 Noam Rudich1
Noam Rudich1
- 1 Department of Cell and Developmental Biology, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
- 2 Department of Medicine, Boston University School of Medicine, Boston, MA, USA
- 3 Department of Biochemistry, Boston University School of Medicine, Boston, MA, USA
- 4 Whitaker Cardiovascular Institute, Boston University School of Medicine, Boston, MA, USA
- 5 Evans Center for Interdisciplinary Biomedical Research, Boston University School of Medicine, Boston, MA, USA
Adenosine is a metabolite, which has long been implicated in a variety of inflammatory processes. Inhaled adenosine provokes bronchoconstriction in asthmatics or chronic obstructive pulmonary disease patients, but not in non-asthmatics. This hyper responsiveness to adenosine appears to be mediated by mast cell activation. These observations have marked the receptor that mediates the bronchoconstrictor effect of adenosine on mast cells (MCs), as an attractive drug candidate. Four subtypes (A1, A2a, A2b, and A3) of adenosine receptors have been cloned and shown to display distinct tissue distributions and functions. Animal models have firmly established the ultimate role of the A3 adenosine receptor (A3R) in mediating hyper responsiveness to adenosine in MCs, although the influence of the A2b adenosine receptor was confirmed as well. In contrast, studies of the A3R in humans have been controversial. In this review, we summarize data on the role of different adenosine receptors in mast cell regulation of inflammation and pathology, with a focus on the common and distinct functions of the A3R in rodent and human MCs. The relevance of mouse studies to the human is discussed.
The Mast Cell
Mast cells (MCs) are hematopoietic-derived cells that play important physiological roles in innate and adaptive immunity, as well as in wound healing (Galli, 1997; Metcalfe et al., 1997; Noli and Miolo, 2001; Weller et al., 2006; Metz and Maurer, 2007; Shelburne and Abraham, 2011). Arising from committed progenitors (CD34+, c-kit positive cells) in the bone marrow (Rodewald et al., 1996), MCs progenitors pass through the vascular space, entering the tissues, where they complete their differentiation and maturation process in situ (Metcalfe et al., 1981, 1997; Galli, 1997). Mature MCs typically present widespread plasma membrane processes, round nuclei, and numerous electron-dense cytoplasmic secretory granules. Growth, survival, differentiation, and homing are dependent on SCF-dependent c-kit signaling (Metcalfe et al., 1981, 1997) and accordingly, gain of functions mutations in the c-kit receptor result in mastocytosis (Lim et al., 2008; Fritsche-Polanz et al., 2010; Valent et al., 2011).
Human and rodent mature MCs are classified into one of two categories, based on phenotypic, biochemical, and functional differences. In rodents, MCs are classified as connective tissue type MCs (CTMCs), and include primarily skin MCs and cells of the peritoneal cavity, and mucosal MCs (MMCs), which are associated with the mucosa of the digestive tract or lungs (Welle, 1997; Bischoff and Krämer, 2007). In human, two types of MCs have been categorized based on their neutral protease compositions: MCTC contain the neutral proteases tryptase and chymase, whereas MCT contain only tryptase (Irani et al., 1986; Welle, 1997). Both types of MCs express on their plasma membrane FcεRI receptors, the high-affinity receptors for the Fc region of monomeric Immunoglobulin E (IgE). In tissues, MCs are distributed at interfaces between the outside world and the internal milieu, consistent with their immune tasks in host defense mechanisms. MCs are also found near blood and lymphatic vessels (Kunder et al., 2011) as well as in close proximity to nerves (Bienenstock et al., 1991; Theoharides et al., 2006).
Mature MCs store in their cytoplasmic secretory granules inflammatory mediators, including vasoactive amines such as histamine and serotonin, multiple proteases such as tryptase and chymase, mentioned above, and lysosomal hydrolases such as β-hexosaminidase and cathepsin D (Schwartz and Austen, 1980). MCs secretory granules also contain proteoglycans, such as heparin and chondroitin sulfate E, and polyamines, which play a role in proteases storage in secretory granules (Garcia-Faroldi et al., 2010). Some cytokines, including tumor necrosis factor (TNF-α) and basic fibroblast growth factor (bFGF) are also prestored in the secretory granules (Ribatti et al., 2002; Olszewski et al., 2007).
MCs as Mediators of Allergic and Inflammatory Diseases
Despite their beneficial physiological roles in immunity and wound healing, MCs are best known for their involvement in allergic and inflammatory diseases, including autoimmune and neurodegenerative diseases and cancer, where they contribute significantly to the complexity of these diseases (Galli, 1997; Theoharides and Kalogeromitros, 2006; Bischoff, 2007). Allergy involves the activation and subsequent degranulation of MCs, where activation can be triggered by a variety of external stimuli (Figure 1). The immune trigger involves production of IgE class antibodies, specific for a wide range of allergens, IgE binding to the FcεRI, followed by their allergen-induced cross-linking and receptor aggregation leading to cell activation (Turner and Kinet, 1999; Siraganian, 2003; Gilfillan and Tkaczyk, 2006; Rivera and Gilfillan, 2006; Wilson et al., 2011). In addition, MCs can be triggered in an IgE-independent fashion, by a variety of soluble stimuli (see below), as well as through cell interactions with neighboring cells including fibroblasts, eosinophils, T cells, and nerve cells (Askenase et al., 1980; Baram et al., 2001; Garbuzenko et al., 2002; Puxeddu et al., 2005; Figure 1). These interactions are complex involving feedback loops. For example, MCs activate eosinophils, which in turn release proteins that further activate MCs (Puxeddu et al., 2005).
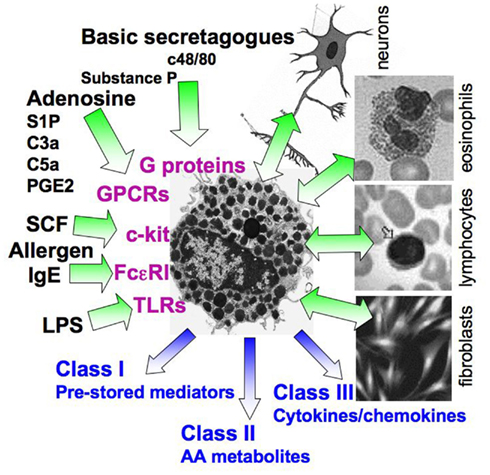
Figure 1. Complexity of MC activation. A scheme illustrating the multiple stimuli that can activate MCs. The latter include the immunological, IgE-mediated pathway, interactions with neighboring cells, such as eosinophils, T cells, and fibroblasts, pathogens that act through TLRs, the c-kit ligand SCF, and numerous stimuli that activate G-proteins, either directly (e.g., basic secretagogues such as the synthetic c48/80) or by binding to GPCRs (e.g., adenosine, complement derived peptides, prostaglandins such as PGE2, cytokines, and more). Depending on the stimulus type, activated MCs may either release preformed mediators, packaged in secretory granules (class I mediators) as well newly synthesized mediators, including metabolites of arachidonic acid (AA, class II mediators, such as prostaglandins and leukotrienes), cytokines, and chemokines (class III mediators), or may only release a subset of mediators. In some cases, the distinct stimuli interact synergistically resulting in an amplified response.
MCs Mechanisms of Secretion
When activated, MCs release the contents of their secretory granules within seconds to minutes of trigger. Depending on the strength of the external signal or the stimulus type, release may occur by kiss-and-run exocytosis that partially releases the secretory granule cargo through a relative narrow and transient fusion pore; full exocytosis, when fusion of plasma membrane docked secretory granules, with the plasma membrane, allows complete expulsion of their contents, and thirdly, compound exocytosis, also termed anaphylactic degranulation, the most extensive mode of cargo release, that involves fusion of the secretory granule membrane with the plasma membrane and release of secretory granule contents, as well as homotypic fusion between secretory granules by mechanisms whose details are still largely unresolved (Alvarez De Toledo and Fernandez, 1990; Blank, 2011; Woska and Gillespie, 2012). In addition, upon activation, MCs also produce and release arachidonic acid (AA) metabolites including leukotrienes and prostaglandins as well as multiple cytokines and chemokines (Metcalfe et al., 1981, 1997; Wasserman, 1983; Galli et al., 1991; Ogawa and Grant, 2007). Also formed are growth factors such as Vascular endothelial growth factor (VEGF), a cytokine crucial to angiogenesis, and the growth of blood vessels (Detoraki et al., 2009; García-Román et al., 2010) and NGF (Xiang and Nilsson, 2000; Cantarella et al., 2011). Notably, the repertoire of mediators released by MCs as well as their responsiveness to external ligands may vary dependently on the MC type or stimulus nature. Indeed, MCs are tunable cells (Galli et al., 2005) and under specified conditions can be programmed to selectively release a subset of mediators with no massive degranulation. Such selective secretion, termed piecemeal degranulation (PMD), is primarily linked with chronic inflammation and accounts for MCs involvement in inflammatory diseases and cancer (Dvorak and Kissell, 1991; Crivellato et al., 2003; Theoharides et al., 2007; Ribatti, 2011). Unlike exocytosis, PMD involves budding of vesicles, carrying the selective cargo to be released, from the secretory granule, and their transport and fusion with the plasma membrane. This type of secretion is also characterized by crosstalk with the endocytic system, whereby following their fusion with the plasma membrane and release of contents, these vesicles recycle to the secretory granule by means of endocytosis and subsequent fusion (Dvorak et al., 1992; Crivellato et al., 2003; Ribatti and Crivellato, 2009; Ribatti, 2011). The molecular mechanisms of PMD remain largely elusive.
Upon their release to the extracellular milieu, preformed and newly synthesized mediators affect multiple target organs and cells giving rise to an immediate inflammatory response followed by the progression of a tremendously amplified late-phase inflammatory response, which may acquire a chronic nature. The wide spectrum of biologically active substances produced and released by MCs thus contributes to the complexity of the allergic diseases. Moreover, an appreciable fraction of mediators released by activated MCs, acts in an autocrine manner to activate MCs and amplify their signaling outputs (Gilfillan et al., 2009). Such is also the case of adenosine, whose function in MCs is the focus of the present review.
