

 Christopher B. Fox1
Christopher B. Fox1
- 1 Pre-clinical Biology, Infectious Disease Research Institute, Seattle, WA, USA
- 2 Department of Microbiology and Immunology, McGill University, Montreal, QC, Canada
Two million new cases of leishmaniasis occur every year, with the cutaneous leishmaniasis (CL) presentation accounting for approximately two-thirds of all cases. Despite the high incidence rates and geographic expansion of the disease, CL remains a neglected tropical disease without effective intervention strategies. Efforts to address this deficit have given rise to the experimental murine model of CL. By virtue of its simplicity and pliability, the CL model has been used to provide substantial information regarding cellular immunity, as well as in the discovery and evaluation of various vaccine adjuvants. The CL model has facilitated in vivo studies of the mechanism of action of many adjuvants, including the TLR4 agonist monophosphoryl lipid A, the TLR7/8 agonist imiquimod, the TLR9 agonist CpG, adenoviral vectors, and the immunostimulatory complexes. Together, these studies have helped to unveil the requirement for certain types of immune responses at specific stages of CL disease and provide a basis to aid the design of effective second-generation vaccines for human CL. This review focuses on adjuvants that have been tested in experimental CL, outlining how they have helped advance our understanding of the disease and ultimately, how they have performed when applied within clinical trials against human CL.
Introduction
The leishmaniases, a group of diseases caused by infection with various species of the protozoan parasite genus Leishmania, are endemic in 88 countries. Together, ~350 million people are at risk of infection with these parasites with the leishmaniases having an overall prevalence of 12 million cases worldwide and accounting for about 60,000 deaths annually (Desjeux, 2001). Expansion of disease and the sharp rise in prevalence has been linked to migration of non-immune people into endemic areas as well as environmental changes resulting in the expansion of the insect vector habitats (WHO, 2010). The latter, in particular, has the potential to widen the geographic reach of the vector and introduce leishmania transmission into previously unaffected areas. Although an estimated 2 million new cases occur annually – 1.5 million cases of cutaneous leishmaniasis (CL) and 500,000 of visceral leishmaniasis (VL) – leishmaniases still represent a neglected tropical disease and have not been considered as attractive vaccine targets by industry. Despite efforts that now span more than a century, a licensed vaccine for use in humans is not yet available.
First-Generation Vaccines for Leishmaniases
The earliest records of vaccination against leishmaniasis date back several hundred years, when leishmanization was practiced in the Middle East. Bedouin tribal societies traditionally exposed their children to sandfly bites in order to protect them from facial lesions. In some cases pus from active lesions was also inoculated by excoriation of the recipient’s arm or thigh (Greenblatt, 1980). This technique was refined in the early 1900s when culture conditions were established for leishmania promastigotes, which were then used to both prevent and treat active disease (Row, 1912; Wenyon, 1928; Greenblatt, 1980). These studies formed the basis for large-scale vaccination trials that were carried out in the Soviet Union, Israel, and Iran (Greenblatt, 1980; Nadim et al., 1983). The success in these trials depended critically on the viability and infectivity of the injected organisms. Organisms which had lost virulence were shown to induce delayed-type hypersensitivity but did not protect from subsequent natural infection. Additionally use of live vaccines had many problems, including the development of large uncontrolled skin lesions, exacerbation of psoriasis, and other skin diseases, which paved the way to trials using killed parasites but conflicting results made this approach unpopular (Handman, 2001). However, this strategy regained popularity in the late 1970s after the route of immunization was found to be crucial for its efficacy (Mayrink et al., 1979; Greenblatt, 1980; Handman, 2001). Convit and colleagues used a combination of killed L. mexicana or L. braziliensis promastigotes and Mycobacterium bovis Bacille Calmette-Guérin (BCG) therapeutically against leishmaniasis in South America. Vaccination of clinically defined patients appeared to induce a high cure rate even in severe cases and was accompanied by the development of T helper type 1 (Th1) immune responses in the recipients (Castes et al., 1989; Cabrera et al., 2000). Prophylactic vaccinations with killed parasites formulated with or without BCG, however, have been less successful (Armijos et al., 1998; Sharifi et al., 1998; Momeni et al., 1999).
Although two of the most successful vaccines used to date (against small pox and polio, respectively) are live pathogens, that strategy does not appear feasible for leishmaniasis. Results from several clinical trials using whole Leishmania (involving either live or killed parasites) vary from 0 to 75% efficacy against CL (Coler and Reed, 2005; The Working Group on Research Priorities for Development of Leishmaniasis Vaccines, 2011). Several investigators are investigating the potential of live-attenuated vaccines for CL. Some genetically or chemically attenuated parasites appear to persist indefinitely without pathology and confer long lasting protection in mice (Titus et al., 1995; Papadopoulou et al., 2002; Uzonna et al., 2004; Davoudi et al., 2005; Daneshvar et al., 2010; Birnbaum et al., 2012). Although not yet evaluated in clinical trials, it is conceivable that these could offer lasting protection in humans. Given their complex and heterogeneous nature, however, it is unclear how they will overcome current vaccine regulations and release criteria. Studies using these crude vaccine approaches, though not ideal, have however proved that it is possible to induce protection and suggest that this is also achievable with parasite components. Several investigators have addressed this issue using the experimental murine model of CL. The simplicity and pliable nature of this model (differential outcomes dependent upon mouse strain and infective dose) has permitted researchers to address basic questions of immunity and have provided significant insight regarding the development of cellular immune responses.
Insight Into Cellular Immunity: The CL Mouse Model
Experimental murine infection with L. major has facilitated our understanding of the Th1/Th2 paradigm, the development of helper T cells during an infection and factors that regulate their maintenance (Locksley and Scott, 1991; Scott, 1991; Reed and Scott, 1993). Additionally, this model has revealed genetic mechanisms of protection conferred by Th1 cells in resistant strains and susceptibility induced by Th2 cells in the BALB/c model (Reed and Scott, 1993; Scharton-Kersten et al., 1995; Park et al., 2000; Hutchins et al., 2005). Most inbred mouse strains (e.g., C57BL/6, CBA/J, and C3H) are considered resistant to infection with L. major. Upon intradermal/sub-cutaneous injection of L. major, these strains develop as mall lesion that subsides within 6–8 weeks (Scott, 1998). Healing in resistant mice infected with L. major is associated with the development of Th1 cells that produce IFN-γ, which is necessary for activation of macrophages and the production of nitric oxide (NO), the key effector molecule for destroying intracellular amastigotes (Liew et al., 1990a,b; Stenger et al., 1994). These mice are also protected against additional L. major challenges. In contrast, BALB/c mice are highly susceptible to L. major infection; parasite burdens can become extremely high, with lesions progressing until ulceration. Deconstructing the failure of BALB/c mice to protect against infection helped to identify the crucial roles of IL-4 and IL-13 in the promotion and expansion of Th2 cells. Both the initial development and maintenance of Th1 cells in infected mice is dependent on the presence of IL-12 produced by dendritic cells. Treatment of BALB/c mice with recombinant IL-12 helps to protect against infection while, conversely, treatment of resistant strains with anti-IL-12 promotes the development of a Th2 response and susceptibility (Heinzel et al., 1993, 1998; Hondowicz et al., 1997, 2000; Fernandez-Botran et al., 1999; Park et al., 2000; Kedzierski et al., 2008). Another key cytokine that has been shown to regulate disease outcome is IL-10. Disruption of the IL-10 gene confers resistance while over-expression of this gene in resistant strains renders them susceptible. IL-10 appears to mediate its effect by blocking the activating effects of IFN-γ. In addition to deactivating macrophages and inhibiting intracellular parasite killing, IL-10 can also directly inhibit Th1 cell development and their production of IFN-γ (Kane and Mosser, 2001). Furthermore, IL-10 production by natural CD4+CD25+ regulatory T cells plays an important role in disease pathology (Belkaid et al., 2002a; Mendez et al., 2004).
Importantly, given its flexibility and broad understanding, the CL mouse model has helped construct a syllogism that supports a platform for adjuvant discovery/selection. By virtue of its simplicity, it has also facilitated the study the mechanism of action of these adjuvants in vivo (Mendez et al., 2003; Darrah et al., 2007, 2010). Among the adjuvants investigated using this model are the TLR4 agonist-monophosphoryl lipid A (MPL®), the TLR7/8 agonist – imiquimod, the TLR9 agonist – CpG, and the immunostimulatory complexes (ISCOM; Figure 1). There is some irony that very few of these adjuvants have actually been evaluated against human CL, although this lack of progression is at least partially attributable to the fact that human CL is a much more complex disease than the experimental model. These adjuvants have, however, helped scientists understand the need for certain types of immune responses at specific stages of disease, which in turn, could aid in designing an effective vaccine for human CL. This review focuses on adjuvants that have been tested in experimental CL, their effects when applied in human CL, and how they have helped advance our understanding of the disease.
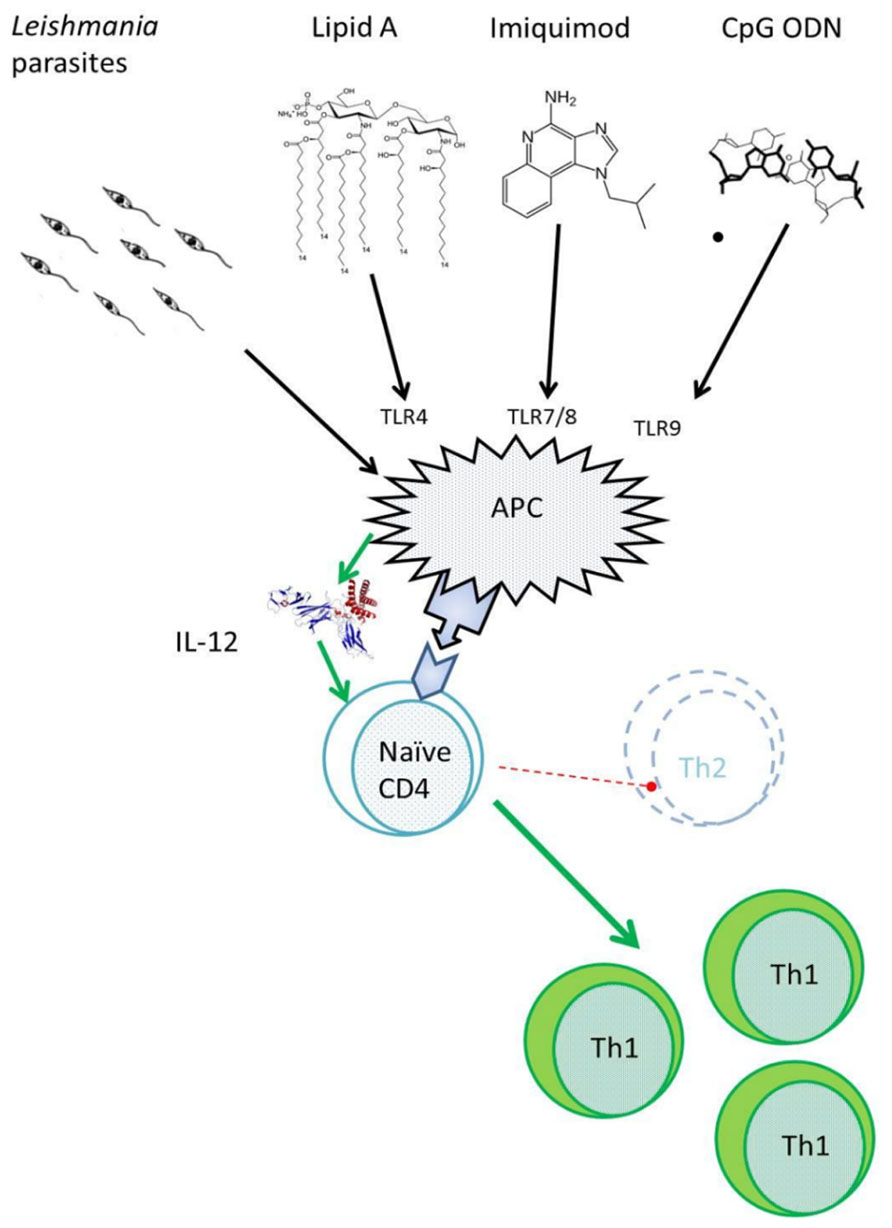
FIGURE 1. Toll-like receptor agonists as adjuvants. All pathogens including bacteria, viruses, and protozoans induce activation of antigen presenting cells (APC) resulting in the production of IL-12. Adjuvant biology relies on this same approach by using TLR agonists that have been derived from these organisms to induce IL-12 production. The induction of IL-12 helps generate a strong Th1 response and inhibit Th2 responses, a key component in battling intra-cellular infections such as Leishmania.
Adjuvant Biology and the Advancement Toward Second Generation Vaccines
Innate immunity forms the host’s first line of defense against microbial invasion. Targeting innate immune receptors with adjuvants is a logical strategy, particularly for infections of innate cells such as leishmaniasis. Studies of adjuvants began more than a hundred years ago with the discovery that an acute bacterial infection could induce regression in a concurrent malignant tumor (Tokunaga, 1980; Bickels et al., 2002). Surprisingly however, the “immunologist’s dirty little secret” (or rather the killed M. tuberculosis) present in Freund’s adjuvant was almost taken for granted until 1989 when Charles Janeway Jr. declared that “the innate immune response (it induced) was prerequisite for an adaptive immune response” (Janeway, 1989). Janeway’s prediction was eventually proven by a number of labs using classical genetic methods, most clearly when TLR4 was identified as the receptor for lipopolysaccharide (LPS), and TLR9 was shown to recognize bacterial DNA (Poltorak et al., 1998; Hemmi et al., 2000; Duthie et al., 2011). The identification of pathogen associated molecular patterns (PAMPs; a group of lipid, carbohydrate, peptide, and nucleic-acid structures, broadly expressed by different microorganisms) and their associated pathogen-recognition receptors (PRRs), has revolutionized vaccine research (Janeway, 1989; Tokunaga et al., 1999; Bickels et al., 2002; Janeway and Medzhitov, 2002; Medzhitov and Janeway, 2002; Pashine et al., 2005; Medzhitov, 2009; Reed et al., 2009). Directed stimulation of PRR, which include the Toll-like receptors (TLRs), C-type lectin-like receptors (CLRs), cytosolic nucleotide oligomerization domain-like receptors, and retinoic acid inducible gene based (RIG)-I-like receptors (RLRs), can trigger different types of immune responses (Janeway and Medzhitov, 2002; Medzhitov and Janeway, 2002; McGreal et al., 2005; Carneiro et al., 2007; Kawai and Akira, 2007; Onomoto et al., 2007). Interaction between PRRs and their cognate PAMPs initiates a complex cascade of intracellular signaling pathways leading to the production of cytokines, chemokines, and type I IFNs that mediate the induction of inflammatory and antimicrobial responses. This process activates an adaptive immune response tailor-made to the invading microbe. These PAMPs, specifically those binding the TLRs, are the basis of many adjuvants. In addition, cytokines, bacterial toxins, and glycolipids released during or after PRR engagement are also being used as adjuvants to elicit immune responses (Pashine et al., 2005; Reed et al., 2009; Duthie et al., 2011). Advances in molecular and cellular biology are rapidly revealing the nature of innate immunity and adjuvant activity. The discovery of the ability of adjuvants to enhance disease-specific immune responses using recombinant antigens has led many researchers to re-focus their vaccine development programs (Reed et al., 2009). Effective adjuvants and adjuvant formulations can utilize multiple compounds and mechanisms to achieve the desired immunological enhancement (Schijns, 2000; Pashine et al., 2005; Reed et al., 2009). These mechanisms can enhance antigen presentation, leading to induction of effective humoral (B cell) and cellular (CD4 and/or CD8 T cell) responses.
Despite the dramatic advances in our understanding of defined PRR, the majority of the vaccines that have been licensed and made available for clinical use remain live, attenuated, or killed vaccines (WHO, 2004; The European Medicines Agency, 2005; WHO Department of Immunization, Vaccines and Biologicals and UNICEF Programme Division, Health Section, 2005; National Advisory Committee on Immunization, 2008; Central Drugs Standard Control Organization, 2010; U.S. Food and Drug Administration, 2011). An ideal vaccine against leishmaniasis must be safe, effective, and given its recognition as a neglected disease, affordable. As addressed earlier, it appears that a live or killed parasite vaccine will not be feasible for leishmaniasis. An inherent and major advantage of vaccines based on attenuated pathogens and inactivated viruses or bacteria is that they are sufficiently immunogenic without additional adjuvants, likely through the undefined inclusion of intrinsic PAMPs. Such activity has been revealed for the highly effective Yellow Fever vaccine (Querec et al., 2006). By contrast, protein-based vaccines, although offering considerable advantages over traditional vaccines in terms of safety and cost of production, are often less immunogenic.
Over the past few decades, many researchers have searched for genes encoding leishmanial proteins that are linked to the protective response and can induce protection against CL in experimental models (Handman et al., 1995; Mougneau et al., 1995; Skeiky et al., 1995a,b, 1997, 1998; Webb et al., 1996, 1997; Probst et al., 1997; Campos-Neto et al., 2002; Coler et al., 2002). Identifying candidate antigens alone, however, is not sufficient as they need to be characterized to ensure quality and stability. More importantly, candidate antigens need to be delivered with appropriate adjuvants to elicit an appropriate immune response (Coler and Reed, 2005; Reed et al., 2009). A significant research effort has focused on adjuvants in CL (Table 1). This review outlines the efforts of several labs, including our own, regarding the use of adjuvants both in CL animal models and their impact on efficacy in the clinic.
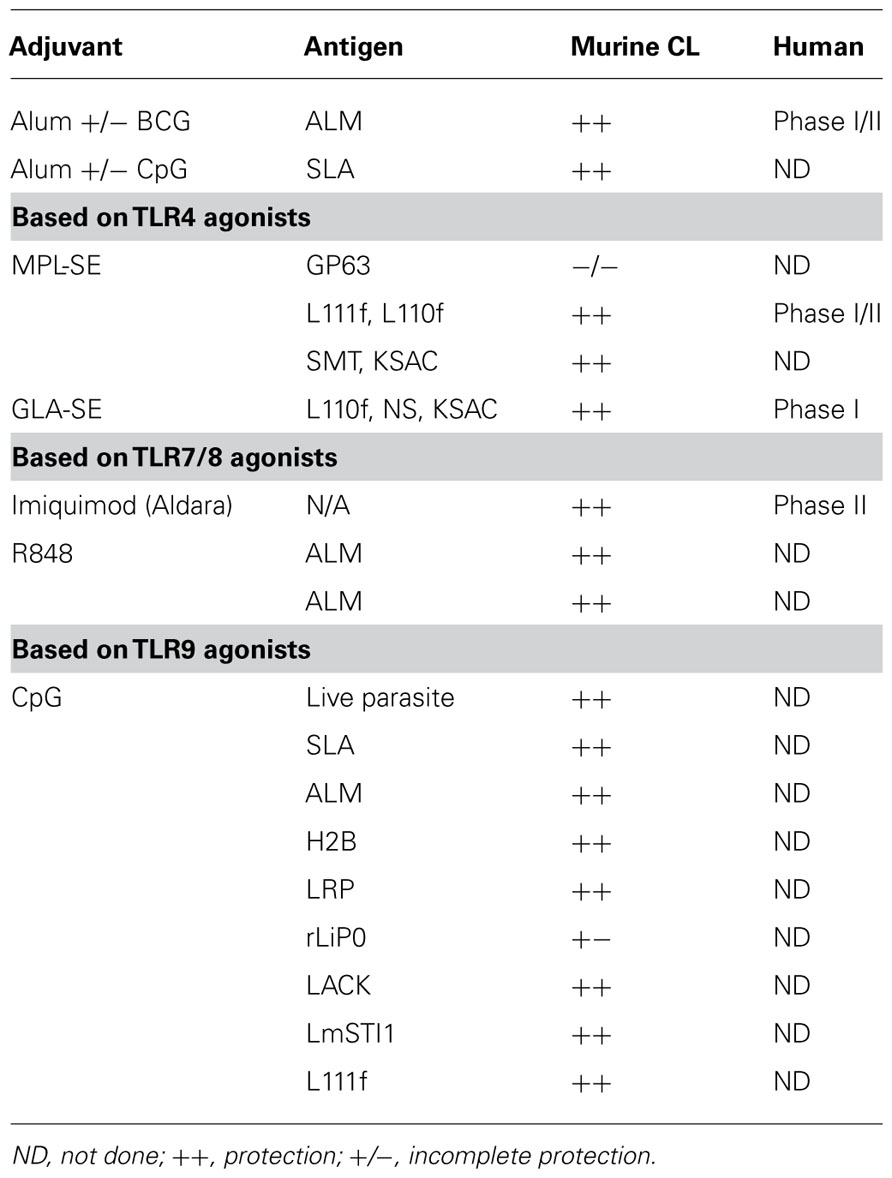
Table 1. List of adjuvants that have been used in experimental and/or clinical CL.
Aluminum Salts (Alum) as Adjuvant
Introduced in the 1920s, alum became the first adjuvant to be approved by the U.S. Food and Drug Administration for use in humans (White et al., 1955; Marrack et al., 2009; Reed et al., 2009). Although the initial and long-held theory was that alum facilitated an immune response by providing a depot, more recent studies have challenged this theory and suggest otherwise (White et al., 1955; Gupta et al., 1996). Studies by Brewer et al. (1999) indicated that alum boosted humoral immunity by providing Th2 cell help to follicular B cells. Additional properties of alum were revealed recently by the demonstration that it can induce the production of monocyte and granulocyte chemoattractants and enhance monocyte differentiation into dendritic cells (Kool et al., 2008; Seubert et al., 2008). It is now apparent that alum can act through both NLRP3-dependent and -independent pathways to induce type 2 inflammatory responses, characterized by the accumulation of eosinophils at the site of injection and the enhanced production of antigen-specific IgE and IgG1 (Li et al., 2007, 2008; Eisenbarth et al., 2008; Reed et al., 2009; Kuroda et al., 2011; Marichal et al., 2011). A number of vaccines, including whole parasite vaccines and recombinant vaccines such as those for Hepatitis B and human papilloma virus, have been developed using alum with the intent of eliciting protective antibody responses (WHO Department of Immunization, Vaccines and Biologicals and UNICEF Programme Division, Health Section, 2005). A major advantage of alum is its ability to enhance humoral immunity with minimal side-effects. Additional benefits include, stabilization of the associated antigens with alum (because formulation is achieved through adsorption of antigens onto highly charged aluminum particles) and the relative simplicity to produce on a large scale. To date, the alum adjuvant delivery system has been used in over 80% of vaccines administered worldwide. The inability of alum to elicit cell-mediated Th1 or CTL responses required for the control of most intracellular pathogens (such as Leishmania) is a major limitation. Alum has, however, been used as a carrier/vehicle for a number of new age adjuvants including IL-12, GM-CSF, CpG, and MPL (Kenney et al., 1999; Stacey and Blackwell, 1999; Tonui et al., 2004; Kundi, 2007; Frazer and Levin, 2011). Most importantly, the recent approval of MPL-Alum® for two different vaccines, Fendrix and Cervarix, has set precedence for the use of TLR4 agonists within human vaccines (Kundi, 2007; Duthie et al., 2011; Frazer and Levin, 2011).
In relation to leishmaniasis, alum has been used in combination with BCG or IL-12 and autoclaved L. major (ALM) to protect against experimental murine CL (Kenney et al., 1999; Soudi et al., 2011). The first clinical study in leishmaniasis that used alum in conjunction with ALM and BCG was conducted for the treatment of VL. Data indicated that the approach was not only safe but could induce strong delayed type hypersensitivity (DTH) reactions in study volunteers (Kamil et al., 2003). More recently, alum-precipitated ALM and BCG was used as part of an immunochemotherapeutic approach to treat post-kala-azar dermal leishmaniasis (PKDL) in Sudan (Musa et al., 2008). This randomized and double-blind study aimed to assess safety, immunogenicity, and curative potential of this regimen in patients with persistent PKDL. Patients were treated with sodium stibogluconate (SSG) + alum-precipitated ALM and BCG. SSG + alum/ALM + BCG was found to be safe and immunogenic, and demonstrated significant healing potential in persistent PKDL lesions.
The applicability of alum-based formulations for leishmania vaccines is neither supported by current knowledge regarding alum’s mechanism of action nor by clinical or experimental experience.
Interleukin-12
Interleukin-12, originally called natural killer cell stimulatory factor, was identified by Trinchieri and colleagues, who discovered that it can stimulate the production of IFN-γ from T cells and natural killer cells (Kobayashi et al., 1989; Chan et al., 1991; Manetti et al., 1993). Further studies demonstrated the ability of several pathogens to induce a Th1 response correlated with their capacity to stimulate the production of IL-12 (Hsieh et al., 1993; Scott, 1993). The critical involvement of IL-12 in promoting Th1 responses in vivo was demonstrated using the mouse model of CL (Sypek et al., 1993). The use of IL-12 as an adjuvant was subsequently examined in various models including the murine CL model (Sypek et al., 1993; Scott and Trinchieri, 1997; Wright et al., 2008; Metzger, 2010). Effective vaccination against L. major in susceptible mouse strains was achieved using recombinant IL-12 with soluble leishmanial antigen (SLA) and with a recombinant LACK protein (Afonso et al., 1994; Mougneau et al., 1995). Several studies have demonstrated the short-lived nature of protection in mice following immunization with recombinant LACK plus IL-12 protein. Protection waned 2 weeks after immunization, whereas protection was sustained up to 12 weeks later when the IL-12 and LACK genes were delivered within a DNA construct (Gurunathan et al., 1997, 1998; Stobie et al., 2000; Mendez et al., 2001). These studies suggested that the continued presence of IL-12 as a requirement for long-lived vaccine-induced immunity.
The safety, immunogenicity, and efficacy of a vaccine combining heat-killed L. amazonensis with recombinant human (rh) IL-12 and alum (aluminum hydroxide gel) as adjuvant, has been assessed in rhesus macaques. When lesion development was followed after intradermal challenge, an IL-12 dose response was evident and protective immunity was demonstrated in all monkeys (12 total) that received 2 μg rhIL-12 at all Ag/alum doses (Kenney et al., 1999). When vervet monkeys were immunized with ALM with or without rhIL-12, despite the generation of substantial Leishmania-specific immune responses, no protection was observed against subsequent challenge with L. major parasites (Gicheru et al., 2001). The transition to use of rhIL-12 in humans has proven difficult, however, as parenteral administration over several days was found to be highly toxic (Cohen, 1995; Wright et al., 2008).
TLR4 Agonists – Lipid A and Derivatives
Lipopolysaccharide, a complex amphipathic molecule that covers the outer surface of Escherichia coli and other Gram-negative bacteria such as M. tuberculosis was the first microbial product discovered to be a TLR agonist. Although lipid A had been defined as the hydrophobic moiety of LPS for over 50 years, the elucidation of lipid A biosynthesis was not achieved until the early 1980s. The ability of the “free” lipid A molecule to induce tumor regression, endotoxic shock, interferon production, and macrophage activation generated much pathophysiological interest (Takayama et al., 1983). Determination of the lipid A structure was followed by attempts to chemically modify lipid A, generally with the intent of uncoupling its toxic effects from its potentially useful immunomodulatory properties. This resulted in the identification of an acylated diglucosamine derivative of lipid A from Salmonella minnesota RC595, which was then further refined to derive MPL® with reduced pyrogenicity (Persing et al., 2002).
Even though the existence of LPS receptors had been identified in the 1970s to be dependent on the integrity of a single locus known as Lps, the locus was not defined until 1998, when it was demonstrated through positional cloning to be identical to the TLR4 (Tlr4) locus6 (Skidmore et al., 1976; Poltorak et al., 1998). TLR4’s ability to utilize both the MyD88 and TRIF signaling pathways has been critical to the success of lipid A as an adjuvant. While it is not clear if a single TLR4 molecule can signal through one or both pathways at the same time, it has been suggested that MyD88/MAL is required for the rapid activation of TLR4 signaling and for the production of TNF, while TRAM stimulates a more sustained induction of NF-κB through the MyD88-independent pathway. Studies with MyD88 KO and TRIF KO mice have indicated that signaling through MyD88/MAL is essential for IL-12 production, and the resulting IFN-γ, while signaling through the TRIF/TRAM pathway appears to be crucial for IL-6 production. Further research defining the roles of these two signaling pathways in TLR4 activation is required (O’Neill and Bowie, 2007).
The efficacy of MPL® was first tested in a syngeneic guinea pig cancer model, where it synergistically enhanced the ability of mycobacterial cell wall skeleton (CWS) to regress transplantable, line-10 tumor (hepatocellular carcinoma; Ribi et al., 1984). Later studies demonstrated that MPL can also enhance immune responses to both killed virus and purified proteins (Masihi et al., 1986). Based upon those studies adjuvanting a mixture of well-defined recombinant antigens together with MPL was a logical progression for a vaccine against leishmaniasis.
Monophosphoryl lipid A was first evaluated against leishmania by comparison against alternative adjuvants in a prophylactic study in the murine model of CL with L. mexicana with a subunit vaccine (Aebischer et al., 2000). The ability of MPL to adjuvant antigen-specific protection against experimental CL was contrasted against a panel of five other adjuvants (IL-12, QS-21, M. bovis BCG, Corynebacterium parvum – now known as Propionibacterium acnes, and Detox). By its ability to protect against CL infection, Detox, an adjuvant formulation made up of MPL and mycobacterial CWS, was shown to be almost equal in efficacy to IL-12. MPL alone induced similar results, albeit with a slightly lower efficacy than either Detox or IL-12. In later studies conducted at IDRI, MPL was formulated in an oil-in-water formulation (MPL-SE) which helped improve its efficacy (Coler and Reed, 2005). Several studies have demonstrated that the inclusion of MPL-SE induces a strong type 1 profile, with high levels of IFN-γ and comparatively low levels of type 2 cytokines such as IL-4 and IL-10. Serum immunoglobulin also becomes skewed toward a higher IgG2a/IgG1 ratio when MPL is present in the vaccine formulation (Coler and Reed, 2005). Further, use of MPL within prophylactic vaccines can induce multi-functional, memory T cells, an established correlate of protection in the murine CL model (Darrah et al., 2007; Bertholet et al., 2009). Moreover, this adjuvant has shown equivalent efficacy in both murine CL and VL models (Coler et al., 2007; Goto et al., 2007).
Of the TLR agonists identified, MPL is the most advanced and is currently the only licensed adjuvant commercially available today. MPL adjuvant has proven to be a potent yet apparently non-toxic vaccine adjuvant when administered with heterologous antigens. MPL has been used extensively as an adjuvant in clinical trials for several infectious disease and cancer indications, and to date, over 100,000 doses of MPL have been administered to human subjects. The safety profile is similar to that of alum. The adjuvant system AS04 (GlaxoSmithKline Biologicals), a combination of aluminum salts and MPL has been approved in both Europe and the United States. Licensed AS04 containing vaccines, Fendrix and Cervarix confer protective immunity against hepatitis B virus or human papilloma virus, respectively. Approval of Cervarix in 2009 made MPL the first defined adjuvant to be licensed for use in vaccines in the United States. AS04 has now been shown to induce a transient, localized innate immune response and activation of DCs, leading to activation of antigen-specific T cells and enhanced adaptive immunity (Didierlaurent et al., 2009). As predicted by its ability to induce IL-12, despite the presence of alum, inclusion of MPL in AS04 biases the immune response to a strong Th1 type phenotype.
IDRI has also used MPL to spearhead several clinical trials directed against leishmaniasis. Our first trial investigated a mixture of antigens for the ability to treat drug-refractory patients suffering from mucosal leishmaniasis (ML; Badaro et al., 2006). The trial was successful with five/six patients showing complete clinical remission at the 9-month follow-up and all six becoming asymptomatic at the 5-year follow-up examination. The antigens had been adjuvanted with GM-CSF at that time but adjuvant was subsequently switched to MPL-SE based practical issues and on results from our pre-clinical studies (Coler and Reed, 2005). The antigens are now expressed as a single entity within Leish F1 chimeric fusion protein, rendering the antigen configuration more accessible to cost-effective, large-scale production. This vaccine was tested in both healthy Columbian and Indian volunteers with either no history of leishmaniasis or sub-clinical infection with L. braziliensis/L. panamensis (detection based on the Montenegro skin test). An increase in anti-Leish F1 IgG response was observed in all, and an antigen-specific IFN-γ response was observed in the majority of recipients (Velez et al., 2009; Chakravarty et al., 2011; Duthie et al., 2012). Based on these results we concluded that the vaccine was safe and well-tolerated in subjects with and without evidence of prior infection. Additionally, we have evaluated the LEISH-F1 + MPL-SE vaccine as an adjunct immunotherapy with standard chemotherapy in both CL and ML. The vaccine was found to be safe and immunogenic in both CL and ML patients and appeared to shorten their time to cure when used in combination with meglumine antimoniate chemotherapy. Further treatment of ML patients with vaccine helped induce a memory T cell subset, associated with clinical cure (Llanos-Cuentas et al., 2010). These studies have proven that the vaccine is safe for use in both healthy and infected patients, and is able to induce a memory response that could help protect against future infections.
We have further optimized the adjuvant MPL by developing a synthetic lipid A derivative that eases manufacture and provides a more homogenous final product. Like MPL, formulations of glucopyranosyl lipid adjuvant (GLA) also signals through TLR4 (Coler et al., 2011). When formulated with SE, an oil-in-water emulsion, and mixed with antigen, the resulting Ag + GLA-SE mixture induces potent Th1 immune response in mice. Studies show that GLA-SE and MPL-SE adjuvanted responses to be dose-dependent, skewing toward a Th1-like response in mice that receive either of these TLR4 agonists. We have also established that GLA-SE is a more potent adjuvant compared to MPL-SE in terms of stimulating antigen-presenting cell activation and inducing greater IFN-γ secretion by antigen-specific T cells. Low doses of GLA-SE are capable of inducing a high frequency of multi-functional effector T cells (Bertholet et al., 2009). Recent work has also suggested that either TLR4-based adjuvant (MPL and GLA) represent good candidates for CL immunotherapy when coupled with the TLR9 ligand, CpG (Raman et al., 2010). Together, these data indicate considerable promise for the development of a safe, effective, and inexpensive vaccine for both prophylactic and therapeutic application against leishmaniasis.
TLR7/8 Agonists – Resiquimod and Imiquimod
Initial characterization studies had demonstrated that imiquimod could induce production of antiviral cytokines, including IFN-α, TNF, and IL-1β, from monocytes (Testerman et al., 1995). Imiquimod could also augment the Th1 immune response by mediating the release of IL-12 and IFN-γ from macrophages (Wagner et al., 1999). It was revealed, that treatment of L. donovani-infected macrophages with imiquimod resulted in killing of intracellular amastigotes and that this was dependent on the production of NO by the treated macrophages (Buates and Matlashewski, 1999). Through a gene array approach it was further demonstrated that, consistent with the ability of imiquimod to activate killing of intracellular Leishmania amastigotes, imiquimod’s related compound S-28463 induced gene expression associated with macrophage activation and an inflammatory response including NF-κB, IL-1, iNOS, and MIP-1 (Buates and Matlashewski, 2001). Only later was it discovered that imiquimod structurally resembled single stranded RNA that was capable of activating macrophages through stimulating the TLR7 pathway (Hemmi et al., 2002).
Based on these experimental observations and because topical treatment with imiquimod had obtained FDA approval in 1997 (for the treatment of cutaneous cervical warts caused by human papilloma viruses infections), a small human trial was conducted to treat CL patients (Arevalo et al., 2001). This study, supported by WHO-TDR was conducted in Peru on 12 CL patients infected with L. V. braziliensis that had failed an initial treatment with pentavalent antimony. Study subjects were given a second course of pentavalent antimony plus topical imiquimod every 2 days for 20 days. The topical application of imiquimod on the lesions was well tolerated and 11 out of 12 patients were cured at the 3-month follow-up period (Arevalo et al., 2001). Subsequently a placebo controlled study was conducted on 40 CL patients who had failed treatment with pentavalent antimony and were given a second treatment with the combination of pentavalent antimony plus topical imiquimod or a placebo cream. The outcome was a 72% cure rate for the group receiving imiquimod versus 35% cure for the group receiving placebo at the 3-month follow-up (Miranda-Verastegui et al., 2005). Overall these observations indicated that combination therapy with topical imiquimod plus pentavalent antimony was well tolerated and this represented a good option for treating patients who had failed the first line treatment with pentavalent antimony alone.
A subsequent study was undertaken to determine whether topical imiquimod combined with pentavalent antimony could be considered as a first line therapy for CL in Peru (Miranda-Verastegui et al., 2009). This randomized placebo control study showed that the combination treatment of imiquimod plus pentavalent antimony performed better (75% cure) than placebo plus pentavalent antimony (58% cure) at the 3-month follow-up period, but that the difference was not statistically significant. A study conducted in Iran involving L. tropica infections reported that there was no beneficial effect of combining topical imiquimod with pentavalent antimony (Firooz et al., 2006). It is possible that the topical imiquimod was unable to penetrate to the infected cells because, in comparison to the ulcer lesions typical of New World CL, L. tropica infections are typically nodular.
An interesting recent report describes the treatment of a 7-year-old boy with a lesion on the left cheek caused by a L. infantum infection (Hervas et al., 2011). For more than a year this patient had failed various treatments, including topical paromomycin, intralesion injection of pentavalent antimony, photodynamic therapy, and two courses of intravenous liposomal amphotericin B. The patient was finally treated with topical imiquimod and cured within a month (Hervas et al., 2011). Treatment with topical imiquimod alone was attempted on L. V. braziliensis infections in Peru and although this provided initial lesion regression, complete cure was not obtained (unpublished observation). For this reason, imiquimod has typically been used in combination therapy in the New World. Based on the available published evidence, a World Health Organization appointed expert committee on control of leishmaniasis recently updated it treatment guidelines to now include the use of imiquimod in combination with pentavalent antimony as a second-line therapy for CL in the New World (WHO, 2010).
Given the adjuvant properties of topical imiquimod in treatment of infectious diseases, topical imiquimod has also been tested as a vaccine adjuvant in an experimental model of CL in BALB/c mice. It was found that application of topical imiquimod on the skin prior to subcutaneous immunization with crude Leishmania antigen provided increased protection against challenge infection compared to immunization of crude antigen alone and this was associated with an enhanced Th1 response against the vaccine antigen (Zhang and Matlashewski, 2008). Subsequently, it was likewise shown that topical application of imiquimod at the site of subcutaneous injected Plasmodium falciparum circumsporozoite (CS) peptide elicited a strong Th1 response and high antibody titers that provided protection against a challenge infection (Othoro et al., 2009). These studies show that further consideration should be given to using topical imiquimod as a vaccine adjuvant for any pathogen antigen delivered subcutaneously, particularly where a Th1 response is required for protective immunity.
TLR9 Agonists – CpG and Immunomodulatory Oligos
The concept of immunostimulatory DNA was first identified in the early 1980s during studies on BCG-mediated tumor resistance (Tokunaga et al., 1999). The identification of CpG dinucleotide motifs within prokaryotic DNA that could activate B cells galvanized research efforts (Krieg et al., 1995). The adjuvant properties of CpG were exploited within experimental vaccines for a number of diseases including lymphoma, hepatitis, malaria, influenza, and leishmania well before the discovery of its innate receptor, TLR9, in 2000 (Lipford et al., 1997b; Wooldridge et al., 1997; Brazolot Millan et al., 1998; Davis et al., 1998; Moldoveanu et al., 1998; Zimmermann et al., 1998; Jones et al., 1999; Hemmi et al., 2000). These findings also provided an additional dimension to be explored, i.e., the possibility of intentionally incorporating these motifs within DNA vaccines (Klinman et al., 1997; Li et al., 2004). While it was first believed that a single type of optimal CpG motif would work in all applications, later reports showed that specific motifs of CpG-containing immunostimulatory oligodeoxynucleotides (CpG ODNs) could vary dramatically in their ability to induce individual immune effects (Ballas et al., 2001; Krug et al., 2001). Three classes of CPG ODN are now recognized – A-class ODN promote the highest degree of NK cell stimulation and also IFN-α secretion by plasmacytoid DCs; B-class ODN stimulate strong B cell and NK cell activation, as well as cytokine production; C-class ODN combine the effects of A- and B-class CpG ODN. The ability of CpG ODN to stimulate macrophages and dendritic cells to synthesize several cytokines, including IL-12, IL-18, TNF-α, and IFNs (IFN-α, IFN-β, and IFN-γ), to upregulate costimulatory molecules such as CD40 and MHC class II, and to enhance the ability of dendritic cells to present soluble protein to class I-restricted T cells, make CpG ODN a good option as an adjuvant (Klinman et al., 1996, 2000).
Initially, the CL mouse model was used to showcase the ability of CpG ODNs to switch the Th2 response associated with the disease to a strong Th1 response. This switch has now been associated with IL-12-dependent production of IFN-γ, an attractive quality for a CL vaccine (Lipford et al., 1997b; Zimmermann et al., 1998; Walker et al., 1999; Fukao et al., 2002; Flynn et al., 2005; Hutchins et al., 2005; Yang et al., 2010). The first studies using CpG ODNs as an adjuvant within prophylactic vaccines for CL demonstrated that inclusion of CpG helped to induce long-term immunity that protected against the parasite (Stacey and Blackwell, 1999; Walker et al., 1999). Further studies demonstrated that this protection is reliant on CD4 and CD8 T cell help, both of which had been shown to be important in L. major infection and induced when CpG was used as an adjuvant (Lipford et al., 1997a; Belkaid et al., 2002b; Rhee et al., 2002).
Later studies by Mendez et al. (2003), showed that mice vaccinated with live parasites plus CpG ODNs displayed a transient inflammation at 3–5 weeks, after which time little or no dermal pathology was observed. The resolution of the transient inflammation and parasite-burden in the ears of CpG ODN-treated mice coincided with the decrease of CD4 T cells in the site, whereas these cells began to accumulate in the ears of unvaccinated mice by 6 weeks after L. major infection. These responses are induced via activation of dermal DCs acting in an IL-6 dependent manner (von Stebut et al., 1998; Wu et al., 2006). Activation of these dermal DCs eliminates the silent phase of the infection by altering the natural kinetics of Treg cell accumulation that usually occurs in the skin after L. major challenge (Belkaid et al., 2000, 2002a; Wu et al., 2006). Recently, the same group reported the unusual IL-2 production by CD11c+ DCs in the skin of mice vaccinated with Lm/CpG. This unusual secretion was concomitant with a peak in IFN-γ-producing NK cells and CD4+ T cells and the control of parasite growth (Laabs et al., 2009). It has now been shown that inclusion of CpG ODN in the vaccine also enhances the proliferation of Th17 cells, which in turn aids the development of a protective cell-mediated immunity against the parasite (Wu et al., 2010). The Th17 response was shown to be dependent upon release of vaccine-induced IL-6 identified in earlier studies (Wu et al., 2006). Neutralization of IFN-γ and, in particular, IL-17, caused increased parasite burdens in Lm/CpG-vaccinated mice. IL-17R-deficient Lm/CpG-vaccinated mice developed lesions, and despite displaying normal IL-12 levels, showed a decrease in IFN-γ production. Neutrophil accumulation was also decreased in the IL-17R-deficient Lm/CpG-vaccinated mice but the number of Treg increased. The most striking observation in all these studies was that mice treated with CpG ODNs had increased levels of IL-10 in their draining lymph nodes, and only a transient increase in the number of Tregs at the site of infection, compared to control animals (Mendez et al., 2003; Wu et al., 2006). Thus, vaccination with live parasites and CpG ODNs appears to not only enhance adaptive immunity but also to modulate the response of innate immune cells, thereby inhibiting parasite growth and the development of vaccine-induced pathology.
Interestingly, CpG ODNs induce similar responses even when mixed with recombinant antigens (Rhee et al., 2002; Coler and Reed, 2005; Darrah et al., 2007). Vaccination with CpG ODN induced a high frequency of multi-functional T cells, a positive correlate of protection, especially in the CL model. Responses generated with these defined vaccines were established as early as 10 days after immunization, suggesting CpG ODN could be good adjuvants for both prophylaxis and therapy (Darrah et al., 2007; Raman et al., 2010). These responses were shown to remain consistent over prolonged periods of time, although they did wane after 6 months (Rhee et al., 2002; Darrah et al., 2007). Further investigations showed an increase in IL-10 levels in sera after immunization with vaccines containing CpG ODNs (Darrah et al., 2010). Inhibition of innate IL-10 produced by antigen presenting cells (APCs) at the time of immunization significantly enhanced the magnitude of the response and the extent of Th1 differentiation and, importantly, increased protection. This particular aspect of the adjuvant makes it much more attractive option for immunotherapy, with the possibility of preventing pathology. Studies performed by IDRI demonstrate that treatment of L. major-infected mice with CpG in conjunction with L110f and MPL-SE, decreases both parasite burden and infection-induced pathology (Raman et al., 2010).
CpG ODN have not yet been used as a vaccine adjuvant against leishmaniasis in human subjects. Preliminary studies involving vaccination of non-human primates with vaccine formulations containing CpG ODN have yielded positive results, inducing strong Th1 responses in both prophylactic and therapeutic settings (Walker et al., 1999; Flynn et al., 2005).
Several synthetic ODN are currently in development for the treatment of various cancers (Krieg, 2008). These ODNs use a nuclease-resistant phosphorothioate backbone that improves the half-life in the body from just a few minutes (for unmodified native DNA) to approximately 48 h. Immune modulatory oligonucleotides (IMOs) that stimulate TLR9 signaling have been developed and tested, for anti-tumor activity in mouse models, both as a monotherapy and in combination with chemotherapeutic agents. A number of these IMOs have also entered Phase I trials (Agrawal and Kandimalla, 2007; Dynavax Technologies, 2010; Hennessy et al., 2010). Similar Immunostimulatory sequences (ISS), comprised of short DNA sequences that have been shown to induce production of memory T cells and a Th1 response, and are also under investigation (Agrawal and Kandimalla, 2007). A potential advantage of ISS is that they can be used alone or linked to antigens to suppress Th2 response (Barry and Cooper, 2007).
Coley Pharmaceutical Group (Wellesley, MA, USA; currently Pfizer) developed CPG7909, which has been tested as a vaccine adjuvant in human clinical trials in the context of several vaccines (including flu, hepatitis, and malaria) with promising results (Cooper et al., 2004a,b; Mullen et al., 2008). The immunostimulatory effect of CpG7909 in these vaccines was shown to depend on the association of the CpG and antigen to alum (Aebig et al., 2007; Mullen et al., 2007). Addition of CpG 7909 to the malaria vaccine, AMA1-C1/alum vaccine in naïve (not exposed to malaria) individuals was shown to elicit significantly higher AMA1 specific immune IgG compared to individuals who only received AMA1-C1/alum (Mullen et al., 2008). Later studies on these samples showed that addition of CpG 7909 enhanced the kinetics, magnitude, and longevity of responses (Crompton et al., 2009). In contrast the same CpG-containing malaria vaccine did not enhance the acquisition of memory in semi-immune adults living in Mali (Traore et al., 2009). Understanding the molecular basis of this apparent refractoriness to TLR9 agonist in the endemic population is pertinent and of significant interest to vaccinology.
Importance of Formulation
Modifications to enhance vaccine potency were made early on in vaccine development history. For instance, “lipovaccines,” or homogenized dried bacterial cells mixed with oil, were first reported in 1916 (Achard and Foix, 1916; Pinoy, 1916a,b). This vaccine formulation increased the potency of killed bacterial vaccines in human subjects, reducing the high doses of bacterial cells and number of immunizations required, making them more comparable in efficacy to vaccines based on live-attenuated organisms such as smallpox, which only required one dose (Whitmore, 1919; Lewis and Dodge, 1920).
Formulation effects on vaccine antigen and adjuvant efficacy continue to remain critical. For example, CpGs formulated with particle-based platforms (i.e., alum, oil-in-water emulsion, or polymeric particles) induces increased total antibodies and Th1 responses in a variety of disease and animal models compared to either formulation component alone (Brazolot Millan et al., 1998; Ott et al., 2002; Ioannou et al., 2003; Linghua et al., 2006; Singh et al., 2007; Wack et al., 2008; Salem and Weiner, 2009; Fox et al., 2010a,b). The most abundant data regarding the importance of seemingly subtle formulation variations involve TLR4 agonists. For instance, AS01 (a liposomal formulation containing MPL and the saponin adjuvant QS21) induced higher anti-parasite antibodies (with less reactogenicity) compared to an emulsion formulation of the same molecules in a malaria vaccine clinical trial (Owusu-Agyei et al., 2009). Other TLR4 agonist formulation comparisons include oil-in-water emulsion vs. aqueous suspension or alum-adsorbed; the emulsion formulation was generally superior, inducing qualitative and quantitative differences in immune responses (Hui and Hashimoto, 2008; Lousada-Dietrich et al., 2011).
Several leishmaniasis vaccine studies employed formulation technologies to improve immune responses, especially liposomes. Lipid composition clearly influences immune responses, with synthetic saturated long-chain phosphatidylcholines inducing more protective responses and greater Th1-bias than unsaturated egg phosphatidylcholine in BALB/c mice or hamsters immunized with liposomal leishmania antigens (Kahl et al., 1989; Mazumdar et al., 2005; Badiee et al., 2009a). Liposome size, charge, and stability can be modified by different manufacturing conditions and additional components such as charged or PEGylated headgroups and cholesterol. Interestingly, liposomes were shown to reduce the toxicity of incorporated Lipid A structures (Richards et al., 1989). Another study showed that co-encapsulation of leishmania antigens and adjuvant (pDNA) in liposomes increased protective efficacy by twofold compared to encapsulation of only the antigen (Mazumder et al., 2007). Likewise, coencapsulation of leishmania antigen and CpG in polymeric nanospheres showed the strongest protective responses in BALB/c mice compared to other formulation variations (Tafaghodi et al., 2011). Shimizu et al. (2007) confirmed the importance of leishmania antigen encapsulation in liposomes and augmented protective immune responses further by targeting the liposome to mannose receptors on macrophages. Liposome surface charge was also found to affect immune responses to leishmania antigen, with neutral surface charge resulting in smallest footpad swelling, lowest splenic parasite burden, highest IgG2a/IgG1 ratio, highest IFN-γ production, and lowest IL-4 production compared to cationic or anionic liposomes (Badiee et al., 2009b). The above studies indicate that particulate formulations such as liposomes can serve as adjuvants simply through improved antigen delivery mechanisms, although co-formulation with additional immunostimulatory molecules may further improve immune responses (Kasturi et al., 2011).
Other important vaccine adjuvant formulations include ISCOMs, ISCOmatrix, and virosomes. ISCOMs, or immune-stimulating complexes, are particulate antigen delivery system that consists of antigen, phospholipid, cholesterol, and saponin structures (Morein et al., 2007). ISCOMs have shown promising adjuvant activity, and induced protective immunity in a number of disease models, including murine CL although reactogenicity and inefficacy was reported in some studies (Papadopoulou et al., 1998; Sjolander et al., 1998a,b; Morein et al., 2007; Middleton et al., 2009; Clements et al., 2010). ISCOMATRIX, is a particulate adjuvant that has essentially the same structure of ISCOMs but without antigen. Antigens can be formulated with the ISCOMATRIX to produce ISCOMATRIX vaccines that can provide the similar antigen presentation and immunomodulatory properties as the ISCOMs but with much broader application as they are not limited to hydrophobic membrane proteins (Sun et al., 2009). ISCOMATRIX, is a more well-defined platform that under controlled conditions, form cage-like structures typically 40 nm in diameter. The ISCOMATRIX™ adjuvant has been formulated with a wide range of antigens to produce ISCOMATRIX™ vaccines with great success but has not been tested in CL yet (McKenzie et al., 2010). Virosomes, like liposomes, consist of phospholipid vesicles. However, virosomes also include fusogenic proteins (hemagglutinin and neuraminidase) for enhanced antigen processing (Metcalfe and Gluck, 2006). Inflexal® V (influenza) and Epaxal® (hepatitis A) are virosome-based vaccines manufactured by Crucell and approved for use in many countries.
In order to optimize vaccine formulations, an array of parameters must be investigated. Formulation manufacturability and cost are often taken for granted or not considered in early development stages. However, these factors can play a critical role in ultimate product success (Hilleman, 1966, 1999). Other important factors include particle size, degradability, shape, stability, and release kinetics (Mueller et al., 2004; Garidel et al., 2005; Mohanan et al., 2010). Numerous challenges remain related to adjuvant development, especially in the context of neglected tropical diseases such as leishmaniasis, but these can be overcome by using a systematic approach.
Summary
Experimental CL has become a standard for investigating the development of T helper cell response and is now well established as the classic textbook example of the Th1/Th2 paradigm. Alongside the rapid advances in PRR discovery, this model has served to significantly advance our understanding of vaccine adjuvants. In the current era of translational biology, it is therefore ironic that few agonists (only those engaging TLR4 and TLR 7/8) have advanced into clinical trials against leishmania infection of humans (Table 1). Regardless, it is our hope that the advancement of vaccine adjuvants and improved understanding of the clinical situation will ultimately combine to develop effective tools to control leishmaniases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential con-flict of interest.
Acknowledgments
We would like to thank Dr. Anthony Desbien for critically reviewing the manuscript and the Clinical group at IDRI, especially Dr. Franco Piazza, Mr. Zachary Sagawa, and Ms. Jill Ashman for their constructive comments. Leishmania vaccine and adjuvant development work at IDRI has been supported in part by Grants 39129 and 42387 from the Bill & Melinda Gates Foundation and Grant R01-AI025038 from the National Institutes of Health. Greg Matlashewski acknowledges support from the Canadian Institutes of Health Research, WHO Special Program for Research and Training in Tropical Diseases (TDR) and Drugs for Neglected Diseases Initiative.
References
Achard, C., and Foix, C. (1916). Sur l’emploi des corps gras comme vehicules des vaccines microbiens. Comp. Rend. Soc. Biol. 79, 209–211.
Aebig, J. A., Mullen, G. E., Dobrescu, G., Rausch, K., Lambert, L., Ajose-Popoola, O., Long, C. A., Saul, A., and Miles, A. P. (2007). Formulation of vaccines containing CpG oligonucleotides and alum. J. Immunol. Methods 323, 139–146.
Aebischer, T., Wolfram, M., Patzer, S. I., Ilg, T., Wiese, M., and Overath, P. (2000). Subunit vaccination of mice against new world cutaneous leishmaniasis: comparison of three proteins expressed in amastigotes and six adjuvants. Infect. Immun. 68, 1328–1336.
Afonso, L. C., Scharton, T. M., Vieira, L. Q., Wysocka, M., Trinchieri, G., and Scott, P. (1994). The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science 263, 235–237.
Agrawal, S., and Kandimalla, E. R. (2007). Synthetic agonists of Toll-like receptors 7, 8 and 9. Biochem. Soc. Trans. 35(Pt 6), 1461–1467.
Arevalo, I., Ward, B., Miller, R., Meng, T. C., Najar, E., Alvarez, E., Matlashewski, G., and Llanos-Cuentas, A. (2001). Successful treatment of drug-resistant cutaneous leishmaniasis in humans by use of imiquimod, an immunomodulator. Clin. Infect. Dis. 33, 1847–1851.
Armijos, R. X., Weigel, M. M., Aviles, H., Maldonado, R., and Racines, J. (1998). Field trial of a vaccine against New World cutaneous leishmaniasis in an at-risk child population: safety, immunogenicity, and efficacy during the first 12 months of follow-up. J. Infect. Dis. 177, 1352–1357.
Badaro, R., Lobo, I., Munos, A., Netto, E. M., Modabber, F., Campos-Neto, A., Coler, R. N., and Reed, S. G. (2006). Immunotherapy for drug-refractory mucosal leishmaniasis. J. Infect. Dis. 194, 1151–1159.
Badiee, A., Jaafari, M. R., Khamesipour, A., Samiei, A., Soroush, D., Kheiri, M. T., Barkhordari, F., McMaster, W. R., and Mahboudi, F. (2009a). Enhancement of immune response and protection in BALB/c mice immunized with liposomal recombinant major surface glycoprotein of Leishmania (rgp63): the role of bilayer composition. Colloids Surf. B Biointerfaces 74, 37–44.
Badiee, A., Jaafari, M. R., Khamesipour, A., Samiei, A., Soroush, D., Kheiri, M. T., Barkhordari, F., McMaster, W. R., and Mahboudi, F. (2009b). The role of liposome charge on immune response generated in BALB/c mice immunized with recombinant major surface glycoprotein of Leishmania (rgp63). Exp. Parasitol. 121, 362–369.
Ballas, Z. K., Krieg, A. M., Warren, T., Rasmussen, W., Davis, H. L., Waldschmidt, M., and Weiner, G. J. (2001). Divergent therapeutic and immunologic effects of oligodeoxynucleotides with distinct CpG motifs. J. Immunol. 167, 4878–4886.
Barry, M., and Cooper, C. (2007). Review of hepatitis B surface antigen-1018 ISS adjuvant-containing vaccine safety and efficacy. Expert Opin. Biol. Ther. 7, 1731–1737.
Belkaid, Y., Mendez, S., Lira, R., Kadambi, N., Milon, G., and Sacks, D. (2000). A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J. Immunol. 165, 969–977.
Belkaid, Y., Piccirillo, C. A., Mendez, S., Shevach, E. M., and Sacks, D. L. (2002a). CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420, 502–507.
Belkaid, Y., von Stebut, E., Mendez, S., Lira, R., Caler, E., Bertholet, S., Udey, M. C., and Sacks, D. (2002b). CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J. Immunol. 168, 3992–4000.
Bertholet, S., Goto, Y., Carter, L., Bhatia, A., Howard, R. F., Carter, D., Coler, R. N., Vedvick, T. S., and Reed, S. G. (2009). Optimized subunit vaccine protects against experimental leishmaniasis. Vaccine 27, 7036–7045.
Bickels, J., Kollender, Y., Merinsky, O., and Meller, I. (2002). Coley’s toxin: historical perspective. Isr. Med. Assoc. J. 4, 471–472.
Birnbaum, R., Haskell, J., Vanchinathan, V., