 Alessio Montresor
Alessio Montresor
- Division of General Pathology, Department of Pathology, University of Verona, Verona, Italy
Integrin-mediated adhesion is a general concept referring to a series of adhesive phenomena including tethering–rolling, affinity, valency, and binding stabilization altogether controlling cell avidity (adhesiveness) for the substrate. Arrest chemokines modulate each aspect of integrin activation, although integrin affinity regulation has been recognized as the prominent event in rapid leukocyte arrest induced by chemokines. A variety of inside-out and outside-in signaling mechanisms have been related to the process of integrin-mediated adhesion in different cellular models, but only few of them have been clearly contextualized to rapid integrin affinity modulation by arrest chemokines in primary leukocytes. Complex signaling processes triggered by arrest chemokines and controlling leukocyte integrin activation have been described for ras-related rap and for rho-related small GTPases. We summarize the role of rap and rho small GTPases in the regulation of rapid integrin affinity in primary leukocytes and provide a modular view of these pro-adhesive signaling events. A potential, albeit still speculative, mechanism of rho-mediated regulation of cytoskeletal proteins controlling the last step of integrin activation is also discussed. We also discuss data suggesting a functional integration between the rho- and rap-modules of integrin activation. Finally we examine the universality of signaling mechanisms regulating integrin triggering by arrest chemokines.
Introduction
Leukocytes spend the majority of their life circulating into blood and lymphoid vessels until local environmental cues claim their presence into sites of immune response. The capability to resist to extreme hemodynamic stress and turbulence within high diameter vessels and to avoid cell–cell aggregation in the circulation are prerequisites to allow leukocytes to embrace blood and lymph vessels like highways leading to the widest possible distribution of the immune system in the organism. However, there are adverse implications. Indeed, nothing can be more difficult for a cell than trying to stop its motion under the frantic flow conditions generated by the circulation. These conditions are important to regulate cell trafficking, by imposing physical thresholds to leukocyte recruitment, with the flow itself providing mechano-chemical signals regulating leukocyte trafficking (Zhu et al., 2000; McEver and Zhu, 2010). But at the end, to fulfill their duties, leukocytes must be able to counteract the pushing force generated by the flow, arresting on the surface of endothelial cells and transmigrating into tissues. Everything must be done within few second or less to cope with the timing imposed by flow dynamics. In the past two decades consistent efforts have been made to understand the physiology and molecular bases of the leukocyte recruitment process and a general, widely validated, model describing the entire process has been generated (Laudanna and Alon, 2006; Ley et al., 2007; Alon and Shulman, 2011). A critical step in this process is the transition from rolling to stable arrest, which is the moment when leukocytes become fully resistant to the flow and definitively stop on the vessel wall. This critical phase is mediated by a family of leukocyte-expressed cytoskeleton-regulated adhesive receptors, called integrins (Takada et al., 2007). Integrins are capable of establishing sudden and very stable adhesive interactions with endothelial ligands expressed on the inner surface of the vessels and belonging to the immunoglobulin superfamily. The strong adhesive interaction between integrins and their ligands supporting arrest of circulating leukocytes is, de facto, the primum movens of the immune response.
Integrins basally interact with the ligand with rather low affinity. To increase binding efficiency integrins must undergo dramatic structural and topological modifications consisting of extensive conformational changes leading to increased affinity for the ligand, along with concurrent spatial rearrangement on the cell plasma membrane. This phenomenon is globally indicated as integrin activation and is mandatory to rapid arrest of circulating cells. The step of integrin activation is finely regulated to allow diversity of leukocyte recruitment, but its most distinguishing property is the speediness. This implies the existence of environmental factors capable of activating integrins with corresponding dynamics. The original discovery of a role for a PTX-sensitive Gai-protein linked signaling in regulation of lymphocyte homing (Bargatze and Butcher, 1993) prompted the search for microvessel-presented agonists capable of triggering integrin-dependent arrest within seconds. It is now established that arrest chemokines, a subgroup of chemotactic cytokines capable of rapid integrin activation (Rot and von Andrian, 2004; Bromley et al., 2008), are the most potent physiological activator of leukocyte adhesiveness. They do that by triggering complex signaling transduction mechanisms leading to extremely rapid activation of integrins and, ultimately, of adhesion. In this context, the most studied and best-known signaling events are represented by the signaling networks regulated by the small GTP binding proteins of the ras-like rap and rho family (Caron, 2003; Scheele et al., 2007; Tybulewicz and Henderson, 2009). More than 670 interacting proteins belong to these signaling networks, including upstream regulators and downstream effectors (see http://www.pathwaycommons.org/pc/), with specific sub-sets of these interactions devoted to modulation of leukocyte integrin activation and dependent adhesion. Here we will summarize the available data about the signaling mechanisms triggered by arrest chemokines and controlling rapid integrin affinity transitions critical to leukocyte arrest.
Integrin-Dependent Adhesion: An Overview
At least two distinct modalities of integrin activation are known, namely conformational changes, leading to increased affinity, and lateral mobility leading to increased valency, both concurrently enhancing cell avidity (adhesiveness; Arnaout et al., 2005). The most detailed information about integrin structural rearrangement during affinity up-regulation in leukocytes comes from studies of LFA-1. Recent structural and biophysical data predict that LFA-1 exists in at least three conformational states, which differ both in their overall extension over the plasma membrane as well as in the arrangement of their headpiece (Carman and Springer, 2003; Luo et al., 2007; Springer and Dustin, 2011; Figure 1). Inside-out signaling events trigger integrins to undergo a dramatic transition from a bent low-affinity conformation to an extended intermediate-affinity to a high-affinity conformation, characterized by a complete opening of the ligand-binding pocket (Nishida et al., 2006; Luo et al., 2007). Extended β2 integrin conformations with high topographical availability of the ligand-binding headpiece but low affinity for the ligand have been also postulated (Salas et al., 2002, 2006). This extended but low/intermediate-affinity conformation may increase the capability of LFA-1 to mediate rolling on ICAM-1 upon selectin triggering (Chesnutt et al., 2006; Zarbock et al., 2007; Miner et al., 2008).
