
 Esther Kohler1
Esther Kohler1

- 1 Institute of Molecular Cancer Research, University of Zurich, Zurich, Switzerland
- 2 Department of Molecular Biology and Genetics, Istanbul Technical University, Istanbul, Turkey
Chronic infection with the gastric bacterial pathogen Helicobacter pylori causes gastric adenocarcinoma in a particularly susceptible fraction of the infected population. The intestinal type of gastric cancer is preceded by a series of preneoplastic lesions that are of immunopathological origin, and that can be recapitulated by experimental infection of C57BL/6 mice with Helicobacter species. Several lines of evidence suggest that specific T cell subsets and/or their signature cytokines contribute to the control of Helicobacter infections on the one hand, and to the associated gastric preneoplastic pathology on the other. Here, we have used virulent H. pylori and H. felis isolates to infect mice that lack α/β T cells due to a targeted deletion of the T cell receptor β-chain, or are deficient for the unique p35 and p19 subunits of the Th1- and Th17-polarizing cytokines interleukin (IL)-12 and IL-23, respectively. We found that α/β T cells are absolutely required for Helicobacter control and for the induction of gastric preneoplastic pathology. In contrast, neither IL-12-dependent Th1 nor IL-23-dependent Th17 cells were essential for controlling the infection; IL-12p35-/- and IL-23p19-/- mice did not differ significantly from wild type animals with respect to Helicobacter colonization densities. Gastritis and gastric preneoplastic pathology developed to a similar extent in all three strains upon H. felis infection; in the H. pylori infection model, IL-23p19-/- mice exhibited significantly less gastritis and precancerous pathology. In summary, the results indicate that neither Th1 nor Th17 cells are by themselves essential for Helicobacter control; the associated gastric pathology is reduced only in the absence of Th17-polarizing IL-23, and only in the H. pylori, but not the H. felis infection model. The results thus suggest the involvement of other, as yet unknown T cell subsets in both processes.
Introduction
Persistent colonization of the human gastric mucosa with the Gram-negative bacterial pathogen Helicobacter pylori causes gastritis (Marshall and Warren, 1984) and predisposes carriers to a high risk of developing gastric and duodenal ulcers, gastric mucosa-associated lymphoid tissue lymphoma, and gastric adenocarcinoma (Parsonnet et al., 1991, 1994; Parsonnet and Isaacson, 2004; Pritchard and Crabtree, 2006). H. pylori infection-associated gastric cancer develops via a sequence of histologically defined preneoplastic lesions, all of which have been linked independently to gastric cancer risk (Correa, 1995; Fox and Wang, 2007). Experimental infection of Mongolian gerbils (Rieder et al., 2005; Wiedemann et al., 2009) or C57BL/6 mice (Fox et al., 2002, 2003) with Helicobacter species recapitulates the sequential development of chronic gastritis, atrophic gastritis, epithelial hyperplasia, and intestinal metaplasia, all of which are hallmark lesions in a subset of particularly susceptible human H. pylori carriers (Correa, 1995; Fox and Wang, 2007). H. pylori patient isolates and the closely related species H. felis have been used successfully to induce gastric preneoplastic lesions in mice (Fox et al., 2002, 2003; Sayi et al., 2009; Arnold et al., 2011). Several lines of experimental evidence suggest that precancerous lesions in the infected stomach are immunopathological in origin, i.e., they arise as a consequence of T cell-driven adaptive immune responses to the infection. (1) Mice lacking a B and/or T lymphocyte compartment (Rag-1-/-, TCR-β-/-) are resistant to gastritis induced by Helicobacter infection, and the adoptive transfer of CD4+ effector T cells from infected wild type (WT) donors to infected TCR-β-/- recipients is in itself sufficient to restore the full range of preneoplastic pathology (Roth et al., 1999; Smythies et al., 2000; Sayi et al., 2009, 2011; Arnold et al., 2011). (2) Prior immunization with a cholera toxin-adjuvanted, whole cell vaccine aggravates, rather than prevents, infection-induced preneoplastic pathology (Mueller et al., 2009; Sayi et al., 2009). (3) Finally, mice that have been infected during the neonatal period, i.e., at a time when their immune system is not yet fully developed and inherently biased toward immune tolerance rather than immunity, are largely protected against infection-associated pathology (Arnold et al., 2011). Observational studies in human carriers support the view that pathogenic T cells are instrumental in inducing H. pylori-associated lesions: Robinson et al. (2008) showed for instance that patients with peptic ulcer disease exhibited stronger Th1 and Th2 responses to H. pylori than asymptomatic carriers. Conversely, the asymptomatic carriers were characterized by a regulatory T cell (Treg)-predominant response to the infection. Tregs expressing the anti-inflammatory cytokine interleukin (IL)-10 were particularly abundant in the gastric mucosa of the normal carriers compared to the peptic ulcer disease patients (Robinson et al., 2008). In a similar study conducted by Harris et al. (2008), the relatively mild gastritis typical of H. pylori-infected children could also be linked to Treg-predominant T cell responses and higher levels of IL-10 and TGF-β.
