 Katherine M. Buckley1,2 and Jonathan P. Rast1,2*
Katherine M. Buckley1,2 and Jonathan P. Rast1,2*- 1 Department of Immunology, Sunnybrook Research Institute, University of Toronto, Toronto, ON, Canada
- 2 Department of Medical Biophysics, Sunnybrook Research Institute, University of Toronto, Toronto, ON, Canada
The genome sequence of the purple sea urchin, Strongylocentrotus purpuratus, a large and long-lived invertebrate, provides a new perspective on animal immunity. Analysis of this genome uncovered a highly complex immune system in which the gene families that encode homologs of the pattern recognition receptors that form the core of vertebrate innate immunity are encoded in large multigene families. The sea urchin genome contains 253 Toll-like receptor (TLR) sequences, more than 200 Nod-like receptors and 1095 scavenger receptor cysteine-rich domains, a 10-fold expansion relative to vertebrates. Given their stereotypic protein structure and simple intron-exon architecture, the TLRs are the most tractable of these families for more detailed analysis. A role for these receptors in immune defense is suggested by their similarity to TLRs in other organisms, sequence diversity, and expression in immunologically active tissues, including phagocytes. The complexity of the sea urchin TLR multigene families is largely derived from expansions independent of those in vertebrates and protostomes, although a small family of TLRs with structure similar to that of Drosophila Toll can be traced to an ancient eumetazoan ancestor. Several other echinoderm sequences are now available, including Lytechinus variegatus, as well as partial sequences from two other sea urchin species. Here, we present an analysis of the invertebrate deuterostome TLRs with emphasis on the echinoderms. Representatives of most of the S. purpuratus TLR subfamilies and homologs of the mccTLR sequences are found in L. variegatus, although the L. variegatus TLR gene family is notably smaller (68 TLR sequences). The phylogeny of these genes within sea urchins highlights lineage-specific expansions at higher resolution than is evident at the phylum level. These analyses identify quickly evolving TLR subfamilies that are likely to have novel immune recognition functions and other, more stable, subfamilies that may function more similarly to those of vertebrates.
Introduction
The discovery of an immune function for Drosophila Toll (Lemaitre et al., 1996) and the subsequent identification of immune recognition roles for mammalian Toll-like receptor (TLR) 4 (Medzhitov et al., 1997; Poltorak et al., 1998) catalyzed an intensely renewed interest in innate immunity and more generally an appreciation for the potential of invertebrate models in mainstream immunology. As genome sequences from an increasing number of animal phyla are resolved, it has become clear that TLRs are present in virtually all eumetazoans (Messier-Solek et al., 2010). In the genome of the purple sea urchin, Strongylocentrotus purpuratus, these receptors are encoded in a very large multigene family that contrasts sharply with the small families of insects and vertebrates (Hibino et al., 2006). Recently sequenced genomes from several animals, including another invertebrate deuterostome, amphioxus, and the annelid Capitella capitata suggest that large TLR repertoires may be widespread throughout Bilateria (e.g., Davidson et al., 2008; Huang et al., 2008). An understanding of the function of these TLRs may provide a new perspective on this important family of innate immune receptors.
It is far from settled whether or not these TLRs function in immunity (Leulier and Lemaitre, 2008). In insects and mammals, the two animals groups for which function is well-understood, the mechanisms by which TLRs recognize non-self and the systems in which they operate differ considerably. In mammals, where all TLRs function as immune receptors that interact directly with non-self factors, defense is the primary role. In contrast, Drosophila Toll signals far downstream of immune recognition and thus its role in immune recognition is indirect (Lemaitre and Hoffmann, 2007). The remaining eight Drosophila TLRs have not been associated with immunity and, where their function is defined, are more closely associated with development and other cellular processes. In Drosophila, Toll-9 is the single member of the Toll family that structurally resembles vertebrate TLRs. Although early work suggested that Toll-9 may be responsible for maintaining constitutive expression of antimicrobial peptides (Ooi et al., 2002), more recent studies analyzing Toll-9 mutants reveal that this protein is not required to mount an efficient antibacterial response (Narbonne-Reveau et al., 2011). The central role of Drosophila Toll signaling in mesoderm patterning (Huang et al., 1997) has not been demonstrated outside of insects. While mammalian TLRs have relatively modest roles in modulating cell differentiation (e.g., in the gut), these are sequential to their function in immune recognition and are not counterparts to the developmental function of Drosophila Toll. Ancient homologs of TLRs are also present within the genomes of the cnidarians Nematostella vectensis and Hydra magnipapillata (Miller et al., 2007). The single TLR in Nematostella structurally resembles Toll, although its function has not been investigated. In contrast, the Hydra genome encodes four Toll-related proteins (HyTRR-1/HyLRR-1 and Hy-TRR2/Hy-LRR2) that interact to form two receptors that have been shown to play a role in epithelial immunity (Bosch et al., 2009). Thus it remains difficult to make definitive statements about function across animal phyla and inference of ancestral function remains elusive, although there is some indication of an ancient immune role.
Despite these difficulties, other characteristics of the genes that encode TLRs in the sea urchin and other animal genomes may shed light on their function and thus on TLR evolution. Here we present an analysis of TLR multiplicity, phylogeny, diversity, and expression in the purple sea urchin against the background of new sequence information from other sea urchin species. We find that the unique characteristics of TLRs in the purple sea urchin are present also in other sea urchin species. The multiplicity, apparently rapid gene turnover, and sequence diversity of the TLRs within this complex gene family, in addition to enriched expression in immunologically active tissues are consistent with a role in immunity. Most notably the evolutionary patterns of family member diversification suggest rapid changes in binding potential that are unlike those seen in the TLRs of vertebrates or Drosophila. Thus, TLRs in the sea urchin, and possibly other Bilateria, may have been co-opted for use in an immune recognition strategy that is more evolutionarily dynamic than the pathogen-associated molecular pattern (PAMP)-based systems of vertebrates and insects. In contrast to the paradigm of vertebrate TLRs, in which conserved receptors recognize static microbial elements, in sea urchins, closely related but rapidly diversifying variants of receptors may respond to quickly evolving pathogens.
Materials and Methods
Sequence Analysis
The S. purpuratus genome sequence (v3.1; released July, 2011) was obtained from SpBase1 (Cameron et al., 2009). Additional echinoderm genome sequences and unassembled genomic traces were obtained from the Sea Urchin Genome Project website of the Human Genome Sequencing Center at Baylor College of Medicine (HGSC-BCM2) and the National Center for Biotechnology Information (NCBI; Lytechinus variegatus GenBank Assembly ID: GCA_000239495.1; L. variegatus 454 sequence: SRX112894, SRX112895, SRX112896). The Saccoglossus kowalevskii genome (Skow_1.0) was obtained from the HGSC-BCM website3.
Genome sequences were translated and open reading frames were identified using tools within the EMBOSS package.4 All potential open reading frames greater than 75 amino acids in length, without requirement to start with a methionine were analyzed. Domain searches were performed with HMMER 3.05 and leucine-rich repeats (LRRs) were identified using LRRfinder (Offord et al., 2010). TLR sequences were classified into three categories: (1) complete genes that were uninterrupted by a stop codon in the translated sequence; (2) pseudogenes that were characterized by an in frame stop codon or frame shift leading to missense sequence; or (3) partial genes in which the sequences were truncated by either the end of a scaffold or indeterminate sequences (N’s). Genomic coordinates and descriptions of the TLR sequences can be found in Tables S1 and S2 in Supplementary Material. Sequences were aligned using ClustalX (Larkin et al., 2007), and alignments were manually edited in Bioedit (Hall, 1999). Sequence entropy was calculated based on described methods (Durbin et al., 1998).
Phylogenetic analyses of the Toll/Interleukin-1 Receptor (TIR) domains were done in MEGA5.0 (Tamura et al., 2011). Neighbor-joining trees were constructed using evolutionary distances calculated with the Poisson correction method. Alignment positions containing gaps were removed from the entire analysis. Bootstrap support was calculated based on 1,000 replicates.
An analysis of evolutionary selection was performed for each TLR subfamily that contained eight or more complete, non-pseudogene sequences. Sequence alignments for that were used for these analyses are in Files S1–S6 in Supplementary Material. A maximum likelihood tree built in PHYLIP (Felsenstein, 2005) served as the working topology for the analyses. Selection within the sequences was analyzed in CODEML within PAML (Yang, 2007) under two models: the M7 model, which allows neutral or purifying selection, and the M8 model, which also includes a class of sites that evolve under positive selection. The two models were compared using a likelihood ratio test (Yang, 1998). Residues under positive selection were identified using the Bayes empirical Bayes approach under the M8 model (Yang, 2007).
To validate the multiplicity of the TLR gene families within the S. purpuratus and L. variegatus assembled genomes and also to estimate the gene family sizes in Allocentrotus fragilis and Strongylocentrotus franciscanus, we analyzed the unassembled genomic traces. The amino acid sequences of the TIR domains from the S. purpuratus and L. variegatus TLRs were used as queries in a tblastn search against the unassembled traces from A. fragilis, S. franciscanus, and L. variegatus. All traces that matched with an e-value of less than 0.01 were collected and used as queries in a blastx search against the TIR domains to classify the partial sequences by subfamily and to enumerate the sequences.
Larval Culture, Infection Model, and Coelomocytes
S. purpuratus larvae were maintained at a concentration of four larvae per mL in artificial seawater (ASW; Instant Ocean) at 15°C and fed Rhodomonas lens (5,000/mL) starting at 5 days post-fertilization (dpf). For some of measurements of TLR transcript prevalence S. purpuratus larvae were exposed to Vibrio diazotrophicus (ATCC strain 33466). Samples were collected at 0, 6, 12, and 24 h of exposure to bacteria and used in RNA-Seq analysis. The larvae in these four samples were derived a single fertilization of eggs from one female.
To induce an immune response, a single adult animal was injected intracoelomically with complex microbiota isolated from the gut of another adult animal (4.8 × 106 total bacteria). After 12 h, whole coelomocytes and gut tissue were collected for RNA-Seq experiments. Phagocytic coelomocytes were isolated using discontinuous gradient density centrifugation (Gross et al., 2000). Gut tissue was homogenized for RNA extraction and consisted of mixed samples from the entire length of the gut.