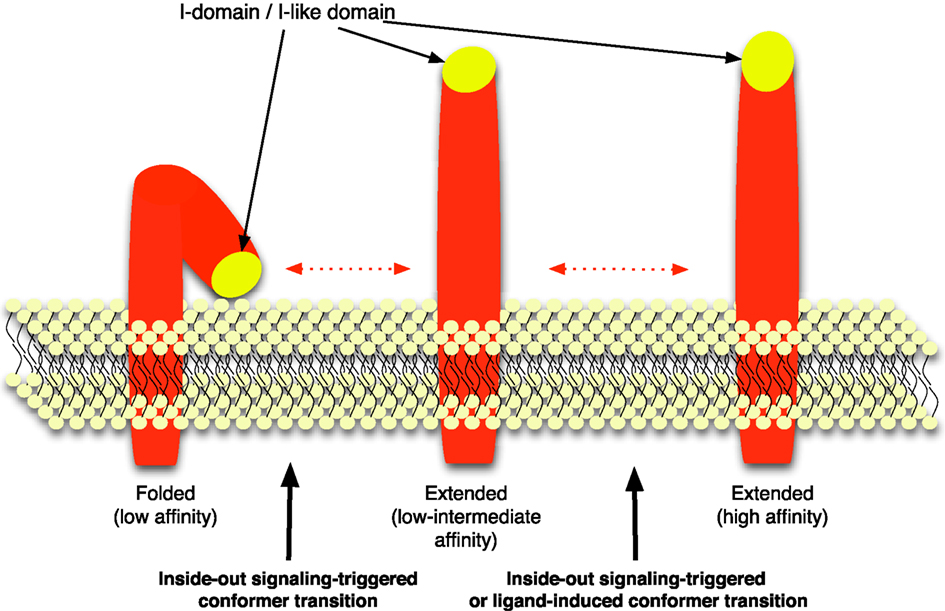
Figure 1. Integrin affinity triggering. The diagram refers to the LFA-1 affinity-triggering model. Shown is the dynamic equilibrium between three conformers displaying low-, low/intermediate-, and high-affinity for ICAM-1. The progressive extension of the heterodimer is accompanied by increasing topological availability of the I-domain and I-like domain (in yellow), which are involved in ligand binding with increasing affinity.
It is important to emphasize that low-, intermediate-, and high-affinity integrins likely represent discrete, reversible, states in a continuum of integrin conformational changes (Figure 1). Thus, the correct interpretation of the integrin activation process is a dynamic equilibrium between different conformers, corresponding to inside-out triggered conformational changes of the heterodimer displaying increasing affinity (binding energy) for the ligand (Carman and Springer, 2003; Shamri et al., 2005). Importantly, it has been demonstrated that LFA-1 integrin conformational changes are critical to the in vivo arrest of lymphocytes on the high endothelial venules (HEV) of secondary lymphoid organs (Giagulli et al., 2004). Thus, modulation of integrin affinity is now recognized as the critical step to leukocyte arrest in vivo.
Notably, integrins may undergo not only rapid inside-out regulation of affinity but also structural stabilization transmitted to the cytosolic tail by the bound ligand. The two processes are believed to cooperate to enhance integrin-mediated adhesiveness in a rapid and reversible manner. For instance, the induction of conformational changes transmitted to the cytosolic tail of the LFA-1 heterodimer upon ICAM-1 binding (Kim et al., 2003) may have a role in stabilizing leukocytes arrest under flow. However, this bi-directional regulation of integrin binding may be insufficient to support prolonged shear-resistant firm adhesions, and so post ligand occupancy events leading to integrin anchorage to the cytoskeleton are apparently required to further increase mechanical stability of individual ligand-occupied integrins (Cairo et al., 2006). Notably, these phenomena can be concurrent to the other modality of integrin activation, that is valency up-regulation. Heterodimer lateral mobility and valency increase is also regulated by interaction with the cytoskeleton, which could behave as mobility restrain (Stewart et al., 1998; van Kooyk and Figdor, 2000; Svensson et al., 2010; Bakker et al., 2012). Thus, it seems that, to stabilize the adhesion, a shuttling between restraining and stabilizing cytoskeletal proteins must occur. Valency up-regulation can be directly triggered by chemokine signaling leading to formation of multivalent complex on the plasma membrane. This may have a role under specific conditions by facilitating the encountering of activated mobile integrins with the immobilized ligand (Constantin et al., 2000). Furthermore, increase of integrin valency may also contribute to the initiation of outside-in signaling cascades, leading to the efficient recruitment of protein tyrosine kinases (PTKs; Berton et al., 2005) and the initiation of the full repertoire of outside-in signaling pathways leading to adhesion stabilization. Notably, it has been shown that lacking of integrin signaling capability leads to accelerated leukocyte detachment (Giagulli et al., 2006). Thus, chemoattractant-triggered inside-out and integrin-initiated outside-in signaling events concurrently cooperate to increase integrin affinity for the ligand and to stabilize and prolong the arrest of circulating leukocytes.
The Role of rap and rho in Integrin Affinity Modulation
A plethora of signaling events have been implicated in the regulation of various kinetic aspects of integrin-mediated adhesion. Overall, at least 65 signaling proteins are possibly involved in the regulation of integrin-mediated adhesion by chemoattractants and other agonists (Table 1). However, it is important to emphasize that only a subset of this group of signaling molecules has been validated under physiological conditions significant to chemokine-modulated rapid arrest of circulating leukocytes. These conditions, which we may call “the four criteria,” should include: (a) evaluation of signaling events in primary leukocytes; (b) evaluation of adhesion under flow conditions; (c) measurement of rapid kinetics of adhesion triggering (seconds or less); and (d) direct detection of heterodimer conformational changes. The rationale for adopting such criteria is based of the following considerations: (a) Signaling studies should be always accurately contextualized, by first focusing on physiological, standard, conditions followed by comparative analysis in more specific contexts. For instance, neoplastic leukocyte cell lines are not appropriate models of physiologic leukocyte adhesion since the neoplastic transformation may alter the signaling machinery with respect to normal primary cells, thus affecting response to the agonists and data interpretation. (b) Flow is the natural condition during cell recruitment by generating a shear stress, which imposes a mechanistic threshold to adhesion activation; thus, the efficacy of signaling events in regulating leukocyte arrest should be always challenged by applying flow conditions. (c) Integrin activation under-flow occurs in the range of seconds or less (likely milliseconds); thus, to correlate signaling events to integrin activation relevant to leukocyte recruitment, the kinetics of the two events must be coherent. Such kinetics cannot be studied in static assays. (d) As stated above, integrin-mediated adhesion is a general concept. To precisely assess whether a signaling event specifically regulates integrin conformational changes leading to affinity increase it is necessary to be able to directly detect integrin structural rearrangements by means of soluble ligand-binding assays or reporter monoclonal antibodies detecting activation epitopes. These four criteria should be always satisfied in order to correlate signaling events to affinity triggering controlling leukocyte rapid arrest in physiological contexts. Unfortunately, these experimental criteria are not systematically applied in the literature and this may affect the correct interpretation of the regulatory role of pro-adhesive signal transduction events.