Whereas a causal role for T cells in H. pylori infection-associated pathology has thus been rather well established, the contribution of the various T helper-cell subsets (defined by their cytokine profiles and lineage-determining transcription factors) is less thoroughly understood. Th1 and Th17 cell subsets have both been alternatively implicated in infection control on the one hand, and preneoplasia on the other (Algood et al., 2009; Otani et al., 2009; Sayi et al., 2009; Stoicov et al., 2009; Sheh et al., 2010; Shi et al., 2010). However, most of these studies examined one Th1 or Th17 cytokine in isolation, rather than analyzing mice that have defects in generating Th1 and/or Th17 subsets. We have recently exploited mouse strains with targeted deletions of the genes encoding the IL-12 subunit p35 and the IL-23 subunit p19, respectively, to elucidate the contribution of Th subsets to H. pylori-specific, vaccine-induced protective immunity (Hitzler et al., 2011). p19 and p35 represent the unique subunits of the Th17- and Th1-inducing cytokines IL-23 and IL-12, respectively, covalently linked heterodimers which are each composed of a unique (p19 or p35) chain and the shared p40 chain (Oppmann et al., 2000). IL-23p19-/- and IL-12p35-/- mice exhibit defects in generating (pathogenic) Th17 and Th1 responses to foreign or autoantigens, respectively (Cua et al., 2003; McGeachy and Cua, 2008). Interestingly, both subsets were absolutely required for vaccine-induced protection; both IL-23p19-/- and IL-12p35-/- mice exhibited significant defects with respect to control of a challenge infection (Hitzler et al., 2011).
In the present study, we have utilized the two strains to examine the relative contribution of IL-12-dependent Th1 and IL-23-dependent Th17 cells to long term Helicobacter infection control and to infection-associated gastric immunopathology in two complementary H. pylori and H. felis infection models. To our surprise, we found that, despite the fact that T cells expressing the α/β T cell receptor were clearly required for infection control and gastric preneoplasia, neither Th1 nor Th17 cells were alone responsible for either of the two processes under all conditions tested. IL-23p19-/- and IL-12p35-/- mice were colonized at similar levels as WT animals, and also exhibited similar degrees of gastric immunopathology upon H. felis infection. IL-23p19-/- but not IL-12p35-/- mice were somewhat less susceptible to H. pylori-induced immunopathology. In conclusion, IL-12-dependent Th1 and IL-23-dependent Th17 subsets are dispensable for infection control and also to some extent for the induction of infection-associated pathology despite a clear requirement for α/β T cells in both processes, and despite a documented role for both subsets in vaccine-induced protective immunity.
Materials and Methods
Animal Experimentation
C57BL/6 WT, BL/6.TCR-β-/-, and BL/6.IL-12p35-/- mice were originally purchased from Charles River Laboratories. IL23p19-/- mice were kindly provided by Regeneron Pharmaceuticals (Tarrytown, NY, USA) and backcrossed for >10 generations to C57BL/6 genetic background. All mice were bred at a University of Zurich specific pathogen-free facility. Mixed gender groups were included in studies at 7 weeks of age. All animal experimentation was reviewed and approved by the Zurich cantonal veterinary office. Mice were infected with 108 H. pylori PMSS1 (CFU estimated based on OD) or H. felis (strain CS1, ATCC 49179) grown as described previously (Sayi et al., 2009; Arnold et al., 2011). Mice were sacrificed 3 months after infection by CO2 inhalation.
Quantitative Assessment of Helicobacter Colonization and Gastric Histopathology
Bacterial colonization was assessed by colony count assay (H. pylori) or flaB-specific real time PCR (H. felis) as described earlier (Sayi et al., 2009; Arnold et al., 2011). Briefly, for H. pylori quantification, one-sixth of each stomach was transferred to a tube containing Brucella broth and homogenized with an Ultra Turrax homogenizer (John Morris Scientific Ltd, Chatswood, Australia). Serial dilutions were plated on horse blood plates to determine bacterial loads (Arnold et al., 2011). For the qPCR-based assessment of H. felis colonization, genomic DNA was isolated using the DNeasy blood and tissue kit (Qiagen, Hilden, Germany) and subjected to qPCR analysis of the flaB gene as described (Sayi et al., 2009). For the quantitative assessment of gastric histopathology, Giemsa-stained paraffin-embedded stomach sections were scored on a scale of 0–6 for the parameters chronic inflammation, atrophy, epithelial hyperplasia, and metaplasia as described in detail previously (Sayi et al., 2009). All pictures were taken with a Leica Leitz DM RB microscope equipped with a DFC 420C camera. Images were acquired at 100× final magnification using the Leica Application Suite 3.3.0 software.
Cytokine qPCR
For real-time RT-PCR of mucosal cytokine expression, total RNA was isolated from the scraped gastric mucosa of one-sixth of every stomach (antrum and corpus) using NucleoSpin RNA II kits (Macherey-Nagel, Düren, Germany). The corresponding cDNA served as a template for real-time PCR performed using the LightCycler 480 SYBR Green I master kit (Roche, Basel, Switzerland). Absolute values of IFN-γ and IL-17 expression were normalized to GAPDH expression for the relative quantification of cytokine gene expression. The efficiency of each primer–template combination was assessed using the standard curve method, i.e., 1:5 fold serial titrations were included for the gene of interest and the GAPDH housekeeping gene on every microtiter plate (conditions: temperature 55°C, 50 cycles; primers: GAPDH forward GAC ATT GTT GCC ATC AAC GAC C/GAPDH reverse CCC GTT GAT GAC CAG CTT CC; IFN-γ forward CAT GGC TGT TTC TGG CTG TTA CTG/IFN-γ reverse GTT GCT GAT GGC CTG ATT GTC TTT; IL-17 forward GCT CCA GAA GGC CCT CAG A/IL-17 reverse AGC TTT CCC TCC GCA TTG A).
Statistical Analysis
All P-values were calculated using Graph Pad prism 5.0 or R software. The significance of categorical differences in histopathology scores was calculated by Mann–Whitney test and the significance of numerical differences was calculated by Student’s t-test. In all graphs showing colonization data and histopathology scores, the medians are indicated by horizontal bars. In column bar graphs, standard errors of the mean are indicated by vertical bars. n.a. denotes “not applicable,” and n.s. stands for “not significant.”
