
- 1 Department of Microbiology and Immunology, Tohoku University Graduate School of Medicine, Sendai, Japan
- 2 Division of Immune Regulation, La Jolla Institute for Allergy and Immunology, La Jolla, CA, USA
Antigen primed T lymphocytes need to expand and persist to promote adaptive immunity. The growth and survival signals that control this are in large part provided by the NF-κB pathway in activated or effector/memory T cells. Although several membrane receptors impact NF-κB activation, signaling from OX40 (CD134, TNFRSF4), a member of the tumor necrosis factor receptor (TNFR) superfamily, has proven to be important for T cell immunity and a strong contributor to NF-κB activity. PKCθ directs the T cell receptor (TCR) and CD28-dependent assembly of a CBM complex (CARMA1, BCL10, and MALT1) for efficient activation of NF-κB, raising the question of whether other membrane bound receptors that activate NF-κB also require this PKCθ-CBM axis to control TCR-independent T cell activity. We discuss here our recent data demonstrating that after ligation by OX40L (CD252, TNFSF4) expressed on antigen-presenting cells, OX40 translocates into detergent-insoluble membrane lipid microdomains (DIM or lipid rafts) in T cells irrespective of TCR signals, and assembles into a signaling complex containing PKCθ, together with TRAF2, RIP1, the CBM complex, and the IKKα/β/Γ complex. PKCθ is required for optimal NF-κB activation mediated by OX40 and thus works as an essential component of this OX40 signalosome. We also discuss the likelihood that other TNFR superfamily molecules might complex with PKCθ in T cells, and whether PKC isoforms may be critical to the function of TNFR molecules in general.
Introduction
Interactions between several members of the tumor necrosis factor (TNF) superfamily and the TNF receptor (TNFR) superfamily are vital for regulation of T cell activation, differentiation, and survival (Croft, 2003, 2009). Many TNFR molecules, such as TNFR2 (TNFRSF1B), OX40 (TNFRSF4), CD27 (TNFRSF7), CD30 (TNFRSF8), 4-1BB (TNFRSF9), HVEM (TNFRSF14), GITR (TNFRSF18), and DR3 (TNFRSF25) are constitutively or inducibly expressed on T cells. They can be viewed as major sources of nuclear factor κB (NF-κB) signals, through TNF ligand-dependent recruitment of adaptors (TNF receptor-associated factors or TRAFs), making them important components of the T cell signaling machinery (Croft, 2003, 2009, 2010; Sugamura et al., 2004; Watts, 2005; So et al., 2006; Nocentini et al., 2007; Nolte et al., 2009; Faustman and Davis, 2010). Although the molecular machinery through which TNFR1 (TNFRSF1A) regulates signaling has been intensively studied in non-T cells and has become a framework to understand the classical or canonical NF-κB (NF-κB1) pathway as well as induction of apoptosis, it is unclear how other members of the TNFR superfamily organize their signaling complexes on the membrane, especially in T cells, and whether the respective complexes are comparable to that recruited by TNFR1.
Activation of NF-κB1 is initiated by signal-dependent phosphorylation, ubiquitination, and subsequent degradation of inhibitory IκB. This allows cytoplasmic NF-κB1/RelA to stably translocate to the nucleus and activate gene transcription. IκB phosphorylation is catalyzed by the IκB kinase (IKK) complex that contains two homologous catalytic subunits, IKKα and IKKβ, and the regulatory subunit IKKΓ. Activation of IKKβ is essential for NF-κB1 in response to all pro-inflammatory stimuli (Hayden and Ghosh, 2008; Vallabhapurapu and Karin, 2009). Although all stimuli leading to NF-κB1 activation appear to converge on IKKβ-mediated IκB phosphorylation, the upstream events controlling activation of the IKK complex are possibly distinct and specific to the individual type of NF-κB-activating stimulus. In T cells, the T cell receptor (TCR) and the Ig superfamily costimulatory molecule CD28 are capable of synergizing together and activating NF-κB1. CARMA1 [caspase-recruitment domain (CARD)-membrane-associated guanylate kinase (MAGUK) protein 1] has been shown to control this NF-κB1 activation by forming a complex with B cell lymphoma 10 (BCL10), and mucosa-associated-lymphoid-tissue lymphoma-translocation gene 1 (MALT1), termed CBM (Gaide et al., 2002; Wang et al., 2002; Thome, 2004). Importantly, PKCθ (protein kinase C θ) is also recruited after cross-linking the TCR and CD28 (Bi et al., 2001). Phosphorylation of CARMA1 by PKCθ induces the assembly of the CBM complex that then activates IKKβ (Matsumoto et al., 2005; Sommer et al., 2005).
The question was then raised as to whether this PKCθ-CBM module to activate IKKβ was specific to cooperation between the TCR and CD28 or might be a shared pathway with other molecules, either in T cells or non-T cells. Initial studies of CARMA1 suggested the former. TNF binding with TNFR, largely on non-T cells such as embryonic fibroblasts, has been extensively characterized, and shown to recruit TRAF2 that links the serine/threonine kinase RIP1 (receptor interacting protein kinase-1) to activation of IKKβ (Chen and Goeddel, 2002; Wajant et al., 2003; Muppidi et al., 2004; Kovalenko and Wallach, 2006). In contrast, TNF was found to induce NF-κB activation equivalently in CARMA1-deficient T cells (Gaide et al., 2002; Wang et al., 2002), implying that TNFR superfamily members may not use or require the PKCθ-CBM pathway for their activities. We have now recently defined a novel signaling complex of OX40, which does contains PKCθ and the CBM complex, as well as TRAF2, RIP1, and the IKK complex (IKKα, IKKβ, and IKKΓ), but not TCR, CD28, or other TCR-proximal signaling kinases (So et al., 2011b). Upon OX40L (TNFSF4) stimulation, this signaling module is organized in detergent-insoluble membrane lipid microdomains (DIM or lipid rafts) and regulates TCR-independent NF-κB1 activation. This review focuses on these new findings regarding the OX40 complex and discusses its relevance to other TNFR members in terms of regulation of PKCθ and other PKC isoforms.