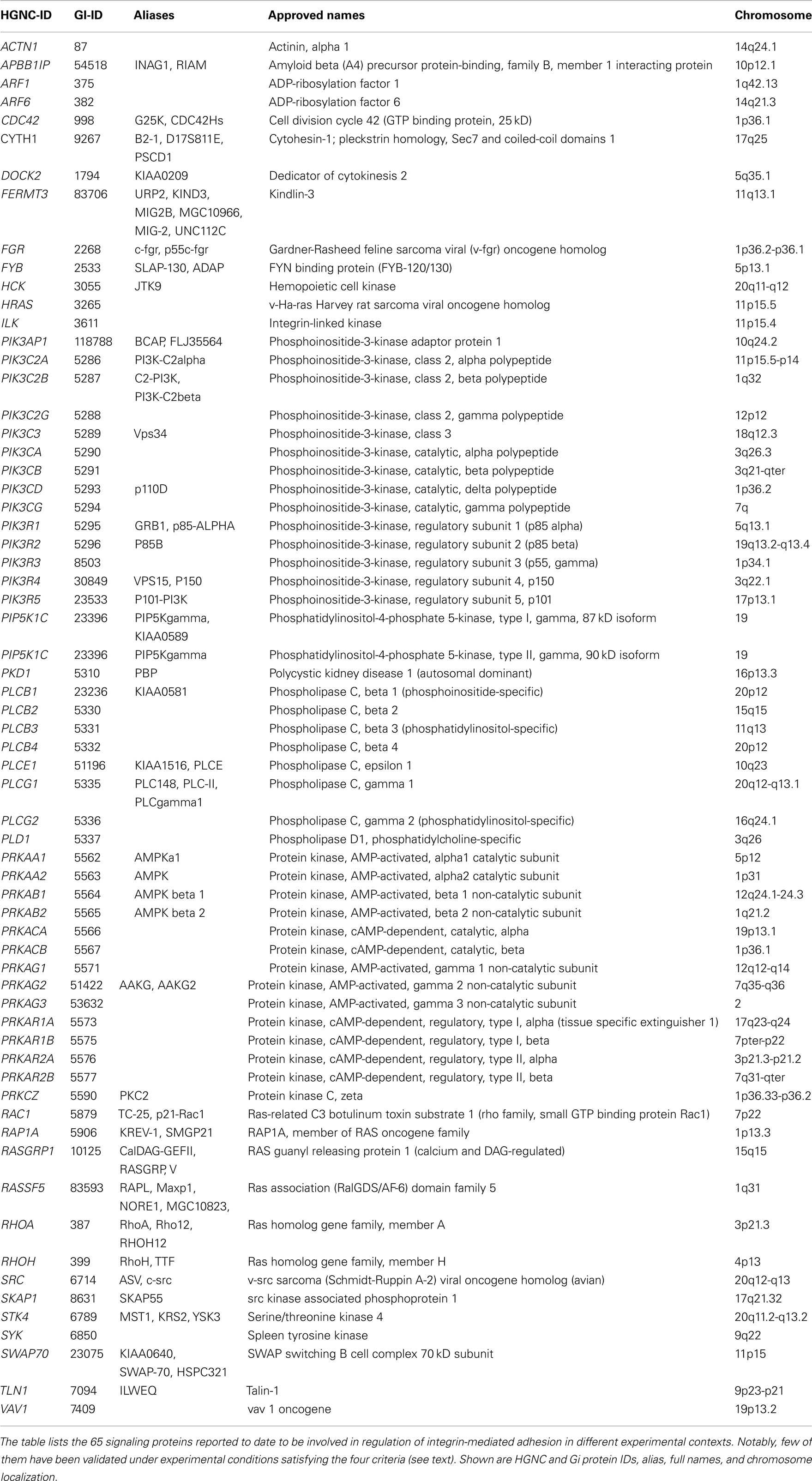
Table 1. The signaling molecules involved in the regulation of integrin-dependent adhesion in leukocytes.
The intracellular signaling cascade from arrest chemokines, such as CCL19, CCL21, or CXCL12, to integrin affinity modulation is still incompletely understood. The two most studied and validated signaling mechanisms activated by arrest chemokines and leading to up-regulation of integrin affinity, especially in the context of the β2 integrin LFA-1, are related to signaling delivered by the small GTP binding proteins of the rap and rho family. Both rap and rho are capable of activating a variety of downstream effectors and are, in turn, activated by several upstream regulators (Caron, 2003; Scheele et al., 2007; Tybulewicz and Henderson, 2009). However, in the specific context of integrin activation by arrest chemokines, rap and rho regulate restricted signaling modules devoted to the specific control of integrin affinity.
The role of the rap isoform Rap1A in integrin activation by arrest chemokines has been extensively studied and it is now clearly demonstrated that Rap1A regulates rapid integrin-dependent adhesion either in the context of the β2 integrin LFA-1 as well as β1 integrin VLA-4 (Duchniewicz et al., 2006). Rap1A was shown to control arrest of rolling lymphocytes as well as in vivo homing to secondary lymphoid organs (Ebisuno et al., 2010). Rap1A is activated by arrest chemokines by means of an upstream signaling mechanism involving phospholipase C (PLC). Indeed, ligation of chemokine receptors, and more in general of all chemoattractants Gαi-protein coupled receptors (GPCRs), results in rapid intracellular calcium influx and activation of PLC (particularly of the β isoforms), which leads to generation of inositol-1,4,5-trisphosphate (IP3, which further increases intracellular calcium from intracellular stores) and diacylglycerol (DAG). Calcium and DAG are, in turn, critical to activate the rap-specific guanine-nucleotide-exchange factor (GEF) calcium- and DAG-regulated GEF (CALDAG–GEF), also known as RAPGEF2; Crittenden et al., 2004; Bergmeier et al., 2007), which, ultimately, activates Rap1A. The role of other rap GEFs, such as C3G (RAPGEF1) and EPAC (RAPGEF3), in the context of chemokine signaling leading to integrin affinity regulation, is still not addressed. Once activated, Rap1A transmits downstream signals through different effectors, including RAPL (RASSF5; Ebisuno et al., 2010) RIAM (APBB1IP; Lafuente and Boussiotis, 2006), MST1 (STK4; Katagiri et al., 2006), SKAP55 (SKAP1), and ADAP (FYB; Menasche et al., 2007; see below). Although these signaling events lead to pro-adhesive events, the role of these signaling molecules in chemokine-triggered integrin affinity regulation mediating leukocyte arrest is still not clarified. Notably, in monocytes, PLC-mediated calcium signaling is required for induction of high-affinity α4-integrin ligation and monocyte arrest (Hyduk et al., 2007). However, an important recent study showed that Rap1A is, de facto, unable to modulate LFA-1 affinity conformeric transitions, thus apparently implicating Rap1A and its effectors in adhesive events other than integrin affinity regulation (Ebisuno et al., 2010), such as removal of cytosolic constrains or post-binding stabilization (Ebisuno et al., 2010).
