Abstract
In 2005 a newly discovered T helper cell subset that secreted interleukin (IL)-17 became the center of attention in immunology. Initial studies painted Th17 cells as the culprit for destruction in many different autoimmune and auto-inflammatory diseases. Subsequently, the discovery of patients with primary immunodeficiencies in the IL-17 pathway taught us that Th17 cells have a critical role in defense against certain fungal and bacterial infections. Moreover, the paradoxical exacerbation of Crohn’s disease in the clinical trials of a Secukinumab (AIN457), a fully human neutralizing antibody to IL-17A, has cast into doubt a universal pro-inflammatory and harmful role for Th17 cells. Evidence now suggests that depending on the environment Th17 cells can alter their differentiation program, ultimately giving rise to either protective or pro-inflammatory cells. In this review we will summarize the evidence from patients with immunodeficiencies, autoimmune, or auto-inflammatory diseases that teaches us how the pro-inflammatory versus protective function of Th17 cells varies within the context of different human diseases.
Introduction
In rare cases, a mutation in an essential gene can disrupt immune homeostasis, leading to clinical immunodeficiency. More commonly, when individuals with a genetic predisposition are exposed to environmental triggers, a failure of immune homeostasis can lead to autoimmunity. In this review, we will discuss how parallel studies of immunodeficiencies and autoimmune diseases have advanced our knowledge of a CD4+ T cell lineage first characterized by production of IL-17A, Th17 cells.
Th17 cells were identified in 2005 (Harrington et al., 2005; Langrish et al., 2005; Park et al., 2005) and, as for other CD4+ T cell lineages, their development, is controlled by a combination of cytokines which initiate a program of transcription factor expression and epigenetic re-modeling (van der Gast et al., 2011). In humans, the cytokines which instruct Th17 cell lineage development likely include IL-6, IL-21, IL-23, and IL-1β (Acosta-Rodriguez et al., 2007a; Chen et al., 2007; Evans et al., 2007; Wilson et al., 2007; Liu and Rohowsky-Kochan, 2008), with a potential synergistic role for TGF-β (Manel et al., 2008; Volpe et al., 2008; Yang et al., 2008a) via its ability to suppress Th1 cell lineage commitment (Santarlasci et al., 2009). Cytokine-driven activation of the signal transducer and activator of transcription (STAT) 3 pathway is an essential step in Th17 cell differentiation (Holland et al., 2007; Yang et al., 2007; Ma et al., 2008), ultimately leading to expression of their lineage-defining transcription factor: retinoid orphan receptor (ROR)C2 (Acosta-Rodriguez et al., 2007a; Annunziato et al., 2007; Wilson et al., 2007; Manel et al., 2008; Crome et al., 2009). Although the IL-17 cytokine family includes six members (Kolls and Linden, 2004), Th17 cells are thought to only produce IL-17A and IL-17F, which are 55% identical (Kolls and Linden, 2004). IL-17A can combine with IL-17F to form a heterodimer and both can form homodimers (Wright et al., 2007).
Th17 cells have many phenotypic characteristics that distinguish them from other Th cell lineages. In addition to IL-17A and IL-17F, Th17 cells secrete other signature cytokines including IL-21 and IL-22 (Bending et al., 2011). They have also been reported to produce IFN-γ (Annunziato et al., 2007), IL-4 (Cosmi et al., 2010), IL-10 (McGeachy et al., 2007), IL-9 (Beriou et al., 2010), IL-26, CXCL8, and CCL20 (Boniface et al., 2008). They are poor producers of IL-2, which may result in their poor proliferative potential in vitro (Santarlasci et al., 2012). They constitutively co-express CCR4 and CCR6, but not CXCR3 (Acosta-Rodriguez et al., 2007b), and are derived from CD161+ precursors (Cosmi et al., 2008). The effects of Th17 cells on other cells have recently been highlighted in many reviews (Annunziato and Romagnani, 2011; Gaffen, 2011; Gaffen et al., 2011; Ghoreschi et al., 2011; Milner, 2011; Pappu et al., 2011; Wilke et al., 2011).
Th17 cells initially developed a reputation as a destructive element in several diseases including multiple sclerosis (MS), rheumatoid arthritis (RA), psoriasis, and inflammatory bowel disease (IBD). In animal models this reputation, came from evidence that the lack of IL-17-producing cells ameliorates experimental autoimmune encephalitis (EAE) and collagen induced arthritis (CIA; Cua et al., 2003). In humans, the reputation was due to correlative data documenting an increase in IL-17-producing cells, particularly at sites of tissue inflammation (Wilke et al., 2011). However, these original conclusions were over-simplified and as discussed below in some diseases Th17 cells clearly have a protective role.
The Role of Th17 Cells in Primary Immunodeficiencies
Much of what we know about human Th17 cells comes from the study of a rare primary immunodeficiency called Hyperimmunoglobulin E (Job’s, syndrome). This disease is caused by mutations in STAT3 (Holland et al., 2007) but the underlying cellular basis for the characteristic phenotype of severe pneumonias, mucocutaneous candidiasis, and Staphylococcus aureus abscesses (Buckley et al., 1972; Grimbacher et al., 1999) remained unknown until several groups found that these patients lack Th17 cells in their peripheral blood (de Beaucoudrey et al., 2008; Ma et al., 2008; Milner et al., 2008; Renner et al., 2008). In addition, naïve Th cells from Job’s syndrome patients have low levels of RORC2 expression and cannot be differentiated into Th17 cells in vitro (de Beaucoudrey et al., 2008; Ma et al., 2008; Milner et al., 2008; Renner et al., 2008).
One complication when interpreting data from Job’s syndrome patients is that STAT3 is activated downstream of other cytokines, making it difficult to attribute a clinical phenotype to one pathway. Recently, other immunodeficiencies have been described which involve more specific defects in the IL-17 pathway. For example, two patients with chronic mucocutaneous candidiasis (CMC) disease, characterized by chronic or persistent infection with Candida albicans and S. aureus, were found to have an IL-17RA autosomal recessive deficiency or an IL-17F autosomal dominant deficiency (Puel et al., 2011). In addition, patients with a deficiency in the intracellular adaptor molecule CARD9, which is essential for dectin-1 signaling, also suffer from systemic Candidiasis infection (Glocker et al., 2009) and have low numbers of Th17 cells in their peripheral blood.
Together, these data have led to the hypothesis that in humans Th17 cells have an essential role in protective immunity the specific pathogens C. albicans and S. aureus. In accordance with this conclusion, Sallusto et al. have characterized different subsets of human Th17 cells that can be differentiated in vitro with antigen specific stimulation by C. albicans and S. aureus (Zielinski et al., 2012).
Th17 cells have an intriguing close developmental link with FOXP3+CD4+ regulatory T cells (Tregs). FOXP3 and RORC2 can directly interact via a DNA-independent mechanism, and during Th17 cell development FOXP3 is transiently expressed (Zhou et al., 2008). Moreover, upon activation fully differentiated human Th17 cells preferentially express FOXP3 in comparison to Th1 cells (McMurchy and Levings, unpublished data). Indeed there is increasing evidence for the existence of cells that co-express IL-17 and FOXP3 (Ayyoub et al., 2009; Beriou et al., 2009; Miyara et al., 2009; Voo et al., 2009; Kryczek et al., 2011; Ye et al., 2011).
Immune dysregulation, polyendocrinopathy, enteropathy X-linked syndrome (IPEX) is a triad of autoimmune syndromes including enteropathy, type 1 diabetes, and hyper-IgE (McMurchy et al., 2010), resulting from mutations in FOXP3. The cellular basis for this disease has been attributed to Treg dysfunction (Bacchetta et al., 2006; D’Hennezel et al., 2009), but recently Bacchetta et al. investigated whether part of the cellular defect in IPEX patients may not only relate to Treg dysfunction, but also to changes in Th17 cells. Indeed IPEX patients possessed an increased frequency of cells with a surface profile characteristic of Th17 cells (CD4+CCR6+CD161+), expressing RORC2, and producing IL-17 (Passerini et al., 2011). Interestingly, in patients with point mutations that do not abrogate FOXP3 expression, there was an increased frequency of FOXP3+ cells within the CD161+ Th17 cell gate. These data suggest that dysfunctional Tregs may preferentially differentiate into Th17 cells and that an expansion of this subset may underlie some of the clinical phenotype of IPEX. An alternative interpretation is that the Th17 cells in IPEX patients are highly activated and that in this case FOXP3 expression is a consequence of T cell activation and not Treg lineage commitment (Ziegler, 2006). Regardless, in this immunodeficiency there is strong correlative evidence that Th17 cells may have a detrimental pro-inflammatory effect.
Evidence for the Pro-Inflammatory Role of Th17 in Human Autoimmunity
The first recognition of the importance of Th17 cells came from studies of EAE. The notion of EAE as a Th1-mediated disease was challenged when mice deficient in the p40 subunit of IL-12 were found to be resistant to EAE whereas mice deficient in the p35 subunit were actually more susceptible to disease (Becher et al., 2002). Cua et al. (2003) solved this paradox by using genetically deficient mice to show that IL-23p19 and IL-12p40, but not IL-12p35, were essential for EAE development. IL-23, which shares the IL-12p40 subunit with IL-12, was subsequently found to stabilize Th17 cells, and these cells were found to be the main contributing factor in EAE (Langrish et al., 2005). Subsequently, a correlation between Th17 cells and human autoimmunity was sought. Below we discuss the evidence for a pro-inflammatory role in autoimmunity and describe attempts to target this axis therapeutically.
Psoriasis is an auto-inflammatory skin disease characterized by recurrent demarcated red and scaly skin plaques. These plaques include infiltrating T cells (mainly Th cells) and dendritic cells in the dermis as well as cytotoxic T cells and neutrophil in the epidermis (Lowes et al., 2007). The resulting inflammatory process results in rapid keratinocyte proliferation, abnormal keratinocyte differentiation, and angiogenesis (Lowes et al., 2007). Initially, increased levels of IFN-γ, TNF-α, and IL-12 in the serum and lesions of psoriasis patients labeled this as a Th1-mediated disease (Di Cesare et al., 2009). However, RORC, IL-1β, IL-6, and IL-23 are also increased in psoriatic skin lesions (Di Cesare et al., 2009) leading to the possibility that Th17 and Th1 act in synergy to produce psoriatic inflammation.
Th17 cells are thought to be recruited to the skin by expression of CCL20, the ligand for CCR6, then locally stabilized by IL-1 and IL-23. Since both IL-17 and IFN-γ cause keratinocytes and antigen presenting cells (APCs) to produce more IL-1, IL-23, and CCL20, a positive feedback loop causing keratinocyte proliferation is established (Kryczek et al., 2008; Zaba et al., 2009). Several monoclonal antibodies targeting TNF-α and the p40 subunit shared by IL-12 and IL-23 (Ustekinumab) have been approved for clinical use in psoriasis. Since IL-17 can act synergistically with TNF-α to induce keratinocytes to express inflammatory proteins (Chiricozzi et al., 2011), it is possible that anti-TNF-α acts in part by inhibiting Th17 cell-driven inflammation. Targeting IL-17 alone with Secukinumab (AIN457) or Ixekizumab, both fully human neutralizing antibodies to IL-17A, is also effective in psoriasis (Hueber et al., 2010; Leonardi et al., 2012), confirming that this is likely a major pathogenic cytokine in this skin disease. Since Th17 cells are not the sole producers of IL-17 [other possible sources of this cytokine in psoriatic plaques include γδ T cells (Cai et al., 2011), mast cells (Lin et al., 2011), neutrophils (Lin et al., 2011), and Tregs (Bovenschen et al., 2011)], whether or not Th17 cells are the major source of this cytokine in skin remains to be determined.
Another disease with strong links to Th17 cells is RA, a chronic autoimmune disease that leads to joint destruction. T cells infiltrating the synovial fluid of RA patients produce high amounts of IL-17A, IL-1β, and IL-6 (Cascao et al., 2010), especially during early disease and pre-treatment (Chabaud et al., 1999; Ziolkowska et al., 2000; Hwang and Kim, 2005; Leipe et al., 2010). Notably, the levels of IL-17 in the synovium correlate with joint damage, whereas those of IFN-γ correlate with protection (Kirkham et al., 2006). Recent evidence supports a role for IL-17F as well as IL-17A in RA, with the two related cytokines acting in synergy to induce other pro-inflammatory cytokines and chemokines in synoviocytes, myeloid cells, and synovial fibroblasts (Lundy et al., 2007; Tran et al., 2007; Hot and Miossec, 2011; van Hamburg et al., 2011). Hence, analogous to the process in psoriasis, a positive pro-inflammatory feedback loop encourages more Th17 differentiation and maintenance in the joint (reviewed in Sarkar and Fox, 2010). Direct clinical evidence for the role of IL-17 in RA comes from recent clinical trials which found that Secukinumab and another anti-IL-17A therapeutic known as LY2439821 significantly benefit these patients (Genovese et al., 2010; Morrison et al., 2011).
Multiple sclerosis is a neurological disease that results from auto-inflammatory damage to the myelin sheaths surrounding nerves in the brain and spinal cord. This disease has historically been associated with the discovery of Th17 cells since, as discussed above, they have a major pathogenic role in EAE (Bettelli et al., 2008; Dong, 2008; Dubin and Kolls, 2008; Weaver and Hatton, 2009). MS patients have increased IL-17 mRNA in their blood as well as cerebrospinal fluid (Matusevicius et al., 1999), and expression of miRNA326 in their peripheral blood mononuclear cells promotes Th17 cell differentiation and correlates with disease severity (Du et al., 2009). Blood-brain barrier endothelial cells layers are more permeable to in vitro polarized Th17 cells, especially if the monolayer is pre-treated with IL-17 or IL-22 (Kebir et al., 2007). These data led to the hypothesis that in MS Th17 cells weaken the blood-brain barrier and enable the migration of immune cells into the normally immune privileged sites within the central nervous system. If this is the case, then Th17 cells may have more of a facilitative than directly pathogenic role in the nervous system, distinct from their clear role in the positive feedback loop of inflammation in psoriasis and RA. Recruitment has begun for a phase II clinical trials of Secukinumab in patients with relapsing-remitting MS. This trial will provide significant insight into the question of whether IL-17 blockade in MS can induce a clinically relevant protective function.
