 Julia Riewaldt1
Julia Riewaldt1 Sandra Düber2
Sandra Düber2 Marie Boernert1 Martina Krey2 Marcin Dembinski1 Siegfried Weiss2 Annette I. Garbe3
Marie Boernert1 Martina Krey2 Marcin Dembinski1 Siegfried Weiss2 Annette I. Garbe3 Karsten Kretschmer1*
Karsten Kretschmer1*- 1 Center for Regenerative Therapies Dresden, Technical University Dresden, Dresden, Germany
- 2 Helmholtz Centre for Infectious Research, Braunschweig, Germany
- 3 Institute of Physiological Chemistry, Technical University Dresden, Dresden, Germany
The role of Foxp3-expressing regulatory T (Treg) cells in tolerance and autoimmunity is well-established. However, although of considerable clinical interest, the role of Treg cells in the regulation of hematopoietic homeostasis remains poorly understood. Thus, we analysed B and T lymphopoiesis in the scurfy (Sf) mouse model of Treg cell deficiency. In these experiments, the near-complete block of B lymphopoiesis in the BM of adolescent Sf mice was attributed to autoimmune T cells. We could exclude a constitutive lympho-hematopoietic defect or a B cell-intrinsic function of Foxp3. Efficient B cell development in the BM early in ontogeny and pronounced extramedullary B lymphopoietic activity resulted in a peripheral pool of mature B cells in adolescent Sf mice. However, marginal zone B and B-1a cells were absent throughout ontogeny. Developmental B lymphopoietic defects largely correlated with defective thymopoiesis. Importantly, neonatal adoptive Treg cell therapy suppressed exacerbated production of inflammatory cytokines and restored thymopoiesis but was ineffective in recovering defective B lymphopoiesis, probably due to a failure to compensate production of stroma cell-derived IL-7 and CXCL12. Our observations on autoimmune-mediated incapacitation of the BM environment in Foxp3-deficient mice will have direct implications for the rational design of BM transplantation protocols for patients with severe genetic deficiencies in functional Foxp3+ Treg cells.
Introduction
In mice and humans, B cells are continuously generated throughout life from self-renewing, hematopoietic stem cells (HSCs). During development, the major anatomical location of hematopoietic activity transits from liver to bone marrow (BM) in late fetal life. Thus, the BM represents the primary site of B lymphopoiesis in the adult (Dorshkind and Montecino-Rodriguez, 2007). During B cell ontogeny, HSC-derived early B cell progenitors undergo a series of highly ordered maturation steps to ultimately give rise to immature B cells with surface IgM expression (sIgM; Hardy et al., 2000; Hardy and Hayakawa, 2001). After exit from the BM, such newly formed sIgM+ B cells continue their maturation in the spleen to complete development as immunocompetent mature B cells (Chung et al., 2003). With regard to surface markers and anatomical location, the overall population of mature B cells in peripheral lymphoid tissues consists of subpopulations with distinctly different effector functions (Allman and Pillai, 2008). Recirculating IgMlowIgDhighCD23high CD21int B-2 cells are located in follicles of spleen and lymph nodes, whereas non-recirculating IgMhighIgDlowCD23low/–CD21high B cells are enriched in the splenic marginal zone (MZ). In addition, IgMhighIgDlowMac-1+ B-1 cells, which can originate from both fetal and adult B cell developmental pathways (Montecino-Rodriguez et al., 2006; Tung et al., 2006; Duber et al., 2009; Esplin et al., 2009), represent the predominant B cell subset in body cavities, such as the peritoneal cavity.
It has been a longstanding observation that acute and chronic inflammatory immune responses modulate hematopoietic activity, including central and peripheral B cell development (Nagasawa, 2006; Cain et al., 2009). In recent years, considerable advances have been made in unraveling molecular and cellular mechanisms of inflammatory lympho-hematopoiesis (Nagasawa, 2006; Cain et al., 2009; Trumpp et al., 2010; King and Goodell, 2011). Notably, quiescent HSCs have been demonstrated to respond directly to inflammatory cytokines such as IFN-α (Essers et al., 2009) and IFN-γ (Baldridge et al., 2010) by proliferative activation. Additional examples of mechanisms that may impinge on B lymphopoiesis include the redirection of lymphoid progenitor differentiation into the myeloid lineage e.g. by Toll-like receptor stimulation (Nagai et al., 2006), the induction of apoptosis in early B cell progenitors by the direct action of IFN-γ (Grawunder et al., 1993; Garvy and Riley, 1994) and resident BM macrophage-derived type I interferons (Wang et al., 1995), as well as the mobilization of BM-derived B cell precursors by TNF-α-mediated modulation of the CXCL12/CXCR4 axis (Nagasawa et al., 1996; Nagasawa, 2006).
CD4+CD25+ regulatory T (Treg) cells expressing the forkhead family transcription factor Foxp3 play an indispensable role in controlling inflammatory responses and maintaining immune homeostasis under physiological conditions (Sakaguchi, 2005; Sakaguchi et al., 2008). However, a possible role of Treg cells in maintaining homeostasis of the lympho-hematopoietic system remains poorly understood. Scurfy (Sf) mice develop a fatal multi-organ autoimmune syndrome due to a spontaneous loss-of-function mutation in the gene encoding Foxp3 and a concomitant genetic deficit of functional Treg cells (Brunkow et al., 2001; Fontenot et al., 2003). Early studies in this mouse model indicated a decrease in the proportions of splenic B220+ cells that coincided with an increase of Mac-1+ cells (Clark et al., 1999). This appeared consistent with systemically elevated GM-CSF levels in Sf mice (Kanangat et al., 1996) and the notion that GM-CSF administration inhibits B lymphopoiesis in normal mice (Dorshkind, 1991). However, the impact of Foxp3+ Treg cell deficiency on lympho-hematopoietic homeostasis, and in particular B lymphopoiesis, has only recently begun to be explored further (Chen et al., 2010; Leonardo et al., 2010; Chang et al., 2012). Studies during ontogeny of Sf mice revealed progressive hypocellularity in BM, as compared to Foxp3-proficient mice (Chen et al., 2010). The concomitant acquisition of an enlarged HSC compartment with increased proportions of actively dividing cells in the BM of Sf mice has been associated with altered HSC differentiation (Chen et al., 2010). Indeed, sustained HSC proliferation is well known to compromise long-term HSC quality (Passegue et al., 2005; Wilson et al., 2008). However, the analysis of BM irradiation chimeras provided ambiguous results with regard to the repopulation capacity of Sf-derived HSCs (Chen et al., 2010; Chang et al., 2012). Repopulation of lethally irradiated recipient mice with total BM cells from adolescent Sf mice resulted either in substantially compromised reconstitution of hematopoietic cells of the T, B, and myeloid lineages (Chen et al., 2010), or in efficient, although somewhat delayed reconstitution of the T and B lymphoid compartments (Chang et al., 2012). While the reason underlying these apparent discrepancies among studies is at present still unclear, an overall defect in HSC function appears inconsistent with the notion that Sf mice exhibit greatly enhanced myelopoietic activity (Lee et al., 2009) and increased numbers of myeloid lineage cells (Lyon et al., 1990; Clark et al., 1999; Brunkow et al., 2001; Lin et al., 2005).
Interestingly, with regard to B cell development in situ, evidence was provided that adolescent Sf mice exhibit severe B lymphopoietic defects in the BM (Leonardo et al., 2010; Chang et al., 2012), coinciding with the virtually complete absence of newly formed B cells in the spleen (Leonardo et al., 2010). Although it appeared tempting to speculate that defective lympho-hematopoiesis in Foxp3-deficient mice is secondary to the development of T cell-mediated autoimmunity in the absence of functional Treg cells (Chen et al., 2010; Leonardo et al., 2010; Chang et al., 2012), this interpretation has not been corroborated by any direct evidence. Furthermore, these studies did not reveal to what extent B cell developmental defects observed in the BM of Foxp3-deficient mice are accompanied by defective T cell development in the thymus. This appears a particularly relevant question, considering that both B and T cell developmental pathways are thought to share a common lymphoid progenitor residing in the BM (Kondo et al., 1997). Furthermore, it is unknown how the remarkable severity of defective B cell development in the BM of adolescent Sf mice (Leonardo et al., 2010; Chang et al., 2012) can be reconciled with the establishment of a peripheral pool of mature B cells, which is characterized by recognition of a broad spectrum of self-antigens (Nguyen et al., 2007; Zhang et al., 2009; Huter et al., 2010; Leonardo et al., 2010) and increased antibody production with a bias toward IgE as well as IgG1/IgG2a (Lin et al., 2005). Here, we report on experiments in the Sf mouse model of Treg cell deficiency that were designed to address these issues.
Materials and Methods
Mice
CD45.2 C57BL/6 mice were purchased from Janvier or Taconic. Congenic CD45.1 C57BL/6 mice and Foxp3-deficient Foxp3ΔEGFP mice (Lin et al., 2007) were kindly provided by H. von Boehmer (Dana-Farber Cancer Institute, Boston, MA, USA) and T. Chatila (The David Geffen School of Medicine, Los Angeles, CA, USA), respectively. Foxp3-deficient Sf, T cell receptor (TCR)α−/− (Mombaerts et al., 1992) and IL-6−/− (Kopf et al., 1994) mice on the C57BL/6 background, as well as Foxp3 bacterial artificial chromosome (BAC) transgenic mice expressing a Cre recombinase-GFP fusion protein (Zhou et al., 2008; hereafter referred to as BAC-Foxp3Cre-GFP) were purchased from the Jackson Laboratory (Bar Harbor, USA). For some experiments, CD45.1 C57BL/6 mice were backcrossed to C57BL/6 Foxp3GFP mice (Fontenot et al., 2005), and Sf mice were backcrossed to TCRα−/− or IL-6−/− mice, as indicated. Additionally, BAC-Foxp3Cre-GFP mice were backcrossed to mice, which expressed a Cre recombination reporter allele of the ubiquitously expressed ROSA26 locus containing a loxP site–flanked STOP cassette followed by a DNA sequence encoding yellow fluorescent protein (YFP; R26-Y; Srinivas et al., 2001) to obtain BAC-Foxp3Cre-GFP × R26-Y mice. All mice were housed and bred at the Experimental Center of the Medizinisch-Theoretisches Zentrum (Dresden University of Technology, Germany) under specific pathogen-free conditions. Animal experiments were performed as approved by the Regierungspräsidium Dresden.
Flow Cytometry and Cell Sorting
All single cell suspensions were prepared in fluorescent activated cell sorting (FACS) buffer (1 × HBSS, 5% FCS, 10 mM HEPES; all Invitrogen). For this, thymus, spleen, liver, mesenteric lymph nodes (mLNs), and a pool of subcutaneous LNs (scLNs; Lnn. mandibularis, Lnn. cervicales superficiales, Lnn. axillares et cubiti, Lnn. inguinales superficiales, and Lnn. subiliaci) were meshed through 70 μm cell strainers (BD). BM cells were harvested from femurs and tibias by flushing either mechanically dissociated bones or cavities of intact bones with FACS buffer, followed by filtration through 70 μm cell strainers. Peritoneal exudate cells (PECs) were obtained by peritoneal cavity lavage with FACS buffer. Single cell suspensions from spleen, liver and BM were subjected to red blood cell lysis (erythrocyte lysis buffer EL, Qiagen, Hilden, Germany). Monoclonal antibodies (mAbs) to B220 (RA3-6B2), CD4 (RM4-5, GK1.5), CD5 (53–7.3), CD8 (53–6.7), CD11c (N418), CD19 (1D3), CD21/CD35 (8D9), CD23 (B3B4), CD25 (PC61), CD45.1 (A20), CD45.2 (104), CD138 (281–2), c-kit (2B8), GL7 (GL-7), Gr-1 (RB6-8C5), IgD (11–26), IgM (II/41), Mac-1 (M1/70), NK1.1 (PK136), Ter119 (TER-119), as well as APC-, FITC-, Pacific Blue-, PE-, and PerCP-conjugated streptavidin were purchased from eBioscience (Frankfurt, Germany) or BD (Heidelberg, Germany). Phosphatidylcholine liposomes were prepared as described (Kretschmer et al., 2003, 2004). Prior to FACS, for some experiments CD4+ cells were enriched from single cell suspensions using biotinylated antibodies directed against CD4, streptavidin-conjugated microbeads, and the AutoMACS magnetic separation system (Miltenyi Biotec). Samples were analyzed on a LSRII or FACSCalibur or sorted using a FACS Aria (all BD). Data were analyzed using FlowJo software (Tree Star Inc.).