RNA-Seq
Total RNA was isolated using Trizol (Invitrogen), and mRNA was purified with the Poly(A)Purist kit (Ambion). cDNA sequencing was performed on an Applied Biosystems SOLiD4 and 5500 SOLiD machines at the Sunnybrook Genomics Facility. For the larval and coelomocyte samples, the paired-end reads were 50 and 35 nt long; from the gut, paired-end reads were 75 and 35 nt in length.
Sequences were mapped in color space to the S. purpuratus genome (v3.1) using Bowtie version 0.12.7 (Langmead et al., 2009) with the following parameters that differed from the default: up to 50 alignments reported for each read (-k); reads with greater than 50 alignments suppressed (-m); the maximum number of mismatches in the seed was set at 3 (-n); the maximum sum of the quality scores for mismatches was 900 (-e); five nucleotides were trimmed from the 3′ ends of the reads (-3); and the SNP fraction was set at 0.04 (–snpfrac), which is consistent with estimates of SNPs in the sea urchin genome (Sodergren et al., 2006). Only reads that mapped to the TIR domains were included in the expression analysis of TLR subfamilies and reads that mapped to TLR genes from more than one subfamily were excluded. TIR domain sequences for which >5% of the reads mapped in the incorrect direction with respect to the coding sequence were not included in the analysis.
Results
The Expanded TLR Family in the Purple Sea Urchin
Toll-like receptors are type-1 transmembrane proteins with a solenoid-like ectodomain structure composed of a series of LRRs that is responsible for ligand-binding (Jin and Lee, 2008). The hydrophobic core of this structure is capped on either end by specialized cysteine-rich LRR-NT and LRR-CT domains that are distinct in sequence and structure from the central LRRs. Hereafter, “LRRs” refers only to the central repeats. C-terminal to the single transmembrane region is a TIR domain that mediates interactions with downstream signaling factors (Gay and Keith, 1991; O’Neill and Bowie, 2007). Our previous analysis of the S. purpuratus genome (v2.1) identified 222 genes that encode TLR homologs (Hibino et al., 2006). This genome assembly contained 114,222 scaffolds with an N50 of 123.5 kb. Improvements to the assembly using additional BAC sequencing and high-throughput next-generation sequencing strategies have resulted in the most recent version (v3.1) that is composed of 32,008 scaffolds with an N50 of 401.9 kb (see text footnote 1). To incorporate these updates to the genome sequence into our analysis of the S. purpuratus TLR gene family, we reanalyzed the improved genome (v3.1) to identify open reading frames that contained TIR domains (Pfam domain PF01582.12). The majority of sea urchin TLRs are encoded in a single exon, which enables their identification directly from the translated genome, rather the predicted gene models. The sequence flanking the TIR domains was analyzed for the presence of other protein domains, including a transmembrane region, LRR-CT, central LRRs, LRR-NT, and signal peptides. In total, 284 TIR domains were identified in the genome that were part of authentic genes or pseudogenes. TIR domains are also present in several other molecules, including the TLR adaptors and IL1R family members, which were excluded from the analysis. The remaining TIR domains defined 253 TLR sequences within the sea urchin genome.
Most of the sea urchin TLR proteins (240) are structurally similar to those of vertebrates (Figure 1). The LRRs in the ectodomains of these proteins are flanked by LRR-NT and LRR-CT domains. TLRs with this type of extracellular domain are structurally distinct from Drosophila Toll (Rock et al., 1998) and are known as single cysteine cluster TLRs (sccTLRs; Leulier and Lemaitre, 2008). The sea urchin sccTLRs have between 21 and 25 LRRs. This is the structure of the vertebrate TLRs as well as Drosophila Toll-9 (Table 1). In addition, the sea urchin genome contains 13 TLRs that differ from the sccTLRs both in the structure of the ectodomain and also in the sequence of the TIR domain. Five of these divergent TLRs are characterized by shortened ectodomains that are composed of nine LRRs, rather than the typical 21–25. The LRRs within the ectodomains of these short TLRs are flanked by LRR-NT and LRR-CT domains (Figure 1). Four of the divergent TLRs, which comprise a supported clade, resemble the sccTLRs with respect to domain architecture, but the coding sequence is interrupted by a single intron. Finally, the ectodomains of four of the sea urchin TLRs resemble those of Drosophila Toll, in which LRR-CT and LRR-NT domains interrupt the typical LRRs. This domain organization has been termed multiple cysteine cluster TLRs (mccTLRs; Figure 1; Leulier and Lemaitre, 2008) and is the predominant structure of the Drosophila Toll proteins (Table 1).
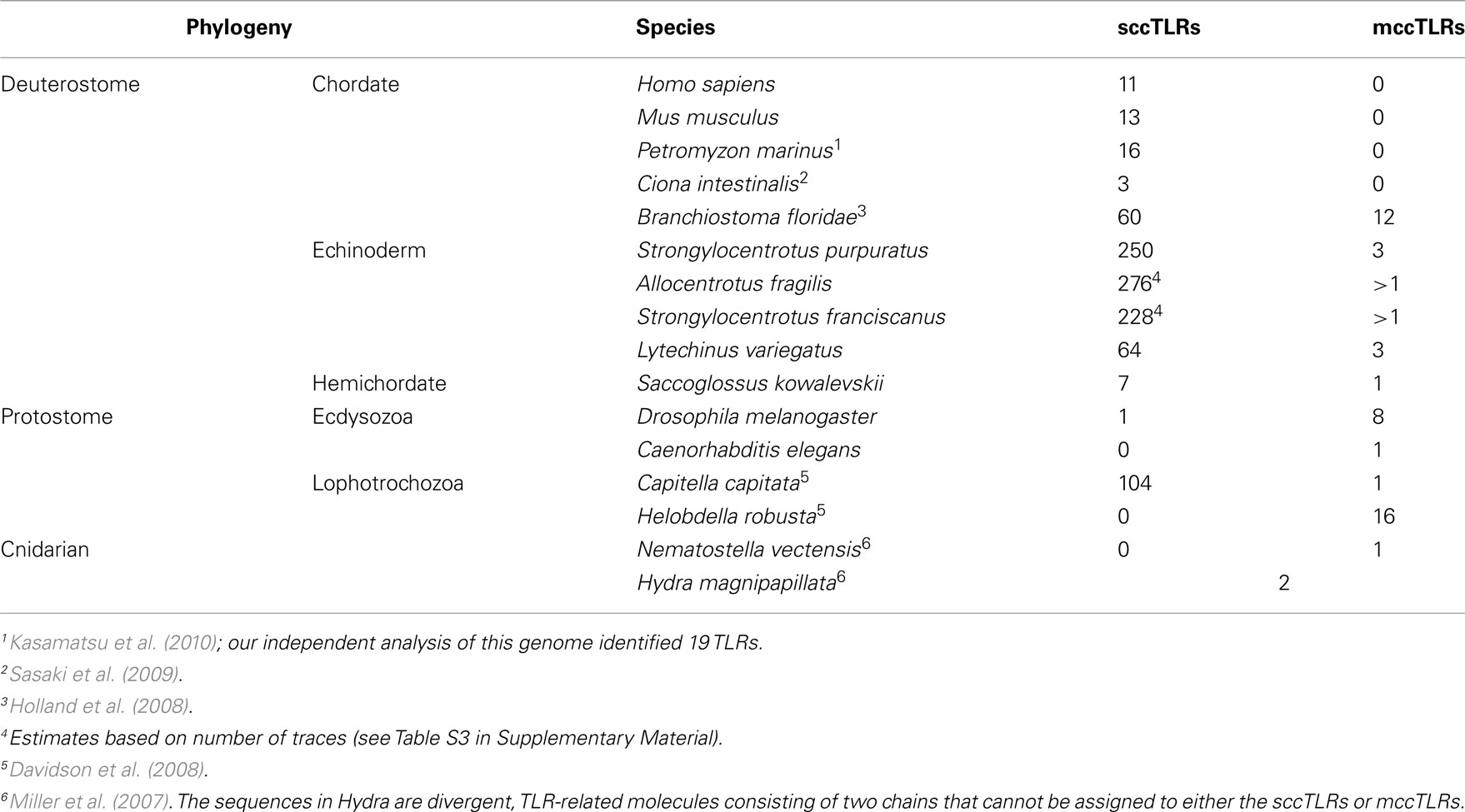
Table 1. Sizes of TLR gene family vary among species.
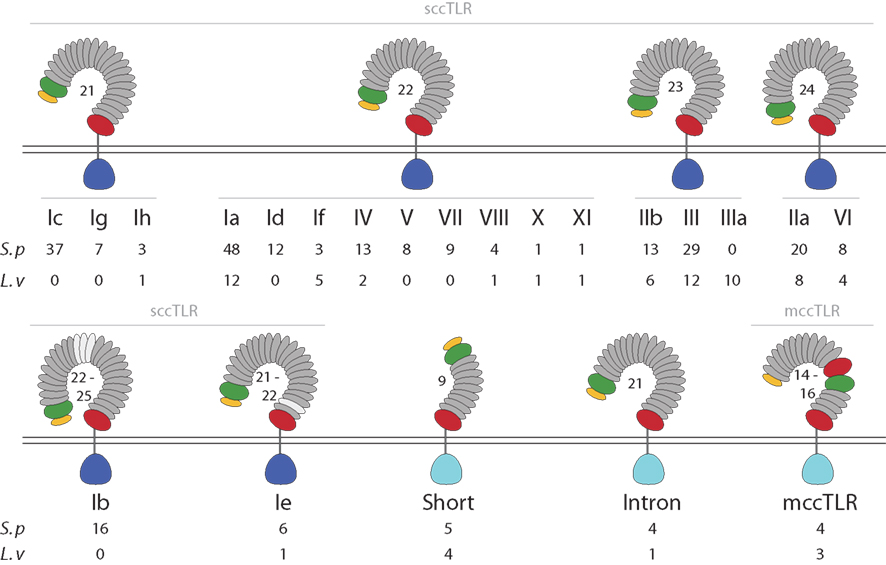
Figure 1. The TLR subfamily in sea urchins. The protein domain structure for each of the TLR subfamilies is shown. Among the sccTLR sequences, the number of LRRs varies from 21 to 25. TLRs in subfamilies Ib and Ie vary in the total number of LRRs (LRRs that are not present in all sequences are shown in light gray). The divergent TIR sequences that are present in the mccTLRs, intron-containing, and short TLRs are shown in light blue (see Figure 2). The numbers of TIR domains from each group and subfamily that are present in the S. purpuratus and L. variegatus genomes are shown below the diagram structures.