Type 1 diabetes (T1D) is characterized by autoimmune destruction of pancreatic islet cells resulting in the loss of insulin production. Murine studies have yielded conflicting results on the role of Th17 in the NOD mouse model of T1D (Vukkadapu et al., 2005; Jain et al., 2008; Emamaullee et al., 2009; van den Brandt et al., 2010; Lee et al., 2011; Liu et al., 2011; Joseph et al., 2012), including a potential ability to convert into Th1 cells in vivo (Bending et al., 2009; Martin-Orozco et al., 2009). However, more recent data suggest some of this apparent plasticity could be related to the study of in vitro polarized Th17 cells, which are not sufficiently stabilized at the epigenetic level (Bending et al., 2011; Cohen et al., 2011). Patients with T1D, have an increase in circulating IL-17-producing cells (Honkanen et al., 2010; Marwaha et al., 2010; Hughson et al., 2011), including FOXP3 expressing Th17 cells (Marwaha et al., 2010), monocytes that secrete Th17 polarizing cytokines (Bradshaw et al., 2009), and islet-antigen specific Th17 cells (Arif et al., 2011). There is also evidence that pancreatic lymph nodes from T1D patients have an increase in Th17 cells (Ferraro et al., 2011) and that islets from T1D patients, who died close to diagnosis, express IL-17A, RORC, and IL-22 (Arif et al., 2011). Mechanistically IL-17 enhances IL-1β, IFN-γ, and TNF-α-induced apoptosis in human islets (Arif et al., 2011). We have also found that significantly elevated levels of CD8+IL-17+ cells are detectable in the peripheral blood of a large subset of patients with T1D at disease onset (Marwaha et al., 2010). In the context of mounting correlative evidence that IL-17-producing cells may be pathogenic in the early stages of T1D onset, clinical trials to test the effects of therapy with agents such as Secukinumab or Ustekinumab are warranted.
A Protective Role for Th17 in the Gut
The success of Ustekinumab, a human IL-12/23 monoclonal antibody, in patients with moderate to severe Crohn’s disease held promise for the targeting of the IL-17 pathway to modulate this disease (Sandborn et al., 2008). However, the paradoxical exacerbation of Crohn’s disease in the clinical trial of a Secukinumab, cast into doubt the pro-inflammatory role of Th17 cells in the gut. Whilst IL-17 cell-producing cells are found in high numbers in inflamed mucosa in Crohn’s disease and Ulcerative colitis patients, more recent data demonstrate that characterization on the basis of IL-17 alone is insufficient to classify these cells as pathogenic. As described above, Th17 can co-secrete IFN-γ (Annunziato et al., 2007; Lee et al., 2009; Cosmi et al., 2011; Hirota et al., 2011) or co-express FOXP3 (Ayyoub et al., 2009; Beriou et al., 2009; Miyara et al., 2009; Voo et al., 2009; Kryczek et al., 2011; Ye et al., 2011), indicating the existence of multiple subsets of Th17 cells with functional specialization (Figure 1).
Figure 1
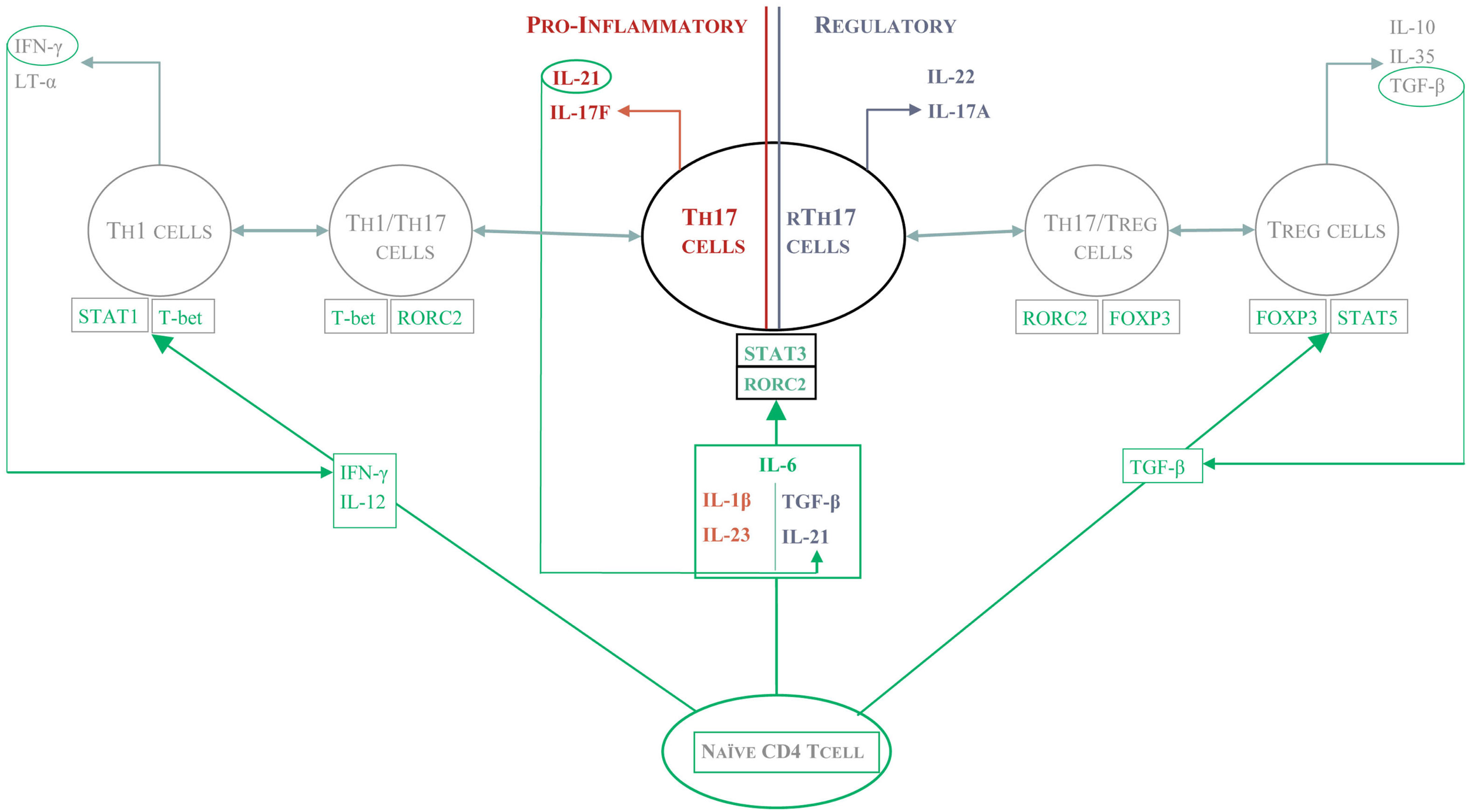
The protective and pathogenic functions of Th17 cells. Depending on the local cytokine environment, different subsets of Th17 cells arise and mediate distinct effector function. In the presence of IL-23, Th17 cells seem to be pro-inflammatory and can either make IFN-γ themselves or work in concert with Th1 cells to drive a positive feedback pathway of tissue damage such as that seen in psoriasis or RA. In Crohn’s disease, Th17 cells can differentiate in the gut into protective or pathogenic Th17 cells. In the presence of TGF-β and IL-21, Th17 are re-programmed into “regulatory” Th17 cells which seem to protect from intestinal inflammation.
In retrospect, data from mouse models of colitis heeded a warning as to the protective role for Th17 cells in Crohn’s disease. In the dextran sulfate sodium (DSS)-induced colitis model, administration of a neutralizing IL-17A antibody (Ogawa et al., 2004), deletion of IL-17A (Yang et al., 2008b), or of IL-22 (Zenewicz et al., 2008) all resulted in a worsening of the colitis. In contrast, IL-17F-deficient (Yang et al., 2008b) and IL-21-deficient mice (Fina et al., 2008) were protected against DSS induced colitis. These data suggest there are non-redundant roles of IL-17A versus F, and that, at least in the gut, IL-21 rather than IL-17A or IL-22 may be a primary Th17-derived pathogenic cytokine.
How could Th17-derived cytokines exert a protective functional role in the intestine? First, IL-17A improves barrier function by strengthening tight junctions after inducing claudin and mucin expression (Kinugasa et al., 2000; Chen et al., 2003). Second, IL-22 improves barrier function by inducing epithelial cell proliferation (Brand et al., 2006) and enhancing goblet cell restoration and mucus production (Sugimoto et al., 2008). Also, a novel suppressive Th17 subset dubbed regulatory Th17 (rTh17) cells has recently been described. When Esplugues et al. (2011) used a CD3-antibody strategy to induce mucosal tolerance, Th17 cells were recruited to the gut but then re-programmed into suppressive, FOXP3-negative, rTh17 cells. The function of rTh17 cells depends on IL-10, TGF-β, and CTLA-4, and does not occur in CCR6-deficient mice where Th17 are not recruited to the gut. The latter data indicate that the mucosal immunity micro-environment is critical for the development of rTh17 cells.
Conclusion
Different flavors of Th17 differentiation, ranging from highly pro-inflammatory to suppressive, result from different cytokine micro-environments in various diseases. Th17 cells can no longer be identified solely on the production of IL-17A since the combination of co-secreted cytokines is key to defining their effector function. Moreover, IL-17 is not only produced by Th17 cells, and under certain conditions γδ T cells (Stark et al., 2005), CD8+ T cells (Shin et al., 1998; He et al., 2006), T follicular helper cells (Cua and Tato, 2010), Lymphoid Tissue induced (LTi) cells (Cupedo et al., 2009), and NKT cells (Michel et al., 2007; Lee et al., 2008; Rachitskaya et al., 2008), can all secrete IL-17. We must therefore start to redefine the partial role that Th17 cells play in IL-17-guided immune response. An additional consideration is their potential for plasticity and co-secretion of cytokines that define other Th cell lineages (e.g., IFN-γ), although this is more likely a transient rather than permanent change based on epigenetic analysis (Bending et al., 2011; Cohen et al., 2011). In summary, the notion that Th17 cells are purely pro-inflammatory cells is mistaken, rather these cells mediate a diverse set of responses in infection, autoimmunity, and immunodeficiency.
Statements
Acknowledgments
The authors’ own work is supported by grants from the Canadian Institutes for Health Research and the Juvenile Diabetes Research Foundation. Megan K. Levings holds a Canada Research Chair in Transplantation. Alicia N. McMurchy is a Canada Vanier Scholar. Ashish K. Marwaha holds a Michael Smith Research Trainee award and a Radcliffe Travelling Fellowship at the University of Oxford.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Acosta-RodriguezE. V.NapolitaniG.LanzavecchiaA.SallustoF. (2007a). Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol.8, 942–949.10.1038/ni1467
2
Acosta-RodriguezE. V.RivinoL.GeginatJ.JarrossayD.GattornoM.LanzavecchiaA.SallustoF.NapolitaniG. (2007b). Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol.8, 639–646.10.1038/ni1467
3
AnnunziatoF.CosmiL.SantarlasciV.MaggiL.LiottaF.MazzinghiB.ParenteE.FilìL.FerriS.FrosaliF.GiudiciF.RomagnaniP.ParronchiP.TonelliF.MaggiE.RomagnaniS. (2007). Phenotypic and functional features of human Th17 cells. J. Exp. Med.204, 1849–1861.10.1084/jem.20070663
4
AnnunziatoF.RomagnaniS. (2011). Mouse T helper 17 phenotype: not so different than in man after all. Cytokine56, 112–115.10.1016/j.cyto.2011.06.009
5
ArifS.MooreF.MarksK.BouckenoogheT.DayanC. M.PlanasR.Vives-PiM.PowrieJ.TreeT.MarchettiP.HuangG. C.GurzovE. N.Pujol-BorrellR.EizirikD. L.PeakmanM. (2011). Peripheral and islet interleukin-17 pathway activation characterizes human autoimmune diabetes and promotes cytokine-mediated beta-cell death. Diabetes60, 2112–2119.10.2337/db10-1643
6
AyyoubM.DeknuydtF.RaimbaudI.DoussetC.LevequeL.BioleyG.ValmoriD. (2009). Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc. Natl. Acad. Sci. U.S.A.106, 8635–8640.10.1073/pnas.0900621106
7
BacchettaR.PasseriniL.GambineriE.DaiM.AllanS. E.PerroniL.Dagna-BricarelliF.SartiranaC.Matthes-MartinS.LawitschkaA.AzzariC.ZieglerS. F.LevingsM. K.RoncaroloM. G. (2006). Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J. Clin. Invest.116, 1713–1722.10.1172/JCI25112
8
BecherB.DurellB. G.NoelleR. J. (2002). Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J. Clin. Invest.110, 493–497.10.1172/JCI15751
9
BendingD.De La PenaH.VeldhoenM.PhillipsJ. M.UyttenhoveC.StockingerB.CookeA. (2009). Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J. Clin. Invest.119, 565–572.10.1172/JCI37865
10
BendingD.NewlandS.KrejciA.PhillipsJ. M.BrayS.CookeA. (2011). Epigenetic changes at Il12rb2 and Tbx21 in relation to plasticity behavior of Th17 cells. J. Immunol.186, 3373–3382.10.4049/jimmunol.1003216
11
BeriouG.BradshawE. M.LozanoE.CostantinoC. M.HastingsW. D.OrbanT.ElyamanW.KhouryS. J.KuchrooV. K.Baecher-AllanC.HaflerD. A. (2010). TGF-beta induces IL-9 production from human Th17 cells. J. Immunol.185, 46–54.10.4049/jimmunol.1000356
12
BeriouG.CostantinoC. M.AshleyC. W.YangL.KuchrooV. K.Baecher-AllanC.HaflerD. A. (2009). IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood113, 4240–4249.10.1182/blood-2008-10-183251
13
BettelliE.KornT.OukkaM.KuchrooV. K. (2008). Induction and effector functions of T(H)17 cells. Nature453, 1051–1057.10.1038/nature07036
14
BonifaceK.BlomB.LiuY. J.De Waal MalefytR. (2008). From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol. Rev.226, 132–146.10.1111/j.1600-065X.2008.00714.x
15
BovenschenH. J.Van De KerkhofP. C.Van ErpP. E.WoestenenkR.JoostenI.KoenenH. J. (2011). Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J. Invest. Dermatol.131, 1853–1860.10.1038/jid.2011.139
16
BradshawE. M.RaddassiK.ElyamanW.OrbanT.GottliebP. A.KentS. C.HaflerD. A. (2009). Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J. Immunol.183, 4432–4439.10.4049/jimmunol.0900576
17
BrandS.BeigelF.OlszakT.ZitzmannK.EichhorstS. T.OtteJ. M.DiepolderH.MarquardtA.JaglaW.PoppA.LeclairS.HerrmannK.SeidererJ.OchsenkuhnT.GokeB.AuernhammerC. J.DambacherJ. (2006). IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol.290, G827–G838.10.1152/ajpgi.00513.2005
18
BuckleyR. H.WrayB. B.BelmakerE. Z. (1972). Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics49, 59–70.
19
CaiY.ShenX.DingC.QiC.LiK.LiX.JalaV. R.ZhangH. G.WangT.ZhengJ.YanJ. (2011). Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity35, 596–610.10.1016/j.immuni.2011.10.006
20
CascaoR.MouraR. A.PerpetuoI.CanhaoH.Vieira-SousaE.MouraoA. F.RodriguesA. M.Polido-PereiraJ.QueirozM. V.RosarioH. S.Souto-CarneiroM. M.GracaL.FonsecaJ. E. (2010). Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res. Ther.12, R196.10.1186/ar3168
21
ChabaudM.DurandJ.BuchsN.FossiezF.PageG.FrappartL.MiossecP. (1999). Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum.42, 963–970.10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E
22
ChenY.ThaiP.ZhaoY. H.HoY. S.DesouzaM. M.WuR. (2003). Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J. Biol. Chem.278, 17036–17043.10.1074/jbc.M208661200
23
ChenZ.TatoC. M.MuulL.LaurenceA.O’SheaJ. J. (2007). Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum.56, 2936–2946.10.1002/art.22316
24
ChiricozziA.Guttman-YasskyE.Suarez-FarinasM.NogralesK. E.TianS.CardinaleI.ChimentiS.KruegerJ. G. (2011). Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J. Invest. Dermatol.131, 677–687.10.1038/jid.2010.340
25
CohenC. J.CromeS. Q.MacdonaldK. G.DaiE. L.MagerD. L.LevingsM. K. (2011). Human Th1 and Th17 cells exhibit epigenetic stability at signature cytokine and transcription factor loci. J. Immunol.187, 5615–5626.10.4049/jimmunol.1101058
26
CosmiL.CimazR.MaggiL.SantarlasciV.CaponeM.BorrielloF.FrosaliF.QuerciV.SimoniniG.BarraG.PiccinniM. P.LiottaF.De PalmaR.MaggiE.RomagnaniS.AnnunziatoF. (2011). Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum.63, 2504–2515.10.1002/art.30332
27
CosmiL.De PalmaR.SantarlasciV.MaggiL.CaponeM.FrosaliF.RodolicoG.QuerciV.AbbateG.AngeliR.BerrinoL.FambriniM.CaproniM.TonelliF.LazzeriE.ParronchiP.LiottaF.MaggiE.RomagnaniS.AnnunziatoF. (2008). Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J. Exp. Med.205, 1903–1916.10.1084/jem.20080397
28
CosmiL.MaggiL.SantarlasciV.CaponeM.CardilicchiaE.FrosaliF.QuerciV.AngeliR.MatucciA.FambriniM.LiottaF.ParronchiP.MaggiE.RomagnaniS.AnnunziatoF. (2010). Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL-17A and IL-4. J. Allergy Clin. Immunol.125, 222–230.10.1016/j.jaci.2009.10.012
29
CromeS. Q.WangA. Y.KangC. Y.LevingsM. K. (2009). The role of retinoic acid-related orphan receptor variant 2 and IL-17 in the development and function of human CD4+ T cells. Eur. J. Immunol.39, 1480–1493.10.1002/eji.200838908
30
CuaD. J.SherlockJ.ChenY.MurphyC. A.JoyceB.SeymourB.LucianL.ToW.KwanS.ChurakovaT.ZurawskiS.WiekowskiM.LiraS. A.GormanD.KasteleinR. A.SedgwickJ. D. (2003). Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature421, 744–748.10.1038/nature01355
31
CuaD. J.TatoC. M. (2010). Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol.10, 479–489.10.1038/nri2800
32
CupedoT.CrellinN. K.PapazianN.RomboutsE. J.WeijerK.GroganJ. L.FibbeW. E.CornelissenJ. J.SpitsH. (2009). Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol.10, 66–74.10.1038/ni.1668
33
de BeaucoudreyL.PuelA.Filipe-SantosO.CobatA.GhandilP.ChrabiehM.FeinbergJ.Von BernuthH.SamarinaA.JanniereL.FieschiC.StephanJ. L.BoileauC.LyonnetS.JondeauG.Cormier-DaireV.Le MerrerM.HoarauC.LebranchuY.LortholaryO.ChandesrisM. O.TronF.GambineriE.BianchiL.Rodriguez-GallegoC.ZitnikS. E.VasconcelosJ.GuedesM.VitorA. B.MarodiL.ChapelH.ReidB.RoifmanC.NadalD.ReichenbachJ.CaragolI.GartyB. Z.DoguF.CamciogluY.GulleS.SanalO.FischerA.AbelL.StockingerB.PicardC.CasanovaJ. L. (2008). Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J. Exp. Med.205, 1543–1550.10.1084/jem.20080321
34
D’HennezelE.Ben-ShoshanM.OchsH. D.TorgersonT. R.RussellL. J.LejtenyiC.NoyaF. J.JabadoN.MazerB.PiccirilloC. A. (2009). FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. N. Engl. J. Med.361, 1710–1713.10.1056/NEJMc091749
35
Di CesareA.Di MeglioP.NestleF. O. (2009). The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J. Invest. Dermatol.129, 1339–1350.10.1038/jid.2009.59
36
DongC. (2008). TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat. Rev. Immunol.8, 337–348.10.1038/nri2295
37
DuC.LiuC.KangJ.ZhaoG.YeZ.HuangS.LiZ.WuZ.PeiG. (2009). MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat. Immunol.10, 1252–1259.10.1038/ni.1798
38
DubinP. J.KollsJ. K. (2008). Th17 cytokines and mucosal immunity. Immunol. Rev.226, 160–171.10.1111/j.1600-065X.2008.00703.x
39
EmamaulleeJ. A.DavisJ.MeraniS.TosoC.ElliottJ. F.ThiesenA.ShapiroA. M. (2009). Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes58, 1302–1311.10.2337/db08-1113
40
EspluguesE.HuberS.GaglianiN.HauserA. E.TownT.WanY. Y.O’ConnorW.Jr.RongvauxA.Van RooijenN.HabermanA. M.IwakuraY.KuchrooV. K.KollsJ. K.BluestoneJ. A.HeroldK. C.FlavellR. A. (2011). Control of TH17 cells occurs in the small intestine. Nature475, 514–518.10.1038/nature10228
41
EvansH. G.SuddasonT.JacksonI.TaamsL. S.LordG. M. (2007). Optimal induction of T helper 17 cells in humans requires T cell receptor ligation in the context of Toll-like receptor-activated monocytes. Proc. Natl. Acad. Sci. U.S.A.104, 17034–17039.10.1073/pnas.0707719104
42
FerraroA.SocciC.StabiliniA.ValleA.MontiP.PiemontiL.NanoR.OlekS.MaffiP.ScaviniM.SecchiA.StaudacherC.BonifacioE.BattagliaM. (2011). Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes60, 2903–2913.10.2337/db11-0090
43
FinaD.SarraM.FantiniM. C.RizzoA.CarusoR.CaprioliF.StolfiC.CardoliniI.DottoriM.BoirivantM.PalloneF.MacdonaldT. T.MonteleoneG. (2008). Regulation of gut inflammation and th17 cell response by interleukin-21. Gastroenterology134, 1038–1048.10.1053/j.gastro.2008.01.041
44
GaffenS. L. (2011). Recent advances in the IL-17 cytokine family. Curr. Opin. Immunol.23, 613–619.10.1016/j.coi.2011.07.006
45
GaffenS. L.Hernandez-SantosN.PetersonA. C. (2011). IL-17 signaling in host defense against Candida albicans. Immunol. Res.50, 181–187.10.1007/s12026-011-8226-x
46
GenoveseM. C.Van Den BoschF.RobersonS. A.BojinS.BiaginiI. M.RyanP.Sloan-LancasterJ. (2010). LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum.62, 929–939.10.1002/art.27612
47
GhoreschiK.LaurenceA.YangX. P.HiraharaK.O’SheaJ. J. (2011). T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol.32, 395–401.10.1016/j.it.2011.06.007
48
GlockerE. O.HennigsA.NabaviM.SchafferA. A.WoellnerC.SalzerU.PfeiferD.VeelkenH.WarnatzK.TahamiF.JamalS.ManguiatA.RezaeiN.AmirzargarA. A.PlebaniA.HannesschlagerN.GrossO.RulandJ.GrimbacherB. (2009). A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med.361, 1727–1735.10.1056/NEJMoa0810719
49
GrimbacherB.HollandS. M.GallinJ. I.GreenbergF.HillS. C.MalechH. L.MillerJ. A.O’ConnellA. C.PuckJ. M. (1999). Hyper-IgE syndrome with recurrent infections – an autosomal dominant multisystem disorder. N. Engl. J. Med.340, 692–702.10.1056/NEJM199903043400904
50
HarringtonL. E.HattonR. D.ManganP. R.TurnerH.MurphyT. L.MurphyK. M.WeaverC. T. (2005). Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol.6, 1123–1132.10.1038/ni1254
51
HeD.WuL.KimH. K.LiH.ElmetsC. A.XuH. (2006). CD8+ IL-17-producing T cells are important in effector functions for the elicitation of contact hypersensitivity responses. J. Immunol.177, 6852–6858.
52
HirotaK.DuarteJ. H.VeldhoenM.HornsbyE.LiY.CuaD. J.AhlforsH.WilhelmC.TolainiM.MenzelU.GarefalakiA.PotocnikA. J.StockingerB. (2011). Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol.12, 255–263.10.1038/ni.1993
53
HollandS. M.DeleoF. R.ElloumiH. Z.HsuA. P.UzelG.BrodskyN.FreemanA. F.DemidowichA.DavisJ.TurnerM. L.AndersonV. L.DarnellD. N.WelchP. A.KuhnsD. B.FruchtD. M.MalechH. L.GallinJ. I.KobayashiS. D.WhitneyA. R.VoyichJ. M.MusserJ. M.WoellnerC.SchafferA. A.PuckJ. M.GrimbacherB. (2007). STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med.357, 1608–1619.10.1056/NEJMoa073687
54
HonkanenJ.NieminenJ. K.GaoR.LuopajarviK.SaloH. M.IlonenJ.KnipM.OtonkoskiT.VaaralaO. (2010). IL-17 immunity in human type 1 diabetes. J. Immunol.185, 1959–1967.10.4049/jimmunol.1000788
55
HueberW.PatelD. D.DryjaT.WrightA. M.KorolevaI.BruinG.AntoniC.DraelosZ.GoldM. H.Psoriasis StudyG.DurezP.TakP. P.Gomez-ReinoJ. J.Rheumatoid Arthritis Study GroupFosterC. S.KimR. Y.SamsonC. M.FalkN. S.ChuD. S.CallananD.NguyenQ. D.Uveitis Study GroupRoseK.HaiderA.Di PadovaF. (2010). Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med.2, 52ra72.10.1126/scitranslmed.3001107
56
HotA.MiossecP. A. (2011). Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Ann. Rheum. Dis.70, 727–732.10.1136/ard.2010.132233
57
HughsonA.BrombergI.JohnsonB.QuataertS.JospeN.FowellD. J. (2011). Uncoupling of proliferation and cytokines from suppression within the CD4+CD25+Foxp3+ T-cell compartment in the 1st year of human type 1 diabetes. Diabetes60, 2125–2133.10.2337/db10-1661
58
HwangS. Y.KimA. H. (2005). Expression of IL-17 homologs and their receptors in the synovial cells of rheumatoid arthritis patients. Mol. Cells19, 180–184.
59
JainR.TartarD. M.GreggR. K.DivekarR. D.BellJ. J.LeeH. H.YuP.EllisJ. S.HoemanC. M.FranklinC. L.ZaghouaniH. (2008). Innocuous IFNgamma induced by adjuvant-free antigen restores normoglycemia in NOD mice through inhibition of IL-17 production. J. Exp. Med.205, 207–218.10.1084/jem.20071878
60
JosephJ.BittnerS.KaiserF. M.WiendlH.KisslerS. (2012). IL-17 silencing does not protect nonobese diabetic mice from autoimmune diabetes. J. Immunol.188, 216–221.10.4049/jimmunol.1101215
61
KebirH.KreymborgK.IferganI.Dodelet-DevillersA.CayrolR.BernardM.GiulianiF.ArbourN.BecherB.PratA. (2007). Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med.13, 1173–1175.10.1038/nm1651
62
KinugasaT.SakaguchiT.GuX.ReineckerH. C. (2000). Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology118, 1001–1011.10.1016/S0016-5085(00)84329-2
63
KirkhamB. W.LassereM. N.EdmondsJ. P.JuhaszK. M.BirdP. A.LeeC. S.ShnierR.PortekI. J. (2006). Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort). Arthritis Rheum.54, 1122–1131.10.1002/art.21749
64
KollsJ. K.LindenA. (2004). Interleukin-17 family members and inflammation. Immunity21, 467–476.10.1016/j.immuni.2004.08.018
65
KryczekI.BruceA. T.GudjonssonJ. E.JohnstonA.AphaleA.VatanL.SzeligaW.WangY.LiuY.WellingT. H.ElderJ. T.ZouW. (2008). Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J. Immunol.181, 4733–4741.
66
KryczekI.WuK.ZhaoE.WeiS.VatanL.SzeligaW.HuangE.GreensonJ.ChangA.RolinskiJ.RadwanP.FangJ.WangG.ZouW. (2011). IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J. Immunol.186, 4388–4395.10.4049/jimmunol.1003251
67
LangrishC. L.ChenY.BlumenscheinW. M.MattsonJ.BashamB.SedgwickJ. D.McclanahanT.KasteleinR. A.CuaD. J. (2005). IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med.201, 233–240.10.1084/jem.20041257
68
LeeK. A.KangM. H.LeeY. S.KimY. J.KimD. H.KoH. J.KangC. Y. (2008). A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity. Cell. Immunol.251, 50–55.10.1016/j.cellimm.2008.03.004
69
LeeS. M.YangH.TartarD. M.GaoB.LuoX.YeS. Q.ZaghouaniH.FangD. (2011). Prevention and treatment of diabetes with resveratrol in a non-obese mouse model of type 1 diabetes. Diabetologia54, 1136–1146.10.1007/s00125-011-2083-y
70
LeeY. K.TurnerH.MaynardC. L.OliverJ. R.ChenD.ElsonC. O.WeaverC. T. (2009). Late developmental plasticity in the T helper 17 lineage. Immunity30, 92–107.10.1016/j.immuni.2008.11.005
71
LeipeJ.GrunkeM.DechantC.ReindlC.KerzendorfU.Schulze-KoopsH.SkapenkoA. (2010). Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum.62, 2876–2885.10.1002/art.27622
72
LeonardiC.MathesonR.ZachariaeC.CameronG.LiL.Edson-HerediaE.BraunD.BanerjeeS. (2012). Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med.366, 1190–1199.10.1056/NEJMoa1109997
73
LinA. M.RubinC. J.KhandpurR.WangJ. Y.RiblettM.YalavarthiS.VillanuevaE. C.ShahP.KaplanM. J.BruceA. T. (2011). Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol.187, 490–500.10.4049/jimmunol.1100174
74
LiuH.Rohowsky-KochanC. (2008). Regulation of IL-17 in human CCR6+ effector memory T cells. J. Immunol.180, 7948–7957.