Non-Immunological Activation of MCs
Multiple and diverse stimuli can activate MCs independently of IgE (Lagunoff et al., 1983; Bhattacharyya et al., 1998; Theoharides et al., 2007). Already mentioned are neighboring cells and SCF, that by binding to the c-kit receptor, a member of the receptor tyrosine kinase (RTK) family of receptors, transmits signals for MC survival (Iemura et al., 1994), induces direct degranulation of rat peritoneal MCs (RPMCs; Taylor et al., 1995), and synergizes with the immunological, FcεRI mediated response, in stimulating degranulation, and cytokine release from bone marrow derived MCs (BMMCs) or human MCs (Coleman et al., 1993; Hundley et al., 2004). Toll like receptors (TLRs) serve as pattern-recognition receptors and mediate MCs responses to pathogens (Qiao et al., 2006; Mrabet-Dahbi et al., 2009; Avila and Gonzalez-Espinosa, 2011; Lorentz, 2011; Wesolowski and Paumet, 2011; Chan et al., 2012). Different TLRs are expressed in the different MCs types (reviewed in Novak et al., 2010), that dependently on the ligand/receptor complex type, display distinct modulator functions on the FcεRI mediated responses (Qiao et al., 2006). Finally, and particularly intriguing is the large number of non-immunological pathways that involve activation of G-proteins, either directly (Repke and Bienert, 1987; Aridor and Sagi-Eisenberg, 1990; Aridor et al., 1990, 1993; Mousli et al., 1990), or through binding to G-protein coupled receptors (GPCRs; Okayama et al., 2008; Druey, 2009).
Receptor mimetic activation of G-proteins was demonstrated in RPMCs that seem to display unique membrane characteristics that allow molecules comprising a hydrophobic stretch combined with a positively charged domain, to penetrate the membrane (Ortner and Chingell, 1981) and interact directly with G-proteins (Higashijima et al., 1988; Aridor et al., 1990). This group of molecules, collectively known as the basic secretagogues of MCs, were recognized as early as 1951 as potent IgE-independent activators of MCs that triggered exocytosis in a pertussis toxin (Ptx) sensitive manner, therefore implicating Gi protein(s) as essential mediators of their triggered exocytosis (Nakamura and Ui, 1984, 1985). Indeed, the large repertoire of molecules that constitute this family of stimuli, including neuropeptides, opiates, and the synthetic polyamine compound 48/80 (c48/80; Lagunoff et al., 1983), their common structural features and the fact that micromolar concentrations are required to evoke their biological activity, have suggested that members of this family trigger MC activation in a receptor-independent manner (Repke and Bienert, 1987; Aridor and Sagi-Eisenberg, 1990; Aridor et al., 1990, 1993; Mousli et al., 1990). More recent data demonstrated expression of a low-specificity activation site for basic secretagogues also in human MCs (LAD-2 and CD34-derived) and have identified this site as MrgX2, a member of the Mrg class of GPCRs (Tatemoto et al., 2006; Kashem et al., 2011; Subramanian et al., 2011). Whether or not RPMCs express MrgX2 is presently unknown. However, it is noteworthy that while the basic secretagogues-stimulated signaling networks and outputs, including degranulation, Ca2+ rise and activation of the MAP kinases, are Ptx sensitive, Ptx inhibits MrgX2 mediated degranulation in human MCs, but has no effect on Ca2+ influx, suggesting that in sharp contrast to the exclusive activation of Gi proteins by basic secretagogues in RPMCs, the MrgX2 couples to both Gi and presumably to Gq in human MCs (Shefler et al., 1999; Shefler and Sagi-Eisenberg, 2002; Subramanian et al., 2011).
We identified the G-protein Gi3 as the principal mediator of basic secretagogues-induced exocytosis in RPMCs by demonstrating that a peptide comprising the 10 C-terminal amino acids of Gαi3 (KNNLKECGLY), introduced into permeabilized cells, could inhibit c48/80-induced histamine release (Aridor et al., 1993). We then engineered a cell permeable version of this peptide, in which an importation sequence derived from the signal sequence of the Kaposi fibroblast growth factor (AAVALLPAVLLALLAP) was fused to Gαi3 C-terminal sequence, giving rise to a 16 amino acids peptide, we termed ALL1. We could show that ALL1 efficiently penetrates into intact RPMCs and blocks c48/80-induced histamine secretion, protein tyrosine phosphorylation, and release of Prostaglandin D2 (PGD2) in a dose dependent fashion (Shefler et al., 2008).
Mast cells can also be activated by ligands that bind to classical, high-affinity, GPCRs. Included are some basic secretagogues, such as the neuropeptide substance P, that activates human or MMCs by binding to a tachykinin class receptor (Asadi et al., 2012), as well as prostaglandins, sphingosine-1-phosphate, histamine, cytokines, adenine nucleotides, complement, and adenosine (reviewed in Okayama et al., 2008; Druey, 2009; Gilfillan et al., 2009). This list includes mediators that are also released by activated MCs, or whose concentrations are elevated consequently to tryptase release (e.g., C3a, generated upon complement processing; Ali, 2009). These ligands may thus act in a paracrine or autocrine fashion to amplify or modulate the MC cellular inflammatory responses (reviewed in Okayama et al., 2008; Druey, 2009; Gilfillan et al., 2009).
Adenosine and MC-Mediated Inflammation
Non-immunological activation of MCs is primarily linked with chronic inflammation and accounts for MC involvement in autoimmune and degenerative diseases and cancer (Theoharides and Kalogeromitros, 2006; Theoharides et al., 2007; Ribatti and Crivellato, 2009; Ribatti, 2011). In most cases, the underlying mechanism involves stimulation of selective release of a subset of mediators, with no massive degranulation by PMD, described above. For example, such is the case of corticotropin-releasing hormone (CRH) or prostaglandin E2 (PGE2), that stimulate release of VEGF without degranulation from the human mast cell line (HMC-1) or human umbilical cord blood derived MCs (HUCBMCs; Cao et al., 2006). Adenosine, the focus of this review, is produced both intracellularly and extracellularly under conditions of increased energy consumption such as stress or hypoxia that develop at sites of inflammation and injury (Linden, 2001; Bours et al., 2006). Hence, during systemic inflammation, the circulating adenosine concentration increases rapidly (Ramakers et al., 2011). Adenosine then functions in an autocrine or paracrine fashion through binding to four GPCRs: the A1, A2a, A2b, and A3 receptors that display distinct pharmacological characteristics. The A1R and A2a subtypes have the highest affinity for adenosine, whereas the affinity for adenosine of the A2b and A3 receptors is significantly lower (Linden, 2001). Individual receptors can be specifically activated or blocked by type specific agonists or antagonists (Table 1). Adenosine receptors also couple to different G-proteins, whereby the A1 and A3 receptors couple to Ptx sensitive Gi proteins, while the A2aR couples to Gs and the A2bR to Gs and Gq/G11 (reviewed in Brown et al., 2008). Accordingly, engagement of the adenosine receptor subtypes evokes distinct cellular responses (Table 1).
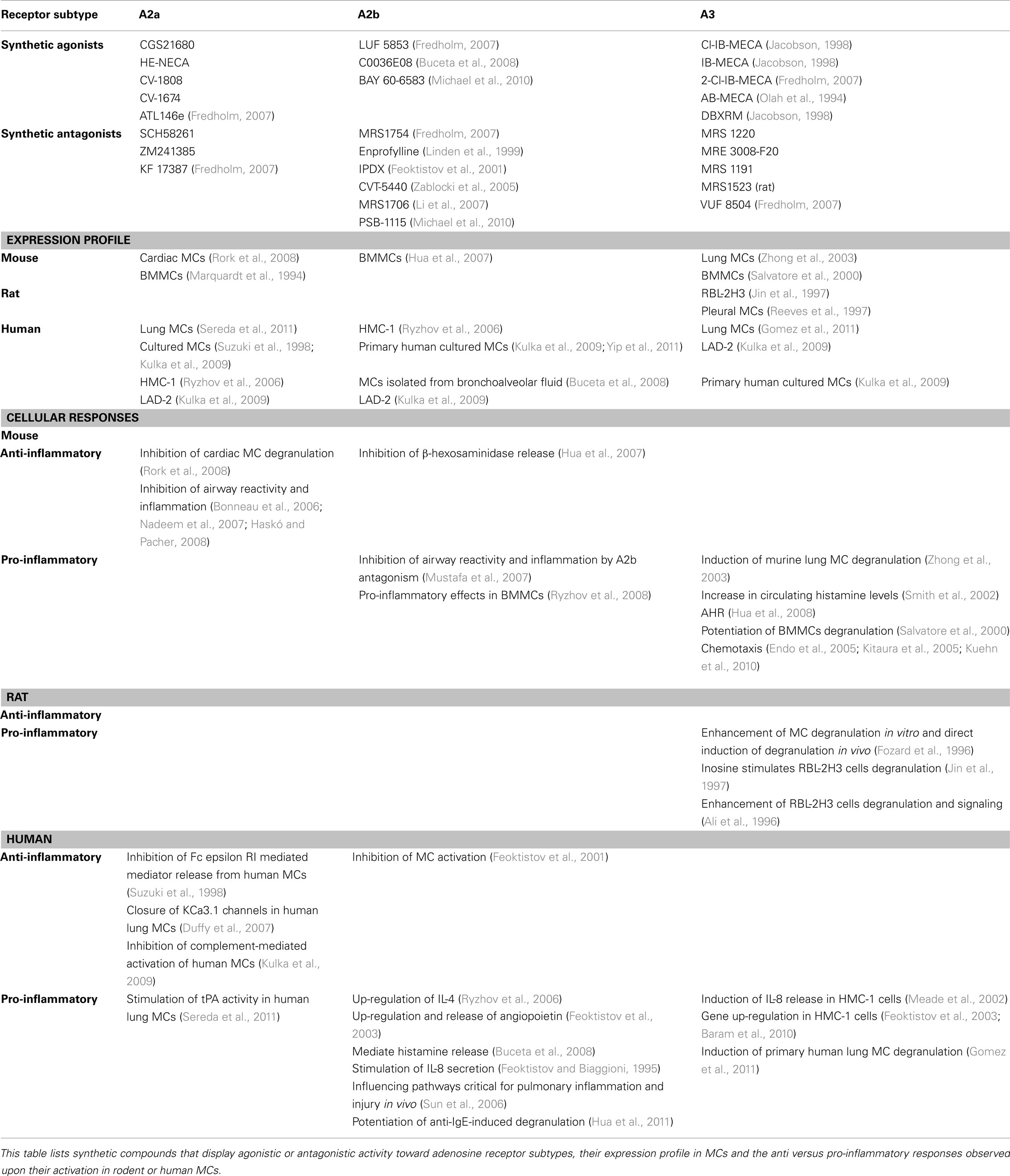
Table 1. Adenosine receptor subtypes and their cellular distribution and functions in MCs.