NF-κB1 Signaling Through OX40 is Essential for Activated/Effector T Cell Responses
The TNF receptor OX40 is induced on activated CD4+ and CD8+ T cells and the TNF ligand OX40L is induced on activated antigen-presenting cells (APCs; Croft, 2010). Signaling through OX40 dominantly regulates T cell turnover at the peak of the expansion phase of many immune responses and the subsequent survival of activated/effector T cells when antigen becomes limiting (Gramaglia et al., 2000; Rogers et al., 2001; Bansal-Pakala et al., 2004). OX40-deficient T cells cannot persist well and exhibit decreased survival rates, resulting in reduced accumulation of memory cells with time (Gramaglia et al., 2000; Murata et al., 2000; Humphreys et al., 2007; Soroosh et al., 2007; Mousavi et al., 2008). The signaling mechanisms by which OX40 contributes to T cell survival are reasonably well defined in CD4+ T cells. Little has been done in terms of signaling in CD8 T cells but the targets and molecules involved are likely similar. One critical pathway that regulates CD4+ T cell survival mediated by OX40 is NF-κB1 (Song et al., 2008). Phosphorylation of IκB, nuclear translocation of NF-κB1/RelA, and NF-κB1 activities, are impaired in antigen-responding CD4+ T cells which lack OX40. In accordance with this, OX40-deficient CD4+ T cells cannot maintain high levels of several anti-apoptotic Bcl-2 family members that are under the control of NF-κB1. Correspondingly, retroviral transduction of a constitutively active form of IKKβ into OX40-deficient CD4+ T cells rescues the poor survival phenotype and increases the expression of Bcl-2 family members (Song et al., 2008).
The TNF ligand OX40L is a type II transmembrane and homotrimeric protein composed of three TNF homology domains, whereas OX40 is a type I transmembrane protein monomer and is trimerized through binding with OX40L, resulting in formation of a quaternary organized hexamer complex. OX40 has four cysteine-rich domains (CRDs) and the first three CRDs from the N-terminus interact with OX40L in the extracellular space (Compaan and Hymowitz, 2006). OX40 has the potential to recruit TRAF2, TRAF3, and TRAF5 to a QEE motif existing in its ∼40 amino acid cytoplasmic tail (Arch and Thompson, 1998; Kawamata et al., 1998; Table 1). However, whether all TRAFs are recruited in vivo is not clear and the downstream signaling that is controlled by these TRAFs has not been investigated in detail. To easily visualize and uncover the signaling modules induced by OX40 ligation, we established an MCC-specific T cell hybridoma cell from OX40-deficient and TCR transgenic mice, and introduced cMyc-tagged-OX40 into this T cell (So et al., 2011b). Although the cMyc-tag is attached to the N-terminus of OX40, this cMyc-OX40 can interact normally with OX40L and induce strong NF-κB1 activity in the T cell. Furthermore, the cMyc-tagged OX40 can be efficiently precipitated from this cell (So et al., 2011b). After triggering OX40 with membrane bound OX40L expressed on a fibroblast cell (Gramaglia et al., 1998), we observed recruitment of the canonical TRAF2, RIP1, and IKK complex, and also PKCθ and the CBM complex (Table 1). Importantly, this signalosome did not require TCR signals, and was formed without antigen recognition and in the complete absence of a TCR. Moreover, an anti-OX40 agonist antibody immobilized on a plate induced the same signaling complex (So et al., 2011b).
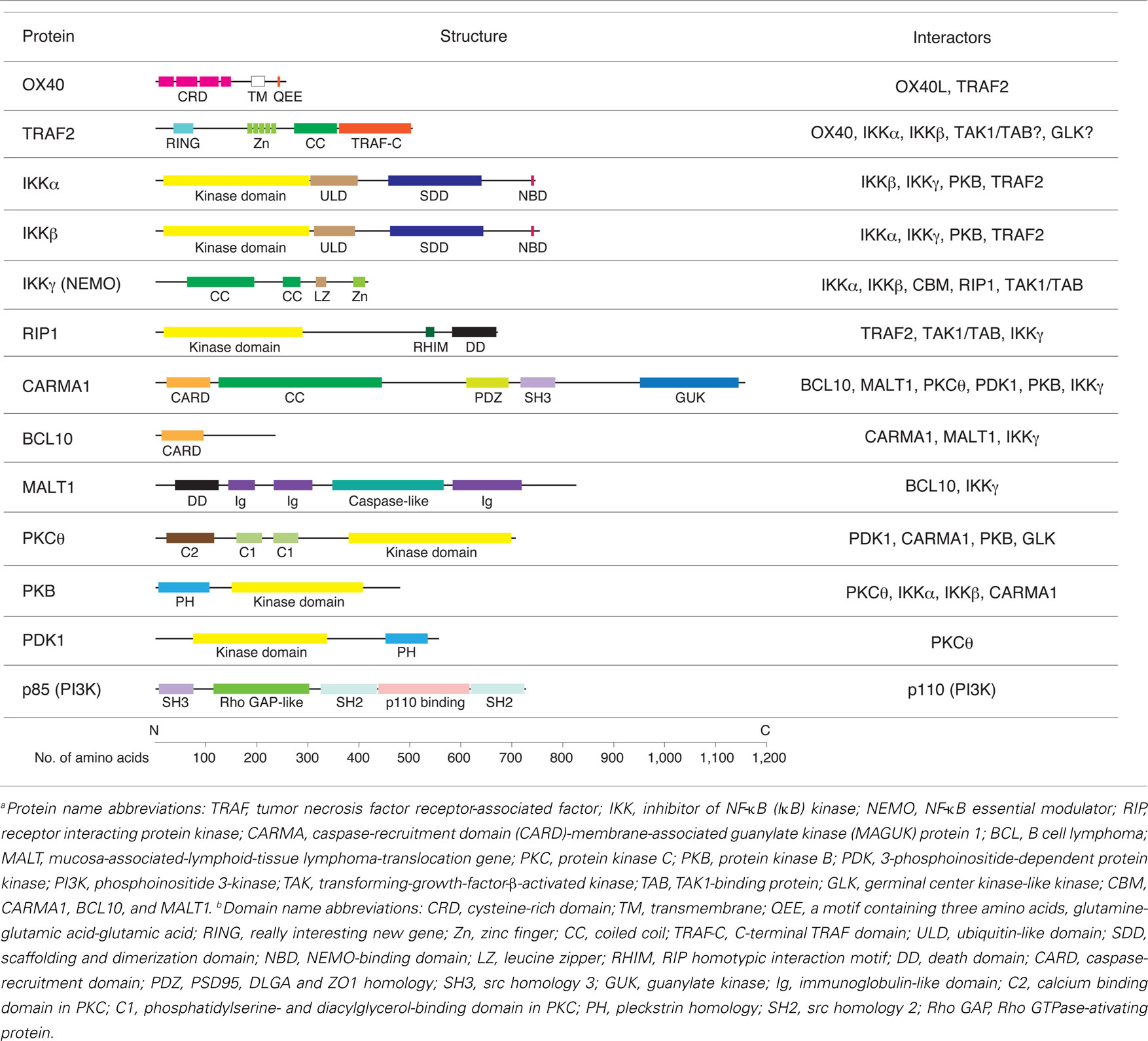
Table 1. Proteins involved in the OX40 signalosomea, b.