75
LiuS. M.LeeD. H.SullivanJ. M.ChungD.JagerA.ShumB. O.SarvetnickN. E.AndersonA. C.KuchrooV. K. (2011). Differential IL-21 signaling in APCs leads to disparate Th17 differentiation in diabetes-susceptible NOD and diabetes-resistant NOD.Idd3 mice. J. Clin. Invest.121, 4303–4310.10.1172/JCI60243
76
LowesM. A.BowcockA. M.KruegerJ. G. (2007). Pathogenesis and therapy of psoriasis. Nature445, 866–873.10.1038/nature05663
77
LundyS. K.SarkarS.TesmerL. A.FoxD. A. (2007). Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res. Ther.9, 202.10.1186/ar2107
78
MaC. S.ChewG. Y.SimpsonN.PriyadarshiA.WongM.GrimbacherB.FulcherD. A.TangyeS. G.CookM. C. (2008). Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med.205, 1551–1557.10.1084/jem.20080218
79
ManelN.UnutmazD.LittmanD. R. (2008). The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol.9, 641–649.10.1038/ni.1610
80
Martin-OrozcoN.ChungY.ChangS. H.WangY. H.DongC. (2009). Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur. J. Immunol.39, 216–224.10.1002/eji.200838475
81
MarwahaA. K.CromeS. Q.PanagiotopoulosC.BergK. B.QinH.OuyangQ.XuL.PriatelJ. J.LevingsM. K.TanR. (2010). Cutting edge: increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J. Immunol.185, 3814–3818.10.4049/jimmunol.1001860
82
MatuseviciusD.KivisäkkP.HeB.KostulasN.OzenciV.FredriksonS.LinkH. (1999). Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult. Scler.5, 101–104.10.1191/135245899678847275
83
McGeachyM. J.Bak-JensenK. S.ChenY.TatoC. M.BlumenscheinW.McclanahanT.CuaD. J. (2007). TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol.8, 1390–1397.10.1038/ni1539
84
McMurchyA. N.GilliesJ.AllanS. E.PasseriniL.GambineriE.RoncaroloM. G.BacchettaR.LevingsM. K. (2010). Point mutants of forkhead box P3 that cause immune dysregulation, polyendocrinopathy, enteropathy, X-linked have diverse abilities to reprogram T cells into regulatory T cells. J. Allergy Clin. Immunol.126, 1242–1251.10.1016/j.jaci.2010.09.001
85
MichelM. L.KellerA. C.PagetC.FujioM.TrotteinF.SavageP. B.WongC. H.SchneiderE.DyM.Leite-De-MoraesM. C. (2007). Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J. Exp. Med.204, 995–1001.10.1084/jem.20061551
86
MilnerJ. D. (2011). IL-17 producing cells in host defense and atopy. Curr. Opin. Immunol.23, 784–788.10.1016/j.coi.2011.09.006
87
MilnerJ. D.BrenchleyJ. M.LaurenceA.FreemanA. F.HillB. J.EliasK. M.KannoY.SpaldingC.ElloumiH. Z.PaulsonM. L.DavisJ.HsuA.AsherA. I.O’SheaJ.HollandS. M.PaulW. E.DouekD. C. (2008). Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature452, 773–776.10.1038/nature06764
88
MiyaraM.YoshiokaY.KitohA.ShimaT.WingK.NiwaA.ParizotC.TaflinC.HeikeT.ValeyreD.MathianA.NakahataT.YamaguchiT.NomuraT.OnoM.AmouraZ.GorochovG.SakaguchiS. (2009). Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity30, 899–911.10.1016/j.immuni.2009.03.019
89
MorrisonP. J.BallantyneS. J.KullbergM. C. (2011). Interleukin-23 and T helper 17-type responses in intestinal inflammation: from cytokines to T-cell plasticity. Immunology133, 397–408.10.1111/j.1365-2567.2011.03454.x
90
OgawaA.AndohA.ArakiY.BambaT.FujiyamaY. (2004). Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin. Immunol.110, 55–62.10.1016/j.clim.2003.09.013
91
PappuR.Ramirez-CarrozziV.SambandamA. (2011). The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. Immunology134, 8–16.10.1111/j.1365-2567.2011.03465.x
92
ParkH.LiZ.YangX. O.ChangS. H.NurievaR.WangY. H.WangY.HoodL.ZhuZ.TianQ.DongC. (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol.6, 1133–1141.10.1038/ni1261
93
PasseriniL.OlekS.Di NunzioS.BarzaghiF.HambletonS.AbinunM.TommasiniA.VignolaS.CipolliM.AmendolaM.NaldiniL.GuidiL.CecconiM.RoncaroloM. G.BacchettaR. (2011). Forkhead box protein 3 (FOXP3) mutations lead to increased TH17 cell numbers and regulatory T-cell instability. J. Allergy Clin. Immunol.128, 1376–1379.10.1016/j.jaci.2011.09.010
94
PuelA.CypowyjS.BustamanteJ.WrightJ. F.LiuL.LimH. K.MigaudM.IsraelL.ChrabiehM.AudryM.GumbletonM.ToulonA.BodemerC.El-BaghdadiJ.WhittersM.ParadisT.BrooksJ.CollinsM.WolfmanN. M.Al-MuhsenS.GalicchioM.AbelL.PicardC.CasanovaJ. L. (2011). Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science332, 65–68.10.1126/science.1200439
95
RachitskayaA. V.HansenA. M.HoraiR.LiZ.VillasmilR.LugerD.NussenblattR. B.CaspiR. R. (2008). Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J. Immunol.180, 5167–5171.
96
RennerE. D.RylaarsdamS.Anover-SombkeS.RackA. L.ReichenbachJ.CareyJ. C.ZhuQ.JanssonA. F.BarbozaJ.SchimkeL. F.LeppertM. F.GetzM. M.SegerR. A.HillH. R.BelohradskyB. H.TorgersonT. R.OchsH. D. (2008). Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J. Allergy Clin. Immunol.122, 181–187.10.1016/j.jaci.2008.04.037
97
SandbornW. J.FeaganB. G.FedorakR. N.ScherlE.FleisherM. R.KatzS.JohannsJ.BlankM.RutgeertsP.Ustekinumab Crohn’s Disease Study Group. (2008). A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology135, 1130–1141.10.1053/j.gastro.2008.09.053
98
SantarlasciV.MaggiL.CaponeM.FrosaliF.QuerciV.De PalmaR.LiottaF.CosmiL.MaggiE.RomagnaniS.AnnunziatoF. (2009). TGF-beta indirectly favors the development of human Th17 cells by inhibiting Th1 cells. Eur. J. Immunol.39, 207–215.10.1002/eji.200838748
99
SantarlasciV.MaggiL.CaponeM.QuerciV.BeltrameL.CavalieriD.D’AiutoE.CimazR.NebbiosoA.LiottaF.De PalmaR.MaggiE.CosmiL.RomagnaniS.AnnunziatoF. (2012). Rarity of human T helper 17 cells is due to retinoic acid orphan receptor-dependent mechanisms that limit their expansion. Immunity36, 201–214.10.1016/j.immuni.2011.12.013
100
SarkarS.FoxD. A. (2010). Targeting IL-17 and Th17 cells in rheumatoid arthritis. Rheum. Dis. Clin. North Am.36, 345–366.10.1016/j.rdc.2010.02.006
101
ShinH. C.BenbernouN.FekkarH.EsnaultS.GuenounouM. (1998). Regulation of IL-17, IFN-gamma and IL-10 in human CD8(+) T cells by cyclic AMP-dependent signal transduction pathway. Cytokine10, 841–850.10.1006/cyto.1998.0375
102
StarkM. A.HuoY.BurcinT. L.MorrisM. A.OlsonT. S.LeyK. (2005). Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity22, 285–294.10.1016/j.immuni.2005.01.011
103
SugimotoK.OgawaA.MizoguchiE.ShimomuraY.AndohA.BhanA. K.BlumbergR. S.XavierR. J.MizoguchiA. (2008). IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest.118, 534–544.