Adenosine has been long implicated in a variety of inflammatory processes, including allergy and especially asthma (Bours et al., 2006; Spicuzza et al., 2006; Matera and Polosa, 2007; Brown et al., 2008; Polosa and Blackburn, 2009). That MCs mediate adenosine responses in allergic inflammation was first indicated by the ability of adenosine to potentiate IgE-induced mediator release from rodent MCs (Holgate et al., 1980). The physiological relevance to human was then advocated by observations documenting the ability of adenosine to provoke bronchoconstriction in atopic and asthmatic individuals, but not in normal subjects, a phenomenon referred to as adenosine hyper responsiveness (AHR; Cushley and Holgate, 1985). Furthermore, elevated levels of adenosine are found in bronchoalveolar lavage fluid and exhaled breath condensates of allergic inflammation patients (Driver et al., 1993; Huszar et al., 2002; Vass et al., 2006) and inhalation of adenosine by asthmatics leads to increased levels of MC mediators such as histamine and tryptase in bronchoalveolar fluid. Finally, blockers of mediators released by activated MCs inhibit the bronchoconstrictor response to adenosine in asthma (Meade et al., 2001; Spicuzza et al., 2006). This notion was supported further by the findings that MCs express the two ecto-enzymes, CD73, a nucleoside triphosphate phosphohydrolase, and CD39, an ecto-5′-nucleotidase, that by concerted conversion of adenine nucleotides to adenosine increase its extracellular concentrations (Linden, 2001); prominent secretion of adenosine is observed in activated MCs (Marquardt et al., 1984) as well as the rat derived mast cell line, RBL-2H3 (Lloyd et al., 1998); and that MCs express adenosine receptors (Carruthers and Fozard, 1993). However, the identity of the specific adenosine receptor(s) that mediate(s) adenosine triggered MCs responses remained elusive.
The A3R as Mediator of MCs-Dependent Inflammation
Animal models have substantiated the central role played by activated MCs in mediating AHR and have identified the A3R as the major contributor. Specifically, airway responses elicited by adenosine are significantly attenuated in A3R- or MC-deficient mice (Tilley et al., 2003). Furthermore, aerosolized adenosine-5′N-ethylcarboxamide (NECA; a non-selective ligand) induces hyper responsiveness in wt or A1R-deficient mice, but not in A3R knockout mice (Hua et al., 2008) and AHR develops in MC-deficient mice that are reconstituted with wt, but not with A3R−/− MCs (Hua et al., 2008). In vitro studies employing isolated MCs and selective receptor agonists have substantiated this notion further by demonstrating that adenosine directly stimulates murine lung MCs degranulation by activating the A3R (Zhong et al., 2003). In a similar fashion, intravenous application of N6-2-(4-aminophenyl)ethyladenosine (APNEA; Fozard et al., 1996) or intradermal introduction of the A3R agonist 2-(1-Hexynyl)-N-methyladenosine (IB-MECA; Reeves et al., 1997) to rats, respectively induced MC degranulation or plasma protein extravasation (PPE), confirming a key role for the A3R in mediating MCs responses. However, exposure of rat isolated pleural MCs, RBL-2H3 cells, or BMMCs to IB-MECA, enhanced FcεRI-induced secretion, but failed to induce directly such degranulation (Ramkumar et al., 1993; Reeves et al., 1997; Yamano et al., 2005, 2006). Therefore, collectively these studies have firmly established the involvement of the A3R in MC degranulation. Yet, they have also indicated that whether or not engagement of the A3R suffices to promote MCs degranulation, or whether additional signals, stemming from the FcεRI or the other adenosine receptors (i.e., A2bR, see below), are required, depends on the MC type, the tissue it resides in, and perhaps even the species or the mouse strain (Meade et al., 2001).
In addition to direct or synergistic effects on MC degranulation, occupied A3R mediates the stimulatory impacts of adenosine and the FcεRI on MCs migration (Endo et al., 2005; Kitaura et al., 2005; Kuehn et al., 2010). Since MC migration is an important mechanism toward their accumulation at allergic inflammatory sites, this activity of the A3R further adds to its pro-inflammatory functions.
Genetic and pharmacological approaches have assigned the A2aR anti-inflammatory properties (Bonneau et al., 2006; Nadeem et al., 2007; Haskó and Pacher, 2008), and have revealed a complex function of the A2bR (Table 1). Hence, BMMCs derived from A2bR knockout mice demonstrated increased sensitivity to IgE-mediated anaphylaxis (Hua et al., 2007), but work in the adenosine deaminase (ADA)-deficient model, pharmacological studies employing the A2bR antagonist CVT-6883 in a mouse model of ragweed sensitization and challenge, and in vitro studies on BMMCs derived from A2bR knockout mice, supported a pro-inflammatory role of this receptor in mediating MC functions, and have particularly placed this receptor in playing a role in late-stages or chronic features of airway diseases such as asthma (Sun et al., 2006; Mustafa et al., 2007; Ryzhov et al., 2008; Polosa and Blackburn, 2009).
Studies in Human MCs
Studies employing human lung MCs demonstrated that at low concentrations, adenosine potentiated degranulation of immunologically activated MCs, whereas at higher concentrations a counteractive inhibitory process took place (Peachell et al., 1988, 1991). These responses were interpreted as being mediated by the A2 type receptors. Consistent with these results, studies with HMC-1, a human mast cell line, confirmed the expression of both the A2a and A2b receptors, where A2bR mRNA was eightfold more abundant than A2a mRNA (Meade et al., 2002). A2bR stimulation resulted in the release of IL-4, IL-8, and IL-13, cytokines linked with allergic inflammation and asthma (Feoktistov and Biaggioni, 1995; Ryzhov et al., 2004, 2006). Taken together with the demonstration that enprofylline and theophylline, two anti asthmatic drugs, block adenosine-induced release of IL-8 from HMC-1 cells, these results implicated the A2bR as the principal mediator of AHR in humans (Haskû et al., 2009). Thus, while the studies in rodent MCs have provided unequivocal evidence for the involvement of the A3R in mediating the adenosine bronchoconstrictor response, this role was questioned in human MCs and was rather assigned to the A2bR. Moreover, studies in humans demonstrating A3R-mediated inhibition of neutrophils degranulation (Bouma et al., 1997) and eosinophils chemotaxis (Knight et al., 1997; Walker et al., 1997), have suggested that the A3R may play an anti-inflammatory role. However, against this dogma are results obtained in HMC-1 cells, where the data clearly indicate that IL-8 release can also be stimulated by IB-MECA (Meade et al., 2002). The latter was approximately fivefold less potent than the A2bR agonist, but this lower potency was consistent with A3R being eightfold less abundant than the A2bR in this cell line (Meade et al., 2002). Further analyses revealed that activation of A3R in HMC-1 cells induces the expression, though not secretion, of angiopoietin-2, by a mechanism, which did not involve inhibition of adenylyl cyclase or activation of PLC (Feoktistov et al., 2003). Activation of A2b could enhance secretion of the angiogenic factor, but a maximal response depended on the simultaneous activation of both the A2b and A3 receptors (Feoktistov et al., 2003).
Employing the HMC-1 cell line, we found no evidence for A3R stimulated degranulation or cytokine release (Baram et al., 2010). However, a wide-genome screen demonstrated up-regulation of genes that play important roles in allergic inflammation and asthma upon A3R activation (Baram et al., 2010). The latter included cytokines such as IL-8, chemokines, growth factors, and angiogenic factors (Baram et al., 2010). Therefore, these studies supported a pro-inflammatory, modulator role of this receptor in human MCs. Consistent with this premise we also obtained evidence for the activation of the A3R, in an autocrine manner, in cells activated through contact with T cell derived membranes (Baram et al., 2010).
Because HMC-1 cells do not express the FcεRI, the modulator action of adenosine on immunologically stimulated MCs could not be investigated in this model. However, this question was addressed using HUCBMCs, in which only an inhibitory A2aR-mediated function could be detected (Suzuki et al., 1998). Specifically, exposure of HUCBMCs to 4-[2-[[6-Amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl]benzene propanoic acid hydrochloride (CGS21680), a specific agonist of the A2aR, resulted in the inhibition of tryptase release (Suzuki et al., 1998). Accordingly, RT-PCR analysis indicated the dominant expression of A2aR mRNA, though A3R mRNA was detected as well (Suzuki et al., 1998). Using the same model of HUCBMCs, a biphasic effect of adenosine was noted on anti-IgE-induced degranulation, where potentiation was assigned to the A1R and inhibition of the anti-IgE-induced responses to the A2bR (Yip et al., 2009). Finally, in a recent study, where HUCBMCs were treated for 3 days with IL-4 and IgE, addition of adenosine potentiated anti-IgE-induced degranulation and this enhancement was reduced or abolished by A2bR antagonists or A2b specific siRNA implicating the A2bR in mediating the adenosine synergistic response (Hua et al., 2011). In same study, neither selective A3R agonist, nor an A3R antagonist displayed any activity. In fact, in human MC cultures derived from CD34+ peripheral blood progenitor cells (HuMCs) or the LAD-2 cells, adenosine failed to effect degranulation (Kuehn et al., 2011). But then contradictory to these observations, a recent study employing human lung derived MCs demonstrated potentiation of IgE-induced degranulation by adenosine in a Ptx sensitive fashion. Furthermore, an A3R-specific agonist recapitulated this potentiation (Gomez et al., 2011). Notably, these lung derived MCs were shown to express threefold more A3R mRNA than do skin MCs (Gomez et al., 2011).