A role for the rho isoform RhoA in chemoattractant-induced rapid integrin activation was originally suggested in the context of the β1 integrin VLA-4 (Laudanna et al., 1996), although those studies did not fully satisfied the four criteria described above. However, more recently it was clearly demonstrated that, under physiological conditions, RhoA and Rac1 mediate LFA-1 affinity triggering by arrest chemokines (Giagulli et al., 2004; Bolomini-Vittori et al., 2009). The role of rho-specific GEF in mediating RhoA and/or Rac1 activation by arrest chemokines is still not fully clarified, although recent data show that DOCK2 (dedicator of cytokinesis 2; Garcia-Bernal et al., 2006) and VAV1 (Gakidis et al., 2004; Garcia-Bernal et al., 2005) may participate to leukocyte integrin affinity modulation. In other studies, however, VAV1 seems to have a negative regulatory role on VLA-4 affinity regulation (Garcia-Bernal et al., 2009). Once activated, RhoA and Rac1 activate a variety of downstream effectors, but only few of them have been tested as effectors to integrin affinity modulation by chemokines. In this context, PLD1 and the 87 kD isoform of PIP5K1C have been demonstrated to play a critical role in LFA-1 affinity modulation by chemokines. Thus, RhoA- and Rac1-activated PLD1 was shown to control chemokine triggering of LFA-1 extensions corresponding to both intermediate and high affinity states. Moreover, by leading to plasma membrane accumulation of phosphatidic acid, PLD1 mediates the activation of PIP5K1C. However, PIP5K1C was shown to control LFA-1 affinity triggering by chemokines in a conformer-selective manner, with transition from intermediate to high affinity, but not from low to intermediate affinity, states controlled by the kinase activity of PIP5K1C (Bolomini-Vittori et al., 2009). These findings have important implication for our comprehension of LFA-1 affinity modulation. Indeed, these data show that the complete LFA-1 conformeric transition from a bent to a fully extended structure is accurately controlled at the level of inside-out signal transduction, even in absence of ICAM-1 interaction with LFA-1 in extended intermediate affinity state (Bolomini-Vittori et al., 2009). Thus, arrest chemokines are fully competent to trigger a complete LFA-1 affinity transition supporting arrest of rolling leukocytes. In contrast, in the context of rho-mediated LFA-1 affinity modulation, CDC42 was shown to negatively regulate LFA-1 affinity triggering by chemokines, thus establishing a sharp dichotomy with respect to the other two most homologous rho small GTPases (Bolomini-Vittori et al., 2009). CDC42 seems to affect LFA-1-mediated adhesion by blocking PIP5K1C activation (Bolomini-Vittori et al., 2009). Moreover, it was recently shown that activated CDC42 also inhibits Rap1A activation by chemoattractants (Kempf et al., 2011), thus highlighting a complex negative regulatory role for CDC42 in adhesion-de-adhesion cycling. Overall, arrest chemokines regulate integrin-dependent rapid adhesion by means of two main signaling modules: (a) the rap-module, likely including PLC, CALDAG–GEF, Rap1A, RAPL, RIAM, STK4, SKAP55, and ADAP; (b) the rho-module likely including at least DOCK2, VAV1, RhoA, Rac1, CDC42, PLD1, and PIP5K1C.
Deep in the Modules: The Very Downstream Events
Several cytosolic proteins, either or not of cytoskeletal nature, have been shown to directly interact with the integrin cytoplasmic tails of both alpha and beta chains and to regulate integrin functionality (Alon, 2010; Hogg et al., 2011). The most proximal to the heterodimer (downstream) signaling event leading to integrin affinity triggering is likely represented by interaction with actin-binding proteins. Among them, Talin-1 (TLN1) is the most studied actin-binding protein implicated in triggering integrin affinity up-regulation. Talin-1 is an anti-parallel homodimer. The F3 region of the head domain interacts with the cytoplasmic tail of the β chain of platelet gpIIb/IIIa (αIIbβ3-integrin) and triggers the transition to an increased affinity state (Tadokoro et al., 2003). The idea that the head of Talin-1 wedges between the α and β cytoplasmic tails of integrins (Tadokoro et al., 2003) is consistent with the observation that the α and β tails move apart during LFA-1 activation (Kim et al., 2003). At present, it is not clear whether Talin-1 controls the triggering of LFA-1 to its intermediate- or high-affinity state. Other actin-binding proteins, such as α-actin in and L-plastin have been also suggested to mediate LFA-1 affinity transition (Jones et al., 1998; Sampath et al., 1998). More recently, Kindlin-3 (FERMT3) and Cytohesin-1 (CYTH1), a GEF for ADP-ribosylation factor 6 (Arf6), have been suggested to mediate LFA-1 affinity activation (Weber et al., 2001; Manevich-Mendelson et al., 2009; Moser et al., 2009; Lefort et al., 2012), although not always under fully physiological conditions. Furthermore, the Rap1A effectors RAPL and RIAM have been shown to behave as direct integrin-binding proteins regulating integrin activation. RAPL was shown to directly bind the cytosolic tail of LFA-1 alpha chain (CD11a), but this was related to stabilization events and not to affinity triggering (Ebisuno et al., 2010). On the other hand, RIAM was suggested to directly bind Talin-1, thus potentially bridging Rap1A to Talin-1-controlled integrin affinity triggering. However, the role of these Rap1A effectors in affinity modulation by chemokines in conditions satisfying the four criteria is still not described. Thus, at present, is not clear how the rap-module may control the last steps of integrin activation. A link between the rho-module and the very downstream events of integrin affinity activation demonstrated under conditions satisfying the four criteria is also lacking. However, it is possible to speculate. Indeed, Talin-1, Kindlin-3, Cytohesin-1, and RIAM possess FERM and PH domains critical to support their interaction with plasma membrane phospholipids. Thus, it is likely that the lipid kinase activity of rho/PLD1-activated PIP5K1C, by increasing the local membrane concentration of phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2), may trigger the membrane translocation, activation, and direct integrin interaction of these regulatory proteins. Notably, a functional link between Cytohesin-1 and RhoA in the context of LFA-1 affinity triggering was recently described (Quast et al., 2009). Theoretically, since the 90 kD isoform of PIP5K1C (also activated by RhoA) directly interacts with Talin-1 (Di Paolo et al., 2002), it is also possible that this PIP5K1C isoform physically bridges directly RhoA to integrin affinity activation, independently of PtdIns(4,5)P2 lipid kinase activity. However, a recent report, although obtained under condition not satisfying the four criteria, seems to challenge this hypothesis (Wernimont et al., 2010).
Do rap and rho Talk Each Other?