Results
TCR-β-/- Mice Fail to Control H. felis Infection, but are Protected Against Gastric Preneoplasia
We and others have reported previously that C57BL/6 mice lacking α/β T cells due to a targeted mutation of the gene encoding the β-chain of the TCR are protected against the early infection-associated gastritis that is a hallmark of T cell proficient animals (Roth et al., 1999; Smythies et al., 2000; Sayi et al., 2009). To assess whether the relative resistance to the earliest detectable histopathological symptoms of Helicobacter infection translates into long-term protection from disease, we infected WT C57BL/6 and TCR-β-/- mice with H. felis for 3 months; we chose H. felis due to its documented virulence in mice (Fox et al., 2002, 2003; Sayi et al., 2009). WT mice infected with H. felis were colonized persistently, albeit at rather low levels (Figure 1A) as reported previously (Sayi et al., 2009), and the majority developed gastritis, atrophy, epithelial hyperplasia, and/or intestinal metaplasia (Figures 1B, C). In contrast, TCR-β-/- mice were colonized at higher levels (Figure 1A), and were completely protected from infection-associated gastric preneoplastic lesions (Figures 1B, C). The results indicate that α/β T cells contribute critically to Helicobacter control and to gastric Helicobacter-associated immunopathology and confirm previous results that have used SCID mice (Smythies et al., 2000) and RAG-1-/- and TCR-β-/- mice (Roth et al., 1999; Sayi et al., 2009, 2011) for H. pylori or H. felis infections.
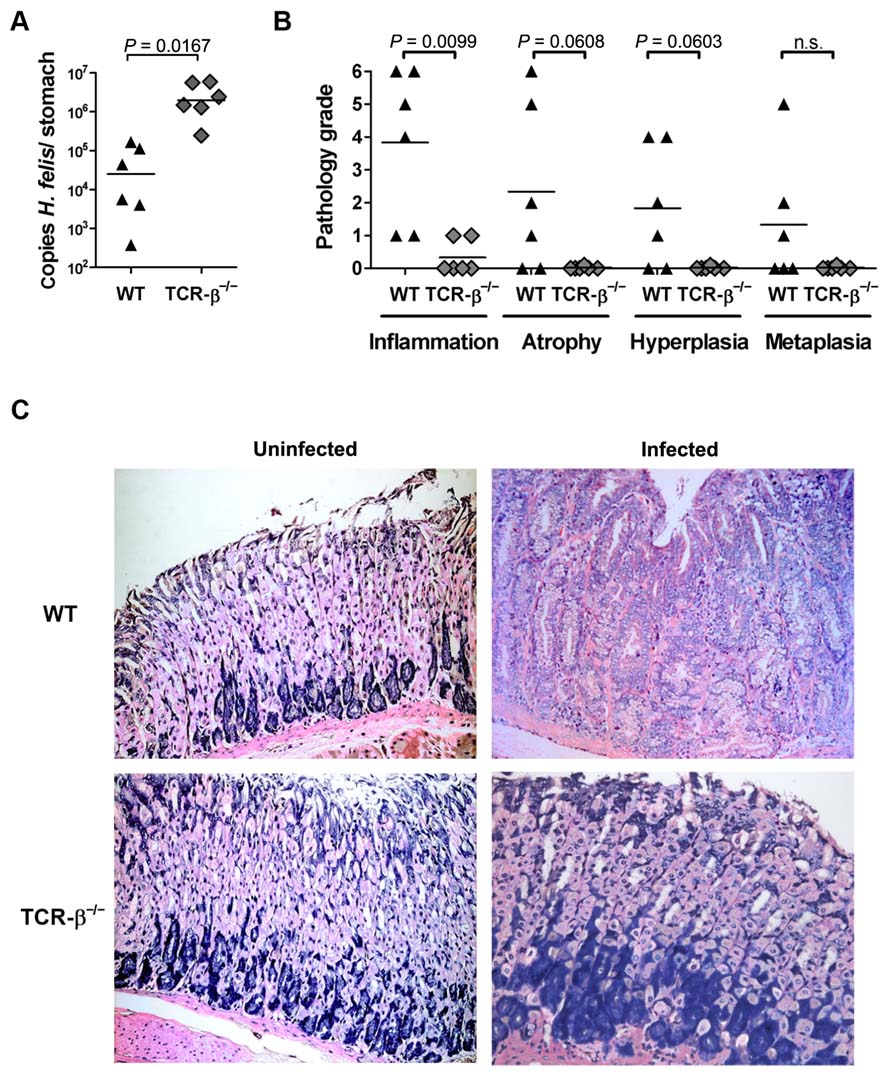
FIGURE 1. The lack of α/β T cells in TCR-β-/- mice impairs spontaneous H. felis infection control, but protects against gastric immunopathology. (A) Colonization levels as determined by quantitative PCR of the H. felis flaB gene of wild type and TCR-β-/- mice infected with H. felis for 3 months. Medians are indicated by horizontal lines; each symbol represents one mouse. (B) Histopathology scores assigned to every mouse shown in (A) for all indicated parameters on a scale of 0–6. (C) Representative micrographs of Giemsa-stained sections of H. felis-infected and uninfected wild type and TCR-β-/- mice, respectively. Data are representative of three independent experiments.
Neither IL-12-Dependent Th1 Nor IL-23-Dependent Th17 Cells are Absolutely Required for the Control of Experimental H. felis Infection
To assess the contribution of Th1 and Th17 cells to H. felis infection control, we infected WT, IL-12p35-/-, and IL-23p19-/- mice with H. felis for 3 months and compared their colonization levels. All three groups were colonized at comparable levels (Figure 2A), and exhibited the wide intragroup variation in H. felis colonization densities that is a hallmark of this infection (see Figure 1A). To assess whether the defect in generating functional Th1 and Th17 subsets that is a well-documented feature of IL-12p35-/- and IL-23p19-/- mice, respectively (Cua et al., 2003; McGeachy and Cua, 2008), would manifest in decreased levels of the respective signature cytokines IFN-γ and IL-17, we isolated RNA from the gastric mucosa of all animals included in the study. To our surprise, neither the gastric mucosal expression of IFN-γ (Figure 2B), nor of IL-17 (Figure 2C), was linked to the genotype of the mice, suggesting either that the bulk of both cytokines is produced by sources other than Th1 and Th17 cells, or that factors other than IL-23 and IL-12 influence Th17 and Th1 polarization in the context of Helicobacter infection. In conclusion, H. felis infection is controlled by T cells, but independently of IL-12-dependent Th1 and IL-23-dependent Th17 cell subsets.
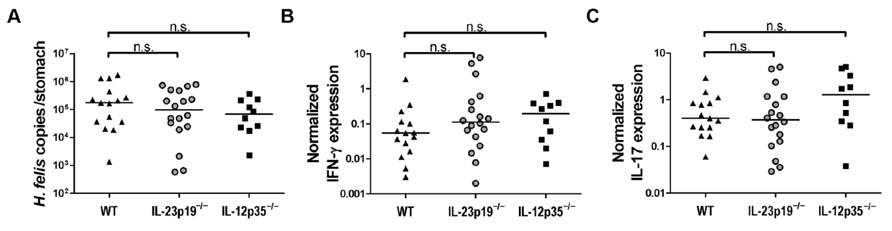
FIGURE 2. IL-23p19-/- and IL-12p35-/- mice do not differ significantly from wild type animals with respect to H. felis infection control. (A) Colonization levels as determined by quantitative PCR of the H. felis flaB gene of wild type (WT; n = 15), IL-23p19-/- (n = 18), and IL-12p35-/- (n = 10) mice infected with H. felis for 3 months. Medians are indicated by horizontal lines; each symbol represents one mouse. (B,C) Real-time RT-PCR results for gastric IFN-γ (B) and IL-17 (C) expression of all mice shown in (A), normalized to GAPDH. Note that uninfected controls of all strains were analyzed in parallel and exhibited normalized cytokine expression levels under 0.01 (IFN-γ and IL-17; data not shown). Data in this figure are pooled from two infection experiments.
IL-12 and IL-23 are not Absolutely Required for the Induction of Gastric Preneoplastic Pathology
To determine a possible contribution of IL-12-dependent Th1 and IL-23-dependent Th17 cell subsets to gastritis and gastric preneoplastic pathology, the gastric histopathology of all H. felis-infected mice of the three genotypes was examined and scored with respect to the severity of gastritis, atrophy, epithelial hyperplasia, and intestinal metaplasia (Figures 3A, B). All WT, IL-12p35-/-, and IL-23p19-/- mice had developed mild to severe gastritis in the course of the infection, and no significant differences were observed between groups in terms of the average severity grade (Figures 3A, B). Virtually all WT, and a majority of IL-12p35-/- and IL-23p19-/- mice had further developed atrophy, hyperplasia, and metaplasia; again, considerable variation was observed within groups (as reported previously; Sayi et al., 2009) and no significant differences were obtained between groups. The combined results suggest that gastric cancer precursor lesions develop readily in H. felis-infected mice, even under conditions where the generation of Th1 or Th17 cells is impaired due to the lack of the respective Th-polarizing cytokines IL-12 and IL-23.
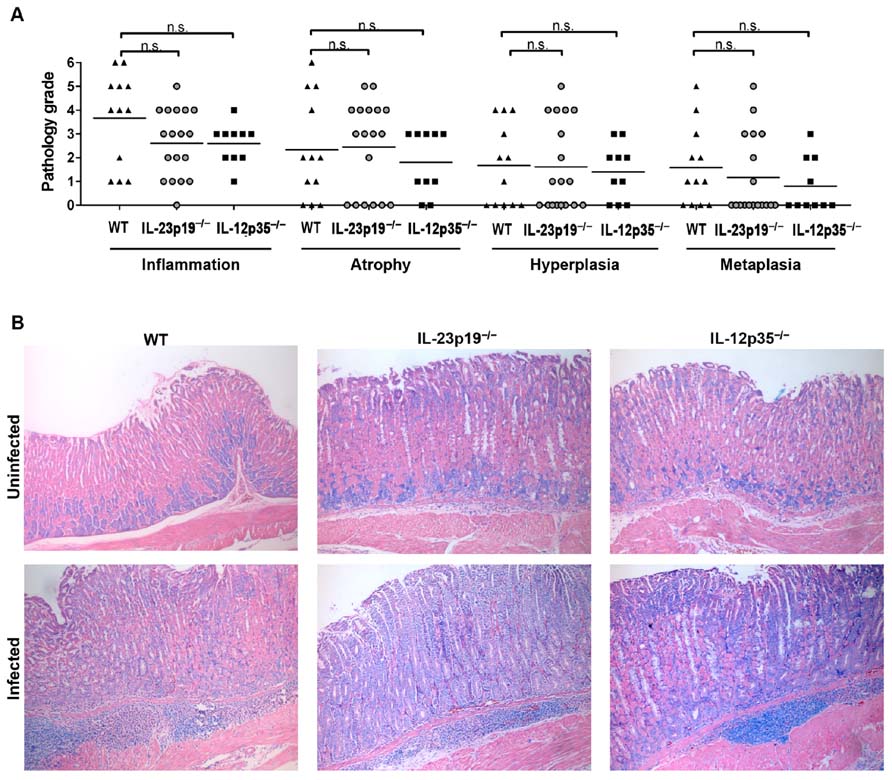
FIGURE 3. The gastric histopathology induced by H. felis infection of IL-23p19-/- and IL-12p35-/- mice is not significantly different from that of wild type animals. (A) Histopathology scores assigned to every mouse shown in Figure 2 for all indicated parameters on a scale of 0–6. Note that five uninfected controls were examined per group and received scores of 0 throughout. Of the 15 infected WT animals, only 12 were available for histopathological evaluation. (B) Representative micrographs of Giemsa-stained sections of H. felis-infected and uninfected wild type, IL-23p19-/-, and IL-12p35-/- mice, respectively.
Neither Th1 Nor Th17 Cells are Absolutely Required for the Control of Experimental Infection With an H. pylori Patient Isolate
To confirm our findings in an alternative model of Helicobacter-induced disease, we infected animals of the same three genotypes with a recently described H. pylori patient isolate, strain PMSS1 (Arnold et al., 2011). This strain is highly virulent in mice due to its Cag pathogenicity island-encoded type IV secretion system; the pathology induced by PMSS1 is qualitatively similar to H. felis-induced lesions, both in terms of the kinetics and severity of disease (Arnold et al., 2011). As observed with H. felis, there were no significant differences in colonization levels between the three host strains (Figure 4A). The evaluation of gastric histopathology revealed relatively minor differences between the strains, which reached statistical significance for some parameters (Figure 4B). Th17-deficient IL-23p19-/- mice appeared to be somewhat protected against advanced preneoplasia, as atrophy, hyperplasia, and metaplasia were less common and less severe in this group. A similar finding was recently reported by another lab utilizing the PMSS1 derivative SS1 in 3-month infection experiments conducted in the same IL-23p19-/- strain (Horvath et al., 2012), supporting the notion that IL-23-dependent Th17 cells contribute to H. pylori-induced gastric immunopathology. In conclusion, the two infection models combined imply that neither Th1 nor Th17 cells are critical for Helicobacter control, but suggest that IL-23 (and Th17 cells) may contribute to the gastric T cell-driven immunopathology associated with this infection.
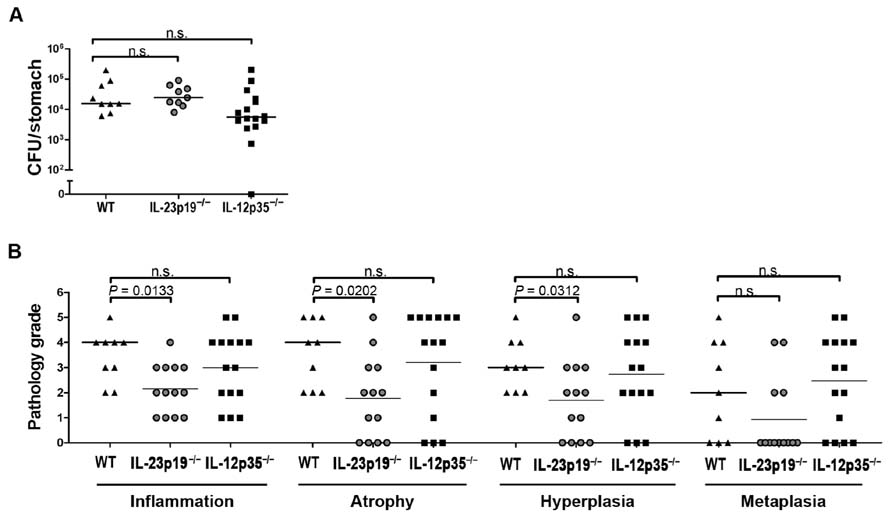
FIGURE 4. IL-23p19-/- and IL-12p35-/- mice do not differ significantly from wild type animals with respect to H. pylori infection control; infection-associated immunopathology is reduced in IL-23p19-/- mice. (A) Colony forming units (CFU) as determined by plating and colony counting of wild type (WT; n = 9), IL-23p19-/- (n = 13), and IL-12p35-/- (n = 15) mice infected with H. pylori strain PMSS1 for 3 months. Note that of the 13 infected IL-23p19-/- animals, only 9 were available for colony counting. (B) Histopathology scores assigned to every mouse shown in (A) for all indicated parameters on a scale of 0–6. Medians are indicated by horizontal lines; each symbol represents one mouse. Data are from two pooled experiments.
Discussion
Several previous studies have addressed the contribution of Th1 and Th17 cells to infection control and gastric preneoplastic pathology. We and others found that IFN-γ production by T cells, and the expression of the Th1 lineage-committing transcription factor T-bet, respectively, are required for H. pylori-induced gastric preneoplasia in mice (Sayi et al., 2009; Stoicov et al., 2009). Similarly, Sheh et al. (2010) noted an increased mutation frequency in the H. pylori-infected murine gastric mucosa that they attributed to H. pylori-specific Th1 responses and prolonged exposure to oxidative stress. The available data on a causal role of Th17 cells in gastric pathology is somewhat conflicting. Whereas Shi et al. (2010) reported that both IL-17 neutralization and IL-17 gene targeting reduced gastric inflammation, the groups of Otani et al. (2009) and Algood et al. (2009) each detected more, rather than less, gastric inflammation upon IL-17 neutralization (Otani et al., 2009) and in IL-17A receptor gene-targeted mice (Algood et al., 2009). All studies available thus far on a possible Th17 contribution to gastric pathology share the caveat of examining one specific Th17 cytokine (IL-17, which, to complicate things further, is also expressed by other cell types such as innate lymphoid tissue inducer cells (Takatori et al., 2009), γ/δ T cells and various non-lymphocyte sources). However, Th17 cells produce two forms of IL-17 – IL-17A and IL-17F – in addition to IL-22 and IL-21, and all cytokines ascribed to the Th17 panel have been linked in one way or another to the pro-inflammatory properties of this Th cell subset (Becher and Segal, 2011). We therefore believe that IL-23p19-/- mice with their complete defect in generating IL-23-dependent Th17 responses (and IL-12p35-/- mice defective for Th1 responses; Cua et al., 2003; McGeachy and Cua, 2008) represent the best currently available models to dissect the contribution of both subsets to immunity and to gastric disease.