A Molecular Framework for the OX40 Signalosome
An important issue is how OX40 builds the functional signaling complex for NF-κB1 in the absence of TCR signals. In the TNFR1-NF-κB1 pathway, a pro-survival complex I is formed by recruitment of TNF receptor-associated death domain (TRADD), RIP1, TRAF2, cellular inhibitor of apoptosis protein 1 and 2 (cIAP and cIAP2), and the linear ubiquitin chain assembly complex (LUBAC; Chen and Goeddel, 2002; Wajant et al., 2003; Muppidi et al., 2004; Kovalenko and Wallach, 2006; Iwai and Tokunaga, 2009; Vallabhapurapu and Karin, 2009; Walczak, 2011). RIP1 and TRAF2 are conjugated with non-degradative Lys-63 (K63)-linked polyubiquitin chains, which are thought to be critical to recruit a transforming-growth-factor-β-activated kinase-1 (TAK1)/TAK1-binding protein (TAB) 2/TAB3 complex and the IKK complex, leading to IKK activation (Wertz and Dixit, 2008; Li et al., 2009; Skaug et al., 2009; Napolitano and Karin, 2010). TRAF2 acts as an adaptor and it may function as part of the E3 ubiquitin ligase for RIP1 in concert with cIAP1/2 (Ea et al., 2006). In contrast, OX40 does not have a death domain (DD) to recruit TRADD but may simply rely on its QEE motif to recruit TRAFs (Table 1; Figure 1). Short hairpin RNA mediated silencing of TRAF2 significantly decreased the association between OX40 and the IKK complex and blocked NF-κB1 activation (So et al., 2011b), showing that TRAF2 is as an essential keystone for the OX40-NF-κB1 axis. RIP1 was ubiquitinated following OX40 triggering, but the deficiency in TRAF2 did not change the level of ubiquitination and did not affect recruitment of RIP1 to OX40 (So et al., 2011b). Although RIP1 is thought to play a role in TNFR1 driven NF-κB signaling as described above, it has been reported that TNF-induced NF-κB1 activation is normal in some RIP1-deficient cells, suggesting that the requirement for RIP1 is cell-type specific (Bertrand and Vandenabeele, 2010). The functional significance of RIP1 in the OX40 complex has yet to be determined, but it is possible that it is not sufficient for recruitment of the IKK complex or IKK phosphorylation. This may explain our finding that PKCθ and the CBM complex associate with OX40.
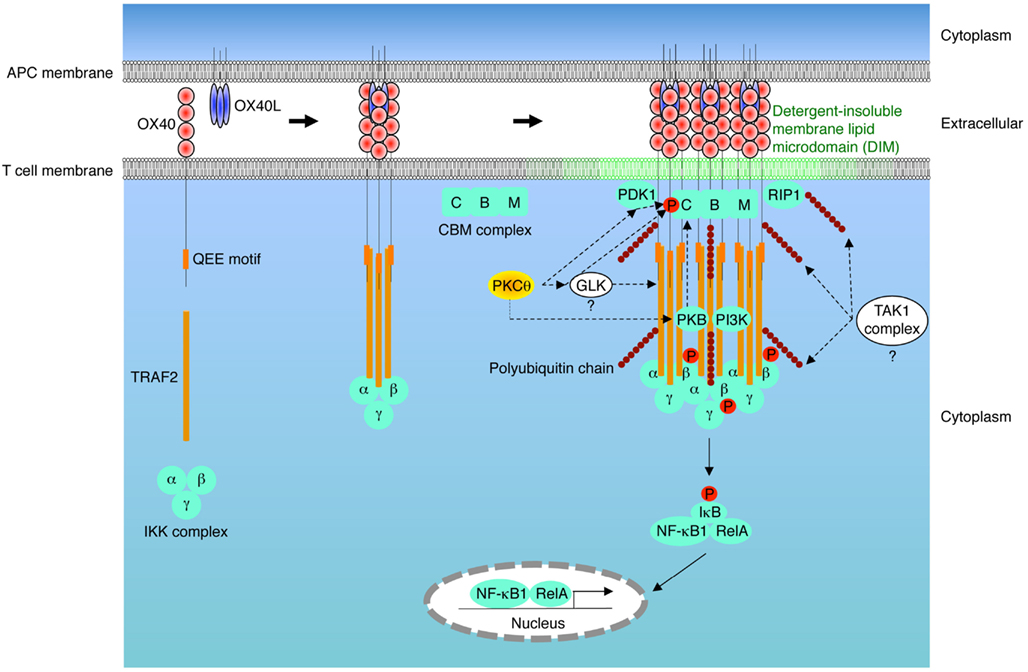
Figure 1. A model of the OX40 complex cascade leading to IKK activation. OX40L binding to OX40 on T cells results in trimerization of OX40 monomers and recruitment of TNFR-associated factor 2 (TRAF2) to the cytoplasmic QEE motif in OX40. The IκB kinase (IKK) complex (IKKα, IKKβ, and IKKΓ) is then recruited to the OX40-TRAF2 module, but this event is not sufficient for IKK activation. The OX40-OX40L hexamer complex translocates into detergent-insoluble membrane lipid microdomains (DIM). This organizes the higher ordered multimolecular receptor-ligand architecture that is required for IKK activation in a T cell receptor (TCR)-independent manner. Phosphoinositide 3-kinase (PI3K), 3-phosphoinositide-dependent protein kinase-1 (PDK1), and protein kinase B (PKB) are recruited to OX40 in the DIM. Although not yet determined, germinal center kinase-like kinase (GLK) is possibly incorporated into the OX40 signalosome through TRAF2. Protein kinase C θ (PKCθ) translocates to the DIM resident OX40, likely through the activities of PDK1, PKB, or GLK, and may be activated by either PDK1 or GLK. Caspase-recruitment domain (CARD)-membrane-associated guanylate kinase (MAGUK) protein 1 (CARMA1) also associates with OX40 in DIM, possibly through PKB. Activated PKCθ phosphorylates CARMA1, and this subsequently induces the CBM complex [CARMA1, B cell lymphoma 10 (BCL10), and mucosa-associated-lymphoid-tissue lymphoma-translocation gene 1 (MALT1)]. Receptor interacting protein 1 (RIP1) is also incorporated into the OX40 complex in DIM in a TRAF2-independent manner. OX40, TRAF2, and RIP1 are highly polyubiquitinated in the OX40 complex, and thus the transforming-growth-factor-β-activated kinase-1 (TAK1) complex might be recruited through these polyubiquitin chains. The IKK complex activated in the OX40 complex phosphorylates and degrades IκB, and then this facilitates entry of NF-κB1/RelA into the nucleus.
PKCθ is highly expressed in T cells and the importance for mature T cell activation is well recognized (Sun et al., 2000; Isakov and Altman, 2002). We had previously observed in one in vivo system that OX40 signaling could not compensate for defective activation of PKCθ-deficient CD4+ T cells even though OX40 was expressed (Salek-Ardakani et al., 2005). This implied that PKCθ was a possible mediator of OX40 signals. Although TRAF2, RIP1, CARMA1, and the IKK complex were pulled down with OX40 under conditions of immunoprecipitation with a stringent buffer (RIPA), the PKCθ-CBM complex was only pulled down using a milder buffer containing n-dodecyl-β-maltoside, a detergent that preserves membrane protein structure. This shows that the PKCθ-CBM compartment of the OX40 complex is weaker in association and may require additional intermediates, and that the membrane environment is required to organize the compartment.
It has been demonstrated that PKCθ specifically interacts with lipids or protein components in DIM (Bi et al., 2001; Melowic et al., 2007; Kong et al., 2011). Phosphoinositide 3-kinase (PI3K) participates in the selective membrane recruitment of PKCθ (Villalba et al., 2002). Protein kinase B (PKB or Akt; Bauer et al., 2001) and 3-phosphoinositide-dependent protein kinase-1 (PDK1; Park et al., 2009) interact with PKCθ, and can also control NF-κB1 activity. The interaction between PKB and CARMA1 additionally may play an important role for NF-κB1 (Narayan et al., 2006). In our experiments, OX40 translocated into DIM after interaction with OX40L and although we found that the interaction between OX40 and the TRAF2-IKK compartment was independent of DIM, depletion of cholesterol or suppression of synthesis of sphingolipid/cholesterol strongly inhibited OX40-dependent NF-κB1 activation (So et al., 2011b). This showed that additional molecular events in the DIM are required for activation of the IKK complex by OX40. In accordance, we observed that PKCθ associated with OX40 in DIM and this association was dependent on TRAF2 (So et al., 2011b). PI3K and PKB, and to a minor extent PDK1, were also inducibly recruited into the OX40 complex (So et al., 2011a). PI3K was phosphorylated in this complex (So et al., 2011a) and thus is probably important for conversion of phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) into phosphatidylinositol-3,4,5-triphosphate (PtdIns(3,4,5)P3) in the neighboring membrane where OX40 translocates in the immune synapse. The localization of PtdIns(3,4,5)P3 at the inner leaflet of the plasma membrane is known to recruit pleckstrin homology (PH) domain containing signaling proteins, such as PDK1 and PKB. Activated PDK1 can phosphorylate PKCθ (Park et al., 2009) and PKB may link PKCθ and CARMA1 (Bauer et al., 2001; Narayan et al., 2006), which in turn could lead to activation of CARMA1 and induction of the CBM complex (Matsumoto et al., 2005; Sommer et al., 2005). Furthermore, PKB can directly or indirectly lead to phosphorylation of IKKα and IKKβ (Ozes et al., 1999; Romashkova and Makarov, 1999). Therefore, it is likely that PKCθ may be recruited to OX40 through PDK1 and/or PKB allowing PKCθ to phosphorylate CARMA1 and providing the maximal stimuli necessary to phosphorylate the IKK complex (Table 1; Figure 1). Consistent with this, PKCθ- or CARMA1-deficient primary CD4+ T cells displayed severely reduced activation of NF-κB1 when stimulated by OX40L in spite of normal expression of OX40 (So et al., 2011b).
It is also possible that the cross-talk between OX40 and PKCθ is mediated through the germinal center kinases (GCKs). Four of the mammalian group I GCKs, GCK, GCK-related (GCKR), GCK-like kinase (GLK), and hematopoietic progenitor kinase-1 (HPK1), have a conserved carboxyl terminal regulatory domain that was suggested to target TRAF proteins (Kyriakis, 1999) and thus these four kinases may be recruited to members of the TNFR superfamily. Both GCK and GCKR can physically associate with TRAF2 (Yuasa et al., 1998; Chin et al., 1999; Shi et al., 1999; Shi and Kehrl, 2003) although the stimuli that may induce this are unclear. Most interestingly, GLK was recently found to directly phosphorylate and activate PKCθ in T cells (Chuang et al., 2011). These data then suggest that OX40 might activate PKCθ through the TRAF2-mediated recruitment of GLK (Figure 1). Whether GCKs are recruited to OX40 and function to control PKCθ activity needs to be addressed in the future.
The OX40 complex is likely to be tightly controlled by polyubiquitin chains. A polyubiquitin chain is formed when one of the eight amino groups within ubiquitin (seven ε-amino groups of internal lysines and one α-amino group of N-terminal methionine) is linked to the C-terminal glycine of another ubiquitin. The best characterized linkages utilize ubiquitin K48 and K63. K48-linked polyubiquitination usually targets substrates for proteosomal degradation, whereas K63-linked polyubiquitin chains can function as scaffolds to assemble signaling complexes, such as the TAK1/TAB2/TAB3 and the IKK complexes (Wertz and Dixit, 2008; Skaug et al., 2009). The cytoplasmic tail of OX40 contains three lysine residues, which might be targets for ubiquitination. Indeed, upon triggering with OX40L, OX40 is highly ubiquitinated and the protein level of OX40 is transiently decreased (So et al., 2011a,b). Disruption of DIM decreases the level of polyubiquitin chains, correlating with reduced complex formation and weak NF-κB1 activity induced by OX40 (So et al., 2011a,b). This suggests that DIM works as a platform to attach polyubiquitin chains to OX40 and that this event plays an essential role for IKK activation. At the present, we do not know how many K48- and K63-linked polyubiquitin chains are conjugated to OX40, but we think that both types of polyubiquitin chains should be critical for regulation of the OX40-NF-κB1 axis. Whether ubiquitination of OX40 will affect recruitment of PKCθ remains to be seen.
