of transition between the peripheral pools of CD4+ CD25- and CD4+ CD25+ T cells. Int. Immunol. 18, 1607–1613.
Almeida, A. R., Zaragoza, B., and Freitas, A. A. (2006b). Indexation as a novel mechanism of lymphocyte homeostasis: the number of CD4+ CD25+ regulatory T cells is indexed to the number of IL-2-producing cells. J. Immunol. 177, 192–200.
Annacker, O., Burlen-Defranoux, O., Pimenta-Araujo, R., Cumano, A., and Bandeira, A. (2000). Regulatory CD4 T cells control the size of the peripheral activated/memory CD4 T cells compartment. J. Immunol. 164, 3573–3580.
Antony, P. A., Piccirillo, C. A., Akpinarli, A., Finkelstein, S. E., Speiss, P. J., Surman, D. R., Palmer, D. C., Chan, C. C., Klebanoff, C. A., Overwijk, W. W., Rosenberg, S. A., and Restifo, N. P. (2005). CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 174, 2591–2601.
Asano, M., Toda, M., Sakaguchi, N., and Sakaguchi, S. (1996). Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 184, 387–396.
Barthlott, T., Kassiotis, G., and Stockinger, B. (2003). T cell regulation as a side effect of homeostasis and competition. J. Exp. Med. 197, 451/460.
Bayer, A. L., Yu, A., Adeegbe, D., and Malek, T. R. (2005). Essential role for interleukin-2 for CD4(+)CD25(+) T regulatory cell development during the neonatal period. J. Exp. Med. 201, 769–777.
Belkaid, Y., and Rouse, B. T. (2005). Natural regulatory T cells in infectious disease. Nat. Immunol. 6, 353–360.
Bennett, C. L., Christie, J., Ramsdell, F., Brunkow, M. E., Ferguson, P. J., Whitesell, L., Kelly, T. E., Saulsbury, F. T., Chance, P. F., and Ochs, H. D. (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21.
Berzins, S. P., Boyd, R. L., and Miller, J. F. (1998). The role of the thymus and recent thymic migrants in the maintenance of the adult peripheral lymphocyte pool. J. Exp. Med. 187, 1839–1848.
Blair, P. J., Bultman, S. J., Haas, J. C., Rouse, B. T., Wilkinson, J. E., and Godfrey, V. L. (1994). CD4+ CD8- T cells are the effector cells in disease pathogenesis in the scurfy (sf) mouse. J. Immunol. 153, 3764–3774.
Bocharov, G., Quiel, J., Luzyanina, T., Alon, H., Chiglintsev, E., Chereshnev, V., Meier-Schellersheim, M., Paul, W. E., and Grossman, Z. (2011). Feedback regulation of proliferation vs. differentiation rates explains the dependence of CD4 T-cell expansion on precursor number. Proc. Natl. Acad. Sci. U.S.A. 108, 3318–3323.
Boyman, O., Kovar, M., Rubinstein, M. P., Surh, C. D., and Sprent, J. (2006). Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311, 1924–1927.
Brocker, T. (1997). Survival of mature CD4 T lymphocytes is dependent on major histocompatibility complex class II-expressing dendritic cells. J. Exp. Med. 186, 1223–1232.
Burchill, M. A., Yang, J., Vogtenhuber, C., Blazar, B. R., and Farrar, M. A. (2007). IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 178, 280–290.
Burroughs, N. J., Ferreira, M. F., Oliveira, B., and Pinto, A. A. (2011). Autoimmunity arising from bystander proliferation of T cells in an immune response model. Math. Comput. Model. 53, 1389–1393.
Busse, D., De La Rosa, M., Hobiger, K., Thurley, K., Flossdorf, M., Scheffold, A., and Hofer, T. (2010). Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc. Natl. Acad. Sci. U.S.A. 107, 3058–3063.
Carneiro, J., Leon, K., Caramalho, I., Van Den Dool, C., Gardner, R., Oliveira, V., Bergman, M. L., Sepulveda, N., Paixao, T., Faro, J., and Demengeot, J. (2007). When three is not a crowd: a Crossregulation model of the dynamics and repertoire selection of regulatory CD4+ T cells. Immunol. Rev. 216, 48–68.