Cell Culture
FACS-purified B220+c-kit+ Pro/Pre-B-I cells from BM were cultured in IMDM supplemented with 10% FCS (v/v), 1 mM sodium pyruvate, 1 mM HEPES, 2 mM Glutamax, 100 U/ml Penicillin-Streptomycin, 0.1 mg/ml Gentamicin, 0.1 mM non-essential amino acids, and 0.55 mM β mercaptoethanol (all Invitrogen). For proliferative expansion (Rolink et al., 1991), 0.5 – 2 × 105 cells were co-cultured with 2 × 104 ST2 stromal cells in 24-well plates (Greiner) in the presence of home-made recombinant murine IL-7 (Hock et al., 1991). For some experiments, titrating amounts (0.01–100 ng/ml) of recombinant murine IL-2, IL-4 (both eBioscience), IL-6, IFN-γ (both Peprotech), IL-3, IL-10, IL-17A, GM-CSF, or TNF-α (all Immunotools) were added, as indicated. For differentiation into sIgM+ cells, 3 × 104 B220+c-kit+ Pro/Pre-B-I cells that had been isolated by flow cytometry either from freshly prepared single cell suspensions of BM from donor mice or from IL-7-supplemented Pro/Pre-B-I cell cultures, were seeded in 96-well round-bottom plates (Greiner) in the absence of stromal cells and exogenous IL-7.
Adoptive Cell Transfer
IgM+ cells were FACS-purified from PECs of CD45.1 C57BL/6 mice and 6 × 105 cells were injected i.p. into 2-week-old CD45.2 Sf recipient mice, as indicated. For adoptive T cell transfers, 1 − 1.5 × 107 CD4 magnetic bead-enriched splenocytes from CD45.1 C57BL/6 Foxp3GFP donor mice were injected i.p. into ≤7-day-old CD45.2 Sf recipient mice.
Analysis of Serum Ig Levels
Serum IgE and IgG titers were measured by means of ELISA following standard methods. Plates were coated with rat anti-mouse IgE (R35-72; BD) or goat anti-mouse IgG (Sigma-Aldrich). Igs were detected with biotinylated rat anti-mouse IgE (R35-118; BD) and streptavidin-horseradish peroxidase (BD) or horseradish peroxidase-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories). For quantification, purified mouse IgE κ antibody (BD) and commercial serum (Bethyl Laboratories) were used as a reference. o-Phenylenediamine and H2O2 (both Sigma-Aldrich) were used as substrate.
Gene Expression Analysis
Single cell suspensions of thymus, BM, spleen, scLNs, or liver from individual donor mice were either kept separate or pooled from three donor mice, as indicated, and used for total RNA extraction, employing the RNeasy Mini kit and DNase I digestion (Qiagen), and cDNA was synthesized according to the manufacturer’s recommendations (SuperScript II reverse transcriptase; Invitrogen). The QuantiFast SYBR Green PCR kit (Qiagen) and a Mastercycler ep realplex thermal cycler (Eppendorf) were used to analyze cDNA in duplicates. The following primers were used: HPRT, 5′-GTCAACGGGGGACATAAAAG-3′ and 5′-AGGGCATATCCAACAAC AAAC3′; IL-2, 5′-CCTGAGCAGGAT GGAGAATTACA-3′ and 5′-TCCAGAACATGC CGCAGAG-3′; IL-4, 5′-TGACGGCACAGAGCTATTGATGGGT-3′ and 5′-TCTCCGT GCATGGCGTCCCTT-3′; IL-6, 5′-TCTCTGCAAGA GACTTCCATCCAGT-3′ and 5′-AGCCTCCGACTTGTGAAGT GGT-3′; IL-7, 5′-TCCCGCAGACCATGTTCCATGTTT C-3′ and 5′-TTCAACTTGCGAGCAGCACGA-3′; IL-17A, 5′-CTCTCCA CCGCAATG AAGACCCTGA-3′ and 5′-GCTGCCTGGCGG ACAATCGAG-3′; IFN-γ, 5′-GGCTG TTACTGCCACGGCACA-3′and 5′-CACCATCCTTTTGCCAGTTCCTCC-3′; TNF-α, 5′-G CCCACGTCGTAGCAAACCACCA-3′ and 5′-ACCTGCCCGG ACTCCGCAAA-3′; CXCL12, 5′-GCTCTGCATCAGTGACGGT AAACCA-3′ and 5′-TGCCCTTGCATCTC CCACGGA-3′. In addition, mRNA expression of type 1 interferons was assessed using the following set of primers: IFN-α4, 5′-CCTGTGTGA TGCAGGAACC-3′and 5′-TCACCTCCCAGGCACAGA-3′; IFN-non-α4, 5′-ARSYTGTSTGATGCARCAGGT-3′and 5′-GGWAC ACAGTGATCCTGTGG-3′; IFN-β, 5′-CTGGCTTCCATCATG AACAA-3′and 5′-CATTTCCGAATGTTCGTCCT-3′, as previously described (Lienenklaus et al., 2008).
Statistical Analysis
Statistical significance was assessed using 5.0c Prism software (GraphPad) and the unpaired Student’s t test or one-way ANOVA, as indicated.
Results
B and T Lymphopoiesis in Adolescent Mice with Foxp3 Deficiency
We initially focused on the assessment of B and T lymphopoietic activity in adolescent Sf mice. In this context, it is important to note that all Sf mice in our colony housed under specific pathogen-free conditions develop external signs of severe autoimmune disease (general failure to thrive, exfoliative dermatitis, etc.) by 4 weeks of age (data not shown), with a median survival of 27 days (see also Figure 5A). Flow cytometric analysis of moribund 4-week-old Sf mice revealed an up to 10-fold reduction in both percentages and numbers of early B220+c-kit+ Pro/Pre-B-I cells (nomenclature according to Rolink et al., 1999), as compared to age-matched wild-type (WT) control animals (Figure 1A, left). In WT mice, developmental progression of Pre-B-I cells gives rise to a pronounced compartment of B220+CD25+ Pre-B-II cells, which ultimately generate substantial populations of immature B220lowsIgM+ B cells (Figure 1A, top). In contrast, in Sf mice, regardless of the existence of a clearly discernable, albeit dramatically reduced population of Pro/Pre-B-I cells, subsequent Pre-B-II and immature sIgM+ B cell stages were consistently found to be below the level of detection (Figure 1A, middle and right). Similar results were obtained with adolescent Foxp3ΔEGFP mice (data not shown) that lack functional Treg cells due to a targeted insertion of GFP-encoding sequence into the Foxp3 locus, resulting in the expression of a non-functional Foxp3 protein fused to GFP (Lin et al., 2007). Overall, these data are largely consistent with previous reports (Leonardo et al., 2010; Chang et al., 2012) in that B cell development in the BM of adolescent Foxp3-deficient mice is severely abrogated.
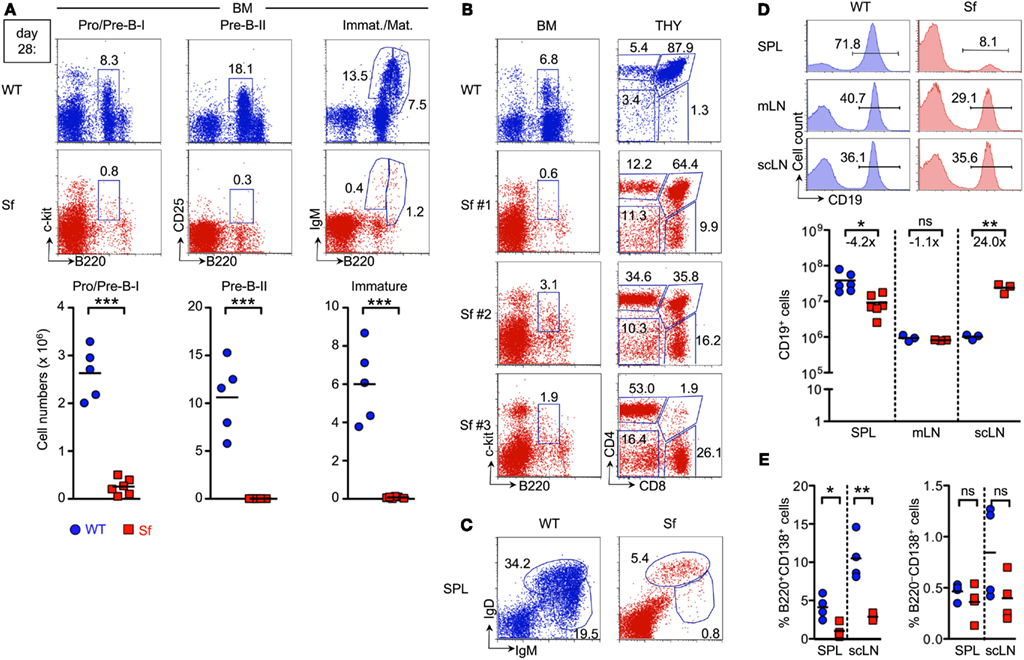
Figure 1. Severe B and T lymphopoietic defects in adolescent Sf mice. B cell compartments in the BM and peripheral lymphoid organs of 4-week-old Sf mice and WT littermate control animals were analyzed by flow cytometry. T cell development in the thymus was concomitantly assessed. (A) Flow cytometry of B220+c-kit+ Pro/Pre-B-I (left), B220+CD25+ Pre-B-II (middle), as well as immature B220lowsIgM+, and mature/recirculating B220highsIgM+ B cells (right) from BM of WT (top) and Sf (bottom) mice. Graphs depict absolute numbers of respective B cell developmental compartments in WT (blue circles) and Sf (red squares) mice, as indicated. (B) Correlation of severely reduced Pro/Pre-B-I cell compartments in the BM (left) of individual Sf mice (Sf #1–3) with T lymphopoietic activity (right) in the thymus (THY). (C,D) Analysis of peripheral B cell compartments. (C) Flow cytometry of newly formed IgDlowIgMhigh and mature IgDhighIgMlow B cells in the spleen. (D) Percentages (histograms) and numbers (graphs) of CD19+ B cells in spleen (SPL), mLNs, and scLNs. Numbers in dot plots and histograms indicate the percentage of gated cells within the respective gates. (E) Percentages of CD138+ plasma cells among B220+ (left) and B220− (right) B cells in peripheral lymphoid tissues of 4-week-old Sf and WT mice, as indicated. Dots and horizontal lines in graphs indicate individual mice and mean values, respectively, and numbers indicate the fold difference in cell numbers observed in WT (blue circles) and Sf (red squares) mice. Data are representative of at least two independent experiments using three to six mice per group. (Student’s t test: ***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; ns: not significant).
Next, we sought to correlate abrogated B lymphopoietic activity in the BM of Sf mice with T cell development in the thymus. Thymic aberrations previously reported for Foxp3-deficient mice include a decrease in thymic cellularity and the proportion of CD4+CD8+ double-positive (DP) cells (Chang et al., 2005; Liston et al., 2007). Proportions of CD4−CD8− double-negative (DN), CD4−CD8+ single-positive (CD8SP), and CD4+CD8− SP (CD4SP) cells were only moderately increased in approximately 2-week-old Sf mice (Chang et al., 2005). Here, we found that B lymphopoietic defects in 4-week-old Sf mice closely correlated with thymic atrophy. Impairment of T cell development varied over a broad range of severity (Figure 1B, Sf #1–3; see also Figure 3D for cell numbers). Thus, despite severely defective B lymphopoiesis, thymopoietic aberrations in individual Sf mice ranged from a somewhat perturbed distribution of DP and SP thymocyte compartments (Figure 1B, Sf #1) to substantially abrogated T lymphopoietic activity, as reflected by the nearly complete absence of DP cells and concomitantly increased proportions of DN and SP cells (Figure 1B, Sf #3).
After exit from the BM, maturation of B cells continues in the periphery (Chung et al., 2003). Consistent with severely reduced numbers of immature B220lowsIgM+ B cell in the BM (Figure 1A, right), the population of newly formed IgDlowIgMhigh B cells in the spleen was substantially reduced in Sf mice (0.6 ± 0.4 × 106 cells), as compared to age-matched WT control animals (4.4 ± 1.4 × 106 cells; Figure 1C). Consequently, splenic B cell numbers with a mature IgDhighIgMlow phenotype were drastically reduced (Sf: 3.0 ± 2.2 × 106 cells; WT: 16.6 ± 3.1 × 106 cells; and Figure 1C). This reduction in B cell numbers was restricted to the spleen (Figure 1D). In LNs, proportions of CD19+ B cells were largely comparable in Sf mice with pronounced lymphadenopathy and healthy WT mice (Figure 1D, histograms). Numbers of CD19+ cells were >20-fold increased in scLNs of Sf mice (Figure 1D, graph). Thus, despite severely abrogated central and peripheral B cell development, which at least partly correlated with defective thymopoiesis, 4-week-old Sf mice exhibit sizable populations of mature B cells in peripheral lymphoid organs. Notwithstanding previously reported severe hyperimmunoglobulinemia E and elevated IgG1 and IgG2a serum levels (Lin et al., 2005), proportions of B cells with an antibody-secreting B220+/−CD138+ plasma cell phenotype in spleen and LNs of adolescent Sf mice appeared somewhat reduced, as compared to WT control mice (Figure 1E).
Mechanisms of Defective B Lymphopoiesis
Previous studies employing mice with Foxp3-dependent fluorochrome reporter activity indicated that high levels of Foxp3 expression are limited to αβ T cells (Fontenot et al., 2005; Wan and Flavell, 2005), with undetectable fluorochrome expression in B220+ cells from BM (Leonardo et al., 2010) and secondary lymphoid organs such as LNs (Wan and Flavell, 2005) and spleen (Leonardo et al., 2010). Whether Foxp3 can be expressed in other hematopoietic (Bernard et al., 2009; Manrique et al., 2011) and non-hematopoietic (Chang et al., 2005; Hinz et al., 2007; Zuo et al., 2007; Chen et al., 2008; Karanikas et al., 2008; Chung et al., 2010) lineages has been actively debated (Liston et al., 2007; Mayer et al., 2012). To formally exclude the possibility that observed alterations in B cell development of Sf mice are due to a B cell-intrinsic function of Foxp3, we tracked B cells in peripheral lymphoid organs of BAC-Foxp3Cre-GFP × R26-Y mice that exhibit YFP expression in cells only after Foxp3-dependent Cre recombinase activity and resultant excision of a loxP-flanked STOP cassette (Zhou et al., 2008). Flow cytometric analysis revealed a minute population of CD19+ B cells (≤0.1%) in spleen and LNs of BAC-Foxp3Cre-GFP × R26-Y mice that appeared to co-express YFP (Figure 2A, left). However, post-sort analysis of FACS-purified cells established that this putative CD19+YFP+ population mainly consisted of doublets of CD19+YFP− and CD19−YFP+ cells, demonstrating that the potential population size of CD19+ cells that are truly YFP+ is negligible (spleen: ≤0.003%; LNs: ≤0.008%; Figure 2A, right).
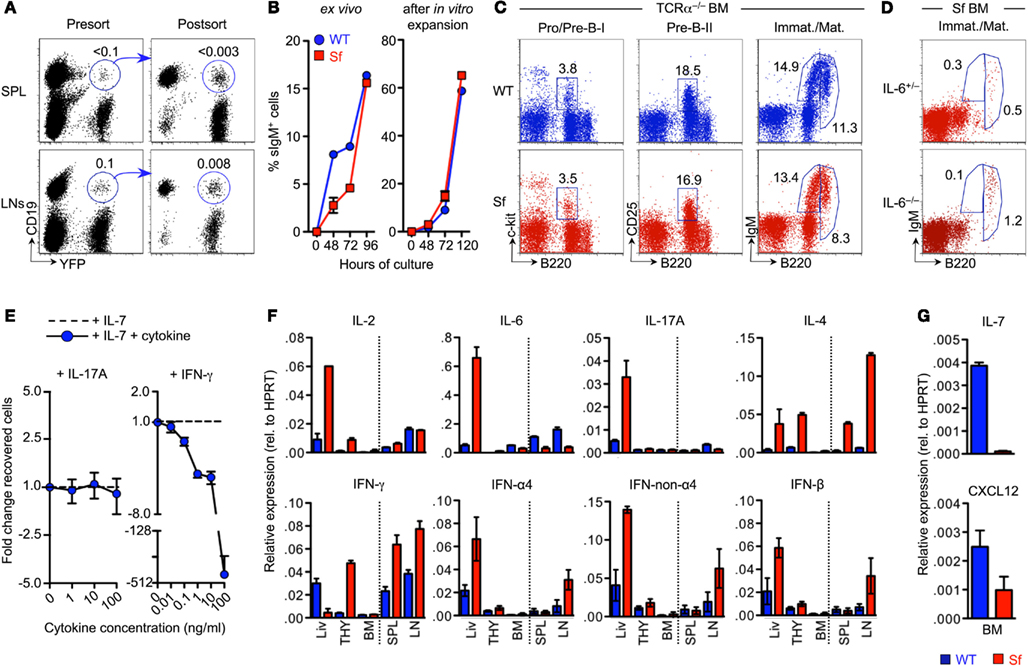
Figure 2. Mechanisms of B cell developmental defects in adolescent Sf mice. (A) Genetic lineage tracing of Foxp3 reporter activity in peripheral B cells. Putative populations of CD19+YFP+ cells were FACS-purified from spleen (top) and pooled mLNs and scLNs (bottom) of adult BAC-Foxp3Cre-GFP × R26-Y mice. Flow cytometric presort analysis (left) and post-sort analysis (right) is depicted. Note that for presort analysis numbers in dot plots indicate percentages of cells within the respective gates, whereas for post-sort analysis numbers indicate percentages of CD19+YFP+ cells among total cell populations prior to FACS purification. (B) Differentiation capacity of Pro/Pre-B-I cells from moribund Sf mice in vitro. Initially sIgM−, B220+c-kit+ Pro/Pre-B-I cells, either freshly isolated from BM of 3-week-old WT (blue circles) or Sf (red squares) mice (left), or isolated from IL-7-supplemented cultures after proliferative expansion of respective Pro/Pre-B-I cell populations (right), were cultured in the absence of added IL-7. The capacity to differentiate into sIgM+ cells was assessed at different time points after initiation of cultures, as indicated. Symbols represent mean values of replicate wells ± SEM. (C) B cell development in Sf mice that lack CD4+ and CD8+ T cells. Flow cytometry of indicated B cell developmental stages in the BM of 4-week-old WT (top) and Sf (bottom) mice that additionally lacked Tcra gene expression (TCRα−/−). Numbers in dot plots indicate the percentage of cells in the respective gate. Data are representative of three independent experiments. (D) Flow cytometric analysis of immature B220lowsIgM+ and mature/recirculating B220highsIgM+ B cell compartments in the BM of 4-week-old Sf mice that were either proficient (IL-6+/−, top) or deficient (IL-6−/−, bottom) for IL-6. Numbers in dot plots indicate the percentage of cells in the respective gate. Data are representative of two independent experiments. (E) Impact of inflammatory cytokines on IL-7-mediated survival of Pro/Pre-B-I cells in vitro. FACS-purified populations of BM-derived B220+c-kit+ Pro/Pre-B-I cells from WT mice were cultured with IL-7, either alone or in the presence of titrating amounts of IL-17A or IFN-γ, as indicated. At day 4 of cultures, numbers of B220+c-kit+ cells recovered from cultures with and without added inflammatory cytokines was calculated as fold difference. Shown are mean and range of duplicate wells. Data are representative of two independent experiments. (F,G) The expression of mRNAs encoding (F) indicated inflammatory cytokines and (G) IL-7 and CXCL12 was assessed by quantitative RT-PCR employing whole tissues harvested from 4-week-old WT (blue) or Sf (red) mice, as indicated. Note that, prior to RNA extraction, prepared single cell suspensions of respective tissues were pooled from three mice per group. HPRT was used for normalization. Shown are mean and range of duplicate samples. Data are representative of at least three independent experiments.
Consistent with the lack of a B cell-intrinsic function of Foxp3, our analysis of differentiation of B cell precursors in vitro in the absence of IL-7 (Rolink et al., 1991; Kretschmer et al., 2002) showed that FACS-purified populations of initially sIgM− Pro/Pre-B-I cells (B220+c-kit+) from moribund Sf mice efficiently differentiated into sIgM+ cells albeit with somewhat delayed kinetics, as compared to cells from WT controls (Figure 2B, left). This subtle delay in efficiency of in vitro differentiation of Pro/Pre-B-I cells from Sf mice was mitigated when sIgM− Pro/Pre-B-I cells were expanded in IL-7-supplemented cultures prior to IL-7 deprivation to initiate differentiation into sIgM+ cells (Figure 2B, right).
To directly assess the impact of T cell-mediated autoimmunity on defective B lymphopoietic homeostasis in the absence of functional Treg cells, we next analyzed B cell developmental compartments in Sf mice that were deficient in the major αβ T cell population due to lack of TCRα chain (Sf × TCRα−/−). As expected from earlier studies in athymic nude mice on the Sf background (Godfrey et al., 1991), Sf × TCRα−/− mice were protected from severe autoimmune pathology and premature death in the absence of CD4+ and CD8+ T cells (data not shown). Furthermore, B cell developmental compartments in the BM of Sf × TCRα−/− mice were found to be comparable with Foxp3-proficient TCRα−/− mice, as judged by proportions of Pro/Pre-B-I, Pre-B-II, and immature B cells (Figure 2C). Thus, αβ T cells play a dominant role in autoimmune-mediated abrogation of B cell development in the BM of Sf mice.
In comparison to WT mice, adolescent Sf mice exhibit largely elevated levels of various inflammatory cytokines like IL-6, IL-17, and IFN-γ that circulate in the blood (Lin et al., 2005; Zhang et al., 2009; Haribhai et al., 2011). The comparative analysis of adolescent Sf mice that were either proficient or deficient in IL-6 revealed a dispensable role of IL-6 and thus IL-6-dependent differentiation of IL-17-producing T effector cells (Bettelli et al., 2006) in mediating external signs of autoimmune-mediated morbidity (data not shown) and defective B lymphopoiesis (Figure 2D). This observation was further corroborated by in vivo administration of neutralizing mAbs to IL-6 (data not shown).
The addition of graded amounts of recombinant IL-6 (data not shown) or IL-17A (Figure 2E, left) to cultures of FACS-purified populations of B220+c-kit+ Pro/Pre-B-I cells from WT mice did also not appreciably impinge on IL-7-dependent proliferation and survival of B cell precursors. Similar results were obtained with IL-2, IL-3, IL-4, IL-10, GM-CSF, and TNF-α (data not shown). However, in agreement with previous observations (Grawunder et al., 1993; Garvy and Riley, 1994), IFN-γ abrogated the survival of Pro/Pre-B-I cells in vitro in a dose-dependent manner (Figure 2E, right). In vivo, long-term administration of neutralizing mAbs to IFN-γ was found to have no appreciable beneficial effect on both defective B lymphopoiesis and premature death of Sf mice (data not shown), suggesting a redundant function of IFN-γ in vivo, possibly due to the compensatory action of other inflammatory cytokines.
We next assessed the in situ production of inflammatory cytokines by quantitative RT-PCR using total RNA extracted from whole tissues of adolescent Sf and WT control animals. These experiments confirmed increased expression levels of various inflammatory cytokines in spleen, LNs, and liver of Sf mice (Zhang et al., 2009), with exacerbated production of IL-6 and IL-17 being mainly restricted to the liver (Figure 2F). The expression of type 1 interferons (IFN-α4, IFN-non-α4, and IFN-β) was found to be moderately increased in liver and LNs of Sf mice (Figure 2F, bottom). In primary lymphoid organs, substantially increased expression levels of IL-2, IL-4, and IFN-γ were found in thymus, whereas inflammatory cytokine expression appeared negligible in BM (Figure 2F).
We then extended our expression analysis to factors with well-established functions in promoting lympho-hematopoiesis and B cell development. It became clear that mRNA expression levels of IL-7 and CXCL12 were consistently decreased in the BM of adolescent Sf mice, as compared to WT control mice (Figure 2G).
Thus, exacerbated production of IFN-γ in peripheral lymphoid tissues, as well as decreased expression of IL-7 and CXCL12 in situ may represent non-mutually exclusive mechanisms that negatively impinge on developmental homeostasis of B cells in the BM of Sf mice.
Origin of Mature B Cells in the Periphery of Adolescent Sf Mice
Adolescent Sf mice exhibit sizable populations of mature B cells in their LNs (Figure 1D). How can the existence of these cells be reconciled with severely abrogated central and peripheral B cell development? Initial BrdU pulse-chase experiments indicated that, in comparison to WT control animals, adolescent Sf mice exhibit moderately increased frequencies (≤2-fold) of cycling BrdU+ cells among peripheral CD19+ B cells (data not shown). This observation raises the possibility that hyperproliferation (e.g., driven by lymphopenia) of B cells, which might originate from minimal B lymphopoietic activity in the BM, may at least partly contribute to the establishment and maintenance of the mature pool of peripheral B cells in adolescent Sf mice. In addition, we addressed the hypothesis that Sf mice might be permissive to efficient B cell production, prior to the manifestation of autoimmune-mediated B cell developmental defects in the BM. To this end, we performed a comparative temporal analysis of B cell development during ontogeny of Sf and WT mice (Figures 3A,B).
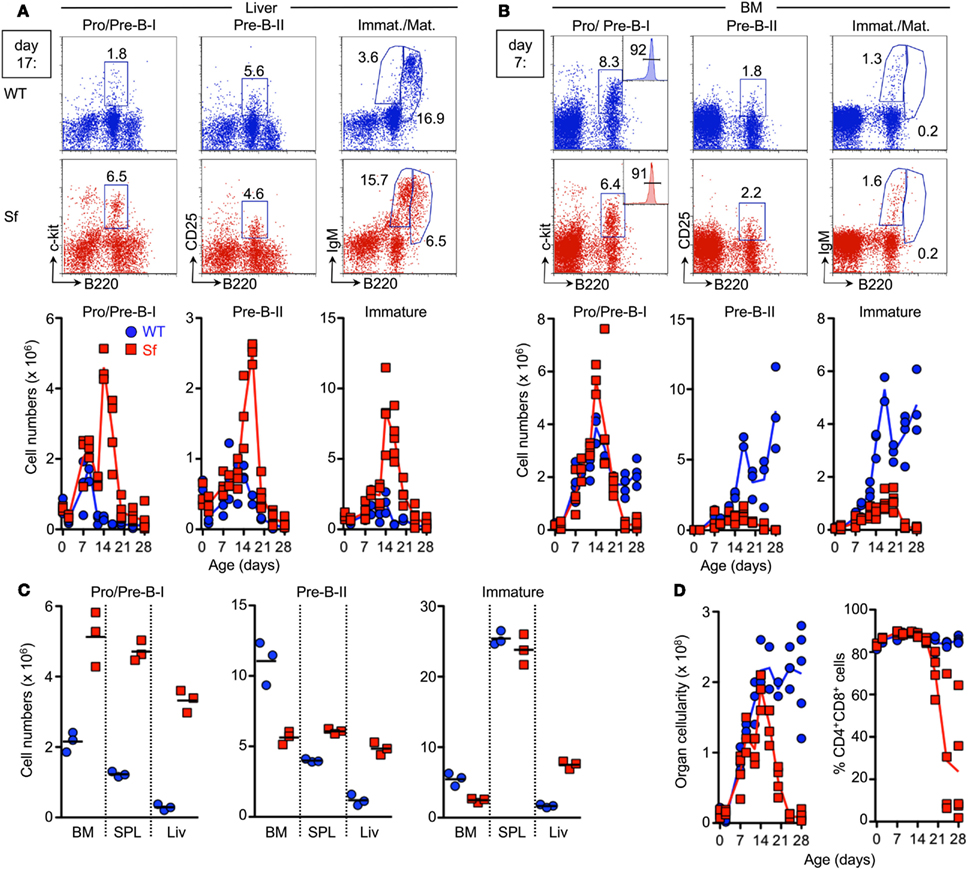
Figure 3. Temporal analysis of lymphopoiesis during ontogeny of Sf mice. B and T lymphopoiesis was analyzed by flow cytometry in newborn pups and at different time points after birth (day 2, 7, 9, 12, 14, 17, 20, 24, and 28). (A) B lymphopoiesis in the liver. Representative flow cytometry of B220+c-kit+ Pro/Pre-B-I (left), B220+CD25+ Pre-B-II (middle), as well as immature B220lowsIgM+ and mature/recirculating B220highsIgM+ (right) cells from liver of 17-day-old WT (top) and Sf (bottom) mice. Graphs depict absolute numbers of respective B cell developmental compartments in WT (blue circles) and Sf (red squares) mice, at indicated time points during ontogeny. (B) B lymphopoiesis in the BM. Representative flow cytometry of B cell development in the BM of 7-day-old WT and Sf mice. CD19 expression on gated B220+c-kit+ Pro/Pre-B-I cells from BM of 17-day-old Sf mice is shown as inserted histograms (left). Graphs depict absolute numbers of respective B cell developmental compartments. (C) Absolute numbers of indicated B cell developmental stages in BM, spleen (SPL), and liver (Liv) of 16-day-old WT (blue circles) and Sf (red squares) mice. Symbols and lines in graphs indicate individual mice and mean values, respectively. (D) Thymic cellularity (left) and proportions of DP thymocytes (right). Numbers in dot plots and histograms indicate the percentages of gated cells within the respective gates. Symbols and lines in graphs indicate individual mice and mean values, respectively. Data were collected from independent experiments using a minimum of three individual mice per age group and strain, as indicated.
Due to postnatal B lymphopoietic activity, residual numbers of early B220+c-kit+ Pro/Pre-B-I cells remained detectable in the liver of WT mice until day 7 postpartum (p.p.), but rapidly declined thereafter (Figure 3A, left). This resulted in an overall decrease of the subsequent Pre-B-II and immature sIgM+ B cell stages below the level of detection within the third week after birth (Figure 3A, middle and right). Cessation of B lymphopoietic activity in the postnatal liver (Figure 3A) coincided with gradually increasing proportions and numbers of Pro/Pre-B-I cells in the BM until the second week p.p. and then apparently reached steady state (Figure 3B, left). The subsequent B220+CD25+ Pre-B-II and immature sIgM+ B cell compartments in BM still continued to increase in size during the observation period (Figure 3B, middle and right).
In striking contrast, Sf mice exhibited aberrantly prolonged and enhanced extramedullary B lymphopoietic activity in the liver (Figure 3A) and spleen (Figure 3C), as indicated by Pro/Pre-B-I, Pre-B-II and immature B cell compartments that continued to markedly increase in size after day 7 p.p. (Figure 3A and data not shown).
Notably, until day 7 p.p., the population size of B cell developmental compartments in the BM was essentially comparable between Sf and WT littermate control animals (Figure 3B). However, severe B cell developmental aberrations became obvious within the second week after birth. This included a substantially increased compartment of CD19-expressing B220+c-kit+ Pro/Pre-B-I cells (Figure 3B, left). Importantly, after day 7 p.p., the population size of all subsequent B cell developmental stages in the BM never reached WT levels (Figure 3B, middle and right), most likely due to impairment in developmental progression and/or imparted proliferative expansion and survival of Pre-B-I/-II cells. In any case, medullary and extramedullary B lymphopoietic activity in Sf mice appeared sufficient to produce significant numbers of immature sIgM+ B cells that peaked within the third week p.p. but dropped below the level of detection thereafter (Figures 3A,B, right).
In contrast to our observation of early B lymphopoietic aberrations in liver, spleen, and BM (Figures 3A,B) and previous reports on thymopoietic aberrations in Sf mice as early as day 7 p.p. (Chang et al., 2005), our concomitant temporal analysis of T cell development during ontogeny of Sf mice revealed normal cellularity of the thymus and thymocyte populations until day 14 p.p. (Figure 3D, and data not shown). Subsequent manifestation of thymic atrophy and abrogated T cell development exemplified by substantially reduced proportions of DP thymocytes (Figure 3D, right; see also Figure 1B), appeared to correlate with progressive autoimmune pathology of Sf mice.
Peripheral B Cell Maturation during Ontogeny of Sf Mice
In the spleen of Sf mice, we found numbers of newly formed IgDlowIgMhigh B cells to be reduced as early as day 7 p.p., as compared to WT littermate control mice (Figure 4A, left). While the compartment size of mature IgDhighIgMlow B cells failed to reach WT levels during ontogenetic progression, significant numbers of CD19+ B cells were maintained until Sf mice succumbed to autoimmune pathology (Figure 4A, middle and right). Thus, reduced splenic B cell maturation in Sf mice is likely to be a consequence of ceasing central B lymphopoiesis and abrogated export of immature sIgM+ B cells to the spleen.
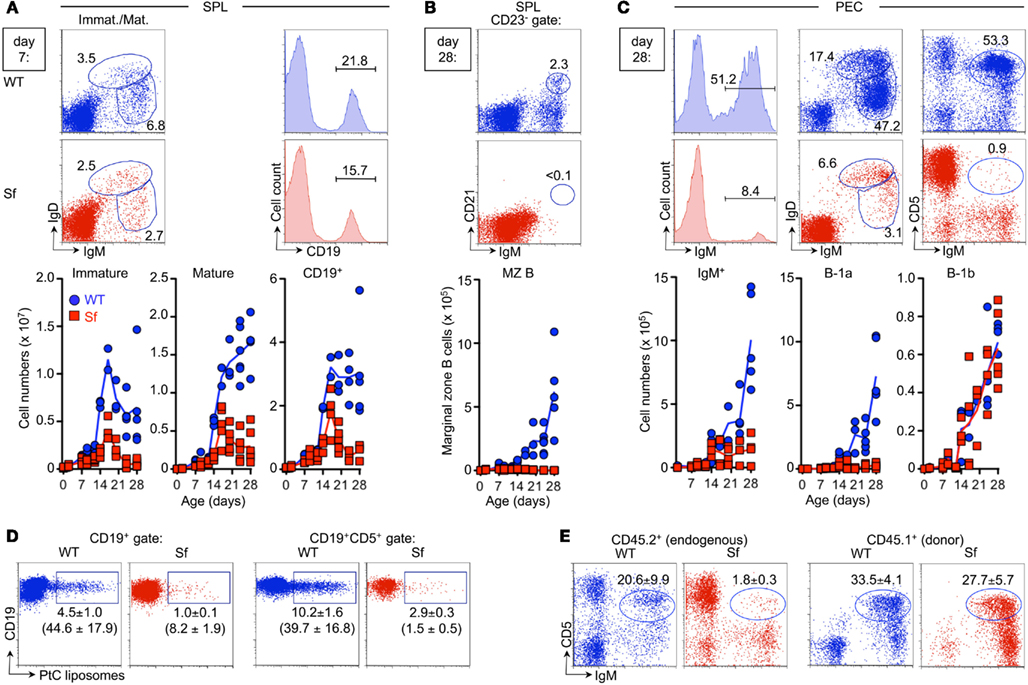
Figure 4. Temporal analysis of peripheral B cell compartments during ontogeny of Sf mice. Cohorts of experimental Sf and WT mice described in Figure 3 were concomitantly employed for the analysis of peripheral B cell compartments. (A) Peripheral B cell maturation in the spleen. Representative flow cytometry of newly formed IgDlowIgMhigh and mature IgDhighIgMlow B cells (dot plots), as well as CD19+ B cells (histograms) in the spleen of 7-day-old WT (top) and Sf (bottom) mice. Graphs depict absolute numbers of respective B cell compartments from WT (blue circles) and Sf (red squares) mice, at indicated time points during ontogeny. (B) IgMhighIgDlow CD23low/−CD21high MZ B cells. Representative flow cytometry of IgMhighCD21high MZ B cells among gated IgDlowCD23low/− splenocytes of 4-week-old mice (top), and absolute numbers of MZ B cells during ontogeny of WT and Sf mice (bottom). (C) B cell subsets in the peritoneal cavity. Representative flow cytometry of IgM, IgD, and CD5 expression among PECs from 4-week-old mice, as well as absolute numbers of peritoneal B cell subsets (B-2: IgDhighIgMlow; B-1: IgDlowIgMhigh; B-1a: IgMhighIgDlowCD5+Mac-1+; B-1b: IgMhighIgDlowCD5−Mac-1+). (D) Flow cytometry of PtC-reactive B cells among gated populations of total CD19+ (left panels) and CD19+CD5+ (right panels) PECs from 3–4-week-old WT (n = 7) and Sf (n = 5) mice, as indicated. (E) Total populations of sIgM+ cells were FACS-purified from PECs of CD45.1+ WT mice and adoptively transferred i.p. into 2-week-old CD45.2+ WT (n = 3) and Sf (n = 3) recipient mice, as indicated. 14 days after cell transfer, recovered PECs were analyzed for the expression of sIgM and CD5 among gated endogenous CD45.2+ cells (left) or CD45.1+ donor cells (right). Data are representative of two independent experiments. Numbers in dot plots and histograms without parentheses indicate the percentage (A–C) and mean percentages ± SEM (D,E) of gated cells within the respective gates. Parenthesized numbers indicate mean absolute numbers (× 103 ± SEM) of PtC+ cells (D). Symbols and lines in graphs indicate individual mice and mean values, respectively.
During normal mouse ontogeny, maturation of the splenic MZ B cell compartment (Figure 4B) is delayed relative to the appearance of conventional B cells (Figure 4A) and coincides with the temporally regulated maturation of the microarchitecture of the MZ until about 3–4 weeks after birth (Pillai et al., 2005). Whereas in WT mice, splenic IgMhighIgDlowCD23low/−CD21high MZ B cells became first apparent at day 14 p.p., Sf mice exhibited a complete lack of this B cell subset throughout life (Figure 4B), probably due to autoimmune-mediated alterations in splenic architecture.
In the peritoneal cavity of Sf mice, the reduction of IgM+ B cell numbers after day 14 p.p. (Figure 4C, left) could be attributed to a selective and nearly complete lack of CD5+ B-1a cells (IgMhighIgDlowMac-1+) throughout ontogeny (Figure 4C). Numbers of CD5− B cells, which comprise the B-1b sister population (IgMhighIgDlowMac-1+) as well as conventional B-2 cells (IgMlowIgDhighMac-1−), were comparable between Sf and WT littermate control mice (Figure 4C, and data not shown). This marked and selective reduction of peritoneal CD5+IgMhigh B-1a cells throughout life of Sf mice was unexpected, considering that B-1a cells can originate from both fetal and adult B cell developmental pathways (Montecino-Rodriguez et al., 2006; Tung et al., 2006; Duber et al., 2009; Esplin et al., 2009). To exclude the possibility that our failure to detect significant populations of CD5+IgMhigh B-1a cells in the peritoneal cavity of Sf mice was due to the mere down-regulation of CD5 expression in an autoimmune environment, we additionally tracked peritoneal B cells whose B cell receptor is reactive to phosphatidylcholine (PtC). PtC represents a well-documented specificity to common bacterial and autoantigens that, in normal mice, is restricted to B-1a cells (Baumgarth et al., 2000; Kretschmer et al., 2003, 2004). Our results showed that proportions and numbers of PtC-specific cells among total CD19+ B cells as well as residual CD5+IgMhigh B-1a cells were markedly decreased in 3–4-week-old Sf mice, as compared to WT littermate controls (Figure 4D).
In addition, we asked whether the autoimmune environment in Sf mice might hamper peritoneal retention and/or survival of B-1a cells. To this end, congenically marked IgM+ B cells that had been FACS-purified from PECs of CD45.1+ WT donor mice were adoptively transferred i.p. into 2-week-old CD45.2+ WT or Sf recipient mice, exhibiting early external signs of autoimmune pathology (data not shown). Flow cytometric analysis of Sf recipient mice, which had to be sacrificed 2 weeks after injection due to severe autoimmune symptoms, confirmed that the population of endogenous CD45.1−CD45.2+ B-1a cells was greatly reduced, as compared to WT recipient mice (Figure 4E, left). In contrast, adoptively transferred CD45.1+CD45.2− B-1a cells were maintained in the peritoneal cavity of Sf mice until the end of the observation period (Figure 4E, right). These findings suggest that the severe and selective reduction of the peritoneal B-1a compartment in Sf mice is due to aberrations in development, migration and recruitment of CD5+IgMhigh B-1a cells, rather than due to abrogated retention and survival signals.
Impact of Adoptive Treg Cell Therapy on Defective B and T Lymphopoiesis
Neonatal adoptive transfer of purified populations of CD4+CD25+Foxp3+ Treg cells ameliorates exacerbated immune cell activation (Fontenot et al., 2003) and prolongs survival of Foxp3-deficient recipient mice (Smyk-Pearson et al., 2003). Interestingly, adoptively transferred bulk populations of CD4+ T cells that include functional Foxp3+ Treg cells have been demonstrated to be far more effective in the suppression of chronic autoimmune inflammation (Smyk-Pearson et al., 2003; Chang et al., 2008; Haribhai et al., 2011). In these studies, the impact of adoptive Treg cell therapy on the establishment of severe lympho-hematopoietic defects in Foxp3-deficient mice had not been addressed. To assess the impact of functional Foxp3+ Treg cells on the development of defective B and T lymphopoiesis during ontogeny of Sf mice, we injected magnetic bead-enriched bulk populations of CD4+CD45.1+ T cells from peripheral lymphoid organs of Foxp3GFP mice into CD45.2+ Sf mice within the first week after birth.
As expected (Smyk-Pearson et al., 2003; Chang et al., 2008; Haribhai et al., 2011), treated Sf mice consistently exhibited long-term survival (Figure 5A) and appeared phenotypically healthy throughout the observation period. Exceptionally, individual Sf mice displayed mild exfoliative dermatitis restricted to the tail (data not shown). Flow cytometric analysis at approximately 8 weeks after adoptive transfer revealed the presence of congenically marked donor CD4+ T cells in all primary (BM and thymus) and secondary (spleen, LNs and peritoneal cavity) lymphoid organs analyzed (Figures 5B,C). Highest numbers of adoptively transferred CD45.1+CD4+ T cells could be recovered from spleen and LNs (Figure 5B). Interestingly, Foxp3GFP+ Treg cells, which initially comprised approximately 15% of the adoptively transferred CD45.1+CD4+ T cell population (data not shown), preferentially accumulated in lymphoid organs of Sf recipient mice, as compared to Foxp3GFP−CD4+ T cells (Figure 5C). This selective enrichment was most pronounced in the BM, with up to 90% of CD45.1+CD4+ T cells expressing Foxp3GFP (Figure 5C).
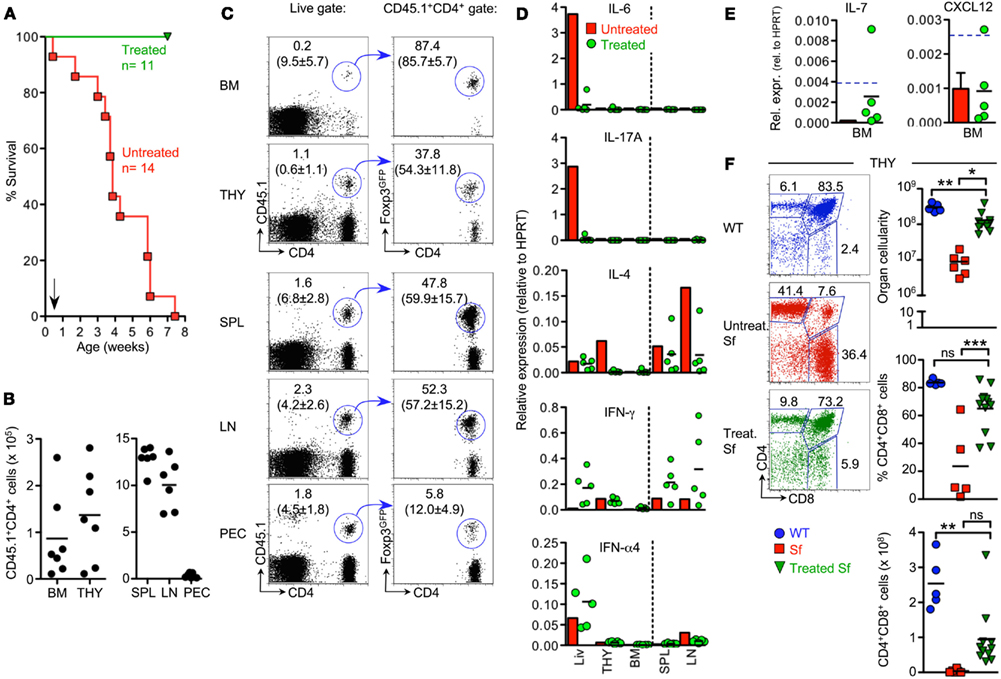
Figure 5. Adoptive Tregcell transfer in Sf mice. (A) Kaplan-Meier survival curves of CD45.2 Sf mice that had either been left untreated (red squares; n = 14) or injected with total populations of CD4+ T cells from Foxp3-proficient CD45.1+ Foxp3GFP mice (green triangles; n = 11), as indicated by the arrow. 6–8 weeks after cell transfer, recipient Sf mice were subjected to analysis by flow cytometry and RT-PCR. Data were collected from 2 (B,C) or 3 (F) independent experiments. (B,C) Congenically marked T cells were tracked in primary and secondary lymphoid organs of recipient mice. (B) Absolute numbers and (C) flow cytometry of CD4+CD45.1+ cells (left) for Foxp3GFP expression among gated CD4+CD45.1+ cells (right) in indicated organs. Lines with arrowheads illustrate the gating scheme. Dots and horizontal lines in graphs indicate individual mice and mean values, respectively. Numbers in dot plots without parentheses indicate the percentage of gated cells within the respective gates. Parenthesized numbers indicate mean percentages ± SD of CD45.1+ cells among gated CD4+ cells (left) or mean percentages ± SD of Foxp3+ cells among gated CD4+CD45.1+ cells (right). (D,E) Impact of adoptive Treg cell transfer on in situ production of cytokines. The expression of mRNAs encoding (D) indicated inflammatory cytokines and (E) IL-7 and CXCL12 was assessed by quantitative RT-PCR employing total RNA extracted from indicated whole tissues of Sf mice that had previously been injected with Treg cells. Each dot represents an individual Sf recipient mouse, and horizontal bars indicate mean values. Bars depict mean values from pooled single cell suspensions of three individual untreated 4-week-old Sf mice. Blue hatched lines indicate mRNA expression levels in WT animals for comparison (see also Figures 2F,G). HPRT was used for normalization. Data are representative of at least three independent experiments. (F) Impact of adoptive Treg cell transfer on T cell development in the thymus of Sf mice. Representative flow cytometry of CD4 and CD8 expression on thymocytes from WT mice (6-week-old; top) and Sf mice that were either left untreated (4-week-old; middle) or treated with adoptive Treg cell transfer (6–8-week-old; bottom). Numbers in dot plots indicate the percentages of gated cells within the respective gates. Graphs depict total thymic cellularity (top), as well as percentages (middle) and numbers (bottom) of DP thymocytes of WT mice (blue circles), as well as Sf mice that were either left untreated (red squares) or treated with adoptive Treg cell transfer (green triangles). Symbols and horizontal lines in graphs indicate individual mice and mean values, respectively. (One-way ANOVA: ***p ≤ 0.01; **p ≤ 0.01; *p ≤ 0.05; ns, not significant).
RT-PCR-based mRNA quantification of inflammatory cytokine expression revealed that adoptively transferred Treg cells effectively suppressed production of IL-4, IL-6, and IL-17 but, unexpectedly, failed to prevent exacerbated production of IFN-γ in primary and secondary lymphoid organs of Sf recipient mice (Figure 5D; see also Figure 2F for comparison). Furthermore, it appeared that individual Sf recipient mice exhibited somewhat increased expression levels of type 1 interferons selectively in the liver (IFN-α4, Figure 5D; IFN-non-α4 and IFN-β, data not shown). In addition, adoptively transferred Treg cells failed to effectively restore expression of IL-7 and CXCL12 in the BM of Sf mice (Figure 5E; see also Figure 2G for comparison).
At approximately 8 weeks of age, total thymic cellularity and the proportional distribution of T cell developmental stages in the thymus of the vast majority of treated Sf mice were largely comparable to age-matched WT control animals (Figure 5F). Consistent with partially rescued IL-7 production in the BM of individual Sf recipient mice (Figure 5E), concomitant analysis of B cell development in the BM revealed that the size of the B220+c-kit+ Pro/Pre-B-I cell compartment was greatly increased in Sf mice that had been treated with WT CD4+ T cells, as compared to untreated, 3- to 4-week-old Sf mice (Figure 6A, left; see also Figures 1A and 3B). However, developmental progression to the Pre-B-II cell stage remained substantially impaired in treated Sf mice (Figure 6A, middle). Minimal production of BM-derived immature sIgM+ B cells (Figure 6A, right) correlated with a considerably reduced splenic compartment of newly formed IgDlowIgMhigh B cells, as compared to age-matched WT animals (Figure 6B). Despite inefficient central and peripheral B lymphopoiesis in treated Sf mice, neonatal Treg cell transfer resulted in the accumulation of robust populations of mature IgDhighIgMlow B cells in peripheral lymphoid tissues (Figure 6B), with substantially reduced proportions of B cells expressing the activation/germinal center B cell marker GL7 (Figure 6C, left panel). Consistently, neonatal Treg cell transfer efficiently prevented the manifestation of hyperimmunoglobulinemia E in Sf mice (Figure 6C, middle). In these experiments, total IgG levels did not vary all that much between WT and moribund Sf mice (both 4-week-old) and neonatally treated Sf mice at the age of >4 months (Figure 6C). Moderately increased IgG levels in treated Sf mice most likely reflect the inevitable accumulation of serum IgGs with aging. Overall, these data suggest that adoptively transferred Treg cells promote the accumulation of mature peripheral B cells by suppression of chronic B cell activation and subsequent differentiation into plasma cells, thereby counteracting exhaustion of the mature peripheral B cell pool.

Figure 6. Impact of adoptive Tregcell transfer on B cell development in Sf mice. Central and peripheral B cell compartments were analyzed in cohorts of experimental mice described in Figure 5. (A) B cell development in the BM. Representative flow cytometry of B220+c-kit+ Pro/Pre-B-I (left), B220+CD25+ Pre-B-II (middle), as well as immature B220lowsIgM+ and mature/recirculating B220highsIgM+ (right) cells from BM of WT (top) and Sf mice that were either left untreated (middle) or treated with adoptive Foxp3+ Treg cell transfer (bottom). Graphs depict percentages of respective B cell developmental compartments from WT (blue circles), as well as untreated (red squares) and treated Sf (green triangles) mice. (B) B cell compartments in the spleen. Representative dot plots of newly formed IgDlowIgMhigh and mature IgDhighIgMlow B cells in mice as in (A), and graphs depicting the ratio of mature and newly formed B cells (top) and absolute numbers of IgMhighIgDlow CD23low/−CD21high MZ B cells (bottom). (C) Percentages of LN-derived B220+ cells expressing the activation/germinal center B cell marker GL7, as revealed by flow cytometry (left), and quantification of IgE and total IgG levels (μg/ml) in serum (middle and right) from WT (blue circles) and untreated Sf (red squares) mice (both 4-week-old), as well as treated Sf (green triangles) mice (>4–7-month-old). (D) Representative flow cytometry of IgM and CD5 expression among PECs from mice as in (A), and graphs depicting absolute numbers of total IgM+ cells (top) and IgMhighCD5+ B-1a cells (bottom). (E) Representative flow cytometry of IgMhighCD21high MZ B cells among gated IgDlowCD23low/− splenocytes (left) and peritoneal IgMhighCD5+ B-1a cells (right) in four-week-old Sf × TCRα+/− and Sf × TCRα−/− mice, as indicated. Numbers in dot plots indicate the percentage of gated cells within the respective gates. Dots and horizontal lines in graphs indicate individual mice and mean values, respectively. For experimental details see also legend to Figure 5. (One-way ANOVA: ***p ≤ 0.001; *p ≤ 0.05; ns, not significant).
However, adoptive Treg cell therapy of Sf mice was ineffective in correcting both the absence of splenic MZ B cells (Figure 6B, bottom right) and the severely reduced peritoneal B-1a compartment (Figure 6D). However, splenic MZ B cells and peritoneal B-1a cells were readily detectable in Sf × TCRα−/− mice (Figure 6E) and in Foxp3-proficient BM irradiation chimeras after reconstitution with HSCs from Sf mice (data not shown). Thus, it appears that neonatal adoptive Treg cell therapy for Sf mice is able to provide long-term survival and thymopoietic homeostasis, but fails to efficiently prevent the severe defects in B lymphopoiesis during ontogeny of Sf mice.
Discussion
In this study, we have shown that the virtually complete block of B lymphopoiesis in the BM of adolescent Sf mice (Figure 1A; Leonardo et al., 2010; Chang et al., 2012) is based on the gradual acquisition of B cell developmental defects during mouse ontogeny, rather than due to a constitutive lympho-hematopoietic deficiency in the absence of functional Foxp3 protein and Foxp3+ Treg cells. This notion is corroborated by multiple lines of evidence, which includes the observation that the BM microenvironment of Sf and WT mice within the first week of life appeared indistinguishable with regard to the capacity to support B cell development in vivo, as well as the demonstration that early B cell precursors derived from the BM of moribund Sf mice efficiently differentiated into sIgM+ B cells in vitro. In these experiments, a putative B cell-intrinsic function of Foxp3 was firmly excluded using genetic cell lineage tracing, thus establishing that CD19+ B cells lack Foxp3 expression throughout their entire development and lifespan.
Our temporal analysis of Sf mice during ontogeny has revealed that both the manifestation of defective B cell development and the concomitant, although delayed manifestation of T cell developmental defects closely correlated with progressive autoimmune pathology. Thus, the apparent discrepancy between previously reported significant residual B lymphopoietic activity in the BM of 4-week-old Sf mice (Leonardo et al., 2010) and the virtually complete block of B cell development observed in the present study is most likely due to variations in the kinetics of autoimmune manifestations, perhaps due to differences in the microbial flora between independent colonies of Foxp3-deficient mice. In accordance with a dominant role of T cell-mediated autoimmunity, genetic ablation of the Tcra gene revealed the potential of the BM from adult Sf × TCRα−/− mice to efficiently support B lymphopoiesis (Figure 2C), which was found to be sufficient to restore peripheral compartments of mature B cells, including MZ B and B-1a cells (Figure 6E). However, these experiments do not formally exclude the possibility of yet to be identified B cell developmental alterations in Sf × TCRα−/− mice. Exacerbated immune responses and autoimmune symptoms have been reported to spontaneously occur in TCRα−/− mice (Mizoguchi et al., 1996).
Clearly, our observation of efficient medullary and extramedullary B lymphopoiesis early in ontogeny of Sf mice (Figure 3) provides a plausible explanation for the existence of a prominent peripheral B cell compartment in adolescent Sf mice, in the absence of de novo production in the BM. Such a scenario is corroborated by previous studies in mice with temporally controllable deletion of the recombination-activating gene-2 (Hao and Rajewsky, 2001), demonstrating that, in the absence of influx from the BM, mature B cells can indeed persist in peripheral lymphoid organs for several weeks.
Overall, markedly enhanced and prolonged B lymphopoietic activity in extramedullary sites such as liver and spleen of young Sf mice may represent an important compensatory mechanism to alleviate compromised medullary B cell production. Notably, extramedullary B lymphopoiesis has previously been implicated in defective negative selection, allowing the survival of developing self-reactive B cells that would otherwise be deleted during B cell development in the BM (Sandel et al., 2001). The existence of self-reactive B cells in Sf mice is evidenced by autoantibodies to a broad spectrum of self-antigens, including keratin 14 (Huter et al., 2010), mitochondria (Zhang et al., 2009), glucose-6-phosphate isomerase (Nguyen et al., 2007), DNA (Leonardo et al., 2010), and insulin (own unpublished observation). Thus, extramedullary B lymphopoiesis as a major source of mature B cells in the periphery of adolescent Sf mice provides a potential mechanism that contributes to the generation of a B cell repertoire that is biased toward self-antigens, thereby raising the potential for B cell-mediated autoimmune pathology. Additional studies are warranted to directly address the relative contributions of extramedullary B lymphopoiesis and self-reactive B cells to autoimmune pathology in the absence of functional Foxp3+ Treg cells.
Rather than a mere consequence of impaired HSC function (Chen et al., 2010), it appears likely that multiple and partly redundant autoimmune mechanisms act in concert to mediate aberrant B lymphopoiesis in Sf mice. Apart from diversion of lymphoid progenitors from the B lineage in favor of enhanced myelopoiesis (Lee et al., 2009), additional mechanisms that directly impinge on B lymphopoiesis in the BM of Sf mice may include the induction of apoptosis in developing B cells by IFN-γ (Figures 2E,F; and Grawunder et al., 1993; Garvy and Riley, 1994) and type 1 IFNs (Figure 2F; Wang et al., 1995), the impairment of developmental progression to Pre-B cells due to decreased expression of IL-7 (Figure 2G) and the mobilization of lympho-hematopoietic progenitors to the periphery due to decreased expression of CXCL12 (Figure 2G). Indeed, in Foxp3-proficient mice, abolishment of CXCL12 retention signals in the BM have been reported to result in a loss of central B lymphopoiesis (Ueda et al., 2004) and reciprocally increased granulopoiesis (Ueda et al., 2005). In addition, and consistent with the observation of reduced BM cellularity (Chen et al., 2010), increasing evidence is pointing toward overall alterations in the BM microenvironment of Foxp3-deficient mice due to aberrations in bone homeostasis owing to exacerbated osteoclast differentiation and activity (Zaiss et al., 2007, 2010a,b). While osteoclasts might be dispensable for HSC maintenance (Miyamoto et al., 2011), their important function in lympho-hematopoietic homeostasis was recently highlighted in studies in an inducible model of osteopetrosis. It was demonstrated that reduced osteoclast activity results in compromised B lymphopoietic activity by indirect mechanisms, involving a decrease in the expression of IL-7 and CXCL12 by BM stromal cells (Mansour et al., 2011).
With regard to extramedullary hematopoiesis, it appears likely that aberrant B lymphopoiesis in Sf mice arises from both mobilization of lympho-hematopoietic progenitors to spleen and liver as a consequence of autoimmune-mediated incapacitation of the BM cavity for hematopoiesis, and from persisting lympho-hematopoietic progenitor cells at sites of normal fetal and neonatal hematopoiesis. In contrast, exacerbated production of IL-6 in the liver of Sf mice (Figure 2F) as potential mechanism to activate excessive extramedullary hematopoiesis (Peters et al., 1997; Schirmacher et al., 1998; Tsantikos et al., 2010) appears to play a minor role, as genetic ablation of IL-6 in Sf × IL-6−/− mice failed to correct extramedullary B lymphopoiesis (data not shown).
While the impact on lympho-hematopoietic homeostasis had not been analyzed, previous studies established that neonatal adoptive transfer of bulk CD4+ T cell populations effectively suppresses autoimmunity in Foxp3-deficient recipient mice (Smyk-Pearson et al., 2003; Chang et al., 2008; Haribhai et al., 2011) through the concerted action of preformed Foxp3+ Treg cells and Foxp3+ Treg cells that peripherally converted from initially Foxp3−CD4+ T cells (Haribhai et al., 2011). Consistently, we found that, in addition to prolonged survival of Sf recipient mice (Figure 5A), adoptive Treg cell therapy potently suppressed both the exacerbated production of T helper (Th) 2 and Th17 cytokines as well as the manifestation of thymic atrophy and abrogated T cell developmental activity. Reminiscent of previous observations under homeostatic conditions in the BM of mice (Lee et al., 2007, 2009) and humans (Zou et al., 2004), Foxp3GFP+ Treg cells among adoptively transferred bulk CD4+ T cell populations were found to be selectively enriched in the BM of Sf recipient mice (Figure 5C). Interestingly, high-resolution in vivo imaging in an experimental setting of allogeneic HSC transplantation recently provided evidence that Foxp3+ Treg cells preferentially accumulate in the HSC niche in the BM of non-irradiated recipient mice and protect HSCs from allogeneic rejection (Fujisaki et al., 2011). It remains to be determined whether this enrichment of Foxp3+ Treg cells is due to preferential recruitment of preformed cells or preferential de novo induction from initially CD4+Foxp3− T cells in the BM microenvironment (Kretschmer et al., 2005, 2006). In fact, such extrathymic induction of Foxp3+ Treg cells has been observed in peripheral lymphoid tissues of mice in the steady state (Schallenberg et al., 2010).
At present, we can only speculate as to why adoptive Treg cell therapy with bulk CD4+ T cell populations is largely ineffective in the attenuation of severely aberrant central and peripheral B cell homeostasis, including a selective failure to suppress exacerbated Th1 cytokine production and to efficiently attenuate BM-derived expression of IL-7 and CXCL12. Apart from a putative function of Foxp3 in non-lymphoid BM-derived cells, this observation raises the interesting possibility that attenuation of individual symptoms of autoimmune pathology in Sf mice requires specialized subsets of Treg cells with distinctly different effector functions (Chaudhry et al., 2009; Koch et al., 2009; Zheng et al., 2009; Feuerer et al., 2010). Importantly, increasing evidence for autoimmune-mediated functional incapacitation of the BM environment in experimental settings of Foxp3 deficiency suggests that transplantation of Foxp3-proficient HSCs prior to the establishment of permanent autoimmune-mediated damage of the BM niche may facilitate efficient HSC engraftment. This should improve efficacy of current therapeutic regimens in human patients with severe genetic deficiencies in functional Foxp3+ Treg cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are indebted to T. Koenig, C. Friebel and M. Naumann for excellent technical assistance. This work was supported by the FZT 111 (Deutsche Forschungsgemeinschaft (DFG), Center for Regenerative Therapies Dresden, Cluster of Excellence), the SFB 655 and an intramural grant to K.K. from the Medical Faculty of the TU Dresden (MeDDrive Program).
Author contributions
Julia Riewaldt designed, performed and analyzed the experiments and contributed to the data interpretation and writing of the manuscript. Marcin Dembinski, Sandra Düber, Martina Krey, Annette I. Garbe, and Marie Boernert performed experiments. Annette I. Garbe and Siegfried Weiss contributed to the research design and the analysis and interpretation of data. Karsten Kretschmer conceived the research, guided its design, analysis and interpretation, and wrote the manuscript.
References
Baldridge, M. T., King, K. Y., Boles, N. C., Weksberg, D. C., and Goodell, M. A. (2010). Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797.
Baumgarth, N., Chen, J., Herman, O. C., Jager, G. C., and Herzenberg, L. A. (2000). The role of B-1 and B-2 cells in immune protection from influenza virus infection. Curr. Top. Microbiol. Immunol. 252, 163–169.
Bernard, J. J., Seweryniak, K. E., Koniski, A. D., Spinelli, S. L., Blumberg, N., Francis, C. W., Taubman, M. B., Palis, J., and Phipps, R. P. (2009). Foxp3 regulates megakaryopoiesis and platelet function. Arterioscler. Thromb. Vasc. Biol. 29, 1874–1882.
Bettelli, E., Carrier, Y., Gao, W., Korn, T., Strom, T. B., Oukka, M., Weiner, H. L., and Kuchroo, V. K. (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238.
Brunkow, M. E., Jeffery, E. W., Hjerrild, K. A., Paeper, B., Clark, L. B., Yasayko, S. A., Wilkinson, J. E., Galas, D., Ziegler, S. F., and Ramsdell, F. (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27, 68–73.
Cain, D., Kondo, M., Chen, H., and Kelsoe, G. (2009). Effects of acute and chronic inflammation on B-cell development and differentiation. J. Invest. Dermatol. 129, 266–277.
Chang, S. E., Guo, L., Tian, J., Liu, Y., Guo, Z., Zheng, B., and Han, S. (2012). Autoimmune bone marrow environment severely inhibits B cell development by inducing extensive cell death and inhibiting proliferation. Autoimmunity 45, 210–217.
Chang, X., Gao, J. X., Jiang, Q., Wen, J., Seifers, N., Su, L., Godfrey, V. L., Zuo, T., Zheng, P., and Liu, Y. (2005). The Scurfy mutation of FoxP3 in the thymus stroma leads to defective thymopoiesis. J. Exp. Med. 202, 1141–1151.
Chang, X., Zheng, P., and Liu, Y. (2008). Homeostatic proliferation in the mice with germline FoxP3 mutation and its contribution to fatal autoimmunity. J. Immunol. 181, 2399–2406.
Chaudhry, A., Rudra, D., Treuting, P., Samstein, R. M., Liang, Y., Kas, A., and Rudensky, A. Y. (2009). CD4 (regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326, 986–991.
Chen, C., Liu, Y., and Zheng, P. (2010). Mammalian target of rapamycin activation underlies HSC defects in autoimmune disease and inflammation in mice. J. Clin. Invest. 120, 4091–4101.
Chen, G. Y., Chen, C., Wang, L., Chang, X., Zheng, P., and Liu, Y. (2008). Cutting edge: Broad expression of the FoxP3 locus in epithelial cells: a caution against early interpretation of fatal inflammatory diseases following in vivo depletion of FoxP3-expressing cells. J. Immunol. 180, 5163–5166.
Chung, H. S., Lee, J. H., Kim, H., Lee, H. J., Kim, S. H., Kwon, H. K., Im, S. H., and Bae, H. (2010). Foxp3 is a novel repressor of microglia activation. Glia 58, 1247–1256.
Chung, J. B., Silverman, M., and Monroe, J. G. (2003). Transitional B cells: step by step towards immune competence. Trends Immunol. 24, 343–349.
Clark, L. B., Appleby, M. W., Brunkow, M. E., Wilkinson, J. E., Ziegler, S. F., and Ramsdell, F. (1999). Cellular and molecular characterization of the scurfy mouse mutant. J. Immunol. 162, 2546–2554.
Dorshkind, K. (1991). In vivo administration of recombinant granulocyte-macrophage colony-stimulating factor results in a reversible inhibition of primary B lymphopoiesis. J. Immunol. 146, 4204–4208.
Dorshkind, K., and Montecino-Rodriguez, E. (2007). Fetal B-cell lymphopoiesis and the emergence of B-1-cell potential. Nat. Rev. Immunol. 7, 213–219.
Duber, S., Hafner, M., Krey, M., Lienenklaus, S., Roy, B., Hobeika, E., Reth, M., Buch, T., Waisman, A., Kretschmer, K., and Weiss, S. (2009). Induction of B-cell development in adult mice reveals the ability of bone marrow to produce B-1a cells. Blood 114, 4960–4967.
Esplin, B. L., Welner, R. S., Zhang, Q., Borghesi, L. A., and Kincade, P. W. (2009). A differentiation pathway for B1 cells in adult bone marrow. Proc. Natl. Acad. Sci. U.S.A. 106, 5773–5778.
Essers, M. A., Offner, S., Blanco-Bose, W. E., Waibler, Z., Kalinke, U., Duchosal, M. A., and Trumpp, A. (2009). IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908.
Feuerer, M., Hill, J. A., Kretschmer, K., Von Boehmer, H., Mathis, D., and Benoist, C. (2010). Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc. Natl. Acad. Sci. U.S.A. 107, 5919–5924.
Fontenot, J. D., Gavin, M. A., and Rudensky, A. Y. (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336.
Fontenot, J. D., Rasmussen, J. P., Williams, L. M., Dooley, J. L., Farr, A. G., and Rudensky, A. Y. (2005). Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22, 329–341.
Fujisaki, J., Wu, J., Carlson, A. L., Silberstein, L., Putheti, P., Larocca, R., Gao, W., Saito, T. I., Lo Celso, C., Tsuyuzaki, H., Sato, T., Cote, D., Sykes, M., Strom, T. B., Scadden, D. T., and Lin, C. P. (2011). In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 474, 216–219.
Garvy, B. A., and Riley, R. L. (1994). IFN-gamma abrogates IL-7-dependent proliferation in pre-B cells, coinciding with onset of apoptosis. Immunology 81, 381–388.
Godfrey, V. L., Wilkinson, J. E., Rinchik, E. M., and Russell, L. B. (1991). Fatal lymphoreticular disease in the scurfy (sf) mouse requires T cells that mature in a sf thymic environment: potential model for thymic education. Proc. Natl. Acad. Sci. U.S.A. 88, 5528–5532.
Grawunder, U., Melchers, F., and Rolink, A. (1993). Interferon-gamma arrests proliferation and causes apoptosis in stromal cell/interleukin-7-dependent normal murine pre-B cell lines and clones in vitro, but does not induce differentiation to surface immunoglobulin-positive B cells. Eur. J. Immunol. 23, 544–551.
Hao, Z., and Rajewsky, K. (2001). Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J. Exp. Med. 194, 1151–1164.
Hardy, R. R., and Hayakawa, K. (2001). B cell development pathways. Annu. Rev. Immunol. 19, 595–621.
Hardy, R. R., Li, Y. S., Allman, D., Asano, M., Gui, M., and Hayakawa, K. (2000). B-cell commitment, development and selection. Immunol. Rev. 175, 23–32.
Haribhai, D., Williams, J. B., Jia, S., Nickerson, D., Schmitt, E. G., Edwards, B., Ziegelbauer, J., Yassai, M., Li, S. H., Relland, L. M., Wise, P. M., Chen, A., Zheng, Y. Q., Simpson, P. M., Gorski, J., Salzman, N. H., Hessner, M. J., Chatila, T. A., and Williams, C. B. (2011). A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 35, 109–122.
Hinz, S., Pagerols-Raluy, L., Oberg, H. H., Ammerpohl, O., Grussel, S., Sipos, B., Grutzmann, R., Pilarsky, C., Ungefroren, H., Saeger, H. D., Kloppel, G., Kabelitz, D., and Kalthoff, H. (2007). Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res. 67, 8344–8350.
Hock, H., Dorsch, M., Diamantstein, T., and Blankenstein, T. (1991). Interleukin 7 induces CD4+ T cell-dependent tumor rejection. J. Exp. Med. 174, 1291–1298.
Huter, E. N., Natarajan, K., Torgerson, T. R., Glass, D. D., and Shevach, E. M. (2010). Autoantibodies in Scurfy mice and IPEX patients recognize keratin 14. J. Invest. Dermatol. 130, 1391–1399.
Kanangat, S., Blair, P., Reddy, R., Daheshia, M., Godfrey, V., Rouse, B. T., and Wilkinson, E. (1996). Disease in the scurfy (sf) mouse is associated with overexpression of cytokine genes. Eur. J. Immunol. 26, 161–165.
Karanikas, V., Speletas, M., Zamanakou, M., Kalala, F., Loules, G., Kerenidi, T., Barda, A. K., Gourgoulianis, K. I., and Germenis, A. E. (2008). Foxp3 expression in human cancer cells. J. Transl. Med. 6, 19.
King, K. Y., and Goodell, M. A. (2011). Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 11, 685–692.
Koch, M. A., Tucker-Heard, G., Perdue, N. R., Killebrew, J. R., Urdahl, K. B., and Campbell, D. J. (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602.
Kondo, M., Weissman, I. L., and Akashi, K. (1997). Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 91, 661–672.
Kopf, M., Baumann, H., Freer, G., Freudenberg, M., Lamers, M., Kishimoto, T., Zinkernagel, R., Bluethmann, H., and Kohler, G. (1994). Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368, 339–342.
Kretschmer, K., Apostolou, I., Hawiger, D., Khazaie, K., Nussenzweig, M. C., and Von Boehmer, H. (2005). Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 6, 1219–1227.
Kretschmer, K., Apostolou, I., Jaeckel, E., Khazaie, K., and Von Boehmer, H. (2006). Making regulatory T cells with defined antigen specificity: role in autoimmunity and cancer. Immunol. Rev. 212, 163–169.
Kretschmer, K., Engel, H., and Weiss, S. (2002). Strong antigenic selection shaping the immunoglobulin heavy chain repertoire of B-1a lymphocytes in lambda 2(315) transgenic mice. Eur. J. Immunol. 32, 2317–2327.
Kretschmer, K., Jungebloud, A., Stopkowicz, J., Stoermann, B., Hoffmann, R., and Weiss, S. (2003). Antibody repertoire and gene expression profile: implications for different developmental and functional traits of splenic and peritoneal B-1 lymphocytes. J. Immunol. 171, 1192–1201.
Kretschmer, K., Stopkowicz, J., Scheffer, S., Greten, T. F., and Weiss, S. (2004). Maintenance of peritoneal B-1a lymphocytes in the absence of the spleen. J. Immunol. 173, 197–204.
Lee, J. H., Kang, S. G., and Kim, C. H. (2007). FoxP3+ T cells undergo conventional first switch to lymphoid tissue homing receptors in thymus but accelerated second switch to nonlymphoid tissue homing receptors in secondary lymphoid tissues. J. Immunol. 178, 301–311.
Lee, J. H., Wang, C., and Kim, C. H. (2009). FoxP3+ regulatory T cells restrain splenic extramedullary myelopoiesis via suppression of hemopoietic cytokine-producing T cells. J. Immunol. 183, 6377–6386.
Leonardo, S. M., Josephson, J. A., Hartog, N. L., and Gauld, S. B. (2010). Altered B cell development and anergy in the absence of Foxp3. J. Immunol. 185, 2147–2156.
Lienenklaus, S., Walisko, R., Boekhorst, A. T., May, T., Samuelsson, C., Michiels, T., and Weiss, S. (2008). PCR-based simultaneous analysis of the interferon-alpha family reveals distinct kinetics for early interferons. J. Interferon Cytokine Res. 28, 653–660.
Lin, W., Haribhai, D., Relland, L. M., Truong, N., Carlson, M. R., Williams, C. B., and Chatila, T. A. (2007). Regulatory T cell development in the absence of functional Foxp3. Nat. Immunol. 8, 359–368.
Lin, W., Truong, N., Grossman, W. J., Haribhai, D., Williams, C. B., Wang, J., Martin, M. G., and Chatila, T. A. (2005). Allergic dysregulation and hyperimmunoglobulinemia E in Foxp3 mutant mice. J. Allergy Clin. Immunol. 116, 1106–1115.
Liston, A., Farr, A. G., Chen, Z., Benoist, C., Mathis, D., Manley, N. R., and Rudensky, A. Y. (2007). Lack of Foxp3 function and expression in the thymic epithelium. J. Exp. Med. 204, 475–480.
Lyon, M. F., Peters, J., Glenister, P. H., Ball, S., and Wright, E. (1990). The scurfy mouse mutant has previously unrecognized hematological abnormalities and resembles Wiskott-Aldrich syndrome. Proc. Natl. Acad. Sci. U.S.A. 87, 2433–2437.
Manrique, S. Z., Correa, M. A., Hoelzinger, D. B., Dominguez, A. L., Mirza, N., Lin, H. H., Stein-Streilein, J., Gordon, S., and Lustgarten, J. (2011). Foxp3-positive macrophages display immunosuppressive properties and promote tumor growth. J. Exp. Med. 208, 1485–1499.
Mansour, A., Anginot, A., Mancini, S. J., Schiff, C., Carle, G. F., Wakkach, A., and Blin-Wakkach, C. (2011). Osteoclast activity modulates B-cell development in the bone marrow. Cell Res. 21, 1102–1115.
Mayer, C. T., Kuhl, A. A., Loddenkemper, C., and Sparwasser, T. (2012). Lack of Foxp3+ macrophages in both untreated and B16 melanoma-bearing mice. Blood 119, 1314–1315.
Miyamoto, K., Yoshida, S., Kawasumi, M., Hashimoto, K., Kimura, T., Sato, Y., Kobayashi, T., Miyauchi, Y., Hoshi, H., Iwasaki, R., Miyamoto, H., Hao, W., Morioka, H., Chiba, K., Yasuda, H., Penninger, J. M., Toyama, Y., Suda, T., and Miyamoto, T. (2011). Osteoclasts are dispensable for hematopoietic stem cell maintenance and mobilization. J. Exp. Med. 208, 2175–2181.
Mizoguchi, A., Mizoguchi, E., Chiba, C., Spiekermann, G. M., Tonegawa, S., Nagler-Anderson, C., and Bhan, A. K. (1996). Cytokine imbalance and autoantibody production in T cell receptor-alpha mutant mice with inflammatory bowel disease. J. Exp. Med. 183, 847–856.
Mombaerts, P., Clarke, A. R., Rudnicki, M. A., Iacomini, J., Itohara, S., Lafaille, J. J., Wang, L., Ichikawa, Y., Jaenisch, R., Hooper, M. L., and Tonegawa, S. (1992). Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature 360, 225–231.
Montecino-Rodriguez, E., Leathers, H., and Dorshkind, K. (2006). Identification of a B-1 B cell-specified progenitor. Nat. Immunol. 7, 293–301.
Nagai, Y., Garrett, K. P., Ohta, S., Bahrun, U., Kouro, T., Akira, S., Takatsu, K., and Kincade, P. W. (2006). Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24, 801–812.
Nagasawa, T. (2006). Microenvironmental niches in the bone marrow required for B-cell development. Nat. Rev. Immunol. 6, 107–116.
Nagasawa, T., Hirota, S., Tachibana, K., Takakura, N., Nishikawa, S., Kitamura, Y., Yoshida, N., Kikutani, H., and Kishimoto, T. (1996). Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382, 635–638.
Nguyen, L. T., Jacobs, J., Mathis, D., and Benoist, C. (2007). Where FoxP3-dependent regulatory T cells impinge on the development of inflammatory arthritis. Arthritis Rheum. 56, 509–520.
Passegue, E., Wagers, A. J., Giuriato, S., Anderson, W. C., and Weissman, I. L. (2005). Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 202, 1599–1611.
Peters, M., Schirmacher, P., Goldschmitt, J., Odenthal, M., Peschel, C., Fattori, E., Ciliberto, G., Dienes, H. P., Meyer Zum Buschenfelde, K. H., and Rose-John, S. (1997). Extramedullary expansion of hematopoietic progenitor cells in interleukin (IL)-6-sIL-6R double transgenic mice. J. Exp. Med. 185, 755–766.
Pillai, S., Cariappa, A., and Moran, S. T. (2005). Marginal zone B cells. Annu. Rev. Immunol. 23, 161–196.
Rolink, A., Kudo, A., Karasuyama, H., Kikuchi, Y., and Melchers, F. (1991). Long-term proliferating early pre B cell lines and clones with the potential to develop to surface Ig-positive, mitogen reactive B cells in vitro and in vivo. EMBO J. 10, 327–336.
Rolink, A. G., Ten Boekel, E., Yamagami, T., Ceredig, R., Andersson, J., and Melchers, F. (1999). B cell development in the mouse from early progenitors to mature B cells. Immunol. Lett. 68, 89–93.
Sakaguchi, S. (2005). Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 6, 345–352.
Sakaguchi, S., Yamaguchi, T., Nomura, T., and Ono, M. (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787.
Sandel, P. C., Gendelman, M., Kelsoe, G., and Monroe, J. G. (2001). Definition of a novel cellular constituent of the bone marrow that regulates the response of immature B cells to B cell antigen receptor engagement. J. Immunol. 166, 5935–5944.
Schallenberg, S., Tsai, P. Y., Riewaldt, J., and Kretschmer, K. (2010). Identification of an immediate Foxp3(−) precursor to Foxp3(+) regulatory T cells in peripheral lymphoid organs of nonmanipulated mice. J. Exp. Med. 207, 1393–1407.
Schirmacher, P., Peters, M., Ciliberto, G., Blessing, M., Lotz, J., Meyer Zum Buschenfelde, K. H., and Rose-John, S. (1998). Hepatocellular hyperplasia, plasmacytoma formation, and extramedullary hematopoiesis in interleukin (IL)-6/soluble IL-6 receptor double-transgenic mice. Am. J. Pathol. 153, 639–648.
Smyk-Pearson, S. K., Bakke, A. C., Held, P. K., and Wildin, R. S. (2003). Rescue of the autoimmune scurfy mouse by partial bone marrow transplantation or by injection with T-enriched splenocytes. Clin. Exp. Immunol. 133, 193–199.
Srinivas, S., Watanabe, T., Lin, C. S., William, C. M., Tanabe, Y., Jessell, T. M., and Costantini, F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4.
Trumpp, A., Essers, M., and Wilson, A. (2010). Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 10, 201–209.
Tsantikos, E., Oracki, S. A., Quilici, C., Anderson, G. P., Tarlinton, D. M., and Hibbs, M. L. (2010). Autoimmune disease in Lyn-deficient mice is dependent on an inflammatory environment established by IL-6. J. Immunol. 184, 1348–1360.
Tung, J. W., Mrazek, M. D., Yang, Y., and Herzenberg, L. A. (2006). Phenotypically distinct B cell development pathways map to the three B cell lineages in the mouse. Proc. Natl. Acad. Sci. U.S.A. 103, 6293–6298.
Ueda, Y., Kondo, M., and Kelsoe, G. (2005). Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J. Exp. Med. 201, 1771–1780.
Ueda, Y., Yang, K., Foster, S. J., Kondo, M., and Kelsoe, G. (2004). Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J. Exp. Med. 199, 47–58.
Wan, Y. Y., and Flavell, R. A. (2005). Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc. Natl. Acad. Sci. U.S.A. 102, 5126–5131.
Wang, J., Lin, Q., Langston, H., and Cooper, M. D. (1995). Resident bone marrow macrophages produce type 1 interferons that can selectively inhibit interleukin-7-driven growth of B lineage cells. Immunity 3, 475–484.
Wilson, A., Laurenti, E., Oser, G., Van Der Wath, R. C., Blanco-Bose, W., Jaworski, M., Offner, S., Dunant, C. F., Eshkind, L., Bockamp, E., Lio, P., Macdonald, H. R., and Trumpp, A. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129.
Zaiss, M. M., Axmann, R., Zwerina, J., Polzer, K., Guckel, E., Skapenko, A., Schulze-Koops, H., Horwood, N., Cope, A., and Schett, G. (2007). Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum. 56, 4104–4112.
Zaiss, M. M., Frey, B., Hess, A., Zwerina, J., Luther, J., Nimmerjahn, F., Engelke, K., Kollias, G., Hunig, T., Schett, G., and David, J. P. (2010a). Regulatory T cells protect from local and systemic bone destruction in arthritis. J. Immunol. 184, 7238–7246.
Zaiss, M. M., Sarter, K., Hess, A., Engelke, K., Bohm, C., Nimmerjahn, F., Voll, R., Schett, G., and David, J. P. (2010b). Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis Rheum. 62, 2328–2338.
Zhang, W., Sharma, R., Ju, S. T., He, X. S., Tao, Y., Tsuneyama, K., Tian, Z., Lian, Z. X., Fu, S. M., and Gershwin, M. E. (2009). Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis. Hepatology 49, 545–552.
Zheng, Y., Chaudhry, A., Kas, A., Deroos, P., Kim, J. M., Chu, T. T., Corcoran, L., Treuting, P., Klein, U., and Rudensky, A. Y. (2009). Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356.
Zhou, X., Jeker, L. T., Fife, B. T., Zhu, S., Anderson, M. S., Mcmanus, M. T., and Bluestone, J. A. (2008). Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J. Exp. Med. 205, 1983–1991.
Zou, L., Barnett, B., Safah, H., Larussa, V. F., Evdemon-Hogan, M., Mottram, P., Wei, S., David, O., Curiel, T. J., and Zou, W. (2004). Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 64, 8451–8455.
Zuo, T., Wang, L., Morrison, C., Chang, X., Zhang, H., Li, W., Liu, Y., Wang, Y., Liu, X., Chan, M. W., Liu, J. Q., Love, R., Liu, C. G., Godfrey, V., Shen, R., Huang, T. H., Yang, T., Park, B. K., Wang, C. Y., and Zheng, P. (2007). FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell 129, 1275–1286.
Keywords: autoimmunity, lymphopoiesis, scurfy, Foxp3, regulatory T cell, B cell
Citation: Riewaldt J, Düber S, Boernert M, Krey M, Dembinski M, Weiss S, Garbe AI and Kretschmer K (2012) Severe developmental B lymphopoietic defects in Foxp3-deficient mice are refractory to adoptive regulatory T cell therapy. Front. Immun. 3:141. doi: 10.3389/fimmu.2012.00141
Received: 01 March 2012; Paper pending published: 19 March 2012;
Accepted: 16 May 2012; Published online: 05 June 2012.
Edited by:
Daniel Hawiger, Saint Louis University, USAReviewed by:
Paulo Vieira, Institut Pasteur de Paris, FranceRichard DiPaolo, Saint Louis University School of Medicine, USA
Copyright: © 2012 Riewaldt, Düber, Boernert, Krey, Dembinski, Weiss, Garbe and Kretschmer. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Karsten Kretschmer, Center for Regenerative Therapies Dresden, Technical University Dresden, Fetscherstr. 105, 01307 Dresden, Germany. e-mail:a2Fyc3Rlbi5rcmV0c2NobWVyQGNydC1kcmVzZGVuLmRl