To our surprise, we found that neither subset was alone required for the control of H. felis or of H. pylori infection, as colonization levels – known to be tightly controlled by T cells – did not differ across the three mouse strains analyzed. The results suggest that fundamentally different mechanisms govern the spontaneous (rather ineffective) control of Helicobacter infections, and the efficient clearance of H. pylori in vaccinated mice. Whereas the development of vaccine-induced protective immunity and the clearance of H. pylori challenge infections by vaccinated mice depends crucially on both Th subsets (Hitzler et al., 2011), this is not the case for spontaneous infection control. The Th17 dependence of vaccine-induced protection could be attributed directly to the signature cytokine IL-17, as administration of an IL-17-neutralizing antibody during the challenge phase reduced bacterial clearance (Velin et al., 2009). Indeed, the appearance of IL-17+ and IFN-γ+ CD4+ T cells in the gastric mucosa of vaccinated, challenged mice and the production of large amounts of both cytokine transcripts directly precedes H. pylori clearance in all studies that have investigated this association (DeLyria et al., 2009; Velin et al., 2009; Hitzler et al., 2011). The recruitment of neutrophils and mast cells, both of which contribute critically to the effector phase of H. pylori clearance (Velin et al., 2005; Sayi et al., 2009; Hitzler et al., 2011), depends directly on Th1 and Th17 cells and their signature cytokines (Hitzler et al., 2011). The fact that we find levels of both IFN-γ and IL-17 to be high irrespective of p19 and p35 expression argues that, at least in the context of Helicobacter infections, other cell types may serve as more important sources of these cytokines, or factors other than IL-12 and IL-23 are more relevant for governing Th subset polarization. In fact, Wong et al. (2009) have shown previously that macrophage migration inhibitory factor (MIF) is essential for Th1 differentiation in the Helicobacter-infected host, as MIF-deficient mice fail to generate Th1 responses to the infection and thus are protected against Th1-mediated immunopathology.
Our findings on the contribution of Th1 and Th17 cells to gastric infection-associated pathology were less clear. Whereas gastritis and gastric preneoplastic pathology developed in all three strains upon H. felis infection irrespective of IL-23 and IL-12 expression, a clear reduction in most pathology scores was seen in IL-23p19-/- mice in the H. pylori infection model. The results obtained in this model corroborate the findings of a recent study that also reported reduced gastritis upon H. pylori infection of IL-23p19-/- mice (Horvath et al., 2012). The differences obtained by Horvath et al. (2012) were particularly pronounced for chronic inflammation, and less strong with respect to overall inflammation. Preneoplastic pathology characterized by atrophic gastritis, hyperplasia, and metaplasia was not observed upon infection with the SS1 strain used by Horvath et al. (2012) and could therefore not be assessed. While the difference in susceptibility of IL-23p19-/- mice toward H. pylori and H. felis infection is intriguing, the findings need to be reproduced with additional isolates of both species suitable for murine infection before definitive conclusions can be drawn.
In humans, Th responses to H. pylori have been known for a long time to be strongly biased toward Th1. This notion was recently verified once again in a longitudinal study by Perez-Perez et al. (2010), which examined IgG subclass responses in paired serum samples obtained in 1973 and in 1994 from H. pylori-infected, healthy donors. Most (89%) donors exhibited IgG1/IgG4 ratios that were consistent with a predominant Th1 response and were remarkably stable over the 21-year period (Perez-Perez et al., 2010). Although the Th1-predominant response to H. pylori is largely ineffective at clearing the infection (Perez-Perez et al., 2010), it may have positive side effects in the prevention or suppression of co-infections. Perry et al. (2010) recently presented evidence from H. pylori-infected humans and cynomolgus macaques showing that the Th1 response to H. pylori may have beneficial bystander effects on a co-existing Mycobacterium tuberculosis (Mtb) infection. The initial hint of an inverse correlation between active tuberculosis and H. pylori came from the observation that IFN-γ responses to Mtb antigen were 1.5-fold stronger in H. pylori-infected individuals with positive tuberculin skin tests (i.e., latent tuberculosis infection) than in their H. pylori-negative counterparts (Perry et al., 2010). Follow-up studies comparing the H. pylori infection status of active tuberculosis cases and non-progressing household contacts revealed a significantly higher H. pylori seroprevalence in the latter group. This finding was corroborated by a significant inverse relationship between natural H. pylori infection and active tuberculosis in cynomolgus macaques challenged with Mtb (Perry et al., 2010).
In humans, observational studies suggest a contribution of H. pylori-specific Th1 (and Th2) responses to gastric disease; as mentioned above, patients with peptic ulcer disease showed stronger gastric Th1 and Th2 responses to H. pylori than asymptomatic carriers, whose responses had a strong regulatory component (Robinson et al., 2008). On the other hand, polymorphisms in the IL-1 gene cluster suspected of enhancing production of IL-1-beta (IL-1β) are associated with an increased risk of both H. pylori-induced hypochlorhydria and gastric cancer (El-Omar et al., 2000). IL-1 signaling in T cells critically regulates early Th17 differentiation (Chung et al., 2009). IL-1 gene cluster polymorphisms may thus potentially contribute to gastric hypochlorhydria and gastric cancer through the induction of Th17 cells; the observation that the stomach-specific expression of human IL-1β is in itself sufficient to induce gastric inflammation and gastric cancer in transgenic mice supports this notion (Tu et al., 2008). In line with this observation, we have recently reported that IL-1 receptor-deficient mice are completely protected against gastritis and preneoplasia in our H. felis-induced disease model, which we attributed to the complete lack of Th1- and Th17-polarized T cell responses to the infection (Hitzler et al., 2012).
In summary, the results presented here suggest that neither Th1 nor Th17 cells are by themselves absolutely essential for the spontaneous control of Helicobacter infections or for the induction of gastric preneoplastic pathology under all circumstances. Whether both Th subsets synergize to fulfill these functions, or other Th subsets are implicated as well, remains an open question for future investigations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Swiss National Science Foundation and the Zurich Cantonal Cancer League. The authors would like to thank Burkhard Becher for providing backcrossed IL-23p19-/- and IL-12p35-/- mouse strains.
References
Algood, H. M., Allen, S. S., Washington, M. K., Peek, R. M. Jr., Miller, G. G., and Cover, T. L. (2009). Regulation of gastric B cell recruitment is dependent on IL-17 receptor A signaling in a model of chronic bacterial infection. J. Immunol. 183, 5837–5846.
Arnold, I. C., Lee, J. Y., Amieva, M. R., Roers, A., Flavell, R. A., Sparwasser, T., and Müller, A. (2011). Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140, 199–209.
Becher, B., and Segal, B. M. (2011). T(H)17 cytokines in autoimmune neuro-inflammation. Curr. Opin. Immunol. 23, 707–712.
Chung, Y., Chang, S. H., Martinez, G. J., Yang, X. O., Nurieva, R., Kang, H. S., Ma, L., Watowich, S. S., Jetten, A. M., Tian, Q., and Dong, C. (2009). Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30, 576–587.
Correa, P. (1995). Helicobacter pylori and gastric carcinogenesis. Am. J. Surg. Pathol. 19(Suppl. 1), S37–S43.
Cua, D. J., Sherlock, J., Chen, Y., Murphy, C. A., Joyce, B., Seymour, B., Lucian, L., To, W., Kwan, S., Churakova, T., Zurawski, S., Wiekowski, M., Lira, S. A., Gorman, D., Kastelein, R. A., and Sedgwick, J. D. (2003). Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748.
DeLyria, E. S., Redline, R. W., and Blanchard, T. G. (2009). Vaccination of mice against H. pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology 136, 247–256.
El-Omar, E. M., Carrington, M., Chow, W. H., Mccoll, K. E., Bream, J. H., Young, H. A., Herrera, J., Lissowska, J., Yuan, C. C., Rothman, N., Lanyon, G., Martin, M., Fraumeni, J. F. Jr., and Rabkin, C. S. (2000). Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404, 398–402.
Fox, J. G., Sheppard, B. J., Dangler, C. A., Whary, M. T., Ihrig, M., and Wang, T. C. (2002). Germ-line p53-targeted disruption inhibits Helicobacter-induced premalignant lesions and invasive gastric carcinoma through down-regulation of Th1 proinflammatory responses. Cancer Res. 62, 696–702.
Fox, J. G., and Wang, T. C. (2007). Inflammation, atrophy, and gastric cancer. J. Clin. Invest. 117, 60–69.
Fox, J. G., Wang, T. C., Rogers, A. B., Poutahidis, T., Ge, Z., Taylor, N., Dangler, C. A., Israel, D. A., Krishna, U., Gaus, K., and Peek, R. M. Jr. (2003). Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology 124, 1879–1890.
Harris, P. R., Wright, S. W., Serrano, C., Riera, F., Duarte, I., Torres, J., Pena, A., Rollan, A., Viviani, P., Guiraldes, E., Schmitz, J. M., Lorenz, R. G., Novak, L., Smythies, L. E., and Smith, P. D. (2008). Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology 134, 491–499.
Hitzler, I., Oertli, M., Becher, B., Agger, E. M., and Müller, A. (2011). Dendritic cells prevent rather than promote immunity conferred by a Helicobacter vaccine using a mycobacterial adjuvant. Gastroenterology 141, 186–196.
Hitzler, I., Sayi, A., Kohler, E., Engler, D. B., Koch, K. N., Hardt, W. D., and Muller, A. (2012). Caspase-1 has both proinflammatory and regulatory properties in Helicobacter infections, which are differentially mediated by its substrates IL-1beta and IL-18. J. Immunol. 188, 3594–3602.
Horvath, D. Jr., Mk, W., Va, C., and Hms, A. (2012). IL-23 contributes to control of chronic Helicobacter pylori infection and the development of T helper responses in a mouse model. Front. Immunol. 3:56. doi: 10.3389/fimmu.2012.00056
Marshall, B. J., and Warren, J. R. (1984). Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311–1315.
McGeachy, M. J., and Cua, D. J. (2008). Th17 cell differentiation: the long and winding road. Immunity 28, 445–453.
Mueller, A., Sayi, A., and Hitzler, I. (2009). Protective and pathogenic functions of T-cells are inseparable during the Helicobacter–host interaction. Discov. Med. 8, 68–73.
Oppmann, B., Lesley, R., Blom, B., Timans, J. C., Xu, Y., Hunte, B., Vega, F., Yu, N., Wang, J., Singh, K., Zonin, F., Vaisberg, E., Churakova, T., Liu, M., Gorman, D., Wagner, J., Zurawski, S., Liu, Y., Abrams, J. S., Moore, K. W., Rennick, D., De Waal-Malefyt, R., Hannum, C., Bazan, J. F., and Kastelein, R. A. (2000). Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725.
Otani, K., Watanabe, T., Tanigawa, T., Okazaki, H., Yamagami, H., Watanabe, K., Tominaga, K., Fujiwara, Y., Oshitani, N., and Arakawa, T. (2009). Anti-inflammatory effects of IL-17A on Helicobacter pylori-induced gastritis. Biochem. Biophys. Res. Commun. 382, 252–258.
Parsonnet, J., Friedman, G. D., Vandersteen, D. P., Chang, Y., Vogelman, J. H., Orentreich, N., and Sibley, R. K. (1991). Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 325, 1127–1131.
Parsonnet, J., Hansen, S., Rodriguez, L., Gelb, A. B., Warnke, R. A., Jellum, E., Orentreich, N., Vogelman, J. H., and Friedman, G. D. (1994). Helicobacter pylori infection and gastric lymphoma. N. Engl. J. Med. 330, 1267–1271.
Parsonnet, J., and Isaacson, P. G. (2004). Bacterial infection and MALT lymphoma. N. Engl. J. Med. 350, 213–215.
Perez-Perez, G. I., Maw, A. M., Feingold-Link, L., Gunn, J., Bowers, A. L., Minano, C., Rautelin, H., Kosunen, T. U., and Blaser, M. J. (2010). Longitudinal analysis of serological responses of adults to Helicobacter pylori antigens. J. Infect. Dis. 202, 916–923.
Perry, S., De Jong, B. C., Solnick, J. V., De La Luz Sanchez, M., Yang, S., Lin, P. L., Hansen, L. M., Talat, N., Hill, P. C., Hussain, R., Adegbola, R. A., Flynn, J., Canfield, D., and Parsonnet, J. (2010). Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS ONE 5, e8804. doi: 10.1371/journal.pone.0008804
Pritchard, D. M., and Crabtree, J. E. (2006). Helicobacter pylori and gastric cancer. Curr. Opin. Gastroenterol. 22, 620–625.
Rieder, G., Merchant, J. L., and Haas, R. (2005). Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in Mongolian gerbils. Gastroenterology 128, 1229–1242.
Robinson, K., Kenefeck, R., Pidgeon, E. L., Shakib, S., Patel, S., Polson, R. J., Zaitoun, A. M., and Atherton, J. C. (2008). Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut 57, 1375–1385.
Roth, K. A., Kapadia, S. B., Martin, S. M., and Lorenz, R. G. (1999). Cellular immune responses are essential for the development of Helicobacter felis-associated gastric pathology. J. Immunol. 163, 1490–1497.
Sayi, A., Kohler, E., Hitzler, I., Arnold, I., Schwendener, R., Rehrauer, H., and Muller, A. (2009). The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J. Immunol. 182, 7085–7101.
Sayi, A., Kohler, E., Toller, I. M., Flavell, R. A., Muller, W., Roers, A., and Müller, A. (2011). TLR-2-activated B cells suppress Helicobacter-induced preneoplastic gastric immunopathology by inducing T regulatory-1 cells. J. Immunol. 186, 878–890.
Sheh, A., Lee, C. W., Masumura, K., Rickman, B. H., Nohmi, T., Wogan, G. N., Fox, J. G., and Schauer, D. B. (2010). Mutagenic potency of Helicobacter pylori in the gastric mucosa of mice is determined by sex and duration of infection. Proc. Natl. Acad. Sci. U.S.A. 107, 15217–15222.
Shi, Y., Liu, X. F., Zhuang, Y., Zhang, J. Y., Liu, T., Yin, Z., Wu, C., Mao, X. H., Jia, K. R., Wang, F. J., Guo, H., Flavell, R. A., Zhao, Z., Liu, K. Y., Xiao, B., Guo, Y., Zhang, W. J., Zhou, W. Y., Guo, G., and Zou, Q. M. (2010). Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J. Immunol. 184, 5121–5129.
Smythies, L. E., Waites, K. B., Lindsey, J. R., Harris, P. R., Ghiara, P., and Smith, P. D. (2000). Helicobacter pylori-induced mucosal inflammation is Th1 mediated and exacerbated in IL-4, but not IFN-gamma, gene-deficient mice. J. Immunol. 165, 1022–1029.
Stoicov, C., Fan, X., Liu, J. H., Bowen, G., Whary, M., Kurt-Jones, E., and Houghton, J. (2009). T-bet knockout prevents Helicobacter felis-induced gastric cancer. J. Immunol. 183, 642–649.
Takatori, H., Kanno, Y., Watford, W. T., Tato, C. M., Weiss, G., Ivanov, Ii, Littman, D. R., and O’Shea, J. J. (2009). Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206, 35–41.
Tu, S., Bhagat, G., Cui, G., Takaishi, S., Kurt-Jones, E. A., Rickman, B., Betz, K. S., Penz-Oesterreicher, M., Bjorkdahl, O., Fox, J. G., and Wang, T. C. (2008). Overexpression of interleukin-1 beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419.
Velin, D., Bachmann, D., Bouzourene, H., and Michetti, P. (2005). Mast cells are critical mediators of vaccine-induced Helicobacter clearance in the mouse model. Gastroenterology 129, 142–155.
Velin, D., Favre, L., Bernasconi, E., Bachmann, D., Pythoud, C., Saiji, E., Bouzourene, H., and Michetti, P. (2009). Interleukin-17 is a critical mediator of vaccine-induced reduction of Helicobacter infection in the mouse model. Gastroenterology 136, 2237–2246.e1.
Wiedemann, T., Loell, E., Mueller, S., Stoeckelhuber, M., Stolte, M., Haas, R., and Rieder, G. (2009). Helicobacter pylori cag-pathogenicity island-dependent early immunological response triggers later precancerous gastric changes in Mongolian gerbils. PLoS ONE 4, e4754. doi: 10.1371/journal.pone.0004754
Keywords: T helper cell subsets, gastric immunopathology, gastric cancer precursor lesions, experimental infection models
Citation: Hitzler I, Kohler E, Engler DB, Yazgan AS and Müller A (2012) The role of Th cell subsets in the control of Helicobacter infections and in T cell-driven gastric immunopathology. Front. Immun. 3:142. doi: 10.3389/fimmu.2012.00142
Received: 28 February 2012; Paper pending published: 05 April 2012;
Accepted: 16 May 2012; Published online: 01 June 2012.
Edited by:
Thomas Blanchard, University of Maryland School of Medicine, USAReviewed by:
Diane Bimczok, University of Alabama at Birmingham, USAHua Ding, University of Maryland School of Medicine, USA
Copyright: © 2012 Hitzler, Kohler, Engler, Yazgan and Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Anne Müller, Institute of Molecular Cancer Research, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. e-mail:bXVlbGxlckBpbWNyLnV6aC5jaA==