Chaudhry, A., Samstein, R. M., Treuting, P., Liang, Y., Pils, M. C., Heinrich, J. M., Jack, R. S., Wunderlich, F. T., Bruning, J. C., Muller, W., and Rudensky, A. Y. (2011). Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566–578.
Chen, Y., Haines, C. J., Gutcher, I., Hochweller, K., Blumenschein, W. M., McClanahan, T., Hammerling, G., Li, M. O., Cua, D. J., and McGeachy, M. J. (2011). Foxp3(+) Regulatory T cells promote T helper 17 cell development in vivo through regulation of interleukin-2. Immunity 34, 409–421.
Cobb, B. S., Hertweck, A., Smith, J., O’Connor, E., Graf, D., Cook, T., Smale, S. T., Sakaguchi, S., Livesey, F. J., Fisher, A. G., and Merkenschlager, M. (2006). A role for Dicer in immune regulation. J. Exp. Med. 203, 2519–2527.
Curotto de Lafaille, M. A., Lino, A. C., Kutchukhidze, N., and Lafaille, J. J. (2004). CD25- T cells generate CD25+ Foxp3+ regulatory T cells by peripheral expansion. J. Immunol. 173, 7259–7268.
de Freitas, A. A. (2009). Tractus Immuno-Logicus: A Brief History of the Immune System. Austin: Landes Bioscience.
de la Rosa, M., Rutz, S., Dorninger, H., and Scheffold, A. (2004). Interleukin-2 is essential for CD4+ CD25+ regulatory T cell function. Eur. J. Immunol. 34, 2480–2488.
Diggle, S. P., Griffin, A. S., Campbell, G. S., and West, S. A. (2007). Cooperation and conflict in quorum-sensing bacterial populations. Nature 450, 411–414.
Fehervari, Z., Yamaguchi, T., and Sakaguchi, S. (2006). The dichotomous role of IL-2: tolerance versus immunity. Trends Immunol. 27, 109–111.
Feinerman, O., Jentsch, G., Tkach, K. E., Coward, J. W., Hathorn, M. M., Sneddon, M. W., Emonet, T., Smith, K. A., and Altan-Bonnet, G. (2010). Single-cell quantification of IL-2 response by effector and regulatory T cells reveals critical plasticity in immune response. Mol. Syst. Biol. 6, 437.
Fontenot, J. D., Gavin, M. A., and Rudensky, A. Y. (2003). Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat. Immunol. 4, 330–336.
Fontenot, J. D., Rasmussen, J. P., Gavin, M. A., and Rudensky, A. Y. (2005a). A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 6, 1142–1151.
Fontenot, J. D., Rasmussen, J. P., Williams, L. M., Dooley, J. L., Farr, A. G., and Rudensky, A. Y. (2005b). Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22, 329–341.
Freitas, A. A., Agenes, F., and Coutinho, G. C. (1996). Cellular competition modulates survival and selection of CD8+ T cells. Eur. J. Immunol. 26, 2640–2649.
Freitas, A. A., and Rocha, B. (2000). Population biology of lymphocytes: the flight for survival. Annu. Rev. Immunol. 18, 83–111.
Freitas, A. A., and Rocha, B. (2009). Homeostasis of naive T cells: the Foxo that fixes. Nat. Immunol. 10, 133–134.
Gavin, M. A., Clarke, S. R., Negrou, E., Gallegos, A., and Rudensky, A. (2002). Homeostasis and energy of CD4(+)CD25(+) suppressor T cells in vivo. Nat. Immunol. 3, 33–41.
Goudy, K. S., Johnson, M. C., Garland, A., Li, C., Samulski, R. J., Wang, B., and Tisch, R. (2011). Inducible adeno-associated virus-mediated IL-2 gene therapy prevents autoimmune diabetes. J. Immunol. 186, 3779–3786.
Gregersen, P. K., and Olsson, L. M. (2009). Recent advances in the genetics of autoimmune disease. Annu. Rev. Immunol. 27, 363–391.