Taken together, these studies illustrate the difficulties faced studying human MC A3R, whose properties might be altered by physiological/environment specific responses not recapitulated in cell lines or even primary MCs (Gessi et al., 2008).
A3R Signaling in Rodent and Human MCs
To gain insights into the molecular mechanisms behind the A3R function, the signaling pathways elicited by this receptor were explored. These studies demonstrated that histamine release induced by the engagement of the A3R in isolated murine lung MCs is associated with a Ptx-and LY294002-sensitive rise in cytosolic Ca2+ (Zhong et al., 2003). These results therefore implicated Gi protein(s), phosphatidylinositol 3 kinase (PI3K) and presumably phospholipase Cβ (PLCβ) as principal mediators of A3R stimulated degranulation in murine lung MCs. Ca2+ rise, PI3K as well as ERK activation were also noted in BMMCs, whose FcεRI-induced release of mediators was potentiated by the A3R (Yamano et al., 2005, 2006). Finally, studies in RBL-2H3 cells demonstrated A3R stimulated and Ptx sensitive activation of PLC (Ramkumar et al., 1993) and PLD (Ali et al., 1996), rise in Ca2+, and activation of PI3K/Akt (Gao et al., 2001). Microinjection of an antibody to either Gαi3 or Gαq inhibited the Ca2+ signal suggesting the involvement of both G-proteins in Ca2+ mobilization (Hoffman et al., 1997). However, since an antibody to Gαi2 was not tested, the involvement of Gi2, that is co-expressed in MCs, cannot be excluded. Consistent with this notion, rat A3R (rA3R), expressed in CHO cells could interact primarily with Gi2 and Gi3 and to a significantly lesser extent with Gq and G11 (Palmer et al., 1995).
The pharmacological evidence for the involvement of PI3K in mediating A3R function was further supported by genetic studies that indicated that adenosine, acting through the A3R, transiently increases PI(3,4,5)P3 exclusively via PI3Kγ (Laffargue et al., 2002). Moreover, PI3Kγ deficient mice did not form edema after intradermal injection of adenosine or when challenged by passive systemic anaphylaxis (Laffargue et al., 2002). Further analyses of the mechanisms by which PI3Kγ relays inflammatory signals revealed that the p84:p110γ complex was specifically required for synergistic adenosine-promoted degranulation and chemotaxis (Bohnacker et al., 2009). Taken together these results are compatible with a model (Figure 2), where by coupling to Gi2/Gi3 and Gq, the rodent A3R transmits signals that effect the activation of PLCβ and PLD, which in turn function to mediate MC degranulation (i.e., in lung MCs) or synergize with the FcεRI-elicited signals (Kuehn et al., 2008). In addition, activation of PI3Kγ promotes the activation of the ERK pathway that mediates gene up-regulation and presumably also the generation of arachidonic acid metabolites (Zhang et al., 1997a,b; Shefler et al., 1999) and mediates cell migration (Bohnacker et al., 2009).
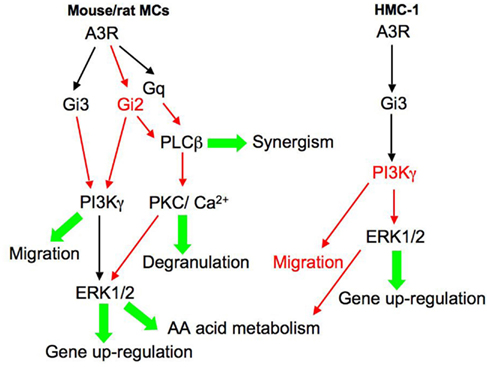
Figure 2. Signaling pathways elicited by the human and rodent A3 adenosine receptor in MCs. This model aims to integrate the signaling pathways reported in the literature, where marked in red are proposed, not yet proven, pathways. According to this model, the rodent A3R couples to Gi2, Gi3, and Gq, leading to the activation of PLD, PLCβ, and PI3Kγ. As a result, the cytosolic concentration of Ca2+ rises and protein kinase C (PKC) and the ERK MAP kinases are activated. According to this model, activation of PLCβ mediates MC degranulation or synergizes with the FcεRI-elicited signals to potentiate degranulation. PI3Kγ and ERK1/2 mediate respectively MC migration and class II and class III mediator formation. In contrast, the human A3R couples mainly to Gi3 leading to activation of ERK1/2 and gene up-regulation.
We have analyzed the signaling patterns associated with gene up-regulation in the A3R-activated HMC-1 cells. By taking advantage of ALL1, the cell permeable Gi3 inhibitory peptide, we also assessed the role of Gi3 in mediating A3R elicited responses. These studies revealed that Cl-IB-MECA enhances phosphorylation of MAP kinases, including ERK1/2, RSK1, p38α,β,γ, JNK, and GSK3α, in an ALL1-sensitive fashion, confirming the involvement of Gi3 (Baram et al., 2010). Though not tested, it is tempting to speculate that the lack of activation of PLCβ by the A3R in the HMC-1 cells prevents them from degranulation and restricts this receptor function to the induction of gene up-regulation and possibly the generation of arachidonic acid metabolites (Figure 2). Interestingly, exploring the signaling patterns of the A3R in BMMCs derived from a humanized mouse, in which the mouse A3R gene was replaced by its human counterpart, revealed that A3R activation evoked a Ptx sensitive rise in Ca2+, but failed to enhance ERK1/2 or Akt phosphorylation, which are stimulated by the murine A3R (Yamano et al., 2005). These results led the authors to conclude that the hA3R couples to different G-proteins for Ca2+ mobilization and PI3K/ERK1/2 activation (Yamano et al., 2005). In contrast, a chimeric receptor, in which the whole intracellular region of the hA3R was substituted by the corresponding mouse A3R, did display identical activity to the murine A3R (Yamano et al., 2006). Yet, the full hA3R was able to activate ERK1/2, via PI3K, when transfected into CHO cells (Graham et al., 2001; Schulte and Fredholm, 2002). Therefore the rodent and human A3 adenosine receptors clearly elicit distinct signaling patterns that are dictated by the G-protein combinations they couple to in a specific cellular setting. Indeed, unlike most GPCRs, the rodent, and human A3Rs share only 72% homology (Linden, 1994). Moreover, these receptors also differ in their down-regulation patterns, reflecting again their variance in partner interactions (Yamano et al., 2005).
Conclusion and Future Perspectives
While the role of the rodent A3R-activated MCs in mediating AHR is firmly established, the role of the hA3R in inflammation and particularly in mediating MC responses remains elusive. Two reasons are likely to account for the difficulties encountered when studying this receptor in human samples. First, it is quite challenging to obtain and culture human MCs, thus enforcing the use of model systems (i.e., HUCBMCs, lung derived MCs, HMC-1, and LAD-2 cell lines), each of which having its own limitations (Hua et al., 2011). Second, it is quite plausible that the human receptor is regulated differently than the rodent counterpart, including by more complex physiological and environmental factors. Consistent with this perception is the up-regulation of the A3R in MCs of chronic obstructive pulmonary disease (COPD) patients (Varani et al., 2006). Indeed, unlike most GPCRs, the rodent and human A3R share only 72% homology, while the Gi proteins are highly conserved. Therefore, these A3Rs may couple to different Gi proteins, thereby yielding differential signaling. In fact, the Gi protein they couple to may vary depending on receptor occupancy or strength of signal, resulting in differential outputs. The variance in their amino acid sequences may also drive distinct interactions with accessory proteins, giving rise to distinct spatio-temporal regulations. The human and rodent A3Rs may also display distinct crosstalk with their co-expressed adenosine receptors. ATP and ADP also activate the MCs by binding to P2 receptors. The P2 receptors are subdivided to P2X and P2Y receptors that are GPCRs whose activation may impact MC responses. Moreover, since various combinations of P2 receptor subtypes are expressed in MCs derived from different sources (e.g., human CBMCs, HMC-1, or BMMCs and RBL-2H3), their contributions to the overall signaling and cellular responses elicited by purinergic receptors may vary accordingly (Bulanova and Bulfone-Paus, 2010). Finally, three isoforms of the A3R have been cloned in human and two in mouse. Whether or not these isoforms play a role in fine-tuning of the receptor is presently unknown. It is noteworthy that the majority of GPCR genes are intronless. However, for GPCRs that are subjected to alternative splicing, the variants have proven to be of physiological relevance (Markovic and Challiss, 2009). Hence, an important lesson from the studies on the adenosine receptors, at least from a therapeutic standpoint, is that the mouse studies do not necessarily predict what is happening in the human. Thus, the question of whether an A3R agonist or rather an A3R antagonist should be considered in asthma treatment still remains unresolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work on mast cell functions has been funded by grants from the Israel Science Foundation, founded by the Israel Academy for Sciences (Ronit Sagi-Eisenberg).
References
Ali, H. (2009). Regulation of human mast cell and basophil function by anaphylatoxins C3a and C5a. Immunol. Lett. 128, 36–45.
Ali, H., Choi, O. H., Fraundorfer, P. F., Yamada, K., Gonzaga, H. M., and Beaven, M. A. (1996). Sustained activation of phospholipase D via adenosine A3 receptors is associated with enhancement of antigen- and Ca2+-ionophore-induced secretion in a rat mast cell line. J. Pharmacol. Exp. Ther. 276, 837–845.
Alvarez De Toledo, G., and Fernandez, J. M. (1990). Compound versus multigranular exocytosis in peritoneal mast cells. J. Gen. Physiol. 95, 397–409.
Aridor, M., Rajmilevich, G., Beaven, M. A., and Sagi-Eisenberg, R. (1993). Activation of exocytosis by the heterotrimeric G protein Gi3. Science 262, 1569–1572.
Aridor, M., and Sagi-Eisenberg, R. (1990). Neomycin is a potent secretagogue of mast cells that directly activates a GTP-binding protein that involved in exocytosis. J. Cell Biol. 111, 2885–2891.
Aridor, M., Traub, L. M., and Sagi-Eisenberg, R. (1990). Exocytosis in mast cells by basic secretagogues: evidence for direct activation of GTP-binding proteins. J. Cell Biol. 111, 909–917.
Asadi, S., Alysandratos, K. D., Angelidou, A., Miniati, A., Sismanopoulos, N., Vasiadi, M., Zhang, B., Kalogeromitros, D., and Theoharides, T. C. (2012). Substance P (SP) induces expression of functional corticotropin-releasing hormone receptor-1 (CRHR-1) in human mast cells. J. Invest. Dermatol. 132, 324–329.
Askenase, P. W., Bursztajn, S., Gershon, M. D., and Gershon, R. K. (1980). T cell-dependent mast cell degranulation and release of serotonin in murine delayed-type hypersensitivity. J. Exp. Med. 152, 1358–1374.
Avila, M., and Gonzalez-Espinosa, C. (2011). Signaling through toll-like receptor 4 and mast cell-dependent innate immunity responses. IUBMB Life 63, 922–929.
Baram, D., Dekel, O., Mekori, Y. A., and Sagi-Eisenberg, R. (2010). Activation of mast cells by trimeric G protein Gi3; Coupling to the A3 adenosine receptor directly and upon T cell contact. J. Immunol. 184, 3677–3688.
Baram, D., Vaday, G. G., Salamon, P., Drucker, I., Hershkoviz, R., and Mekori, Y. A. (2001). Human mast cells release metalloproteinase-9 on contact with activated T cells: juxtacrine regulation by TNFα. J. Immunol. 167, 4008–4016.
Bhattacharyya, S. P., Drucker, I., Reshef, T., Kirshenbaum, A. S., Metcalfe, D. D., and Mekori, Y. A. (1998). Activated T lymphocytes induce degranulation and cytokine production by human mast cells following cell-to-cell contact. J. Leukoc. Biol. 63, 337–341.
Bienenstock, J., Macqueen, G., Sestini, P., Marshall, J. S., Stead, R. H., and Perdue, M. H. (1991). Mast cell/nerve interactions in vitro and in vivo. Am. Rev. Respir. Dis. 143, S55–S588.
Bischoff, S. C. (2007). Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat. Rev. Immunol. 7, 93–104.
Bischoff, S. C., and Krämer, S. (2007). Human mast cells, bacteria, and intestinal immunity. Immunol. Rev. 217, 329–337.
Bohnacker, T., Marone, R., Collmann, E., Calvez, R., Hirsch, E., and Wymann, M. P. (2009). PI3Kγ adaptor subunits define coupling to degranulation and cell motility by distinct PtdIns(3,4,5)P3 pools in mast cells. Sci. Signal. 2, ra27.
Bonneau, O., Wyss, D., Ferretti, S., Blaydon, C., Stevenson, C. S., and Trifilieff, A. (2006). Effect of adenosine A2A receptor activation in murine models of respiratory disorders. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L1036–L1043.
Bouma, M. G., Jeunhomme, T. M., Boyle, D. L., Dentener, M. A., Voitenok, N. N., Van Den Wildenberg, F. A., and Buurman, W. A. (1997). Adenosine inhibits neutrophil degranulation in activated human whole blood: involvement of adenosine A2 and A3 receptors. J. Immunol. 158, 5400–5408.
Bours, M. J., Swennen, E. L., Di Virgilio, F., Cronstein, B. N., and Dagnelie, P. C. (2006). Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 112, 358–404.
Brown, R. A., Spina, D., and Page, C. P. (2008). Adenosine receptors and asthma. Br. J. Pharmacol. 153(Suppl. 1), S446–S456.
Buceta, M., Domínguez, E., Castro, M., Brea, J., Alvarez, D., Barcala, J., Valdés, L., Alvarez-Calderón, P., Domínguez, F., Vidal, B., Díaz, J. L., Miralpeix, M., Beleta, J., Cadavid, M. I., and Loza, M. I. (2008). A new chemical tool (C0036E08) supports the role of adenosine A(2B) receptors in mediating human mast cell activation. Biochem. Pharmacol. 76, 912–921.
Bulanova, E., and Bulfone-Paus, S. (2010). P2 receptor-mediated signaling in mast cell biology. Purinergic Signal. 6, 3–17.
Cantarella, G., Scollo, M., Lempereur, L., Saccani-Jotti, G., Basile, F., and Bernardini, R. (2011). Endocannabinoids inhibit release of nerve growth factor by inflammation-activated mast cells. Biochem. Pharmacol. 82, 380–388.
Cao, J., Cetrulo, C. L., and Theoharides, T. C. (2006). Corticotropin-releasing hormone induces vascular endothelial growth factor release from human mast cells via the cAMP/protein kinase A/p38 mitogen-activated protein kinase pathway. Mol. Pharmacol. 69, 998–1006.
Carruthers, A. M., and Fozard, J. R. (1993). Effect of pertussis toxin treatment on the putative adenosine A3 receptor-mediated hypotensive response in the rat. Eur. J. Pharmacol. 250, 185–188.
Chan, C. Y., St John, A. L., and Abraham, S. N. (2012). Plasticity in mast cell responses during bacterial infections. Curr. Opin. Microbiol. 15, 78–84.
Coleman, J. W., Holliday, M. R., Kimber, I., Zsebo, K. M., and Galli, S. J. (1993). Regulation of mouse peritoneal mast cell secretory function by stem cell factor, IL-3 or IL-4. J. Immunol. 150, 556–562.
Crivellato, E., Nico, B., Mallardi, F., Beltrami, C. A., and Ribatti, D. (2003). Piecemeal degranulation as a general secretory mechanism? Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 274, 778–784.
Cushley, M., and Holgate, S. (1985). Adenosine-induced bronchoconstriction in asthma: role of mast cell-mediator release. J. Allergy Clin. Immunol. 75, 272–278.
Detoraki, A., Staiano, R. I., Granata, F., Giannattasio, G., Prevete, N., De Paulis, A., Ribatti, D., Genovese, A., Triggiani, M., and Marone, G. (2009). Vascular endothelial growth factors synthesized by human lung mast cells exert angiogenic effects. J. Allergy Clin. Immunol. 123, 1142–1149.
Driver, A. G., Kukoly, C. A., Ali, S., and Mustafa, S. J. (1993). Adenosine in bronchoalveolar lavage fluid in asthma. Am. Rev. Respir. Dis. 148, 91–97.
Druey, K. (2009). Regulation of G-protein-coupled signaling pathways in allergic inflammation. Immunol. Res. 43, 62–76.
Duffy, S. M., Cruse, G., Brightling, C. E., and Bradding, P. (2007). Adenosine closes the K+ channel KCa3.1 in human lung mast cells and inhibits their migration via the adenosine A2A receptor. Eur. J. Immunol. 37, 1653–1662.
Dvorak, A. M., and Kissell, S. (1991). Granule changes of human skin mast cells characteristic of piecemeal degranulation and associated with recovery during wound healing in situ. J. Leukoc. Biol. 49, 197–210.
Dvorak, A. M., McLeod, R. S., Onderdonk, A., Monahan-Earley, R. A., Cullen, J. B., Antonioli, D. A., Morgan, E., Blair, J. E., Estrella, P., Cisneros, R. L., Silen, W., and Cohen, Z. (1992). Ultrastructural evidence for piecemeal and anaphylactic degranulation of human gut mucosal mast cells in vivo. Int. Arch. Allergy Immunol. 99, 74–83.
Endo, D., Gon, Y., Nunomura, S., Yamashita, K., Hashimoto, S., and Ra, C. (2005). PI3Kγ differentially regulates FcϵRI-mediated degranulation and migration of mast cells by and toward antigen. Blood 105, 3222–3229.
Feoktistov, I., and Biaggioni, I. (1995). Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J. Clin. Invest. 96, 1979–1986.
Feoktistov, I., Garland, E. M., Goldstein, A. E., Zeng, D., Belardinelli, L., Wells, J. N., and Biaggioni, I. (2001). Inhibition of human mast cell activation with the novel selective adenosine A(2B) receptor antagonist 3-isobutyl-8-pyrrolidinoxanthine (IPDX) (2). Biochem. Pharmacol. 62, 1163–1173.
Feoktistov, I., Ryzhov, S., Goldstein, A. E., and Biaggioni, I. (2003). Mast cell-mediated stimulation of angiogenesis: cooperative interaction between A2B and A3 adenosine receptors. Circ. Res. 92, 485–492.
Fozard, J. R., Pfannkuche, H. J., and Schuurman, H. J. (1996). Mast cell degranulation following adenosine A3 receptor activation in rats. Eur. J. Pharmacol. 298, 293–297.
Fredholm, B. B. (2007). Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 14, 1315–1323.
Fritsche-Polanz, R., Fritz, M., Huber, A., Sotlar, K., Sperr, W. R., Mannhalter, C., Fodinger, M., and Valent, P. (2010). High frequency of concomitant mastocytosis in patients with acute myeloid leukemia exhibiting the transforming KIT mutation D816V. Mol. Oncol. 4, 335–346.
Galli, S. J. (1997). The Paul Kallos Memorial Lecture. The mast cell: a versatile effector cell for a challenging world. Int. Arch. Allergy. Immunol. 113, 14–22.
Galli, S. J., Gordon, J. R., and Wershil, B. K. (1991). Cytokine production by mast cells and basophils. Curr. Opin. Immunol. 3, 865–872.
Galli, S. J., Kalesnikoff, J., Grimbaldeston, M. A., Piliponsky, A. M., Williams, C. M., and Tsai, M. (2005). Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu. Rev. Immunol. 23, 749–786.
Gao, Z., Li, B.-S., Day, Y.-J., and Linden, J. (2001). A3 Adenosine receptor activation triggers phosphorylation of protein kinase B and protects rat basophilic leukemia 2H3 mast cells from apoptosis. Mol. Pharmacol. 59, 76–82.
Garbuzenko, E., Nagler, A., Pickholtz, D., Gillery, P., Reich, R., Maquart, F. X., and Levi-Schaffer, F. (2002). Human mast cells stimulate fibroblast proliferation, collagen synthesis and lattice contraction: a direct role for mast cells in skin fibrosis. Clin. Exp. Allergy 32, 237–246.
Garcia-Faroldi, G., Rodriguez, C. E., Urdiales, J. L., Perez-Pomares, J. M., Davila, J. C., Pejler, G., Sanchez-Jimenez, F., and Fajardo, I. (2010). Polyamines are present in mast cell secretory granules and are important for granule homeostasis. PLoS ONE 5, e15071. doi:10.1371/journal.pone.0015071
García-Román, J., Ibarra-Sánchez, A., Lamas, M., and González Espinosa, C. (2010). VEGF secretion during hypoxia depends on free radicals-induced Fyn kinase activity in mast cells. Biochem. Biophys. Res. Commun. 401, 262–267.
Gessi, S., Merighi, S., Varani, K., Leung, E., Mac Lennan, S., and Borea, P. A. (2008). The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol. Ther. 117, 123–140.
Gilfillan, A. M., Peavy, R. D., and Metcalfe, D. D. (2009). Amplification mechanisms for the enhancement of antigen-mediated mast cell activation. Immunol. Res. 43, 15–24.
Gilfillan, A. M., and Tkaczyk, C. (2006). Integrated signalling pathways for mast-cell activation. Nat. Rev. Immunol. 6, 218–230.
Gomez, G., Zhao, W., and Schwartz, L. B. (2011). Disparity in FcϵRI-induced degranulation of primary human lung and skin mast cells exposed to adenosine. J. Clin. Immunol. 31, 3479–3487.
Graham, S., Combes, P., Crumiere, M., Klotz, K.-N., and Dickenson, J. M. (2001). Regulation of p42/p44 mitogen-activated protein kinase by the human adenosine A3 receptor in transfected CHO cells. Eur. J. Pharmacol. 420, 19–26.
Haskó, G., and Pacher, P. (2008). A2A receptors in inflammation and injury: lessons learned from transgenic animals. J. Leukoc. Biol. 83, 447–455.
Haskû, G., Csûka, B. Z., Nèmeth, Z. N. H., Vizi, E. S., and Pacher, P. L. (2009). A2B adenosine receptors in immunity and inflammation. Trends Immunol. 30, 263–270.
Higashijima, T., Uzu, S., Nakajima, T., and Ross, E. M. (1988). Mastoparan, a peptide toxin from wasp venom, mimics receptors by activating GTP-binding regulatory proteins (G proteins). J. Biol. Chem. 263, 6491–6494.
Hoffman, H. M., Walker, L. L., and Marquardt, D. L. (1997). Mast cell adenosine induced calcium mobilization via Gi3 and Gq proteins. Inflammation 21, 55–68.
Holgate, S., Lewis, R., and Austen, K. F. (1980). Role of adenylate cyclase in immunologic release of mediators from rat mast cells: agonist and antagonist effects of purine- and ribose-modified adenosine analogs. Proc. Natl. Acad. Sci. U.S.A. 77, 6800–6804.
Hua, X., Chason, K. D., Fredholm, B. B., Deshpande, D. A., Penn, R. B., and Tilley, S. L. (2008). Adenosine induces airway hyperresponsiveness through activation of A3 receptors on mast cells. J. Allergy Clin. Immunol. 122, 107–113.
Hua, X., Chason, K. D., Patel, J. Y., Naselsky, W. C., and Tilley, S. L. (2011). IL-4 amplifies the pro-inflammatory effect of adenosine in human mast cells by changing expression levels of adenosine receptors. PLoS ONE 6, e24947. doi:10.1371/journal.pone.0024947
Hua, X., Kovarova, M., Chason, K. D., Nguyen, M., Koller, B. H., and Tilley, S. L. (2007). Enhanced mast cell activation in mice deficient in the A2b adenosine receptor. J. Exp. Med. 204, 117–128.
Hundley, T. R., Gilfillan, A. M., Tkaczyk, C., Andrade, M. V., Metcalfe, D. D., and Beaven, M. A. (2004). Kit and FcϵRI mediate unique and convergent signals for release of inflammatory mediators from human mast cells. Blood 104, 2410–2417.
Huszar, E., Vass, G., Vizi, E., Csoma, Z., Barat, E., Molnar Vilagos, G., Herjavecz, I., and Horvath, I. (2002). Adenosine in exhaled breath condensate in healthy volunteers and in patients with asthma. Eur. Respir. J. 20, 1393–1398.
Iemura, A., Tsai, M., Ando, A., Wershil, B. K., and Galli, S. J. (1994). The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am. J. Pathol. 144, 321–328.
Irani, A. A., Schechter, N. M., Craig, S. S., Deblois, G., and Schwartz, L. B. (1986). Two types of human mast cells that have distinct neutral protease compositions. Proc. Natl. Acad. Sci. U.S.A. 83, 4464–4468.
Jacobson, K. A. (1998). Adenosine A3 receptors: novel ligands and paradoxical effects. Trends Pharmacol. Sci. 19, 184–191.
Jin, X., Shepherd, R. K., Duling, B. R., and Linden, J. (1997). Inosine binds to A3 adenosine receptors, and stimulates mast cell degranulation. J. Clin. Invest. 100, 2849–2857.
Kashem, S. W., Subramanian, H., Collington, S. J., Magotti, P., Lambris, J. D., and Ali, H. (2011). G protein coupled receptor specificity for C3a and compound 48/80-induced degranulation in human mast cells: roles of Mas-related genes MrgX1 and MrgX2. Eur. J. Pharmacol. 668, 299–304.
Kitaura, J., Kinoshita, T., Matsumoto, M., Chung, S., Kawakami, Y., Leitges, M., Wu, D., Lowell, C. A., and Kawakami, T. (2005). IgE- and IgE+ Ag-mediated mast cell migration in an autocrine/paracrine fashion. Blood 105, 3222–3229.
Knight, D., Zheng, X., Rocchini, C., Jacobson, M., Bai, T., and Walker, B. (1997). Adenosine A3 receptor stimulation inhibits migration of human eosinophils. J. Leukoc. Biol. 62, 465–468.
Kuehn, H. S., Beaven, M. A., Ma, H. T., Kim, M. S., Metcalfe, D. D., and Gilfillan, A. M. (2008). Synergistic activation of phospholipases Cγ and Cβ: a novel mechanism for PI3K-independent enhancement of FcϵRI-induced mast cell mediator release. Cell. Signal. 20, 625–636.
Kuehn, H. S., Jung, M. Y., Beaven, M. A., Metcalfe, D. D., and Gilfillan, A. M. (2011). Distinct PGE2-responder and non-responder phenotypes in human mast cell populations: “all or nothing” enhancement of antigen-dependent mediator release. Immunol. Lett. 141, 45–54.
Kuehn, H. S., Rådinger, M., Brown, J. M., Ali, K., Vanhaesebroeck, B., Beaven, M. A., Metcalfe, D. D., and Gilfillan, A. M. (2010). Btk-dependent Rac activation and actin rearrangement following FcϵRI aggregation promotes enhanced chemotactic responses of mast cells. J. Cell Sci. 123, 2576–2585.
Kulka, M., Sheen, C., Tancowny, B. C., and Schleimer, R. P. (2009). Adenosine receptor A2a inhibits complement-mediated activation of human mast cells by activating Gs-proteins. J. Immunol. 182, 139.5.
Kunder, C. A., St John, A. L., and Abraham, S. N. (2011). Mast cell modulation of the vascular and lymphatic endothelium. Blood 118, 5383–5393.
Laffargue, M., Calvez, R., Finan, P., Trifilieff, A., Barbier, M., Altruda, F., Hirsch, E., and Wymann, M. P. (2002). Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity 16, 441–451.
Lagunoff, D., Martin, T. W., and Read, G. (1983). Agents that release histamine from mast cells. Ann. Rev. Pharmacol. Toxicol. 23, 331–351.
Li, Q., Ye, K., Blad, C. C., den Dulk, H., Brouwer, J., Ijzerman, A. P., and Beukers, M. W. (2007). ZM241385, DPCPX, MRS1706 are inverse agonists with different relative intrinsic efficacies on constitutively active mutants of the human adenosine A2B receptor. J. Pharmacol. Exp. Ther. 320, 637–645.
Lim, K. H., Pardanani, A., and Tefferi, A. (2008). KIT and mastocytosis. Acta Haematol. 119, 194–198.
Linden, J. (1994). Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions. Trends Pharmacol. Sci. 15, 298–306.
Linden, J. (2001). Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 41, 775–787.
Linden, J., Thai, T., Figler, H., Jin, X., and Robeva, A. S. (1999). Characterization of human A(2B) adenosine receptors: radioligand binding, western blotting, and coupling to G(q) in human embryonic kidney 293 cells and HMC-1 mast cells. Mol. Pharmacol. 56, 705–713.
Lloyd, H. G., Ross, L., Li, K. M., and Ludowyke, R. I. (1998). Evidence that IgE receptor stimulation increases adenosine release from rat basophilic leukaemia (RBL-2H3) cells. Pulm. Pharmacol. Ther. 11, 41–46.
Lorentz, A. (2011). Corecognition of pathogens: an important trigger for mast cell response? Int. Arch. Allergy. Immunol. 154, 183–184.
Markovic, D., and Challiss, R. (2009). Alternative splicing of G protein-coupled receptors: physiology and pathophysiology. Cell. Mol. Life Sci. 66, 3337.
Marquardt, D. L., Gruber, H. E., and Wasserman, S. I. (1984). Adenosine release from stimulated mast cells. Proc. Natl. Acad. Sci. U.S.A. 81, 6192–6196.
Marquardt, D. L., Walker, L. L., and Heinemann, S. (1994). Cloning of two adenosine receptor subtypes from mouse bone marrow-derived mast cells. J. Immunol. 152, 4508–4515.
Matera, M. G., and Polosa, R. (2007). Adenosine receptors: novel targets for drug development in allergic rhinitis. Clin. Exp. Allergy 37, 4–7.
Meade, C. J., Dumont, I., and Worrall, L. (2001). Why do asthmatic subjects respond so strongly to inhaled adenosine? Life Sci. 69, 1225–1240.
Meade, C. J., Worrall, L., Hayes, D., and Protin, U. (2002). Induction of interleukin 8 release from the HMC-1 mast cell line: synergy between stem cell factor and activators of the adenosine A2b receptor. Biochem. Pharmacol. 64, 317–325.
Metcalfe, D. D., Kaliner, M., and Donlon, M. A. (1981). The mast cell. Crit. Rev. Immunol. 3, 23–74.
Metz, M., and Maurer, M. (2007). Mast cells – key effector cells in immune responses. Trends Immunol. 28, 234–241.
Michael, S., Warstat, C., Michel, F., Yan, L., Müller, C. E., and Nieber, K. (2010). Adenosine A(2A) agonist and A(2B) antagonist mediate an inhibition of inflammation-induced contractile disturbance of a rat gastrointestinal preparation. Purinergic Signal. 6, 117–124.
Mousli, M., Bueb, J.-L., Bronner, C., Rouot, B., and Landry, Y. (1990). G protein activation: a receptor-independent mode of action for cationic amphiphilic neuropeptides and venom peptides. Trends Pharmacol. Sci. 11, 358–362.
Mrabet-Dahbi, S., Metz, M., Dudeck, A., Zuberbier, T., and Maurer, M. (2009). Murine mast cells secrete a unique profile of cytokines and prostaglandins in response to distinct TLR2 ligands. Exp. Dermatol. 18, 437–444.
Mustafa, S. J., Nadeem, A., Fan, M., Zhong, H., Belardinelli, L., and Zeng, D. (2007). Effect of a specific and selective A(2B) adenosine receptor antagonist on adenosine agonist AMP and allergen-induced airway responsiveness and cellular influx in a mouse model of asthma. J. Pharmacol. Exp. Ther. 320, 1246–1251.
Nadeem, A., Fan, M., Ansari, H. R., Ledent, C., and Mustafa, S. J. (2007). Enhanced airway reactivity and inflammation in A2A adenosine receptor-deficient allergic mice. Am. J. Physiol. Lung. Cell. Mol. Physiol. 292, L1335–L1344.
Nakamura, T., and Ui, M. (1984). Islet activating protein, pertussis toxin, inhibits calcium-induced and guanine nucleotide dependent releases of histamine and arachidonic acid from rat mast cells. FEBS Lett. 173, 414–418.
Nakamura, T., and Ui, M. (1985). Simultaneous inhibitions of inositol phospholipid breakdown, arachidonic acid release, and histamine secretion in mast cells by islet-activating protein, pertussis toxin. A possible involvement of the toxin-specific substrate in the Ca2+-mobilizing receptor-mediated biosignaling system. J. Biol. Chem. 260, 3584–3593.
Novak, N., Bieber, T., and Peng, W. M. (2010). The immunoglobulin E-toll-like receptor network. Int. Arch. Allergy Immunol. 151, 1–7.
Ogawa, Y., and Grant, J. A. (2007). Mediators of anaphylaxis. Immunol. Allergy Clin. North Am. 27, 249–260.
Okayama, Y., Saito, H., and Ra, C. (2008). Targeting human mast cells expressing G-protein-coupled receptors in allergic diseases. Allergol. Int. 57, 197–203.
Olah, M. E., Gallo-Rodriguez, C., Jacobson, K. A., and Stiles, G. L. (1994). 125I-4-aminobenzyl-5’-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol. 45, 978–982.
Olszewski, M. B., Groot, A. J., Dastych, J., and Knol, E. F. (2007). TNF trafficking to human mast cell granules: mature chain-dependent endocytosis. J. Immunol. 178, 5701–5709.
Ortner, M. J., and Chingell, C. F. (1981). Spectroscopic studies of rat mast cells, mouse mastocytoma cells and compound 48/80. Biochem. Pharmacol. 30, 283–288.
Palmer, T. M., Gettys, T. W., and Stiles, G. L. (1995). Differential interaction with and regulation of multiple G-proteins by the rat A3 adenosine receptor. J. Biol. Chem. 270, 16895–16902.
Peachell, P. T., Columbo, M., Kagey-Sobotka, A., Lichtenstein, L. M., and Marone, G. (1988). Adenosine potentiates mediator release from human lung mast cells. Am. Rev. Respir. Dis. 138, 1143–1151.
Peachell, P. T., Lichtenstein, L. M., and Schleimer, R. P. (1991). Differential regulation of human basophil and lung mast cell function by adenosine. J. Pharmacol. Exp. Ther. 256, 717–726.
Polosa, R., and Blackburn, M. R. (2009). Adenosine receptors as targets for therapeutic intervention in asthma and chronic obstructive pulmonary disease. Trends Pharmacol. Sci. 30, 528–535.
Puxeddu, I., Ribatti, D., Crivellato, E., and Levi-Schaffer, F. (2005). Mast cells and eosinophils: a novel link between inflammation and angiogenesis in allergic diseases. J. Allergy Clin. Immunol. 116, 531–536.
Qiao, H., Andrade, M. V., Lisboa, F. A., Morgan, K., and Beaven, M. A. (2006). FcepsilonR1 and toll-like receptors mediate synergistic signals to markedly augment production of inflammatory cytokines in murine mast cells. Blood 107, 610–618.
Ramakers, B. P., Riksen, N. P., van der Hoeven, J. G., Smits, P., and Pickkers, P. (2011). Modulation of innate immunity by adenosine receptor stimulation. Shock 36, 208–215.
Ramkumar, V., Stiles, G. L., Beaven, M. A., and Ali, H. (1993). The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J. Biol. Chem. 268, 16887–16890.
Reeves, J. J., Jones, C. A., Sheehan, M. J., Vardey, C. J., and Whelan, C. J. (1997). Adenosine A3 receptors promote degranulation of rat mast cells both in vitro and in vivo. Inflamm. Res. 46, 180–184.
Repke, H., and Bienert, M. (1987). Mast cell activation – a receptor-independent mode of substance P action? FEBS Lett. 221, 236–240.
Ribatti, D., and Crivellato, E. (2009). The controversial role of mast cells in tumor growth. Int. Rev. Cell. Mol. Biol. 275, 89–131.
Ribatti, D., Crivellato, E., Candussio, L., Vacca, A., Nico, B., Benagiano, V., Roncali, L., and Dammacco, F. (2002). Angiogenic activity of rat mast cells in the chick embryo chorioallantoic membrane is down-regulated by treatment with recombinant human alpha-2a interferon and partly mediated by fibroblast growth factor-2. Haematologica 87, 465–471.
Rivera, J., and Gilfillan, A. M. (2006). Molecular regulation of mast cell activation. J. Allergy Clin. Immunol. 117, 1214–1225.
Rodewald, H. R., Dessing, M., Dvorak, A. M., and Galli, S. J. (1996). Identification of a committed precursor for the mast cell lineage. Science 271, 818–822.
Rork, T. H., Wallace, K. L., Kennedy, D. P., Marshall, M. A., Lankford, A. R., and Linden, J. (2008). Adenosine A2A receptor activation reduces infarct size in the isolated, perfused mouse heart by inhibiting resident cardiac mast cell degranulation. Am. J. Physiol. Heart Circ. Physiol. 295, H1825–H1833.
Ryzhov, S., Goldstein, A. E., Biaggioni, I., and Feoktistov, I. (2006). Cross-talk between Gs- and Gq-coupled pathways in regulation of interleukin-4 by A2B adenosine receptors in human mast cells. Mol. Pharmacol. 70, 727–735.
Ryzhov, S., Goldstein, A. E., Matafonov, A., Zeng, D., Biaggioni, I., and Feoktistov, I. (2004). Adenosine-Activated Mast Cells Induce IgE Synthesis by B Lymphocytes: an A2B-mediated process involving Th2 cytokines IL-4 and IL-13 with implications for asthma. J. Immunol. 172, 7726–7733.
Ryzhov, S., Zaynagetdinov, R., Goldstein, A. E., Novitskiy, S. V., Dikov, M. M., Blackburn, M. R., Biaggioni, I., and Feoktistov, I. (2008). Effect of A2B adenosine receptor gene ablation on proinflammatory adenosine signaling in mast cells. J. Immunol. 180, 7212–7220.
Salvatore, C. A., Tilley, S. L., Latour, A. M., Fletcher, D. S., Koller, B. H., and Jacobson, M. A. (2000). Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J. Biol. Chem. 275, 4429–4434.
Schulte, G., and Fredholm, B. B. (2002). Signaling pathway from the human adenosine A3 receptor expressed in Chinese hamster ovary cells to the extracellular signal-regulated kinase 1/2. Mol. Pharmacol. 62, 1137–1146.
Schwartz, L. B., and Austen, K. F. (1980). Enzymes of the mast cell granule. J. Invest. Dermatol. 74, 349–353.
Sereda, M. J., Bradding, P., and Vial, C. (2011). Adenosine potentiates human lung mast cell tissue plasminogen activator activity. J. Immunol. 186, 1209–1217.
Shefler, I., and Sagi-Eisenberg, R. (2002). Gi-mediated activation of the p42/p44 mitogen-activated protein kinases by receptor mimetic basic secretagogues is abrogated by inhibitors of endocytosis. Int. Immunopharmacol. 2, 711–720.
Shefler, I., Seger, R., and Sagi-Eisenberg, R. (1999). Gi-mediated activation of mitogen-activated protein kinase (MAPK) pathway by receptor mimetic basic secretagogues of connective tissue-type mast cells: bifurcation of arachidonic acid-induced release upstream of MAPK. J. Pharmacol. Exp. Ther. 289, 1654–1661.
Shefler, I., Zavaro, O., Raz, T., Baram, D., and Sagi-Eisenberg, R. (2008). Inhibition of basic secretagogue-induced signaling in mast cells by cell permeable Gαi-derived peptides. Int. Arch. Allergy Immunol. 145, 131–140.
Shelburne, C. P., and Abraham, A. S. (2011). The mast cell in innate and adaptive immunity. Adv. Exp. Med. Biol. 716, 162–185.
Siraganian, R. P. (2003). Mast cell signal transduction from the high-affinity IgE receptor. Curr. Opin. Immunol. 15, 639–646.
Smith, S. R., Denhardt, G., and Terminelli, C. (2002). A role for histamine in cytokine modulation by the adenosine A(3) receptor agonist, 2-Cl-IB-MECA. Eur. J. Pharmacol. 457, 57–69.
Spicuzza, L., Di Maria, G., and Polosa, R. (2006). Adenosine in the airways: implications and applications. Eur. J. Pharmacol. 533, 77–88.
Subramanian, H., Gupta, K., Guo, Q., Price, R., and Ali, H. (2011). Mas-related gene X2 (MrgX2) is a novel G protein-coupled receptor for the antimicrobial peptide LL-37 in human mast cells: resistance to receptor phosphorylation, desensitization, and internalization. J. Biol. Chem. 286, 44739–44749.
Sun, C. X., Zhong, H., Mohsenin, A., Morschl, E., Chunn, J. L., Molina, J. G., Belardinelli, L., Zeng, D., and Blackburn, M. R. (2006). Role of A2B receptor signaling in adenosine-dependent pulmonary inflammation and injury. J. Clin. Invest. 116, 1–10.
Suzuki, H., Takei, M., Nakahata, T., and Fukamachi, H. (1998). Inhibitory effect of adenosine on degranulation of human cultured mast cells upon cross-linking of FcϵRI. Biochem. Biophys. Res. Commun. 242, 697–702.
Tatemoto, K., Nozaki, Y., Tsuda, R., Konno, S., Tomura, K., Furuno, M., Ogasawara, H., Edamura, K., Takagi, H., Iwamura, H., Noguchi, M., and Naito, T. (2006). Immunoglobulin E-independent activation of mast cell is mediated by Mrg receptors. Biochem. Biophys. Res. Commun. 349, 1322–1328.
Taylor, A. M., Galli, S. J., and Coleman, J. W. (1995). Stem-cell factor, the kit ligand, induces direct degranulation of rat peritoneal mast cells in vitro and in vivo: dependence of the in vitro effect on period of culture and comparisons of stem-cell factor with other mast cell-activating agents. Immunology 86, 427–433.
Theoharides, T., Kempuraj, D., Tagen, M., Vasiadi, M., and Cetrulo, C. (2006). Human umbilical cord blood-derived mast cells: a unique model for the study of neuro-immuno-endocrine interactions. Stem Cell Rev. 2, 143–153.
Theoharides, T. C., Duraisamy, K., Michael, T., Pio, C., and Dimitris, K. (2007). Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol. Rev. 217, 65–78.
Theoharides, T. C., and Kalogeromitros, D. (2006). The critical role of mast cells in allergy and inflammation. Ann. N. Y. Acad. Sci. 1088, 78–99.
Tilley, S. L., Tsai, M., Williams, C. M., Wang, Z. S., Erikson, C. J., Galli, S. J., and Koller, B. H. (2003). Identification of A3 receptor- and mast cell-dependent and -independent components of adenosine-mediated airway responsiveness in mice. J. Immunol. 171, 331–337.
Turner, H., and Kinet, J. P. (1999). Signalling through the high-affinity IgE receptor FcϵRI. Nature 402, 24–30.
Valent, P., Akin, C., Arock, M., Brockow, K., Butterfield, J. H., Carter, M. C., Castells, M., Escribano, L., Hartmann, K., Lieberman, P., Nedoszytko, B., Orfao, A., Schwartz, L. B., Sotlar, K., Sperr, W. R., Triggiani, M., Valenta, R., Horny, H. P., and Metcalfe, D. D. (2011). Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int. Arch. Allergy Immunol. 157, 215–225.
Varani, K., Caramori, G., Vincenzi, F., Adcock, I., Casolari, P., Leung, E., Maclennan, S., Gessi, S., Morello, S., Barnes, P. J., Ito, K., Chung, K. F., Cavallesco, G., Azzena, G., Papi, A., and Borea, P. A. (2006). Alteration of adenosine receptors in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 173, 398–406.
Vass, G., Huszar, E., Augusztinovicz, M., Baktai, G., Barat, E., Herjavecz, I., and Horvath, I. (2006). The effect of allergic rhinitis on adenosine concentration in exhaled breath condensate. Clin. Exp. Allergy 36, 742–747.
Walker, B. A., Jacobson, M. A., Knight, D. A., Salvatore, C. A., Weir, T., Zhou, D., and Bai, T. R. (1997). Adenosine A3 receptor expression and function in eosinophils. Am. J. Respir. Cell Mol. Biol. 16, 531–537.
Wasserman, S. I. (1983). Mediators of immediate hypersensitivity. J. Allergy Clin. Immunol. 72, 101–119.
Welle, M. (1997). Development, significance, and heterogeneity of mast cells with particular regard to the mast cell-specific proteases chymase and tryptase. J. Leukoc. Biol. 61, 233–245.
Weller, K., Foitzik, K., Paus, R., Syska, W., and Maurer, M. (2006). Mast cells are required for normal healing of skin wounds in mice. FASEB J. 20, 2366–2368.
Wesolowski, J., and Paumet, F. (2011). The impact of bacterial infection on mast cell degranulation. Immunol. Res. 51, 215–226.
Wilson, B. S., Oliver, J. M., and Lidke, D. S. (2011). Spatio-temporal signaling in mast cells. Adv. Exp. Med. Biol. 716, 91–106.
Woska, J. R., and Gillespie, M. E. (2012). SNARE complex-mediated degranulation in mast cells. J. Cell. Mol. Med. 16, 649–656.
Xiang, Z., and Nilsson, G. (2000). IgE receptor-mediated release of nerve growth factor by mast cells. Clin. Exp. Allergy 30, 1379–1386.
Yamano, K., Inoue, M., Masaki, S., Saki, M., Ichimura, M., and Satoh, M. (2005). Human adenosine A3 receptor leads to intracellular Ca2+ mobilization but is insufficient to activate the signaling pathway via phosphoinositide 3-kinase [gamma] in mice. Biochem. Pharmacol. 70, 1487–1496.
Yamano, K., Inoue, M., Masaki, S., Saki, M., Ichimura, M., and Satoh, M. (2006). Generation of adenosine A3 receptor functionally humanized mice for the evaluation of the human antagonists. Biochem. Pharmacol. 71, 294–306.
Yip, K. H., Lau, H. Y., and Wise, H. (2011). Reciprocal modulation of anti-IgE induced histamine release from human mast cells by A1 and A(2B) adenosine receptors. Br. J. Pharmacol. 164, 807–819.
Yip, K. H., Wong, L. L., and Lau, H. Y. (2009). Adenosine: roles of different receptor subtypes in mediating histamine release from human and rodent mast cells. Inflamm. Res. 58(Suppl. 1), 17–19.
Zablocki, J., Kalla, R., Perry, T., Palle, V., Varkhedkar, V., Xiao, D., Piscopio, A., Maa, T., Gimbel, A., Hao, J., Chu, N., Leung, K., and Zeng, D. (2005). The discovery of a selective, high affinity A(2B) adenosine receptor antagonist for the potential treatment of asthma. Bioorg. Med. Chem. Lett. 15, 609–612.
Zhang, C., Hirasawa, N., and Beaven, M. A. (1997a). Antigen activation of mitogen-activated protein kinase in mast cells through protein kinase C-dependent and independent pathways. J. Immunol. 158, 4968–4975.
Zhang, C., Baumgartner, R. A., Yamada, K., and Beaven, M. A. (1997b). Mitogen-activated Protein (MAP) kinase regulates production of tumor necrosis factor- α and release of arachidonic acid in mast cells indications of communication between p38 and p42 Map kinases. J. Biol. Chem. 272, 13397–13402.
Keywords: mast cells, adenosine, A3 adenosine receptor, HMC-1, RBL-2H3
Citation: Rudich N, Ravid K and Sagi-Eisenberg R (2012) Mast cell adenosine receptors function: a focus on the A3 adenosine receptor and inflammation. Front. Immun. 3:134. doi: 10.3389/fimmu.2012.00134
Received: 22 January 2012; Paper pending published: 22 February 2012;
Accepted: 09 May 2012; Published online: 04 June 2012.
Edited by:
Toshiaki Kawakami, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Pablo Pelegrin, Hospital Universitario Virgen Arrixaca, SpainAlasdair Gilfillan, National Institutes of Health, USA
Copyright: © 2012 Rudich, Ravid and Sagi-Eisenberg. This is an openaccess article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ronit Sagi-Eisenberg, Department of Cell and Developmental Biology, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv 69978, Israel. e-mail:aGlzdG9sM0Bwb3N0LnRhdS5hYy5pbA==