Blocking interactions between OX40L and OX40 concomitantly block survival of pathogenic effector T cells and promote clonal expansion and suppressive function of Foxp3+ regulatory T cells. OX40 is therefore a promising drug target for T cell-mediated inflammatory diseases. Mice treated with anti-OX40L blocking mAb or OX40- and OX40L-deficient mice have revealed significantly attenuated inflammation in murine models of colitis, asthma, diabetes, multiple sclerosis, rheumatoid arthritis, atherosclerosis, graft-versus-host disease, sepsis, and uveitis (Croft, 2009, 2010). PKCθ also has a similar dual role in effector and regulatory T cells, i.e., inhibition of PKCθ decreases inflammation mediated by effector T cells, whereas it promotes suppressive functions of regulatory T cells (Zanin-Zhorov et al., 2011). This suggests that inhibitors that target the molecular machinery of OX40 (Table 1; Figure 1) also have a great therapeutic potential with inflammatory and autoimmune diseases.
Regulation of PKC Isoforms by Other Members of the TNFR Superfamily
Of the TNFR family members most closely related to OX40 (TNFR2, HVEM, 4-1BB, CD30, GITR, CD27, DR3), only 4-1BB has been assessed in terms of potentially modulating or requiring PKCθ. Ligation of 4-1BB in activated CD8+ T cells was found to induce translocation of PKCθ into lipid raft domains augmenting PKCθ accumulation in the contact area between a T cell and an APC (Nam et al., 2005). The signaling complex of 4-1BB has not been visualized, but this data implies 4-1BB may recruit a signalosome that is closely related to that recruited by OX40. 4-1BB also binds TRAF2, and given our finding that TRAF2 knockdown inhibited PKCθ association with OX40, it is likely that any TNFR molecule that binds TRAF2 might have the capacity to engage PKCθ. TRAF2 can bind TNFR2, HVEM, CD30, GITR, CD27, and DR3 in transient transfection systems, implying this molecule may be central to the activities of all of these molecules. This remains to be determined, but in this regard, induction of a monocyte inflammatory mediator, TGF-β-inducible gene h3 (βig-h3), by cross-linking DR3 was blocked by several PKC inhibitors that might target PKCθ, although no direct data was provided (Lee et al., 2010).
As discussed above, current ideas suggest that PKCθ is not required for the activity of TNF through TNFR1, however other PKC isoforms may be involved in TNFR family signaling in some situations. It is well known that activation of PKC by phorbol ester can antagonize death (apoptosis) induced by DD containing TNFR members, such as TNFR1, FAS (TNFRSF6), and TRAIL-R1/2 (TNFRSF10A/B; Ruiz-Ruiz et al., 1997; Gomez-Angelats et al., 2000; Meng et al., 2002; Harper et al., 2003). Pretreatment of HeLa cells with phorbol ester inhibits recruitment of key obligatory DD-containing adaptor proteins to the death-inducing signaling complex (DISC) organized by TRAIL-R and TNFR1 (Harper et al., 2003). In the TNFR1 complex, RIP1 may recruit atypical PKCs (PKCζ and λ or ℩) through p62 (Sanz et al., 1999). In human neutrophils, TNFR1 was found to recruit PKCδ to the complex and this counteracted apoptotic signaling mediated by the DISC through activation of NF-κB1 (Kilpatrick et al., 2004). Furthermore, in the TNFR1 complex of mouse embryonic fibroblast, PKCδ and PKCε were recently shown to be responsible for phosphorylation of TRAF2, controlling the introduction of K63-linked polyubiquitin chains into TRAF2, and recruitment of the TAK1/TAB2/TAB3 complex and activation of the IKK complex (Li et al., 2009). In another example, in human peripheral blood lymphocytes and leukemic T cell lines, FAS upon stimulation with FASL (TNFSF6) induced rapid localization of stromal interaction molecule 1 (STIM1) and Orai1 into the membrane receptor cluster and this led to Ca2+ entry and recruitment of PKCβ2 into the DISC. PKCβ2 in turn also delayed DISC formation and prevented induction of the apoptotic pathway (Khadra et al., 2011). Thus, in the apoptosis-inducing members of the TNFR superfamily, PKC recruitment may primarily limit cell death, or function to help molecules like TNFR1 to switch their signaling toward the pro-inflammatory NF-κB pathway (Figure 2).
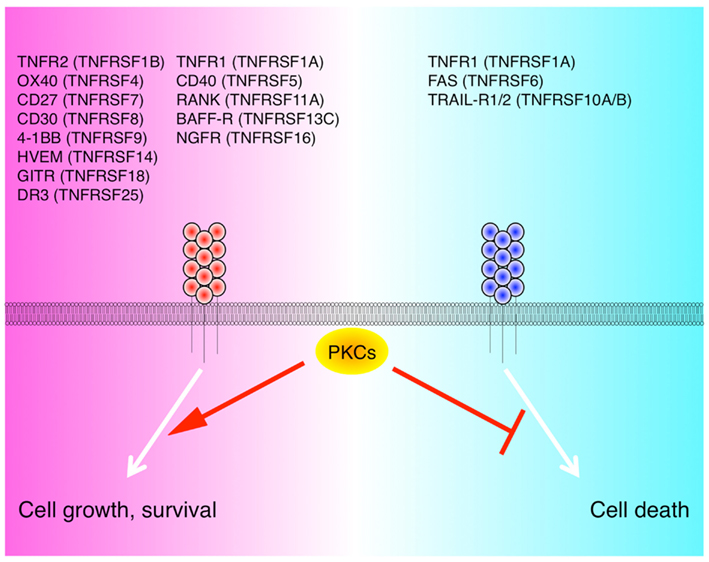
Figure 2. PKC isoforms control life and death induced by TNFRSF. The TNFR-PKC axis activates NF-κB1 or other signaling pathways, which concomitantly promotes cell growth/survival and inhibits cell death.
In other TNFR members that do not contain DD, such as CD40 (TNFRSF5), BAFF-R (TNFRSF13C), RANK (TNFRSF11A), NGFR (TNFRSF16), and GITR, alternate PKC isoforms also appear to play roles in cellular functions. In peritoneal macrophages, strong or weak engagement of CD40 reciprocally regulated PKC isoforms, resulting in differential cellular responsiveness to Leishmania major infection. A higher concentration of anti-CD40 induced phosphorylation and membrane translocation of PKCα, β1, β2, and ε, which favored Th1-type protective immunity effective for the parasite elimination, whereas a lower concentration induced phosphorylation and membrane translocation of PKCδ and ζ/λ, which favored Th2-type immunity and thus permitted parasite growth (Sudan et al., 2012). In mature B cells, triggering of BAFF-R with BAFF (TNFSF13B) also induced membrane translocation of PKCβ which controlled B cell survival through PKB activation (Patke et al., 2006). Stimulation of RANK with RANKL (TNFSF11), in a pre-osteoclast cell line RAW264.7 and in primary bone marrow-derived macrophages, led to recruitment of atypical PKCs through a RANK-TRAF6-p62-PKC linkage. This activated NF-κB1/NFATc1 and played a critical role for osteoclastogenesis (Duran et al., 2004). Moreover, in P12 cells, a rat pheochromocytoma cell line, NGFR stimulation with NGF induced a receptor complex that contained K63-polyubiquitinated TRAF6, p62, IKKβ, and PKC℩, which induced NF-κB1 and was involved in neuronal survival (Wooten et al., 2005). Lastly, stimulation of a macrophage cell line with soluble GITR induced recruitment of PKCδ to the cell membrane fraction (Lee et al., 2004). These data then collectively imply that overall usage of PKC isoforms by members of the TNFR superfamily is likely to be widespread. It is tempting to speculate that the TNFR-PKC axis may be critical for life and death decisions in many different types of cells by inducing NF-κB1 activation or activities of other signaling pathways (Figure 2).
Conclusions
Based on results obtained in our biochemical studies, we present an original model that can explain how PKCθ contributes to the NF-κB1 pathway mediated by the OX40 stimulatory receptor in T cells (Figure 1). Upon interaction with membrane OX40L, OX40 moves into the DIM of T cells and builds a multimolecular complex irrespective of antigen/TCR engagement. This complex provides the molecular machinery that controls IKKβ through PKCθ. PKCθ is recruited to the OX40-TRAF2 compartment, activates CARMA1, and then induces the CBM complex to augment IKK activities. This OX40 complex, which contains several upstream kinases for IKK, is an important source of NF-κB1 in T cells and controls longevity of T cells through induction of pro-survival genes. Although OX40 can provide classical costimulatory signals to T cells in concert with those from antigen and CD28, OX40 also sustains signals initiated by the TCR and CD28 while functioning as an independent signaling unit. PKC is central to signal transduction pathways involved in T cell activation, differentiation, and survival. Our data suggests that PKCθ is an integral component of the complex that allows OX40 to function in this regard, and we speculate that an equivalent signaling complex containing PKCθ is likely to be found in complexes formed by other members of the TNFR superfamily. This may explain the global requirement for many of these molecules in driving T cell responses. It will be very important to compare the molecular mechanisms by which TNFR members control T cell activity in the future and to determine if one or several PKC isoforms are central regulators of TNFR family molecule action.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Michael Croft is supported by grants CA91837, AI49453, AI089624, and AI070535 from the National Institutes of Health. This is publication #1504 from the La Jolla Institute for Allergy and Immunology. Disclosure: Michael Croft has patents on several TNFSF molecules.
References
Arch, R. H., and Thompson, C. B. (1998). 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol. Cell. Biol. 18, 558–565.
Bansal-Pakala, P., Halteman, B. S., Cheng, M. H., and Croft, M. (2004). Costimulation of CD8 T cell responses by OX40. J. Immunol. 172, 4821–4825.
Bauer, B., Krumbock, N., Fresser, F., Hochholdinger, F., Spitaler, M., Simm, A., Uberall, F., Schraven, B., and Baier, G. (2001). Complex formation and cooperation of protein kinase C theta and Akt1/protein kinase B alpha in the NF-kappa B transactivation cascade in Jurkat T cells. J. Biol. Chem. 276, 31627–31634.
Bertrand, M. J., and Vandenabeele, P. (2010). RIP1’s function in NF-kappaB activation: from master actor to onlooker. Cell Death Differ. 17, 379–380.
Bi, K., Tanaka, Y., Coudronniere, N., Sugie, K., Hong, S., van Stipdonk, M. J., and Altman, A. (2001). Antigen-induced translocation of PKC-theta to membrane rafts is required for T cell activation. Nat. Immunol. 2, 556–563.
Chin, A. I., Shu, J., Shan Shi, C., Yao, Z., Kehrl, J. H., and Cheng, G. (1999). TANK potentiates tumor necrosis factor receptor-associated factor-mediated c-Jun N-terminal kinase/stress-activated protein kinase activation through the germinal center kinase pathway. Mol. Cell. Biol. 19, 6665–6672.
Chuang, H. C., Lan, J. L., Chen, D. Y., Yang, C. Y., Chen, Y. M., Li, J. P., Huang, C. Y., Liu, P. E., Wang, X., and Tan, T. H. (2011). The kinase GLK controls autoimmunity and NF-kappaB signaling by activating the kinase PKC-theta in T cells. Nat. Immunol. 12, 1113–1118.
Compaan, D. M., and Hymowitz, S. G. (2006). The crystal structure of the costimulatory OX40-OX40L complex. Structure 14, 1321–1330.
Croft, M. (2003). Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat. Rev. Immunol. 3, 609–620.
Croft, M. (2009). The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol. 9, 271–285.
Croft, M. (2010). Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 28, 57–78.
Duran, A., Serrano, M., Leitges, M., Flores, J. M., Picard, S., Brown, J. P., Moscat, J., and Diaz-Meco, M. T. (2004). The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev. Cell 6, 303–309.
Ea, C. K., Deng, L., Xia, Z. P., Pineda, G., and Chen, Z. J. (2006). Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 22, 245–257.
Faustman, D., and Davis, M. (2010). TNF receptor 2 pathway: drug target for autoimmune diseases. Nat. Rev. Drug Discov. 9, 482–493.
Gaide, O., Favier, B., Legler, D. F., Bonnet, D., Brissoni, B., Valitutti, S., Bron, C., Tschopp, J., and Thome, M. (2002). CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat. Immunol. 3, 836–843.
Gomez-Angelats, M., Bortner, C. D., and Cidlowski, J. A. (2000). Protein kinase C (PKC) inhibits fas receptor-induced apoptosis through modulation of the loss of K+ and cell shrinkage. A role for PKC upstream of caspases. J. Biol. Chem. 275, 19609–19619.
Gramaglia, I., Jember, A., Pippig, S. D., Weinberg, A. D., Killeen, N., and Croft, M. (2000). The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J. Immunol. 165, 3043–3050.
Gramaglia, I., Weinberg, A. D., Lemon, M., and Croft, M. (1998). Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 161, 6510–6517.
Harper, N., Hughes, M. A., Farrow, S. N., Cohen, G. M., and MacFarlane, M. (2003). Protein kinase C modulates tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by targeting the apical events of death receptor signaling. J. Biol. Chem. 278, 44338–44347.
Humphreys, I. R., Loewendorf, A., de Trez, C., Schneider, K., Benedict, C. A., Munks, M. W., Ware, C. F., and Croft, M. (2007). OX40 costimulation promotes persistence of cytomegalovirus-specific CD8 T Cells: a CD4-dependent mechanism. J. Immunol. 179, 2195–2202.
Isakov, N., and Altman, A. (2002). Protein kinase C(theta) in T cell activation. Annu. Rev. Immunol. 20, 761–794.
Iwai, K., and Tokunaga, F. (2009). Linear polyubiquitination: a new regulator of NF-kappaB activation. EMBO Rep. 10, 706–713.
Kawamata, S., Hori, T., Imura, A., Takaori-Kondo, A., and Uchiyama, T. (1998). Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J. Biol. Chem. 273, 5808–5814.
Khadra, N., Bresson-Bepoldin, L., Penna, A., Chaigne-Delalande, B., Segui, B., Levade, T., Vacher, A. M., Reiffers, J., Ducret, T., Moreau, J. F., Cahalan, M. D., Vacher, P., and Legembre, P. (2011). CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase C (PKC) beta2, and prevents death-inducing signaling complex formation. Proc. Natl. Acad. Sci. U.S.A. 108, 19072–19077.
Kilpatrick, L. E., Sun, S., and Korchak, H. M. (2004). Selective regulation by delta-PKC and PI 3-kinase in the assembly of the antiapoptotic TNFR-1 signaling complex in neutrophils. Am. J. Physiol. Cell Physiol. 287, C633–C642.
Kong, K. F., Yokosuka, T., Canonigo-Balancio, A. J., Isakov, N., Saito, T., and Altman, A. (2011). A motif in the V3 domain of the kinase PKC-theta determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat. Immunol. 12, 1105–1112.
Kovalenko, A., and Wallach, D. (2006). If the prophet does not come to the mountain: dynamics of signaling complexes in NF-kappaB activation. Mol. Cell 22, 433–436.
Kyriakis, J. M. (1999). Signaling by the germinal center kinase family of protein kinases. J. Biol. Chem. 274, 5259–5262.
Lee, H. S., Park, S. Y., Lee, H. W., and Choi, H. S. (2004). Secretions of MMP-9 by soluble glucocorticoid-induced tumor necrosis factor receptor (sGITR) mediated by protein kinase C (PKC)delta and phospholipase D (PLD) in murine macrophage. J. Cell. Biochem. 92, 481–490.
Lee, S. H., Kim, E. J., Suk, K., Kim, I. S., and Lee, W. H. (2010). TL1A induces the expression of TGF-beta-inducible gene h3 (betaig-h3) through PKC, PI3K, and ERK in THP-1 cells. Cell. Immunol. 266, 61–66.
Li, S., Wang, L., and Dorf, M. E. (2009). PKC phosphorylation of TRAF2 mediates IKKalpha/beta recruitment and K63-linked polyubiquitination. Mol. Cell 33, 30–42.
Matsumoto, R., Wang, D., Blonska, M., Li, H., Kobayashi, M., Pappu, B., Chen, Y., Wang, D., and Lin, X. (2005). Phosphorylation of CARMA1 plays a critical role in T Cell receptor-mediated NF-kappaB activation. Immunity 23, 575–585.
Melowic, H. R., Stahelin, R. V., Blatner, N. R., Tian, W., Hayashi, K., Altman, A., and Cho, W. (2007). Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Ctheta. J. Biol. Chem. 282, 21467–21476.
Meng, X. W., Heldebrant, M. P., and Kaufmann, S. H. (2002). Phorbol 12-myristate 13-acetate inhibits death receptor-mediated apoptosis in Jurkat cells by disrupting recruitment of Fas-associated polypeptide with death domain. J. Biol. Chem. 277, 3776–3783.
Mousavi, S. F., Soroosh, P., Takahashi, T., Yoshikai, Y., Shen, H., Lefrancois, L., Borst, J., Sugamura, K., and Ishii, N. (2008). OX40 costimulatory signals potentiate the memory commitment of effector CD8+ T cells. J. Immunol. 181, 5990–6001.
Muppidi, J. R., Tschopp, J., and Siegel, R. M. (2004). Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 21, 461–465.
Murata, K., Ishii, N., Takano, H., Miura, S., Ndhlovu, L.C., Nose, M., Noda, T., and Sugamura, K. (2000). Impairment of antigen-presenting cell function in mice lacking expression of OX40 ligand. J. Exp. Med. 191, 365–374.
Nam, K. O., Kang, H., Shin, S. M., Cho, K. H., Kwon, B., Kwon, B. S., Kim, S. J., and Lee, H. W. (2005). Cross-linking of 4-1BB activates TCR-signaling pathways in CD8+ T lymphocytes. J. Immunol. 174, 1898–1905.
Napolitano, G., and Karin, M. (2010). Sphingolipids: the oil on the TRAFire that promotes inflammation. Sci. Signal. 3, pe34.
Narayan, P., Holt, B., Tosti, R., and Kane, L. P. (2006). CARMA1 is required for Akt-mediated NF-kappaB activation in T cells. Mol. Cell. Biol. 26, 2327–2336.
Nocentini, G., Ronchetti, S., Cuzzocrea, S., and Riccardi, C. (2007). GITR/GITRL: more than an effector T cell co-stimulatory system. Eur. J. Immunol. 37, 1165–1169.
Nolte, M. A., van Olffen, R. W., van Gisbergen, K. P., and van Lier, R. A. (2009). Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol. Rev. 229, 216–231.
Ozes, O. N., Mayo, L. D., Gustin, J. A., Pfeffer, S. R., Pfeffer, L. M., and Donner, D. B. (1999). NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 401, 82–85.
Park, S. G., Schulze-Luehrman, J., Hayden, M. S., Hashimoto, N., Ogawa, W., Kasuga, M., and Ghosh, S. (2009). The kinase PDK1 integrates T cell antigen receptor and CD28 coreceptor signaling to induce NF-kappaB and activate T cells. Nat. Immunol. 10, 158–166.
Patke, A., Mecklenbrauker, I., Erdjument-Bromage, H., Tempst, P., and Tarakhovsky, A. (2006). BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. J. Exp. Med. 203, 2551–2562.
Rogers, P. R., Song, J., Gramaglia, I., Killeen, N., and Croft, M. (2001). OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 15, 445–455.
Romashkova, J. A., and Makarov, S. S. (1999). NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 401, 86–90.
Ruiz-Ruiz, M. C., Izquierdo, M., de Murcia, G., and Lopez-Rivas, A. (1997). Activation of protein kinase C attenuates early signals in Fas-mediated apoptosis. Eur. J. Immunol. 27, 1442–1450.
Salek-Ardakani, S., So, T., Halteman, B. S., Altman, A., and Croft, M. (2005). Protein kinase Ctheta controls Th1 cells in experimental autoimmune encephalomyelitis. J. Immunol. 175, 7635–7641.
Sanz, L., Sanchez, P., Lallena, M. J., Diaz-Meco, M. T., and Moscat, J. (1999). The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 18, 3044–3053.
Shi, C. S., and Kehrl, J. H. (2003). Tumor necrosis factor (TNF)-induced germinal center kinase-related (GCKR) and stress-activated protein kinase (SAPK) activation depends upon the E2/E3 complex Ubc13-Uev1A/TNF receptor-associated factor 2 (TRAF2). J. Biol. Chem. 278, 15429–15434.
Shi, C. S., Leonardi, A., Kyriakis, J., Siebenlist, U., and Kehrl, J. H. (1999). TNF-mediated activation of the stress-activated protein kinase pathway: TNF receptor-associated factor 2 recruits and activates germinal center kinase related. J. Immunol. 163, 3279–3285.
Skaug, B., Jiang, X., and Chen, Z. J. (2009). The role of ubiquitin in NF-kappaB regulatory pathways. Annu. Rev. Biochem. 78, 769–796.
So, T., Choi, H., and Croft, M. (2011a). OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. J. Immunol. 186, 3547–3555.
So, T., Soroosh, P., Eun, S. Y., Altman, A., and Croft, M. (2011b). Antigen-independent signalosome of CARMA1, PKCtheta, and TNF receptor-associated factor 2 (TRAF2) determines NF-kappaB signaling in T cells. Proc. Natl. Acad. Sci. U.S.A. 108, 2903–2908.
So, T., Lee, S. W., and Croft, M. (2006). Tumor necrosis factor/tumor necrosis factor receptor family members that positively regulate immunity. Int. J. Hematol. 83, 1–11.
Sommer, K., Guo, B., Pomerantz, J. L., Bandaranayake, A. D., Moreno-Garcia, M. E., Ovechkina, Y. L., and Rawlings, D. J. (2005). Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity 23, 561–574.
Song, J., So, T., and Croft, M. (2008). Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. J. Immunol. 180, 7240–7248.
Soroosh, P., Ine, S., Sugamura, K., and Ishii, N. (2007). Differential requirements for OX40 signals on generation of effector and central memory CD4 + T cells. J. Immunol. 179, 5014–5023.
Sudan, R., Srivastava, N., Pandey, S. P., Majumdar, S., and Saha, B. (2012). Reciprocal regulation of protein kinase C isoforms results in differential cellular responsiveness. J. Immunol. 188, 2328–2337.
Sugamura, K., Ishii, N., and Weinberg, A. D. (2004). Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat. Rev. Immunol. 4, 420–431.
Sun, Z., Arendt, C. W., Ellmeier, W., Schaeffer, E. M., Sunshine, M. J., Gandhi, L., Annes, J., Petrzilka, D., Kupfer, A., Schwartzberg, P. L., and Littman, D. R. (2000). PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature 404, 402–407.
Thome, M. (2004). CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat. Rev. Immunol. 4, 348–359.
Vallabhapurapu, S., and Karin, M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733.
Villalba, M., Bi, K., Hu, J., Altman, Y., Bushway, P., Reits, E., Neefjes, J., Baier, G., Abraham, R. T., and Altman, A. (2002). Translocation of PKC[theta] in T cells is mediated by a nonconventional, PI3-K- and Vav-dependent pathway, but does not absolutely require phospholipase C. J. Cell Biol. 157, 253–263.
Wajant, H., Pfizenmaier, K., and Scheurich, P. (2003). Tumor necrosis factor signaling. Cell Death Differ. 10, 45–65.
Walczak, H. (2011). TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol. Rev. 244, 9–28.
Wang, D., You, Y., Case, S. M., McAllister-Lucas, L. M., Wang, L., DiStefano, P. S., Nunez, G., Bertin, J., and Lin, X. (2002). A requirement for CARMA1 in TCR-induced NF-kappa B activation. Nat. Immunol. 3, 830–835.
Watts, T. H. (2005). TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 23, 23–68.
Wertz, I. E., and Dixit, V. M. (2008). Ubiquitin-mediated regulation of TNFR1 signaling. Cytokine Growth Factor Rev. 19, 313–324.
Wooten, M. W., Geetha, T., Seibenhener, M. L., Babu, J. R., Diaz-Meco, M. T., and Moscat, J. (2005). The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J. Biol. Chem. 280, 35625–35629.
Yuasa, T., Ohno, S., Kehrl, J. H., and Kyriakis, J. M. (1998). Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. Germinal center kinase couples TRAF2 to mitogen-activated protein kinase/ERK kinase kinase 1 and SAPK while receptor interacting protein associates with a mitogen-activated protein kinase kinase kinase upstream of MKK6 and p38. J. Biol. Chem. 273, 22681–22692.
Keywords: OX40, PKCθ, TRAF, NF-κB, IKK, CBM, TNFSF, TNFRSF
Citation: So T and Croft M (2012) Regulation of the PKCθ-NF-κB axis in T lymphocytes by the tumor necrosis factor receptor family member OX40. Front. Immun. 3:133. doi: 10.3389/fimmu.2012.00133
Received: 21 March 2012; Accepted: 08 May 2012;
Published online: 28 May 2012.
Edited by:
Amnon Altman, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Nikolai Petrovsky, Flinders Medical Centre, AustraliaKarsten Sauer, The Scripps Research Institute, USA
Copyright: © 2012 So and Croft. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Takanori So, Department of Microbiology and Immunology, Tohoku University Graduate School of Medicine, 2-1 Seiryo-machi, Aobaku, Sendai 980-8575, Japan. e-mail:dHNvQG1lZC50b2hva3UuYWMuanA=; Michael Croft, Division of Immune Regulation, La Jolla Institute for Allergy and Immunology, La Jolla, CA 92037, USA. e-mail:bWlja0BsaWFpLm9yZw==