Grinberg-Bleyer, Y., Baeyens, A., You, S., Elhage, R., Fourcade, G., Gregoire, S., Cagnard, N., Carpentier, W., Tang, Q., Bluestone, J., Chatenoud, L., Klatzmann, D., Salomon, B. L., and Piaggio, E. (2010). IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J. Exp. Med. 207, 1871–1878.
Guimond, M., Veenstra, R. G., Grindler, D. J., Zhang, H., Cui, Y., Murphy, R. D., Kim, S. Y., Na, R., Hennighausen, L., Kurtulus, S., Erman, B., Matzinger, P., Merchant, M. S., and Mackall, C. L. (2009). Interleukin 7 signaling in dendritic cells regulates the homeostatic proliferation and niche size of CD4+ T cells. Nat. Immunol. 10, 149–157.
Hill, J. A., Feuerer, M., Tash, K., Haxhinasto, S., Perez, J., Melamed, R., Mathis, D., and Benoist, C. (2007). Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800.
Hori, S., Nomura, T., and Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061.
Huang, C. T., Workman, C. J., Flies, D., Pan, X., Marson, A. L., Zhou, G., Hipkiss, E. L., Ravi, S., Kowalski, J., Levitsky, H. I., Powell, J. D., Pardoll, D. M., Drake, C. G., and Vignali, D. A. (2004). Role of LAG-3 in regulatory T cells. Immunity 21, 503–513.
Huber, S., Gagliani, N., Esplugues, E., O’Connor, W. Jr., Huber, F. J., Chaudhry, A., Kamanaka, M., Kobayashi, Y., Booth, C. J., Rudensky, A. Y., Roncarolo, M. G., Battaglia, M., and Flavell, R. A. (2011). Th17 cells express interleukin-10 receptor and are controlled by Foxp3 and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565.
Kalia, V., Sarkar, S., Subramaniam, S., Haining, W. N., Smith, K. A., and Ahmed, R. (2010). Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 32, 91–103.
Kerdiles, Y. M., Beisner, D. R., Tinoco, R., Dejean, A. S., Castrillon, D. H., Depinho, R. A., and Hedrick, S. M. (2009). Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat. Immunol. 10, 176–184.
Khattri, R., Kasprowicz, D., Cox, T., Mortrud, M., Appleby, M. W., Brunkow, M. E., Ziegler, S. F., and Ramsdell, F. (2001). The amount of scurfin protein determines peripheral T cell number and responsiveness. J. Immunol. 167, 6312–6320.
Kim, J. M., Rasmussen, J. P., and Rudensky, A. Y. (2007a). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 8, 191–197.
Kim, P. S., Lee, P. P., and Levy, D. (2007b). Modeling regulation mechanisms in the immune system. J. Theor. Biol. 246, 33–69.
Kirberg, J., Berns, A., and Von Boehmer, H. (1997). Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex-encoded molecules. J. Exp. Med. 186, 1269–1275.
Knoechel, B., Lohr, J., Kahn, E., Bluestone, J. A., and Abbas, A. K. (2005). Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J. Exp. Med. 202, 1375–1386.
Komatsu, N., Mariotti-Ferrandiz, M. E., Wang, Y., Malissen, B., Waldmann, H., and Hori, S. (2009). Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. U.S.A. 106, 1903–1908.
Kundig, T. M., Schorle, H., Bachmann, M. F., Hengartner, H., Zinkernagel, R. M., and Horak, I. (1993). Immune responses in interleukin-2-deficient mice. Science 262, 1059–1061.
Labrecque, N., Whitfield, L. S., Obst, R., Waltzinger, C., Benoist, C., and Mathis, D. (2001). How much TCR does a T cell need? Immunity 15, 71–82.
Lahl, K., Loddenkemper, C., Drouin, C., Freyer, J., Arnason, J., Eberl, G., Hamann, A., Wagner, H., Huehn, J., and Sparwasser, T. (2007). Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 204, 57–63.
Lantz, O., Grandjean, I., Matzinger, P., and Di Santo, J. P. (2000). Gamma chain required for naive CD4+ T cell survival but not for antigen proliferation. Nat. Immunol. 1, 54–58.
Leon, K., Lage, A., and Carneiro, J. (2003). Tolerance and immunity in a mathematical model of T-cell mediated suppression. J. Theor. Biol. 225, 107–126.
Leuchars, E., Wallis, V. J., Doenhoff, M. J., Davies, A. J., and Kruger, J. (1978). Studies of hyperthymic mice. I. The influence of multiple thymus grafts on the size of the peripheral T cell pool and immunological performance. Immunology 35, 801–809.
Link, A., Vogt, T. K., Favre, S., Britschgi, M. R., Acha-Orbea, H., Hinz, B., Cyster, J. G., and Luther, S. A. (2007). Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat. Immunol. 8, 1255–1265.
Liston, A., Lu, L. F., O’Carroll, D., Tarakhovsky, A., and Rudensky, A. Y. (2008). Dicer-dependent microRNA pathway safeguards regulatory T cell function. J. Exp. Med. 205, 1993–2004.
Lu, L. F., Thai, T. H., Calado, D. P., Chaudhry, A., Kubo, M., Tanaka, K., Loeb, G. B., Lee, H., Yoshimura, A., Rajewsky, K., and Rudensky, A. Y. (2009). Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity 30, 80–91.
Malek, T. R., and Castro, I. (2010). Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity 33, 153–165.
Malek, T. R., Porter, B. O., Codias, E. K., Scibelli, P., and Yu, A. (2000). Normal lymphoid homeostasis and lack of lethal autoimmunity in mice containing mature T cells with severely impaired IL-2 receptors. J. Immunol. 164, 2905–2914.
Malek, T. R., Yu, A., Vincek, V., Scibelli, P., and Kong, L. (2002). CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity 17, 167–178.
Maloy, K. J., and Powrie, F. (2001). Regulatory T cells in the control of immune pathology. Nat. Immunol. 2, 816–822.
Miller, J. F. (1965). Effect of thymectomy in adult mice on immunological responsiveness. Nature 208, 1337–1338.
Miller, M. B., and Bassler, B. L. (2001). Quorum sensing in bacteria. Annu. Rev. Microbiol. 55, 165–199.
Moriggl, R., Topham, D. J., Teglund, S., Sexl, V., McKay, C., Wang, D., Hoffmeyer, A., Van Deursen, J., Sangster, M. Y., Bunting, K. D., Grosveld, G. C., and Ihle, J. N. (1999). Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity 10, 249–259.
Nelson, B. H., and Willerford, D. M. (1998). Biology of the interleukin-2 receptor. Adv. Immunol. 70, 1–81.
Ono, M., Yaguchi, H., Ohkura, N., Kitabayashi, I., Nagamura, Y., Nomura, T., Miyachi, Y., Tsukada, T., and Sakaguchi, S. (2007). Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446, 685–689.
Ouyang, W., Beckett, O., Flavell, R. A., and Li, M. O. (2009). An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity 30, 358–371.
Pandiyan, P., Conti, H. R., Zheng, L., Peterson, A. C., Mathern, D. R., Hernandez-Santos, N., Edgerton, M., Gaffen, S. L., and Lenardo, M. J. (2011). CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity 34, 422–434.
Pipkin, M. E., Sacks, J. A., Cruz-Guilloty, F., Lichtenheld, M. G., Bevan, M. J., and Rao, A. (2010). Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90.
Polic, B., Kunkel, D., Scheffold, A., and Rajewsky, K. (2001). How alpha beta T cells deal with induced TCR alpha ablation. Proc. Natl. Acad. Sci. U.S.A. 98, 8744–8749.
Powrie, F., Leach, M. W., Mauze, S., Caddle, L. B., and Coffman, R. L. (1993). Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5, 1461–1471.
Quiel, J., Caucheteux, S., Laurence, A., Singh, N. J., Bocharov, G., Ben-Sasson, S. Z., Grossman, Z., and Paul, W. E. (2011). Antigen-stimulated CD4 T-cell expansion is inversely and log-linearly related to precursor number. Proc. Natl. Acad. Sci. U.S.A. 108, 3312–3317.
Sadlack, B., Lohler, J., Schorle, H., Klebb, G., Haber, H., Sickel, E., Noelle, R. J., and Horak, I. (1995). Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur. J. Immunol. 25, 3053–3059.
Sakaguchi, S. (2000). Regulatory T cells: key controllers of immunologic self-tolerance. Cell 101, 455–458.
Sakaguchi, S. (2004). Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22, 531–562.
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M., and Toda, M. (1995). Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164.
Sallusto, F., Geginat, J., and Lanzavecchia, A. (2004). Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22, 745–763.
Schluns, K. S., Kieper, W. C., Jameson, S. C., and Lefrancois, L. (2000). Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 1, 426–432.
Schorle, H., Holtschke, T., Hunig, T., Schimpl, A., and Horak, I. (1991). Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature 352, 621–624.
Setoguchi, R., Hori, S., Takahashi, T., and Sakaguchi, S. (2005). Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 201, 723–735.
Shevach, E. M. (2009). Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645.
Snow, J. W., Abraham, N., Ma, M. C., Herndier, B. G., Pastuszak, A. W., and Goldsmith, M. A. (2003). Loss of tolerance and autoimmunity affecting multiple organs in STAT5A/5B-deficient mice. J. Immunol. 171, 5042–5050.
Soper, D. M., Kasprowicz, D. J., and Ziegler, S. F. (2007). IL-2Rbeta links IL-2R signaling with Foxp3 expression. Eur. J. Immunol. 37, 1817–1826.
Stahl, H. F., Fauti, T., Ullrich, N., Bopp, T., Kubach, J., Rust, W., Labhart, P., Alexiadis, V., Becker, C., Hafner, M., Weith, A., Lenter, M. C., Jonuleit, H., Schmitt, E., and Mennerich, D. (2009). miR-155 inhibition sensitizes CD4+ Th cells for TREG mediated suppression. PLoS ONE 4, e7158. doi:10.1371/journal.pone.0007158
Suzuki, H., Kundig, T. M., Furlonger, C., Wakeham, A., Timms, E., Matsuyama, T., Schmits, R., Simard, J. J., Ohashi, P. S., Griesser, H., Taniguchi, T., Paige, C. J., and Mak, T. W. (1995). Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science 268, 1472–1476.
Swain, S. L., Hu, H., and Huston, G. (1999). Class II-independent generation of CD4 memory T cells from effectors. Science 286, 1381–1383.
Takeda, K., Tanaka, T., Shi, W., Matsumoto, M., Minami, M., Kashiwamura, S., Nakanishi, K., Yoshida, N., Kishimoto, T., and Akira, S. (1996). Essential role of Stat6 in IL-4 signalling. Nature 380, 627–630.
Tan, J. T., Dudl, E., Leroy, E., Murray, R., Sprent, J., Weinberg, K. I., and Surh, C. D. (2001). IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc. Natl. Acad. Sci. U.S.A. 98, 8732–8737.
Tanchot, C., Lemonnier, F. A., Perarnau, B., Freitas, A. A., and Rocha, B. (1997). Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science 276, 2057–2062.
Tanchot, C., and Rocha, B. (1995). The peripheral T cell repertoire: independent homeostatic regulation of virgin and activated CD8+ T cell pools. Eur. J. Immunol. 25, 2127–2136.
Tang, Q., Adams, J. Y., Tooley, A. J., Bi, M., Fife, B. T., Serra, P., Santamaria, P., Locksley, R. M., Krummel, M. F., and Bluestone, J. A. (2006). Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 7, 83–92.
Tang, Q., Adams, J. Y., Penaranda, C., Melli, K., Piaggio, E., Sgouroudis, E., Piccirillo, C. A., Salomon, B. L., and Bluestone, J. A. (2008). Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28, 687–697.
Vivien, L., Benoist, C., and Mathis, D. (2001). T lymphocytes need IL-7 but not IL-4 or IL-6 to survive in vivo. Int. Immunol. 13, 763–768.
Webster, K. E., Walters, S., Kohler, R. E., Mrkvan, T., Boyman, O., Surh, C. D., Grey, S. T., and Sprent, J. (2009). In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 206, 751–760.
Wildin, R. S., Ramsdell, F., Peake, J., Faravelli, F., Casanova, J. L., Buist, N., Levy-Lahad, E., Mazzella, M., Goulet, O., Perroni, L., Bricarelli, F. D., Byrne, G., McEuen, M., Proll, S., Appleby, M., and Brunkow, M. E. (2001). X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 27, 18–20.
Willerford, D. M., Chen, J., Ferry, J. A., Davidson, L., Ma, A., and Alt, F. W. (1995). Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3, 521–530.
Williams, M. A., Tyznik, A. J., and Bevan, M. J. (2006). Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature 441, 890–893.
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., Nomura, T., and Sakaguchi, S. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275.
Wolf, M., Schimpl, A., and Hunig, T. (2001). Control of T cell hyperactivation in IL-2-deficient mice by CD4(+)CD25(-) and CD4(+)CD25(+) T cells: evidence for two distinct regulatory mechanisms. Eur. J. Immunol. 31, 1637–1645.
Wu, Y., Borde, M., Heissmeyer, V., Feuerer, M., Lapan, A. D., Stroud, J. C., Bates, D. L., Guo, L., Han, A., Ziegler, S. F., Mathis, D., Benoist, C., Chen, L., and Rao, A. (2006). FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126, 375–387.
Yamanouchi, J., Rainbow, D., Serra, P., Howlett, S., Hunter, K., Garner, V. E., Gonzalez-Munoz, A., Clark, J., Veijola, R., Cubbon, R., Chen, S. L., Rosa, R., Cumiskey, A. M., Serreze, D. V., Gregory, S., Rogers, J., Lyons, P. A., Healy, B., Smink, L. J., Todd, J. A., Peterson, L. B., Wicker, L. S., and Santamaria, P. (2007). Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat. Genet. 39, 329–337.
Yao, Z., Kanno, Y., Kerenyi, M., Stephens, G., Durant, L., Watford, W. T., Laurence, A., Robinson, G. W., Shevach, E. M., Moriggl, R., Hennighausen, L., Wu, C., and O’Shea, J. J. (2007). Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood 109, 4368–4375.
Yu, A., Zhu, L., Altman, N. H., and Malek, T. R. (2009). A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity 30, 204–217.
Zaragoza, B., Evaristo, C., Kissenpfennig, A., Libri, V., Malissen, B., Rocha, B., Freitas, A. A., and Almeida, A. R. (2011). Cell-to-cell interactions and signals involved in the reconstitution of peripheral CD8 T(CM) and T(EM) cell pools. PLoS ONE 6, e17423. doi:10.1371/journal.pone.0017423
Keywords: CD4+ T cells, regulatory T cells, IL-2, autoimmunity, immune-therapy, homeostasis, quorum sensing, mathematical modeling
Citation: Almeida ARM, Amado IF, Reynolds J, Berges J, Lythe G, Molina-París C and Freitas AA (2012) Quorum-sensing in CD4+ T cell homeostasis: a hypothesis and a model. Front. Immun. 3:125. doi: 10.3389/fimmu.2012.00125
Afonso R. M. Almeida and Inês F. Amado are the first co-authors.
Received: 13 March 2012; Paper pending published: 06 April 2012;
Accepted: 02 May 2012; Published online: 25 May 2012.
Edited by:
Kendall A. Smith, Weill Medical College of Cornell University, USAReviewed by:
Kendall A. Smith, Weill Medical College of Cornell University, USAShohei Hori, RIKEN, Japan
Copyright: © 2012 Almeida, Amado, Reynolds, Berges, Lythe, Molina-París and Freitas. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Antonio A. Freitas, Institut Pasteur, Département d’Immunologie, Unité de Biologie des Populations Lymphocytaires, 25, rue du Dr. Roux, 75015 Paris, France. e-mail:YW50b25pby5mcmVpdGFzQHBhc3RldXIuZnI=
†Afonso R. M. Almeida and Inês F. Amado are the first co-authors.