Overall, in the context of signal transduction networks controlling chemokine-triggered integrin activation, rap and rho small GTPases may orchestrate the signaling activity of at least 18 signaling proteins and including PLC, CALDAG–GEF, Rap1A, RAPL, RIAM, STK4, SKAP55, ADAP, DOCK2, VAV1, RhoA, Rac1, CDC42, PLD1, PIP5K1C, Talin-1, Kindlin-3, and Cytohesin-1. It is quite likely that other signaling players will be discovered, especially accounting for context-specificity (see below). Moreover, we still need testing the role of some of these molecules under physiological condition, fully satisfying the four criteria proposed above. However, it is unquestionable that rap- and rho-modules represent, at present, our best paradigm of integrin affinity regulation by chemokines. An obvious question is whether these two signaling modules display concurrency and if they work in parallel of serially. The concept of “concurrency” is derived from computer science, where computation of contemporary events often occurs (D’Ambrosio et al., 2004). Thus, in a concurrent model of integrin activation, the final state of the system (fully extended conformation leading to high affinity for the ligand) is achieved only if the regulatory signaling events are delivered simultaneously and integrated at quantitative level. The simple fact that chemokines trigger simultaneously the activation of rap and rho small GTPases with kinetics consistent with rapid integrin activation, clearly suggests that the system displays concurrency (although we are still very far from a quantitative view of the process). This conclusion is supported by recent findings showing cooperation between rap and rho in controlling integrin activation (Vielkind et al., 2005; Kim et al., 2012; Li et al., 2012). An even more interesting question concerns the possibility that rap and rho not only cooperate but also directly influence each other biochemical activity, which corresponds to ask whether rap and rho act in parallel or serially. Although not yet verified in conditions satisfying the four criteria, data from the literature may suggest interesting possibilities. Indeed, RhoA and Rac1 activate PLC isoforms, including the PLCβ, PLCΓ, and PLCε (Thodeti et al., 2002; Illenberger et al., 2003; Wing et al., 2003; Piechulek et al., 2005; Seifert et al., 2008; Walliser et al., 2008; Guo et al., 2010) which, in turn, could determine the activation of Rap1A through CALDAG–GEF. Notably, the lipid kinase activity of PIP5K1C itself may contribute to Rap1A activation by increasing the local concentration of PtdIns(4,5)P2, which, in turn, is substrate of PLCs leading to activation of CALDAG–GEF and, ultimately, of Rap1A. Furthermore, it was recently shown that PLD1 activity is required to Rap1A plasma membrane translocation and activation (Mor et al., 2009), thus establishing a strong functional link between rho signaling activity and rap activation. Finally, as reported above, CDC42 was recently shown to inhibit Rap1A activation (measured as GTP bound state) (Kempf et al., 2011). Altogether, these data suggest that Rap1A signaling activity may be directly influenced by rho, thus controlling a critical arm of the global module of integrin activation, possibly devoted to aspects of integrin-dependent adhesion other than conformational changes and affinity up-regulation (Figure 2).
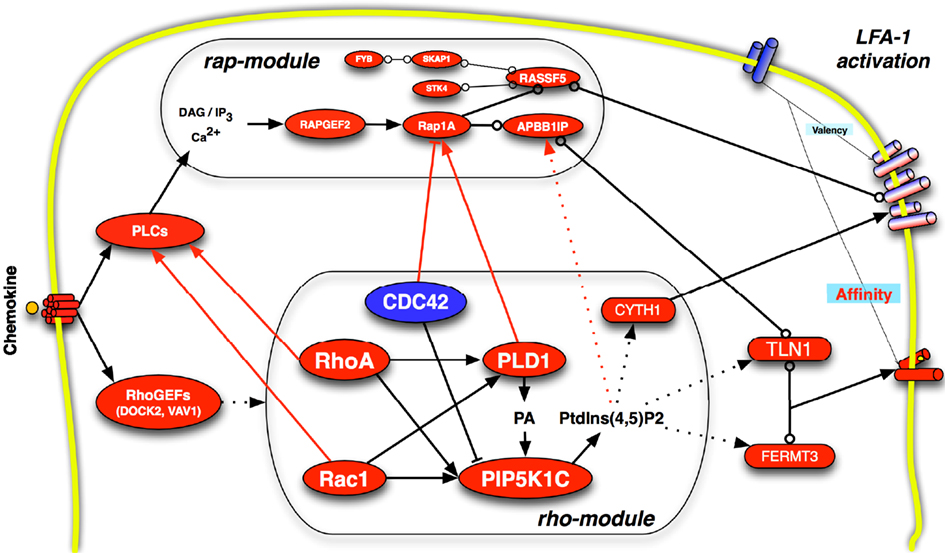
Figure 2. The rap- and rho-modules of integrin affinity modulation by chemokines. The diagram mainly reports data regarding LFA-1 affinity triggering. Dotted lines refer to hypothetic effects. Red lines emphasize the functional relationships between rap and rho modules. Arrowed lines indicate activation. Flat ending lines indicate inhibition. Circle ending lines indicate docking activity. Positive regulators of adhesion are represented as red elliptic shapes; negative regulators are in blue. Notably, although the physical and functional interactions between the different signaling molecules have been demonstrated, the functional role of these interactions in the specific context of integrin affinity regulation under experimental conditions satisfying the four criteria is still not always validated.
Is Chemokine Signaling to Integrin Affinity Modulation Universal?
The complexity of pro-adhesive signaling event triggered by arrest chemokines also imposes more general questions: is the mechanism of integrin affinity regulation conserved among leukocyte subpopulations? Are there universal mechanism of integrin activation or, at least, common relevant proteins activated by chemokines? Some recently published data might provide answers to these questions. For instance, PLD1 does not seem to be crucial to VLA-4 activation (Garcia-Bernal et al., 2009). Moreover, DOCK2 involvement in integrin activation seems cell-specific (Nombela-Arrieta et al., 2004). A chemokine-selective role for RhoA involvement in LFA-1 affinity regulation was also recently suggested (Pasvolsky et al., 2008). Furthermore, surprising data come from a recent study in B cell chronic lymphocytic leukemia (B-CLL; Montresor et al., 2009), a lymphoproliferative disorder characterized by accumulation of immune incompetent B-lymphocytes in the blood, bone marrow, lymph nodes and spleen. In human normal B-lymphocytes, the CXCL12-triggered rho-module of LFA-1 affinity triggering is functionally conserved, with no differences with respect to normal T lymphocytes. However, and in sharp contrast, in B-CLL cells the CXCL12-triggered rho-module of LFA-1 affinity triggering appears no longer fully operational. Specifically, RhoA and PLD1 are fully activated and involved in LFA-1 affinity regulation also in B-CLL cells. In contrast, Rac1 and CDC42 are variably involved in LFA-1 affinity modulation, depending on the studied B-CLL patients, who could be grouped in two cohorts, either showing conserved or absent regulatory role for Rac1 and CDC42 in LFA-1 affinity modulation by CXCL12. Even more surprisingly, PIP5K1C emerges as totally irrelevant to LFA-1 affinity triggering in all studied B-CLL patients. Thus, the neoplastic transformation and progression may completely bypass the role of PIP5K1C and variably affect the Rac1 and CDC42 roles. Since the capability of CXCL12 to trigger LFA-1 affinity states is always fully conserved, altogether these findings show that other proteins regulate the inside-out signaling in leukemic cells, thus highlighting the relative, not universal, nature of the rho-module. Overall, these observations show that universal signaling mechanisms of LFA-1 (and more in general integrin) affinity triggering likely do not exist. An accurate definition of these mechanisms in several different cell-, agonist-, and disease-specific experimental contexts, with particular attention to the four criteria proposed herein, will be mandatory to fully understand the mechanisms by which arrest chemokines regulate cell trafficking.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Associazione Italiana per la Ricerca sul Cancro (AIRC, IG 8690) (Carlo Laudanna); PRIN 2009 grant from the Ministry of Education and Research (MIUR) and Fondazione Cariverona (Carlo Laudanna and Gabriela Constantin); Fondazione Cariverona, project Verona Nanomedicine Initiative (Carlo Laudanna); FP7 Program, European Research Council Grant 261079 “Neurotrafficking” (Gabriela Constantin); Fondazione Italiana Sclerosi Multipla (FISM), Genova, Italy (Gabriela Constantin).