104
TranC. N.LundyS. K.WhiteP. T.EndresJ. L.MotylC. D.GuptaR.WilkeC. M.SheldenE. A.ChungK. C.UrquhartA. G.FoxD. A. (2007). Molecular interactions between T cells and fibroblast-like synoviocytes: role of membrane tumor necrosis factor-alpha on cytokine-activated T cells. Am. J. Pathol.171, 1588–1598.10.2353/ajpath.2007.070004
105
van den BrandtJ.FischerH. J.WalterL.HunigT.KlotingI.ReichardtH. M. (2010). Type 1 diabetes in biobreeding rats is critically linked to an imbalance between Th17 and regulatory T cells and an altered TCR repertoire. J. Immunol.185, 2285–2294.10.4049/jimmunol.1000462
106
van der GastC. J.GoslingP.TiwariB.BendingG. D. (2011). Spatial scaling of arbuscular mycorrhizal fungal diversity is affected by farming practice. Environ. Microbiol.13, 241–249.10.1111/j.1462-2920.2010.02326.x
107
van HamburgJ. P.AsmawidjajaP. S.DavelaarN.MusA. M.ColinE. M.HazesJ. M.DolhainR. J.LubbertsE. (2011). Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum.63, 73–83.10.1002/art.30093
108
VolpeE.ServantN.ZollingerR.BogiatziS. I.HupeP.BarillotE.SoumelisV. (2008). A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol.9, 650–657.10.1038/nrm2446
109
VooK. S.WangY. H.SantoriF. R.BoggianoC.ArimaK.BoverL.HanabuchiS.KhaliliJ.MarinovaE.ZhengB.LittmanD. R.LiuY. J. (2009). Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. U.S.A.106, 4793–4798.10.1073/pnas.0900408106
110
VukkadapuS. S.BelliJ. M.IshiiK.JeggaA. G.HuttonJ. J.AronowB. J.KatzJ. D. (2005). Dynamic interaction between T cell-mediated beta-cell damage and beta-cell repair in the run up to autoimmune diabetes of the NOD mouse. Physiol. Genomics21, 201–211.10.1152/physiolgenomics.00173.2004
111
WeaverC. T.HattonR. D. (2009). Interplay between the TH17 and TReg cell lineages: a (co-)evolutionary perspective. Nat. Rev. Immunol.9, 883–839.10.1038/nri2660
112
WilkeC. M.BishopK.FoxD.ZouW. (2011). Deciphering the role of Th17 cells in human disease. Trends Immunol.32, 603–611.10.1016/j.it.2011.08.003
113
WilsonN. J.BonifaceK.ChanJ. R.MckenzieB. S.BlumenscheinW. M.MattsonJ. D.BashamB.SmithK.ChenT.MorelF.LecronJ. C.KasteleinR. A.CuaD. J.McclanahanT. K.BowmanE. P.De Waal MalefytR. (2007). Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol.8, 950–957.10.1038/nrg2199
114
WrightJ. F.GuoY.QuaziA.LuxenbergD. P.BennettF.RossJ. F.QiuY.WhittersM. J.TomkinsonK. N.Dunussi-JoannopoulosK.CarrenoB. M.CollinsM.WolfmanN. M. (2007). Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J. Biol. Chem.282, 13447–13455.10.1074/jbc.M700499200
115
YangL.AndersonD.Baecher-AllanC.HastingsW. D.BettelliE.OukkaM.KuchrooV. K.HaflerD. A. (2008a). IL-21 and TGF-β are required for differentiation of human Th17 cells. Nature454, 350–352.10.1038/nature07021
116
YangX. O.ChangS. H.ParkH.NurievaR.ShahB.AceroL.WangY. H.SchlunsK. S.BroaddusR. R.ZhuZ.DongC. (2008b). Regulation of inflammatory responses by IL-17F. J. Exp. Med.205, 1063–1075.10.1084/jem.20062027
117
YangX. O.PanopoulosA. D.NurievaR.ChangS. H.WangD.WatowichS. S.DongC. (2007). STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem.282, 9358–9363.10.1074/jbc.M702131200
118
YeJ.SuX.HsuehE. C.ZhangY.KoenigJ. M.HoftD. F.PengG. (2011). Human tumor-infiltrating Th17 cells have the capacity to differentiate into IFN-gamma+ and FOXP3+ T cells with potent suppressive function. Eur. J. Immunol.41, 936–951.10.1002/eji.201040682
119
ZabaL. C.Fuentes-DuculanJ.EungdamrongN. J.AbelloM. V.NovitskayaI.PiersonK. C.GonzalezJ.KruegerJ. G.LowesM. A. (2009). Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J. Invest. Dermatol.129, 79–88.10.1038/jid.2008.194
120
ZenewiczL. A.YancopoulosG. D.ValenzuelaD. M.MurphyA. J.StevensS.FlavellR. A. (2008). Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity29, 947–957.10.1016/j.immuni.2008.11.003
121
ZhouL.LopesJ. E.ChongM. M.IvanovI. I.MinR.VictoraG. D.ShenY.DuJ.RubtsovY. P.RudenskyA. Y.ZieglerS. F.LittmanD. R. (2008). TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature453, 236–240.10.1038/nature06878
122
ZieglerS. F. (2006). FOXP3: of mice and men. Annu. Rev. Immunol.24, 209–226.10.1146/annurev.immunol.24.021605.090547
123
ZielinskiC. E.MeleF.AschenbrennerD.JarrossayD.RonchiF.GattornoM.MonticelliS.LanzavecchiaA.SallustoF. (2012). Pathogen-induced human T(H)17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature484, 514–518.10.1038/nature10957
124
ZiolkowskaM.KocA.LuszczykiewiczG.Ksiezopolska-PietrzakK.KlimczakE.Chwalinska-SadowskaH.MaslinskiW. (2000). High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J. Immunol.164, 2832–2838.
Summary
Keywords
Th17 cells, autoimmunity, T regulatory cells, immunodeficiency, inflammatory bowel disease, psoriasis, type 1 diabetes, secukinumab
Citation
Marwaha AK, Leung NJ, McMurchy AN and Levings MK (2012) TH17 Cells in Autoimmunity and Immunodeficiency: Protective or Pathogenic?. Front. Immun. 3:129. doi: 10.3389/fimmu.2012.00129
Received
10 April 2012
Accepted
04 May 2012
Published
04 June 2012
Volume
3 - 2012
Edited by
Rosa Bacchetta, Fondazione Centro San Raffaele Del Monte Tabor, Italy
Reviewed by
Rosa Bacchetta, Fondazione Centro San Raffaele Del Monte Tabor, Italy; Francesco Annunziato, University of Florence, Italy
Copyright
© 2012 Marwaha, Leung, McMurchy and Levings.
This is an openaccess article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Megan K. Levings, Department of Surgery, University of British Columbia, A4-186, 950 West 28th Avenue, Vancouver, BC, Canada V5Z 4H4. e-mail: mlevings@mail.ubc.ca
This article was submitted to Frontiers in Primary Immunodeficiencies, a specialty of Frontiers in Immunology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.