The TIR domains of the 253 TLR sequences were used in phylogenetic analysis to further classify the genes (Figure 2). The 240 sccTLRs form a strongly supported clade that is distinct from the divergent short, intron-containing, and mccTLR sequences. Our previous analysis of these sequences identified seven groups of sccTLRs (I–VII; Hibino et al., 2006). Here, we describe the presence of an additional four groups (VIII–XI) based on conservation with other sea urchin species and by eliminating the previously named “orphan” sequences (Figure 2; File S7 in Supplementary Material). Some of the groups are also divided into smaller subfamilies. The group I genes fall into eight subfamilies (Ia–Ih) and the group II genes form the IIa and IIb subfamilies. Groups vary considerably in multiplicity and sequence variability. The largest subfamily (Ia) consists of 48 closely related genes. In contrast, the eight TLRs that belong to group VI are on longer branches that may reflect a more ancient evolutionary history (Figure 2). In contrast with our analysis of the TLRs from the previous genome assembly, an additional 31 TLR sequences were identified, the majority of which belong to the Ic subfamily [there were 13 Ic genes in the v2.1 assembly (Hibino et al., 2006), and 37 in v3.1 (Table 3)]. The genes within this subfamily are clustered in large tandem genomic arrays. The larger scaffolds and higher quality sequence in the current assembly enable the identification of these genes.
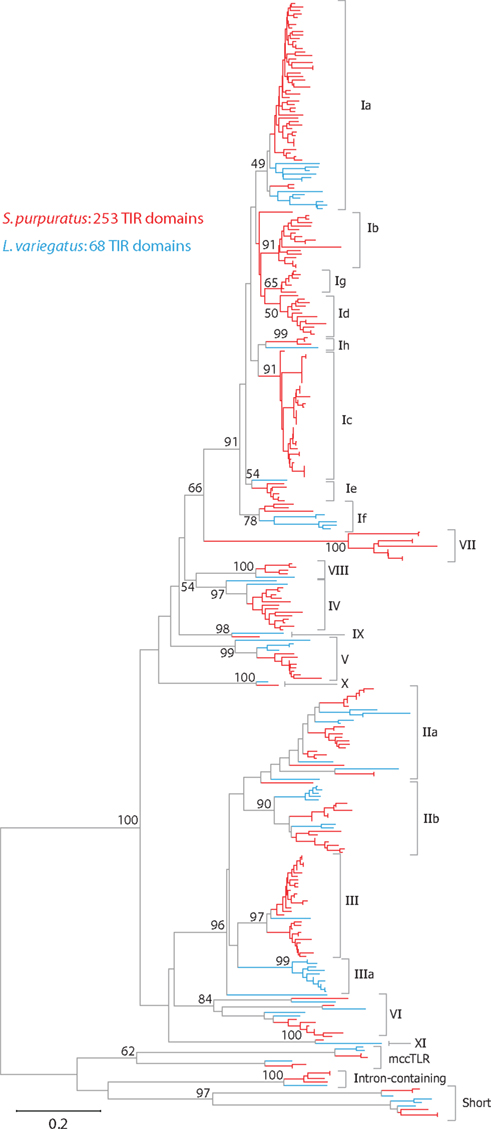
Figure 2. Phylogeny of sea urchin TLR sequences. The TIR domains from the S. purpuratus (red branches) and L. variegatus (blue branches) TLRs were used to construct a neighbor-joining tree in MEGA 5.0 (Tamura et al., 2011) using Poisson corrected distances. Alignment positions containing gaps were eliminated completely from the analysis. Bootstrap values are shown and are based on 1000 replicates. Each of these clades is also reconstructed using other methods (maximum likelihood and maximum parsimony; data not shown). The subfamily for each clade is designated on the right. A more detailed version of this tree is shown in Figure A1 in Appendix.
Additionally, each of the TLR sequences was classified as a complete gene, pseudogene, or partial gene based on the presence of in frame stop codons, and the presence of ambiguous flanking sequence. Given the complexity of this gene family and the similarity among the sequences, it is not surprising that many of the TLR genes are partial due to difficulty in assembling very similar sequence. Overall, 23% of the 253 TLR TIR domains were from partial gene sequences (Table 3). Pseudogenes are identified as those with in frame stop codons or frame shifts that result in missense sequence. Most of the frame shifts and point mutations that were used to designated pseudogenes (80%) could be confirmed by analysis of the genomic trace sequences and chromatographs. However, a few genes that appeared to be pseudogenes in the assembly were shown to be intact genes when the traces were analyzed more carefully (this includes the single group XI gene). Some of the pseudogenes are very similar to complete genes, while others differ substantially in sequence. The proportion of pseudogenes varies among groups (Table 3; Hibino et al., 2006), which is likely a function of varying turnover rates across the subfamilies. In this analysis, we only included sequences that encode intact TIR domains. Thus, this assessment of pseudogenes is incomplete, and many other related sequences that appear to be pseudogenes are present in the genome, varying from almost intact genes to highly divergent sequence fragments.
Although the TIR domains from all the TLRs can be aligned, the LRR portions of these proteins are unalignable across subfamilies. The orthology of individual LRRs cannot be reliably established across groups due to the variation in the number of LRRs and the lack of sequence similarity. Despite the sequence diversity among groups, the ectodomains of TLRs within groups are similar, both with respect to sequence and also the number of LRRs (Figure 1). The exceptions to this are TLRs in subfamilies Ib and Ie, which, although they are similar in sequence, vary in the number of LRRs as a result of discrete deletions or insertions of one or more complete LRRs.
The overall evolution of these groups is difficult to determine. Although each of the subfamilies consistently forms a clade, there is little support for the deeper relationships between the groups. It is notable, however, that the sea urchin sccTLR sequences appear to be the result of an expansion specific to the echinoderm lineage. When TLRs from mammals, other invertebrate deuterostomes, including hemichordates, urochordates, or cephalochordates, or protostomes are included in the analysis, the sea urchin sccTLRs form a strongly supported clade, but support for inter-phyla relationships is not present (data not shown; Messier-Solek et al., 2010).
Sequence Diversity and Signatures of Selection within the TLR Subfamilies
The sea urchin TLR sequences exhibit striking amino acid diversity. There is significant variability within the conserved leucine-rich repeat framework, both with respect to changes in the amino acid sequence and also short indels. To characterize this diversity we analyzed the sequence entropy of each alignment position for the subfamilies that contained eight or more complete sequences excluding pseudogenes (Ia, Ib, Ic, IIa, III, and IV; Figure 3). Sequence entropy is a measure of diversity that is based on the frequency of each amino acid at each position (Durbin et al., 1998). Results indicate that within subfamilies, the TIR domains are much more conserved than the LRR-containing ectodomains. On average, the ectodomain diversity is three times higher than that of the intracellular TIR domain (Figure 3; Table 2). This is consistent with an association between LRR sequence diversity and ligand-binding function. Furthermore, the levels and patterns of diversity vary among the subfamilies. The average diversity of the Ia sequences was over three times that of the Ic sequences, although both groups are composed of a similar numbers of genes (Figure 3). The peak in LRR diversity also varied among subfamilies. In subfamily Ia, the most diverse region of the ectodomains is in LRR16-18, whereas in subfamily IIa, the highest diversity is observed in LRR3 and LRR14. This variation in sequence diversity may reflect differences in ligand-binding mechanisms among the TLR subfamilies.
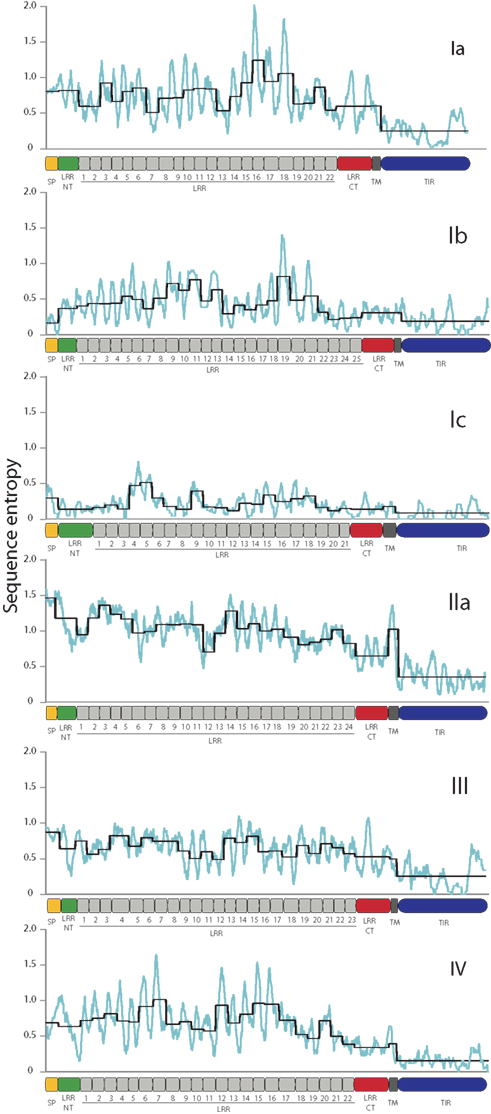
Figure 3. TLR TIR domains are more conserved than the LRR regions. The diversity of the amino acid sequences for each of the subfamilies that contain more than eight complete sequences was analyzed as a measure of sequence entropy (Durbin et al., 1998). In the graphs shown, the light blue line indicates the average diversity over a sliding window of 10 amino acids, and the black line shows the average diversity of each of the protein domains marked on the x-axis. In each of the subfamilies, the TIR domains exhibit greater conservation than the ectodomains, and there is significant sequence variation within the LRR domains.
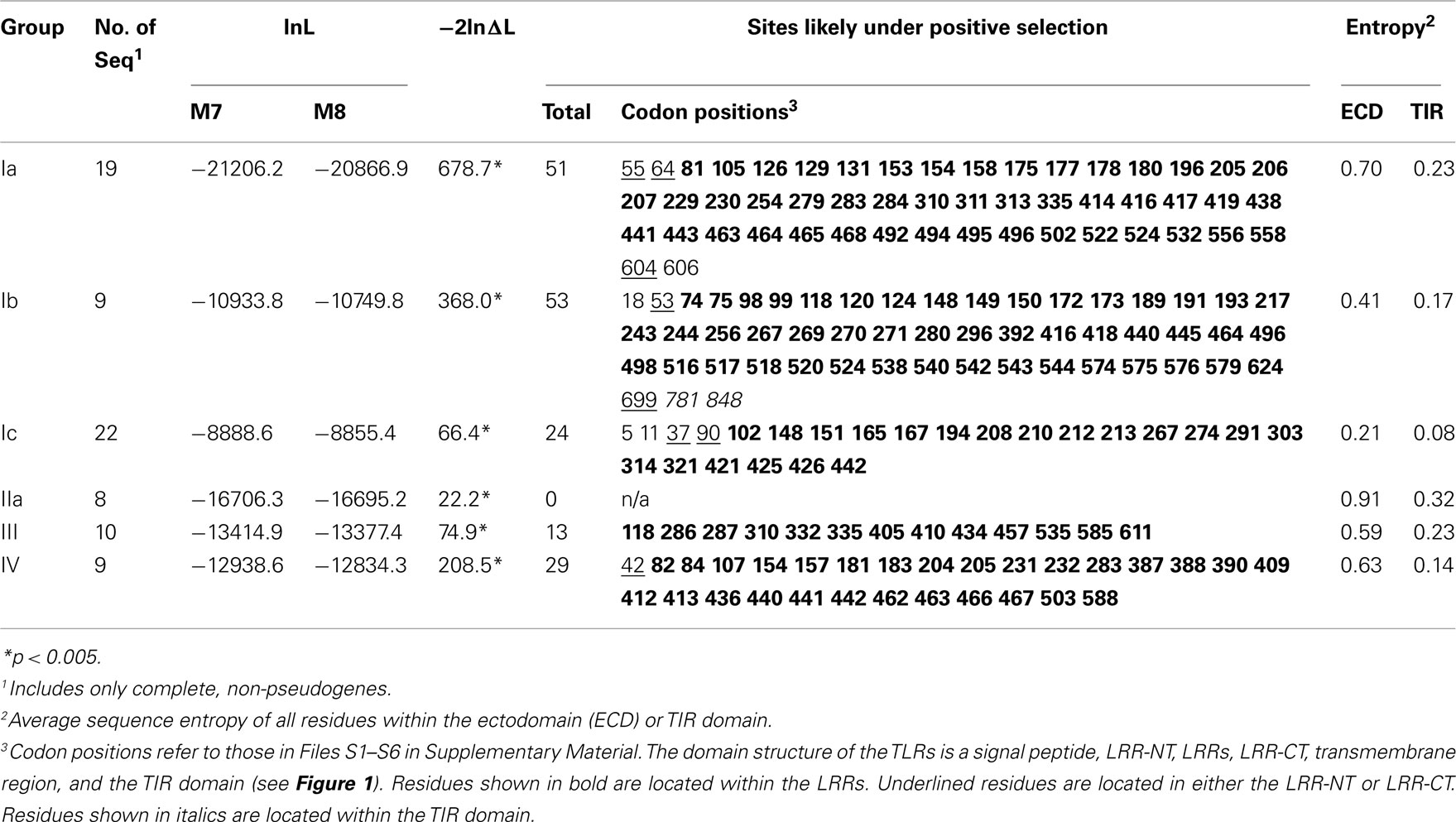
Table 2. Diversity and evolution of the TLR sequences.
We further analyzed the patterns of selection within the S. purpuratus TLRs. Sequence entropy measures the diversity of the amino acids sequences, whereas the selection analyses within PAML take into account the underlying relative frequencies of synonymous and non-synonymous nucleotide substitutions that result in the protein sequence variability. The evolution of the sea urchin TLR sequences was analyzed under two models implemented in PAML that were compared using a likelihood ratio test (Yang, 2007). The first model, M7, allows codons to evolve under only neutral and purifying selection, whereas the second model, M8, also includes a class for residues that evolve under positive selection. For each of the six subfamilies analyzed, the M8 model that incorporated positive selection was a significantly better fit to the data (Table 2), suggesting that at least some of the residues within the TLR genes are subject to positive selection (Yang, 1998).
Specific sites that are likely to be under positive selection were identified and mapped onto a generic structure for the LRR ectodomain that is based on a simple solenoid model (Figure 4). The subfamilies varied in the number and pattern of specific residues under positive selection. Of the 170 total residues likely to be under positive selection from the six TLR subfamilies analyzed, 156 fell within the typical LRRs of the ectodomain, which is a significant enrichment compared to the more conserved TIR domains (Table 2; Figure 4A). Only one of these residues included a conserved amino acids that form the LRR framework (subfamily Ic, LRR15). Two residues were located in the TIR domain (both in subfamily Ib) and the remaining 10 sites were within either the LRR-NT or LRR-CT domains. The TLRs of families Ia and Ib had the greatest number of sites under positive selection (51 and 53, respectively; Table 2). This is in contrast to subfamily IIa, in which no specific residues were identified as significantly likely to be under positive selection.
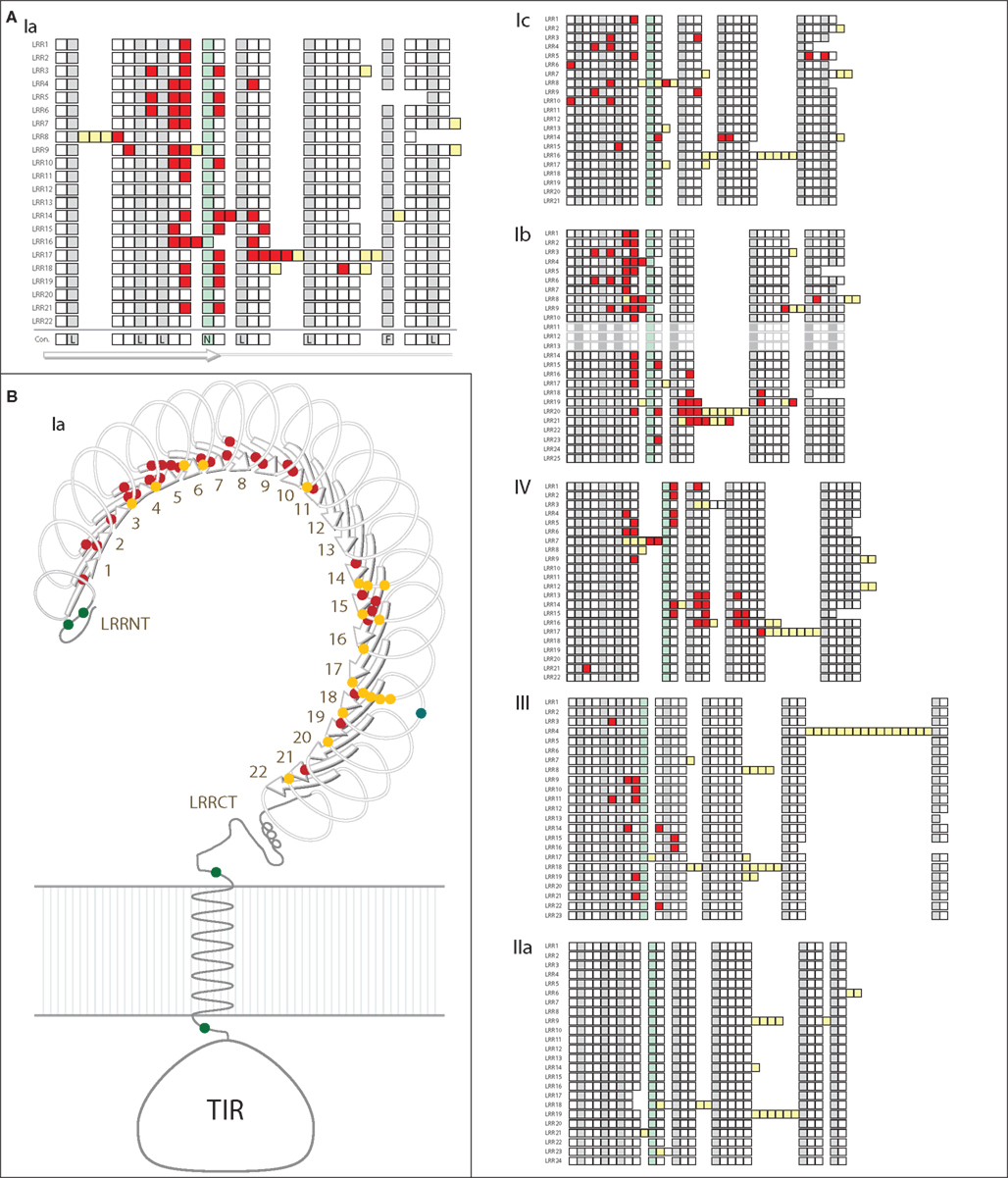
Figure 4. Analysis of positive selection within TLR subfamilies. (A) The majority of residues subject to positive selection are located in the putative convex face of the ectodomain. Residues under positive selection were identified in each subfamily containing more than eight complete, non-pseudogene sequences using PAML (Ia, Ib, Ic, IIa, III, and IV; Yang, 2007). Sequence alignments can be found in Files S1-S6 in Supplementary Material. Each of the LRRs is shown as a single row with individual amino acids indicated as squares. Consensus hydrophobic LRR residues are shown in gray and the conserved asparagines residues are indicated in green (Bell et al., 2003; Kang and Lee, 2011). Amino acids under positive selection are shown in red; insertions into the LRR framework are indicated by yellow squares. The predicted consensus structure is indicated below the squares: the first 10 amino acids form the β-strands on the convex surface of the ectodomains, while the rest of the LRR forms the loop structures. In subfamily IIa, no individual residues were found to be likely under positive selection. (B) Positive selection within the Ia subfamily. A generalized TLR structure is shown based on known solenoid structures of LRR-containing proteins. The majority of positively selected positions are located within the β-strands (red dots), and a few residues are also located on the front face of the TLR just beyond the β-strands (yellow dots). One positively selected residue is found on the outside of the LRRs (blue dot), and there are four residues under positive selection that are not located within the LRRs (two in the LRR-NT domain, and one on either side of the transmembrane domain; green dots).
Notably, the sites under diversifying selection are highly clustered on the three dimensional interpretation of the ectodomain structure (Figure 4). In subfamily Ia, the vast majority of the sites were located within the β-strands that form the concave face of the solenoid ectodomain (red dots; Figure 4B). In contrast, the positively selected residues of subfamily Ic are more scattered throughout the ectodomain (Figure 4A). Subfamilies Ia and IV contain two distinct clusters of residues under positive selection (LRR1-11/LRR14-21 in subfamily Ia and LRR1-9/LRR13-17 for subfamily IV; Figure 4A). In general, the LRRs with greater positive selection also correspond to the more diverse LRRs shown in Figure 3 although these analyses measure different elements of sequence diversity.
TLR Expression
The expression levels of the TLR subfamilies were analyzed in sea urchin larvae and adult immune cells and gut tissue using an RNA-Seq approach (Figure 5). A single batch of sea urchin larvae (9 dpf) was exposed to the marine bacterium V. diazotrophicus, and samples were collected at 0, 6, 12, and 24 h. For each time point, ∼75 million paired-end SOLiD sequencing reads were obtained. Additionally, an adult sea urchin was challenged using bacteria isolated from the digestive tract of another animal to mimic a perforation in the gut and systemic infection. This is intended as a physiologically relevant immune challenge that may be expected to induce a coordinated and complex immune response. Adult phagocytic coelomocytes and gut were isolated 12 h after challenge and used in RNA-Seq experiments, with approximately 130 million and 70 million paired-end reads obtained for each tissue, respectively. From this animal, ∼40 million phagocytes were collected, from which 1.5 μg of polyadenylated mRNA was isolated and used to generate cDNA for sequencing.
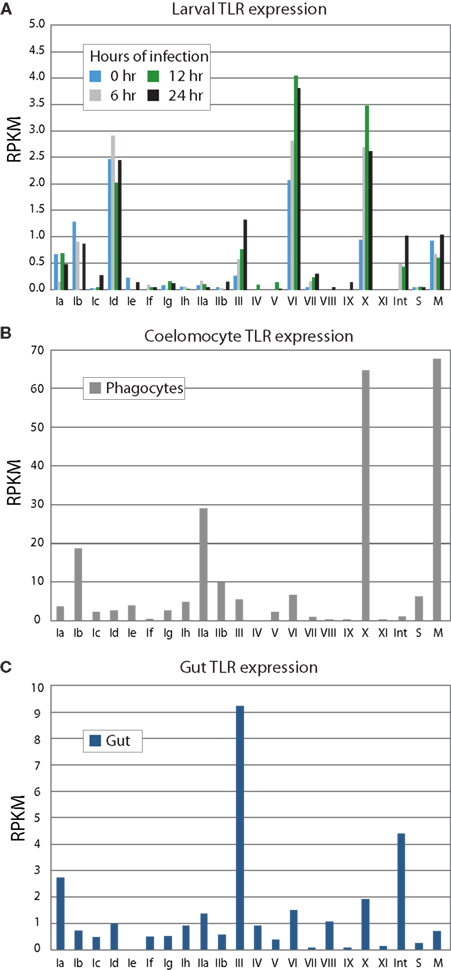
Figure 5. TLR subfamilies are differentially expressed. RNA-Seq was used to analyze gene expression from larvae (A), immune cells (B), and gut (C). Phagocytic coelomocytes and gut were isolated from an animal 12 h after intracoelomic injection of a gut bacteria preparation. Larvae were exposed to V. diazotrophicus for 0, 6, 12, and 24 h, and collected for transcriptome sequencing. RPKM values were calculated for each of the TIR domains; the average RPKM for each group is shown. The sccTLR subfamilies correspond to those shown in Figure 2. Note that the scales are different for each graph. The divergent TLRs are indicated as follows: M, mccTLRs; S, short; Int, intron-containing (Figure 1). In phagocytic coelomocytes, TLRs from the Ib, IIa, and X subfamilies, as well as the mccTLR genes are most highly expressed. In contrast, the III TLRs are primarily expressed in the gut tissue. The TLRs are expressed at lower levels in the larvae, and little change in TLR expression in the larvae is observed in response to bacterial challenge. The predominantly expressed families at the larval stage are Id and VI, are different than those expressed in coelomocytes.
Using RNA-Seq data to analyze the expression of genes from multigene families is not trivial. Standard protocols from RNA-Seq analysis require sequence reads to map uniquely to a reference genome. However, this prevents reads from mapping to closely related paralogs and may artificially lower the expression values for these types of gene. Furthermore, most high-throughput sequence mapping programs are designed for use with the genomes of inbred organisms. Given the similarity of the TLR genes within subfamilies and the relatively high polymorphism among sea urchins (estimated genome heterozygosity is 4–5%; Britten et al., 1978; Pespeni et al., 2011), we have relaxed the stringency of the mapping parameters to analyze the expression of the TLRs. Reads were allowed to map to the genome up to 50 times to accommodate a single read mapping to multiple TLR paralogs, which is slightly larger than the biggest subfamily (Ia, which has 48 sequences). Including only uniquely mapping reads in the analysis disproportionately reduces the expression of the larger subfamilies with closely related genes. Therefore, while we are unable to assess the transcript prevalence of any particular gene relative to its subfamily counterparts, we are able to quantify collective subfamily expression. To clearly assign reads to specific subfamilies, reads that mapped to TLRs from multiple groups, were removed from the analysis (this represented 771 of 20,332 total reads; 3.7%). To account for the high heterozygosity of the sea urchin genome and the expected genetic differences between the experimental animal and that used for the reference genome, we also increased the number of mismatches. These relaxed parameters, however, did not result in a high background of spurious read mapping. Reads that mapped to TLR sequences were directionally specific (<2% of reads mapped in the incorrect orientation for genes that stood above background), which lends additional confidence to our measurements.
Gene expression levels are measured as the number of reads that mapped to the gene per kilobase per million reads mapped to the genome (RPKM; Mortazavi et al., 2008). RPKM is a standard measure of gene expression used in high-throughput sequence analysis that takes into account the length of the gene (longer transcripts produce more sequence fragments), and the total size and quality of the library (poor quality libraries produce reads that do not map to the genome and there is always variation in the number of fragments that are sequenced). These values are comparable across samples and time points. Given the similarity of the TLRs, and the possibility for a read to map to multiple subfamily members, we present the data as the average RPKM for each subfamily (Figure 5).
In sea urchin larvae, the RPKM values of the TLR subfamilies are generally low relative to the expression values of the adult tissues (Figure 5A). However, because the whole animal was used in the sequencing, it may be expected that the TLRs are expressed at relatively higher levels in a small subset of cells within the animal. The highest expression levels are observed for TLR subfamily Id, group VI and the single sequence of group X. This is consistent with qPCR measurements of transcript prevalence (data not shown). Furthermore, while the larvae are able to mount a robust and complex antibacterial response, only modest change is observed in TLR expression in response to bacterial challenge. Many of the TLR subfamilies that are prevalent in adult tissues were not evident in this ontogenetic stage.
We also analyzed RNA-Seq data from phagocytic coelomocytes collected from an immune-challenged animal. There are four primary classes of adult sea urchin coelomocytes: phagocytes, vibratile, colorless spherule cells, and red spherule cells (Smith et al., 2011). Preliminary qPCR data suggests that TLR expression is minimal in the vibratile, colorless spherule, and red spherule cell fractions relative to the phagocytes (data not shown). Analysis of RNA-Seq data from immune-challenged phagocytes indicates that several immune-related genes are expressed in these cells, suggesting that the animal is responding to immune challenge. In contrast to the larval stage, adult coelomocytes express a different suite of TLR subfamilies (Figure 5B). Genes from subfamilies Ib and IIa are expressed highly, as is the TLR gene that comprises the X group, and, notably, the mccTLR genes. Compared to the RPKM values from the larval stage, the average expression levels for the TLR subfamilies in coelomocytes are 20 times higher. Although there may be specific cells within the larvae that express the TLRs at higher levels, this enrichment of RNA-Seq reads that map to the TLR genes in the coelomocyte sample further suggests a role of these proteins in immunity.
Gut tissue was also collected from the immune-challenged animal described above and analyzed by RNA-Seq. The adult gut expresses a suite of TLR genes that is distinct from those expressed in adult coelomocytes and the larval stage (Figure 5C). Here, TLRs from subfamilies III and Ia, as well as the divergent, intron-containing TLRs exhibit the highest levels of expression. Expression of the group X and mccTLRs, which are highly expressed by coelomocytes, was not observed in the gut. These varied expression patterns may point to different roles in the TLR subfamilies in different tissues and at different life stages.
TLRs in Other Sea Urchin Species
As part of the Sea Urchin Genome Project, genome and transcriptome sequencing is underway in several other echinoderm species. A genome sequence is available from L. variegatus, which last shared a common ancestor with S. purpuratus about 50 million years ago (Smith et al., 2006). We analyzed the L. variegatus genome using the same methods as the S. purpuratus genome and identified 68 TLR genes, either as ORFs that contained a TIR domain in addition to the presence of a transmembrane region, LRR-CT domain, and LRRs or by sequence similarity to S. purpuratus TLRs. The TIR domain sequences from these genes were used in phylogenetic analysis with the S. purpuratus TLR TIR domains to classify the L. variegatus genes into subfamilies (Figure 2). The majority of the L. variegatus TLR genes group with the S. purpuratus sccTLR subfamilies (60 of 68). Notably, the L. variegatus genome also encodes orthologs of the intron-containing, mccTLR and short TLR sequences. Although homologous representatives of most of the S. purpuratus sccTLR subfamilies were present in the L. variegatus genome, no sequences were identified that were orthologous to subfamilies Ib, Ic, Id, Ie, Ig or group VII genes (Table 3). In addition to the 10 S. purpuratus sccTLR subfamilies, an additional subfamily, IIIa, is found only within L. variegatus (Figure 2). This strongly supported clade consists of eight sequences and is sister to the S. purpuratus group III sequences. Notably, there are three pairs of orthologous sequences with a single representative in each sea urchin species, the phylogenetic stability of which may suggest a more conserved ligand-binding function relative to the other subfamilies of higher diversity and multiplicity (see groups IX, X, and XI; Figure 2). One of these, subfamily X, which has a single representative in both sea urchin species, is highly expressed in both the S. purpuratus larva and coelomocytes (Figure 5).
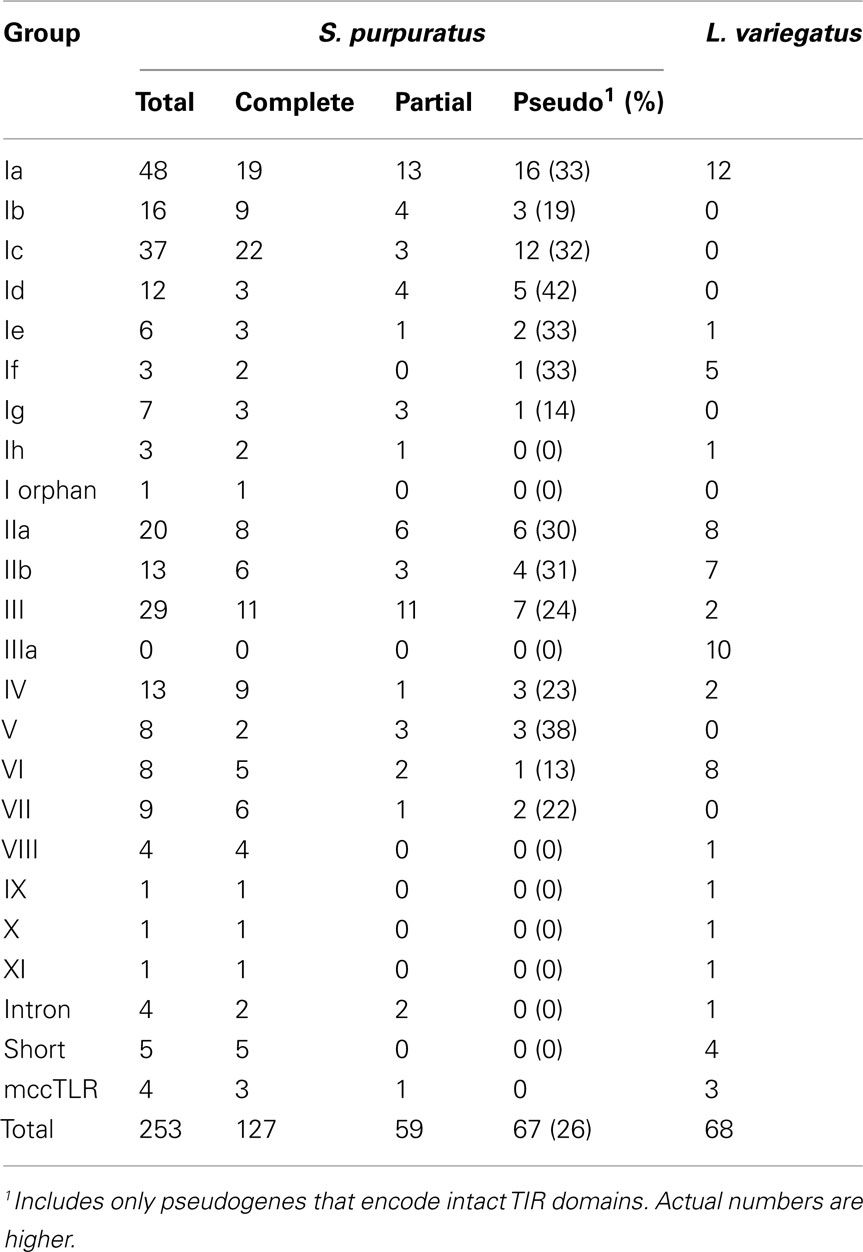
Table 3. Sizes of sea urchin TLR subfamilies.
To estimate gene copy number of the TLRs in L. variegatus in an assembly independent manner, we analyzed the unassembled genomic trace sequences. A conserved region of each of the TIR domains of the 253 S. purpuratus and the 68 L. variegatus TLR sequences was used as a query in a BLAST search against the sequences that were used to assemble the L. variegatus genome. In total, 1054 unique sequences were recovered with similarity to the sea urchin TLRs. Given 19.5× coverage (the 48,120,406 reads had an average length of 340 nt, and the L. variegatus genome is estimated to be 840 MB; Hinegardner, 1974), this indicates that there are 54 TLR TIR domains (Table S3 in Supplementary Material). This is slightly lower than the number of TLR sequences within the assembled genome, which may reflect the presence of allelic copies retained in the assembly. This data is consistent with a L. variegatus TLR gene family that is smaller than that of S. purpuratus, but still expanded relative to vertebrate TLR families.
Low coverage 454 reads are also available for the genomes of two sea urchin species closely related to S. purpuratus: A. fragilis, and S. franciscanus. Despite the nomenclature, A. fragilis is most closely related to S. purpuratus, with an estimated divergence time of 5–7 million years ago. These sister species shared a common ancestor with S. franciscanus about 20 million years ago (Biermann et al., 2003; Lee, 2003). These low coverage reads (∼2×) are insufficient to assemble a complete genome sequence, but allow us to estimate the multiplicity of the TLR gene family. The sequences from both species are an average of ∼235 nt in length, which is shorter than most TIR domains (the average size of the TLR TIR domains from S. purpuratus and L. variegatus is 354 nt). To simplify the analysis and to avoid sequences matching to partial TIR domains, we extracted a conserved region of the S. purpuratus and L. variegatus TLR TIR domains (50 amino acids) to use as queries in a BLAST search against the unassembled traces from A. fragilis and S. franciscanus. Positive reads were isolated and used as queries in a BLAST search against the whole TIR domains from S. purpuratus and L. variegatus to classify the reads by subfamily (Table S3 in Supplementary Material).
In total, 580 unique reads from A. fragilis and 524 reads from S. franciscanus were identified that exhibited similarity to the sea urchin TLR domains, which indicates that the TLR gene families in these species consist of 276 and 228 sequences, respectively (Table S3 in Supplementary Material). The distribution of TLRs among subfamilies is consistent with that in S. purpuratus. There is a reduced number of Ic TLRs in A. fragilis and S. franciscanus (4 and 16, respectively, compared to 37 in S. purpuratus), suggesting that these highly similar and genomically clustered genes may be the product of a very recent expansion in S. purpuratus. Similarly, there is an enrichment in the number of group III TLRs within the A. fragilis genome, which is estimated to have 61 TLR sequences, as compared to 29 in S. purpuratus and 26 in S. franciscanus. There are also homologs of each of the groups that contain a single representative in S. purpuratus and L. variegatus (groups IX, X, and XI), which may point to a conserved function for these receptors. Homologs of each of the divergent S. purpuratus subfamilies are present in both A. fragilis and S. franciscanus, including the mccTLRs. None of the sequences showed similarity to the group IIIa sequences, which appear to be unique to the L. variegatus lineage. As genome sequences become available for additional sea urchin species, as well as other echinoderms, our understanding of the evolution of this complex gene family will be further resolved.
Discussion
The sizes of the gene families that encode TLRs vary substantially among metazoan species (Table 1). Although sea urchin genomes encode the largest of these families, significant expansions have also occurred in the genomes of amphioxus, which has 72 TLR genes (Huang et al., 2008), and the annelid C. capitata, which encodes 105 (Davidson et al., 2008) as well as other invertebrate species that are now being sequenced. Each of these expansions generates a unique suite of TLRs that are not generally orthologous to TLRs in other species. This is not surprising, however, given the apparent rapid turnover of these genes, as suggested by the proportion of pseudogenes and high similarity of some family members. This pattern of species-restricted paralogy is consistent with that seen for other immune multigene families. In all cases where these genes are present as highly expanded multigene families, both in protostomes and deuterostomes, it is the vertebrate-like sccTLRs that are amplified (Table 1).
Although not present in vertebrates, the prototypic Toll-like mccTLR type can be identified in all eumetazoan phyla for which representative genome sequences are available, including the lower chordates. Usually this TLR type is present in single-copy or as very small gene families although moderate expansion is evident in a few species (Table 1). The presence of the mccTLR type as the only TLR gene in a basal eumetazoan, the cnidarian N. vectensis (Miller et al., 2007), as well as in all protostomes and invertebrate deuterostomes suggests that the mccTLR was a primitive component of eumetazoan genomes and that this receptor was lost in the vertebrate lineage. It is notable, however, that this type of receptor is always present in low numbers even in the species with expanded sccTLR gene families. In the sea urchin, mccTLRs are expressed at high levels in activated coelomocytes (Figure 5), which is consistent with an immune-related function.
The members of TLR multigene families in the sea urchin are characterized by apparently rapid sequence divergence within the ectodomain and conservation within the TIR region (Figure 3). This could be explained either by a lack of constraint in the diverging LRRs or by a more active process of diversifying selection. Our analysis suggests that positive selection plays a role in the diversification process and that it does so in spatially restricted regions of the TLR structure. Nearly all residues that are likely under positive selection are located in the LRRs, mainly in the concave region that is formed by the LRR β-strands. Almost no selection is indicated for residues within the TIR domain. This is consistent with observations in Drosophila immune genes, in which proteins involved in immunity, particularly those involved in pathogen recognition, were shown to have a higher proportion of residues under positive selection as compared to non-immune proteins (Sackton et al., 2007). The pathogen-interacting domains of phagocytosis receptors and two peptidoglycan recognition proteins were particularly enriched in codons likely to be under positive selection with respect to the remainder of the proteins (Sackton et al., 2007). This is also the case for many TLRs in analyses of positive selection carried out on the vertebrate sequences (Wlasiuk and Nachman, 2010; Alcaide and Edwards, 2011; Areal et al., 2011; Tschirren et al., 2011). When signatures of positive selection are detected in the vertebrate TLRs with known ligand-binding structure, it tends to be in regions that are known to interact with non-self and in regions that mediate dimerization. Thus the residues likely to be under positive selection in the sea urchin may also correspond to regions that interact with non-self. Notably, not all groups analyzed showed evidence of specific residues under positive selection and, in those that did, there was variation in the pattern of these residues. This may reflect different mechanisms of function within the subfamilies.
Multiplicity and patterns of incremental diversification among members of the major sea urchin sccTLR subfamilies in the ectodomain imply a direct form of ligand recognition, although some of the smaller, more conserved TLR gene families may be specialized to function differently. The sea urchin TLR genes may operate by recognizing non-self molecules that are similar to those recognized by vertebrate TLRs but with greater specialization. Alternatively, they may have evolved to recognize entirely different classes of molecules. The latter possibility is suggested by the spatial distribution and extent of diversity, which is unlike that seen among vertebrate TLRs. Given their multiplicity, the increased variation in LRR regions, the signature of positive selection in the portion of the genes encoding the ectodomain and the range of variation from near identity to high divergence, the sea urchin TLR genes appear to be evolving in response to a changing array of binding requirements.
One problem in analyzing the large families of sea urchin TLRs in the past has been the inability to find any level of orthology among subfamilies in inter-phyla comparisons (Roach et al., 2005; Hibino et al., 2006). The exception to this is the mccTLR-sccTLR division which shows a weak signal of orthology even between sea urchin and Drosophila genes (Hibino et al., 2006; Messier-Solek et al., 2010). The introduction of a second sea urchin genome into this analysis lends considerable insight into this issue. Phylogenetic analysis of the combined sea urchin TLR genes reveals cases of relative conservation in terms of gene number and cases of species-specific expansion or reduction. This can be used as an indicator of which genes may have unique and necessary functions and which genes may have interrelated, evolutionarily labile functions. At the extremes, some groups are encoded in single-copy in L. variegatus and greatly expanded in S. purpuratus (for example the group III genes) while others, such as the group X and mccTLR genes, appear to be more phylogenetically stable with single copies present in the genomes of both species. It is not clear whether the difference in the sizes of the gene families in these species is the result of an expansion in the TLR gene family within the strongylocentrotid lineage or gene loss L. variegatus. As more sea urchin genome sequences from outgroups to this clade are completed, this will become better resolved.
The question of whether or not the sea urchin TLRs are non-self receptors remains open but circumstantial evidence is consistent with an immune function for many of the subfamilies. This includes the following observations. (1) While expression of TLRs is generally low, for some of the largest subfamilies, transcription is greatly enhanced in phagocytic coelomocytes, many-fold over other tissues. (2) Expression of TLRs is not detectable in the embryo when primary developmental processes are unfolding but is initiated in the feeding larva coincident with the transcriptional activation of a suite of immune genes. (3) Multiplicity, variability, and sequence signatures of positive selection are common features of immune multigene families. (4) Finally, while the majority of Drosophila TLRs have not been associated with immunity but are associated with other biological processes (Narbonne-Reveau et al., 2011), all of the mammalian TLRs function as direct immune recognition receptors. The sea urchin is more closely related to vertebrates and the sea urchin TLRs resemble the vertebrate TLRs more closely than they do the Drosophila TLRs with known non-immune functions.
Of course the identification of the ligands for the sea urchin TLRs would answer this question definitively but this is a difficult technical challenge especially if, as may well be the case, the ligands are non-self and diverse. A more tractable path to understand the function of these receptors may be to focus on some of the smaller families which can be experimentally targeted but are nonetheless closely enough related to the expanded subfamilies to imply a similar function. Phylogenetic analysis of TLRs among sea urchins reveals some small TLR subfamilies that fit this pattern and comparative work in species like L. variegatus with relatively smaller TLR families will also be useful.
Whatever the exact biological roles of the large TLR gene families, it is probable that the sea urchin has co-opted this well known receptor to a new variation of function that is more evolutionarily labile than what has been well described in the vertebrates. Some of this reassignment may have taken place within the sea urchins as suggested by species-specific expansions. Nonetheless recent and emerging genome sequences from across the bilaterians indicate that large TLR repertoires may be widespread. It remains to be seen whether these expansions share a common functional purpose or whether they are each the result of a unique reaction to specific evolutionary pressures. While much of the justification for turning to as yet unstudied animal phyla is focused on aspects of host defense that are shared with mammals, in the long run comparative approaches will make a much richer contribution by revealing what is novel across animal immunity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Eric Ho for developing the infection strategy used in the larval RNA-Seq measurements and Yutaka Amemiya and Arun Seth of the Sunnybrook Research Institute Genomics Core Facility. We also thank the reviewers for many helpful comments. This work is supported by grants from the Canadian Institutes for Health Research (MOP74667) and the Natural Sciences and Engineering Research Council of Canada (NSERC 312221) to Jonathan P. Rast.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/Molecular_Innate_Immunity/10.3389/fimmu.2012.00136/abstract
Footnotes
References
Alcaide, M., and Edwards, S. V. (2011). Molecular evolution of the toll-like receptor multigene family in birds. Mol. Biol. Evol. 28, 1703–1715.
Areal, H., Abrantes, J., and Esteves, P. J. (2011). Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evol. Biol. 11, 368.
Bell, J. K., Mullen, G. E., Leifer, C. A., Mazzoni, A., Davies, D. R., and Segal, D. M. (2003). Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. 24, 528–533.
Biermann, C. H., Kessing, B. D., and Palumbi, S. R. (2003). Phylogeny and development of marine model species: strongylocentrotid sea urchins. Evol. Dev. 5, 360–371.
Bosch, T. C., Augustin, R., Anton-Erxleben, F., Fraune, S., Hemmrich, G., Zill, H., Rosenstiel, P., Jacobs, G., Schreiber, S., Leippe, M., Stanisak, M., Grötzinger, J., Jung, S., Podschun, R., Bartels, J., Harder, J., and Schröder, J. M. (2009). Uncovering the evolutionary history of innate immunity: the simple metazoan Hydra uses epithelial cells for host defence. Dev. Comp. Immunol. 33, 559–569.
Britten, R. J., Cetta, A., and Davidson, E. H. (1978). The single-copy DNA sequence polymorphism of the sea urchin Strongylocentrotus purpuratus. Cell 15, 1175–1186.
Cameron, R. A., Samanta, M., Yuan, A., He, D., and Davidson, E. (2009). SpBase: the sea urchin genome database and web site. Nucleic Acids Res. 37, D750–D754.
Davidson, C. R., Best, N. M., Francis, J. W., Cooper, E. L., and Wood, T. C. (2008). Toll-like receptor genes (TLRs) from Capitella capitata and Helobdella robusta (Annelida). Dev. Comp. Immunol. 32, 608–612.
Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. (1998). Biological Sequence Analysis, Probability Models of Proteins and Nucleic Acids. Cambridge: Cambridge University Press.
Felsenstein, J. (2005). PHYLIP (Phylogeny Inference Package) Version 3.6. Seattle: University of Washington.
Gross, P. S., Clow, L. A., and Smith, L. C. (2000). SpC3, the complement homologue from the purple sea urchin, Strongylocentrotus purpuratus, is expressed in two subpopulations of the phagocytic coelomocytes. Immunogenetics 51, 1034–1044.
Hall, T. A. (1999). BioEdit: a user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. (Oxf.) 41, 95–98.
Hibino, T., Loza-Coll, M., Messier, C., Majeske, A. J., Cohen, A. H., Terwilliger, D. P., Buckley, K. M., Brockton, V., Nair, S. V., Berney, K., Fugmann, S. D., Anderson, M. K., Pancer, Z., Cameron, R. A., Smith, L. C., and Rast, J. P. (2006). The immune gene repertoire encoded in the purple sea urchin genome. Dev. Biol. 300, 349–365.
Hinegardner, R. (1974). Cellular DNA content of the echinodermata. Comp. Biochem. Physiol. B 49, 219–226.
Holland, L. Z., Albalat, R., Azumi, K., Benito-Gutiérrez, E., Blow, M. J., Bronner-Fraser, M., Brunet, F., Butts, T., Candiani, S., Dishaw, L. J., Ferrier, D. E., Garcia-Fernàndez, J., Gibson-Brown, J. J., Gissi, C., Godzik, A., Hallböök, F., Hirose, D., Hosomichi, K., Ikuta, T., Inoko, H., Kasahara, M., Kasamatsu, J., Kawashima, T., Kimura, A., Kobayashi, M., Kozmik, Z., Kubokawa, K., Laudet, V., Litman, G. W., McHardy, A. C., Meulemans, D., Nonaka, M., Olinski, R. P., Pancer, Z., Pennacchio, L. A., Pestarino, M., Rast, J. P., Rigoutsos, I., Robinson-Rechavi, M., Roch, G., Saiga, H., Sasakura, Y., Satake, M., Satou, Y., Schubert, M., Sherwood, N., Shiina, T., Takatori, N., Tello, J., Vopalensky, P., Wada, S., Xu, A., Ye, Y., Yoshida, K., Yoshizaki, F., Yu, J. K., Zhang, Q., Zmasek, C. M., de Jong, P. J., Osoegawa, K., Putnam, N. H., Rokhsar, D. S., Satoh, N., and Holland, P. W. (2008). The amphioxus genome illuminates vertebrate origins and cephalochordate biology. Genome Res. 18, 1100–1111.
Huang, A. M., Rusch, J., and Levine, M. (1997). An anteroposterior dorsal gradient in the Drosophila embryo. Genes Dev. 11, 1963–1973.
Huang, S., Yuan, S., Guo, L., Yu, Y., Li, J., Wu, T., Liu, T., Yang, M., Wu, K., Liu, H., Ge, J., Yu, Y., Huang, H., Dong, M., Yu, C., Chen, S., and Xu, A. (2008). Genomic analysis of the immune gene repertoire of amphioxus reveals extraordinary innate complexity and diversity. Genome Res. 18, 1112–1126.
Jin, M. S., and Lee, J. O. (2008). Structures of the toll-like receptor family and its ligand complexes. Immunity 29, 182–191.
Kang, J. Y., and Lee, J. O. (2011). Structural biology of the Toll-like receptor family. Annu. Rev. Biochem. 80, 917–941.
Kasamatsu, J., Oshiumi, H., Matsumoto, M., Kasahara, M., and Seya, T. (2010). Phylogenetic and expression analysis of lamprey toll-like receptors. Dev. Comp. Immunol. 34, 855–865.
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948.
Lee, Y. H. (2003). Molecular phylogenies and divergence times of sea urchin species of Strongylocentrotidae, Echinoida. Mol. Biol. Evol. 20, 1211–1221.
Lemaitre, B., and Hoffmann, J. (2007). The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25, 697–743.
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M., and Hoffmann, J. A. (1996). The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983.
Leulier, F., and Lemaitre, B. (2008). Toll-like receptors – taking an evolutionary approach. Nat. Rev. Genet. 9, 165–178.
Medzhitov, R., Preston-Hurlburt, P., and Janeway, C. A. Jr. (1997). A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397.
Messier-Solek, C., Buckley, K. M., and Rast, J. P. (2010). Highly diversified innate receptor systems and new forms of animal immunity. Semin. Immunol. 22, 39–47.
Miller, D. J., Hemmrich, G., Ball, E. E., Hayward, D. C., Khalturin, K., Funayama, N., Agata, K., and Bosch, T. C. (2007). The innate immune repertoire in cnidaria – ancestral complexity and stochastic gene loss. Genome Biol. 8, R59.
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628.
Narbonne-Reveau, K., Charroux, B., and Royet, J. (2011). Lack of an Antibacterial response defect in Drosophila Toll-9 mutant. PLoS ONE 6, 17470.
Offord, V., Coffey, T. J., and Werling, D. (2010). LRRfinder: a web application for the identification of leucine-rich repeats and an integrative Toll-like receptor database. Dev. Comp. Immunol. 34, 1035–1041.
O’Neill, L. A., and Bowie, A. G. (2007). The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364.
Ooi, J. Y., Yagi, Y., Hu, X., and Ip, Y. T. (2002). The Drosophila Toll-9 activates a constitutive antimicrobial defense. EMBO Rep. 3, 82–87.
Pespeni, M. H., Garfield, D. A., Manier, M. K., and Palumbi, S. R. (2011). Genome-wide polymorphisms show unexpected targets of natural selection. Proc. Biol. Sci. 279, 1412–1420.
Poltorak, A., He, X., Smirnova, I., Liu, M. Y., Van Huffel, C., Du, X., Birdwell, D., Alejos, E., Silva, M., Galanos, C., Freudenberg, M., Ricciardi-Castagnoli, P., Layton, B., and Beutler, B. (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088.
Roach, J. C., Glusman, G., Rowen, L., Kaur, A., Purcell, M. K., Smith, K. D., Hood, L. E., and Aderem, A. (2005). The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. U.S.A. 102, 9577–9582.
Rock, F. L., Hardiman, G., Timans, J. C., Kastelein, R. A., and Bazan, J. F. (1998). A family of human receptors structurally related to Drosophila Toll. Proc. Natl. Acad. Sci. U.S.A. 95, 588–593.
Sackton, T. B., Lazzaro, B. P., Schlenke, T. A., Evans, J. D., Hultmark, D., and Clark, A. G. (2007). Dynamic evolution of the innate immune system in Drosophila. Nat. Genet. 39, 1461–1468.
Sasaki, N., Ogasawara, M., Sekiguchi, T., Kusumoto, S., and Satake, H. (2009). Toll-like receptors of the ascidian, Ciona intestinalis: prototypes with hybrid functionalities of vertebrate Toll-like receptors. J. Biol. Chem. 284, 27336–27343.
Smith, A. B., Pisani, D., Mackenzie-Dodds, J. A., Stockley, B., Webster, B. L., and Littlewood, D. T. (2006). Testing the molecular clock: molecular and paleontological estimates of divergence times in the Echinoidea (Echinodermata). Mol. Biol. Evol. 23, 1832–1851.
Smith, L. C., Ghosh, J., Buckley, K. M., Clow, L. A., Dheilly, N. M., Haug, T., Henson, J. H., Li, C., Lun, C. M., Majeske, A. J., Matranga, V., Nair, S. V., Rast, J. P., Raftos, D. A., Roth, M., Sacchi, S., Schrankel, C. S., and Stensvåg, K. (2011). Echinoderm immunity. Adv. Exp. Med. Biol. 708, 260–301.
Sodergren, E., Weinstock, G. M., Davidson, E. H., Cameron, R. A., Gibbs, R. A., Angerer, R. C., Angerer, L. M., Arnone, M. I., Burgess, D. R., Burke, R. D., Coffman, J. A., Dean, M., Elphick, M. R., Ettensohn, C. A., Foltz, K. R., Hamdoun, A., Hynes, R. O., Klein, W. H., Marzluff, W., McClay, D. R., Morris, R. L., Mushegian, A., Rast, J. P., Smith, L. C., Thorndyke, M. C., Vacquier, V. D., Wessel, G. M., Wray, G., Zhang, L., Elsik, C. G., Ermolaeva, O., Hlavina, W., Hofmann, G., Kitts, P., Landrum, M. J., Mackey, A. J., Maglott, D., Panopoulou, G., Poustka, A. J., Pruitt, K., Sapojnikov, V., Song, X., Souvorov, A., Solovyev, V., Wei, Z., Whittaker, C. A., Worley, K., Durbin, K. J., Shen, Y., Fedrigo, O., Garfield, D., Haygood, R., Primus, A., Satija, R., Severson, T., Gonzalez-Garay, M. L., Jackson, A. R., Milosavljevic, A., Tong, M., Killian, C. E., Livingston, B. T., Wilt, F. H., Adams, N., Bellé, R., Carbonneau, S., Cheung, R., Cormier, P., Cosson, B., Croce, J., Fernandez-Guerra, A., Genevière, A. M., Goel, M., Kelkar, H., Morales, J., Mulner-Lorillon, O., Robertson, A. J., Goldstone, J. V., Cole, B., Epel, D., Gold, B., Hahn, M. E., Howard-Ashby, M., Scally, M., Stegeman, J. J., Allgood, E. L., Cool, J., Judkins, K. M., McCafferty, S. S., Musante, A. M., Obar, R. A., Rawson, A. P., Rossetti, B. J., Gibbons, I. R., Hoffman, M. P., Leone, A., Istrail, S., Materna, S. C., Samanta, M. P., Stolc, V., Tongprasit, W., Tu, Q., Bergeron, K. F., Brandhorst, B. P., Whittle, J., Berney, K., Bottjer, D. J., Calestani, C., Peterson, K., Chow, E., Yuan, Q. A., Elhaik, E., Graur, D., Reese, J. T., Bosdet, I., Heesun, S., Marra, M. A., Schein, J., Anderson, M. K., Brockton, V., Buckley, K. M., Cohen, A. H., Fugmann, S. D., Hibino, T., Loza-Coll, M., Majeske, A. J., Messier, C., Nair, S. V., Pancer, Z., Terwilliger, D. P., Agca, C., Arboleda, E., Chen, N., Churcher, A. M., Hallböök, F., Humphrey, G. W., Idris, M. M., Kiyama, T., Liang, S., Mellott, D., Mu, X., Murray, G., Olinski, R. P., Raible, F., Rowe, M., Taylor, J. S., Tessmar-Raible, K., Wang, D., Wilson, K. H., Yaguchi, S., Gaasterland, T., Galindo, B. E., Gunaratne, H. J., Juliano, C., Kinukawa, M., Moy, G. W., Neill, A. T., Nomura, M., Raisch, M., Reade, A., Roux, M. M., Song, J. L., Su, Y. H., Townley, I. K., Voronina, E., Wong, J. L., Amore, G., Branno, M., Brown, E. R., Cavalieri, V., Duboc, V., Duloquin, L., Flytzanis, C., Gache, C., Lapraz, F., Lepage, T., Locascio, A., Martinez, P., Matassi, G., Matranga, V., Range, R., Rizzo, F., Röttinger, E., Beane, W., Bradham, C., Byrum, C., Glenn, T., Hussain, S., Manning, G., Miranda, E., Thomason, R., Walton, K., Wikramanayke, A., Wu, S. Y., Xu, R., Brown, C. T., Chen, L., Gray, R. F., Lee, P. Y., Nam, J., Oliveri, P., Smith, J., Muzny, D., Bell, S., Chacko, J., Cree, A., Curry, S., Davis, C., Dinh, H., Dugan-Rocha, S., Fowler, J., Gill, R., Hamilton, C., Hernandez, J., Hines, S., Hume, J., Jackson, L., Jolivet, A., Kovar, C., Lee, S., Lewis, L., Miner, G., Morgan, M., Nazareth, L. V., Okwuonu, G., Parker, D., Pu, L. L., Thorn, R., and Wright, R. (2006). The genome of the sea urchin Strongylocentrotus purpuratus. Science 314, 941–952.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
Tschirren, B., Raberg, L., and Westerdahl, H. (2011). Signatures of selection acting on the innate immunity gene Toll-like receptor 2 (TLR2) during the evolutionary history of rodents. J. Evol. Biol. 24, 1232–1240.
Wlasiuk, G., and Nachman, M. W. (2010). Adaptation and constraint at Toll-like receptors in primates. Mol. Biol. Evol. 27, 2172–2186.
Yang, Z. (1998). Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15, 568–573.
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591.
Appendix

Figure A1. Complete phylogeny of the sea urchin TLR sequences. The TIR domains of the TLR sequences from S. purpuratus and L. variegatus were used to construct the tree shown, which is a more detailed version of the tree in Figure 2. Bootstrap values greater than 50 are shown. Red indicates clades that are specific to S. purpuratus, blue clades contain only sequences from L. variegatus, and black clades contain sequences from both species. Each of the boxes (1–4) is shown in greater detail as indicated. The sequences are labeled by scaffold number and the position of the open reading frame (scaffold_start_stop). More information about the sequences can be found in Tables S1 and S2 in Supplementary Material. Subfamily designations are indicated on the right of each tree and correspond to those shown in Figure 2.
Keywords: toll-like receptors, sea urchins, multigene family, evolution, innate immunity
Citation: Buckley KM and Rast JP (2012) Dynamic evolution of toll-like receptor multigene families in echinoderms. Front. Immun. 3:136. doi: 10.3389/fimmu.2012.00136
Received: 21 February 2012; Accepted: 12 May 2012;
Published online: 05 June 2012.
Edited by:
Larry J. Dishaw, University of South Florida, USAReviewed by:
Mike Criscitiello, Texas A&M University, USAZeev Pancer, University of Maryland, USA
Coenraad Adema, University of New Mexico, USA
Copyright: © 2012 Buckley and Rast. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jonathan P. Rast, Department of Medical Biophysics and Department of Immunology, Sunnybrook Research Institute, University of Toronto, 2075 Bayview Avenue, Room S126B, Toronto, ON, Canada M4N 3M5. e-mail:anJhc3RAc3JpLnV0b3JvbnRvLmNh