References
Alon, R. (2010). Chemokine arrest signals to leukocyte integrins trigger bi-directional-occupancy of individual heterodimers by extracellular and cytoplasmic ligands. Cell Adh. Migr. 4, 211–214.
Alon, R., and Shulman, Z. (2011). Chemokine triggered integrin activation and actin remodeling events guiding lymphocyte migration across vascular barriers. Exp. Cell Res. 317, 632–641.
Arnaout, M. A., Mahalingam, B., and Xiong, J. P. (2005). Integrin structure, allostery, and bidirectional signaling. Annu. Rev. Cell Dev. Biol. 21, 381–410.
Bakker, G. J., Eich, C., Torreno-Pina, J. A., Diez-Ahedo, R., Perez-Samper, G., Van Zanten, T. S., Figdor, C. G., Cambi, A., and Garcia-Parajo, M. F. (2012). Lateral mobility of individual integrin nanoclusters orchestrates the onset for leukocyte adhesion. Proc. Natl. Acad. Sci. U.S.A. 109, 4869–4874.
Bargatze, R. F., and Butcher, E. C. (1993). Rapid G protein-regulated activation event involved in lymphocyte binding to high endothelial venules. J. Exp. Med. 178, 367–372.
Bergmeier, W., Goerge, T., Wang, H. W., Crittenden, J. R., Baldwin, A. C., Cifuni, S. M., Housman, D. E., Graybiel, A. M., and Wagner, D. D. (2007). Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J. Clin. Invest. 117, 1699–1707.
Berton, G., Mocsai, A., and Lowell, C. A. (2005). Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. 26, 208–214.
Bolomini-Vittori, M., Montresor, A., Giagulli, C., Staunton, D., Rossi, B., Martinello, M., Constantin, G., and Laudanna, C. (2009). Regulation of conformer-specific activation of the integrin LFA-1 by a chemokine-triggered Rho signaling module. Nat. Immunol. 10, 185–194.
Bromley, S. K., Mempel, T. R., and Luster, A. D. (2008). Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat. Immunol. 9, 970–980.
Cairo, C. W., Mirchev, R., and Golan, D. E. (2006). Cytoskeletal regulation couples LFA-1 conformational changes to receptor lateral mobility and clustering. Immunity 25, 297–308.
Carman, C. V., and Springer, T. A. (2003). Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr. Opin. Cell Biol. 15, 547–556.
Caron, E. (2003). Cellular functions of the Rap1 GTP-binding protein: a pattern emerges. J. Cell. Sci. 116, 435–440.
Chesnutt, B. C., Smith, D. F., Raffler, N. A., Smith, M. L., White, E. J., and Ley, K. (2006). Induction of LFA-1-dependent neutrophil rolling on ICAM-1 by engagement of E-selectin. Microcirculation 13, 99–109.
Constantin, G., Majeed, M., Giagulli, C., Piccio, L., Kim, J. Y., Butcher, E. C., and Laudanna, C. (2000). Chemokines trigger immediate beta2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity 13, 759–769.
Crittenden, J. R., Bergmeier, W., Zhang, Y., Piffath, C. L., Liang, Y., Wagner, D. D., Housman, D. E., and Graybiel, A. M. (2004). CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 10, 982–986.
D’Ambrosio, D., Lecca, P., Constantin, G., Priami, C., and Laudanna, C. (2004). Concurrency in leukocyte vascular recognition: developing the tools for a predictive computer model. Trends Immunol. 25, 411–416.
Di Paolo, G., Pellegrini, L., Letinic, K., Cestra, G., Zoncu, R., Voronov, S., Chang, S., Guo, J., Wenk, M. R., and De Camilli, P. (2002). Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature 420, 85–89.
Duchniewicz, M., Zemojtel, T., Kolanczyk, M., Grossmann, S., Scheele, J. S., and Zwartkruis, F. J. (2006). Rap1A-deficient T and B cells show impaired integrin-mediated cell adhesion. Mol. Cell. Biol. 26, 643–653.
Ebisuno, Y., Katagiri, K., Katakai, T., Ueda, Y., Nemoto, T., Inada, H., Nabekura, J., Okada, T., Kannagi, R., Tanaka, T., Miyasaka, M., Hogg, N., and Kinashi, T. (2010). Rap1 controls lymphocyte adhesion cascade and interstitial migration within lymph nodes in RAPL-dependent and -independent manners. Blood 115, 804–814.
Gakidis, M. A., Cullere, X., Olson, T., Wilsbacher, J. L., Zhang, B., Moores, S. L., Ley, K., Swat, W., Mayadas, T., and Brugge, J. S. (2004). Vav GEFs are required for beta2 integrin-dependent functions of neutrophils. J. Cell Biol. 166, 273–282.
Garcia-Bernal, D., Parmo-Cabanas, M., Dios-Esponera, A., Samaniego, R., Hernan, P. D. L. O. D., and Teixido, J. (2009). Chemokine-induced Zap70 kinase-mediated dissociation of the Vav1-talin complex activates alpha4beta1 integrin for T cell adhesion. Immunity 31, 953–964.
Garcia-Bernal, D., Sotillo-Mallo, E., Nombela-Arrieta, C., Samaniego, R., Fukui, Y., Stein, J. V., and Teixido, J. (2006). DOCK2 is required for chemokine-promoted human T lymphocyte adhesion under shear stress mediated by the integrin alpha4beta1. J. Immunol. 177, 5215–5225.
Garcia-Bernal, D., Wright, N., Sotillo-Mallo, E., Nombela-Arrieta, C., Stein, J. V., Bustelo, X. R., and Teixido, J. (2005). Vav1 and Rac control chemokine-promoted T lymphocyte adhesion mediated by the integrin alpha4beta1. Mol. Biol. Cell 16, 3223–3235.
Giagulli, C., Ottoboni, L., Caveggion, E., Rossi, B., Lowell, C., Constantin, G., Laudanna, C., and Berton, G. (2006). The Src family kinases Hck and Fgr are dispensable for inside-out, chemoattractant-induced signaling regulating beta 2 integrin affinity and valency in neutrophils, but are required for beta 2 integrin-mediated outside-in signaling involved in sustained adhesion. J. Immunol. 177, 604–611.
Giagulli, C., Scarpini, E., Ottoboni, L., Narumiya, S., Butcher, E. C., Constantin, G., and Laudanna, C. (2004). RhoA and zeta PKC control distinct modalities of LFA-1 activation by chemokines: critical role of LFA-1 affinity triggering in lymphocyte in vivo homing. Immunity 20, 25–35.
Guo, Y., Golebiewska, U., D’Amico, S., and Scarlata, S. (2010). The small G protein Rac1 activates phospholipase Cdelta1 through phospholipase Cbeta2. J. Biol. Chem. 285, 24999–25008.
Hogg, N., Patzak, I., and Willenbrock, F. (2011). The insider’s guide to leukocyte integrin signalling and function. Nat. Rev. Immunol. 11, 416–426.
Hyduk, S. J., Chan, J. R., Duffy, S. T., Chen, M., Peterson, M. D., Waddell, T. K., Digby, G. C., Szaszi, K., Kapus, A., and Cybulsky, M. I. (2007). Phospholipase C, calcium, and calmodulin are critical for alpha4beta1 integrin affinity up-regulation and monocyte arrest triggered by chemoattractants. Blood 109, 176–184.
Illenberger, D., Walliser, C., Nurnberg, B., Diaz Lorente, M., and Gierschik, P. (2003). Specificity and structural requirements of phospholipase C-beta stimulation by Rho GTPases versus G protein beta gamma dimers. J. Biol. Chem. 278, 3006–3014.
Jones, S. L., Wang, J., Turck, C. W., and Brown, E. J. (1998). A role for the actin-bundling protein L-plastin in the regulation of leukocyte integrin function. Proc. Natl. Acad. Sci. U.S.A. 95, 9331–9336.
Katagiri, K., Imamura, M., and Kinashi, T. (2006). Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nat. Immunol. 7, 919–928.
Kempf, T., Zarbock, A., Widera, C., Butz, S., Stadtmann, A., Rossaint, J., Bolomini-Vittori, M., Korf-Klingebiel, M., Napp, L. C., Hansen, B., Kanwischer, A., Bavendiek, U., Beutel, G., Hapke, M., Sauer, M. G., Laudanna, C., Hogg, N., Vestweber, D., and Wollert, K. C. (2011). GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat. Med. 17, 581–588.
Kim, J. G., Moon, M. Y., Kim, H. J., Li, Y., Song, D. K., Kim, J. S., Lee, J. Y., Kim, J., Kim, S. C., and Park, J. B. (2012). Ras-related GTPases Rap1 and RhoA collectively induce the phagocytosis of serum-opsonized zymosan particles in macrophages. J. Biol. Chem. 287, 5145–5155.
Kim, M., Carman, C. V., and Springer, T. A. (2003). Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301, 1720–1725.
Lafuente, E., and Boussiotis, V. A. (2006). Rap1 regulation of RIAM and cell adhesion. Methods Enzymol. 407, 345–358.
Laudanna, C., and Alon, R. (2006). Right on the spot. Chemokine triggering of integrin-mediated arrest of rolling leukocytes. Thromb. Haemost. 95, 5–11.
Laudanna, C., Campbell, J. J., and Butcher, E. C. (1996). Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science 271, 981–983.
Lefort, C. T., Rossaint, J., Moser, M., Petrich, B. G., Zarbock, A., Monkley, S. J., Critchley, D. R., Ginsberg, M. H., Fassler, R., and Ley, K. (2012). Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood.
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689.
Li, Y., Kim, J. G., Kim, H. J., Moon, M. Y., Lee, J. Y., Kim, J., Kim, S. C., Song, D. K., Kim, Y. S., and Park, J. B. (2012). Small GTPases Rap1 and RhoA regulate superoxide formation by Rac1 GTPases activation during the phagocytosis of IgG-opsonized zymosans in macrophages. Free Radic. Biol. Med. 52, 1796–1805.
Luo, B. H., Carman, C. V., and Springer, T. A. (2007). Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647.
Manevich-Mendelson, E., Feigelson, S. W., Pasvolsky, R., Aker, M., Grabovsky, V., Shulman, Z., Kilic, S. S., Rosenthal-Allieri, M. A., Ben-Dor, S., Mory, A., Bernard, A., Moser, M., Etzioni, A., and Alon, R. (2009). Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood 114, 2344–2353.
Menasche, G., Kliche, S., Chen, E. J., Stradal, T. E., Schraven, B., and Koretzky, G. (2007). RIAM links the ADAP/SKAP-55 signaling module to Rap1, facilitating T-cell-receptor-mediated integrin activation. Mol. Cell. Biol. 27, 4070–4081.
Miner, J. J., Xia, L., Yago, T., Kappelmayer, J., Liu, Z., Klopocki, A. G., Shao, B., Mcdaniel, J. M., Setiadi, H., Schmidtke, D. W., and McEver, R. P. (2008). Separable requirements for cytoplasmic domain of PSGL-1 in leukocyte rolling and signaling under flow. Blood 112, 2035–2045.
Montresor, A., Bolomini-Vittori, M., Simon, S. I., Rigo, A., Vinante, F., and Laudanna, C. (2009). Comparative analysis of normal versus CLL B-lymphocytes reveals patient-specific variability in signaling mechanisms controlling LFA-1 activation by chemokines. Cancer Res. 69, 9281–9290.
Mor, A., Wynne, J. P., Ahearn, I. M., Dustin, M. L., Du, G., and Philips, M. R. (2009). Phospholipase D1 regulates lymphocyte adhesion via upregulation of Rap1 at the plasma membrane. Mol. Cell. Biol. 29, 3297–3306.
Moser, M., Bauer, M., Schmid, S., Ruppert, R., Schmidt, S., Sixt, M., Wang, H. V., Sperandio, M., and Fassler, R. (2009). Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15, 300–305.
Nishida, N., Xie, C., Shimaoka, M., Cheng, Y., Walz, T., and Springer, T. A. (2006). Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity 25, 583–594.
Nombela-Arrieta, C., Lacalle, R. A., Montoya, M. C., Kunisaki, Y., Megias, D., Marques, M., Carrera, A. C., Manes, S., Fukui, Y., Martinez, A. C., and Stein, J. V. (2004). Differential requirements for DOCK2 and phosphoinositide-3-kinase gamma during T and B lymphocyte homing. Immunity 21, 429–441.
Pasvolsky, R., Grabovsky, V., Giagulli, C., Shulman, Z., Shamri, R., Feigelson, S. W., Laudanna, C., and Alon, R. (2008). RhoA is involved in LFA-1 extension triggered by CXCL12 but not in a novel outside-in LFA-1 activation facilitated by CXCL9. J. Immunol. 180, 2815–2823.
Piechulek, T., Rehlen, T., Walliser, C., Vatter, P., Moepps, B., and Gierschik, P. (2005). Isozyme-specific stimulation of phospholipase C-gamma2 by Rac GTPases. J. Biol. Chem. 280, 38923–38931.
Quast, T., Tappertzhofen, B., Schild, C., Grell, J., Czeloth, N., Forster, R., Alon, R., Fraemohs, L., Dreck, K., Weber, C., Lammermann, T., Sixt, M., and Kolanus, W. (2009). Cytohesin-1 controls the activation of RhoA and modulates integrin-dependent adhesion and migration of dendritic cells. Blood 113, 5801–5810.
Rot, A., and von Andrian, U. H. (2004). Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu. Rev. Immunol. 22, 891–928.
Salas, A., Shimaoka, M., Chen, S., Carman, C. V., and Springer, T. (2002). Transition from rolling to firm adhesion is regulated by the conformation of the I domain of the integrin lymphocyte function-associated antigen-1. J. Biol. Chem. 277, 50255–50262.
Salas, A., Shimaoka, M., Phan, U., Kim, M., and Springer, T. A. (2006). Transition from rolling to firm adhesion can be mimicked by extension of integrin alphaLbeta2 in an intermediate affinity state. J. Biol. Chem. 281, 10876–10882.
Sampath, R., Gallagher, P. J., and Pavalko, F. M. (1998). Cytoskeletal interactions with the leukocyte integrin beta2 cytoplasmic tail. Activation-dependent regulation of associations with talin and alpha-actinin. J. Biol. Chem. 273, 33588–33594.
Scheele, J. S., Marks, R. E., and Boss, G. R. (2007). Signaling by small GTPases in the immune system. Immunol. Rev. 218, 92–101.
Seifert, J. P., Zhou, Y., Hicks, S. N., Sondek, J., and Harden, T. K. (2008). Dual activation of phospholipase C-epsilon by Rho and Ras GTPases. J. Biol. Chem. 283, 29690–29698.
Shamri, R., Grabovsky, V., Gauguet, J. M., Feigelson, S., Manevich, E., Kolanus, W., Robinson, M. K., Staunton, D. E., von Andrian, U. H., and Alon, R. (2005). Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat. Immunol. 6, 497–506.
Springer, T. A., and Dustin, M. L. (2011). Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 24, 107–115.
Stewart, M. P., Mcdowall, A., and Hogg, N. (1998). LFA-1-mediated adhesion is regulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J. Cell Biol. 140, 699–707.
Svensson, L., Mcdowall, A., Giles, K. M., Stanley, P., Feske, S., and Hogg, N. (2010). Calpain 2 controls turnover of LFA-1 adhesions on migrating T lymphocytes. PLoS ONE 5, e15090. doi:10.1371/journal.pone.0015090
Tadokoro, S., Shattil, S. J., Eto, K., Tai, V., Liddington, R. C., De Pereda, J. M., Ginsberg, M. H., and Calderwood, D. A. (2003). Talin binding to integrin beta tails: a final common step in integrin activation. Science 302, 103–106.
Thodeti, C. K., Massoumi, R., Bindslev, L., and Sjolander, A. (2002). Leukotriene D4 induces association of active RhoA with phospholipase C-gamma1 in intestinal epithelial cells. Biochem. J. 365, 157–163.
Tybulewicz, V. L., and Henderson, R. B. (2009). Rho family GTPases and their regulators in lymphocytes. Nat. Rev. Immunol. 9, 630–644.
van Kooyk, Y., and Figdor, C. G. (2000). Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr. Opin. Cell Biol. 12, 542–547.
Vielkind, S., Gallagher-Gambarelli, M., Gomez, M., Hinton, H. J., and Cantrell, D. A. (2005). Integrin regulation by RhoA in thymocytes. J. Immunol. 175, 350–357.
Walliser, C., Retlich, M., Harris, R., Everett, K. L., Josephs, M. B., Vatter, P., Esposito, D., Driscoll, P. C., Katan, M., Gierschik, P., and Bunney, T. D. (2008). rac regulates its effector phospholipase Cgamma2 through interaction with a split pleckstrin homology domain. J. Biol. Chem. 283, 30351–30362.
Weber, K. S., Weber, C., Ostermann, G., Dierks, H., Nagel, W., and Kolanus, W. (2001). Cytohesin-1 is a dynamic regulator of distinct LFA-1 functions in leukocyte arrest and transmigration triggered by chemokines. Curr. Biol. 11, 1969–1974.
Wernimont, S. A., Legate, K. R., Simonson, W. T., Fassler, R., and Huttenlocher, A. (2010). PIPKI gamma 90 negatively regulates LFA-1-mediated adhesion and activation in antigen-induced CD4+ T cells. J. Immunol. 185, 4714–4723.
Wing, M. R., Snyder, J. T., Sondek, J., and Harden, T. K. (2003). Direct activation of phospholipase C-epsilon by Rho. J. Biol. Chem. 278, 41253–41258.
Zarbock, A., Lowell, C. A., and Ley, K. (2007). Spleen tyrosine kinase Syk is necessary for E-selectin-induced alpha(L)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity 26, 773–783.
Keywords: signal transduction, chemokine, integrin activation, integrin affinity, leukocyte recruitment, adhesion, rho small GTPases, rap small GTPases
Citation: Montresor A, Toffali L, Constantin G and Laudanna C (2012) Chemokines and the signaling modules regulating integrin affinity. Front. Immun. 3:127. doi: 10.3389/fimmu.2012.00127
Received: 05 March 2012; Paper pending published: 31 March 2012;
Accepted: 03 May 2012; Published online: 25 May 2012.
Edited by:
Klaus Ley, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Gustavo B. Menezes, Universidade Federal de Minas Gerais, BrazilCraig Lefort, La Jolla Institute for Allergy and Immunology, USA
Copyright: © 2012 Montresor, Toffali, Constantin and Laudanna. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Carlo Laudanna, Division of General Pathology, Department of Pathology, University of Verona, Strada le Grazie 8, Verona, Italy. e-mail:Y2FybG8ubGF1ZGFubmFAdW5pdnIuaXQ=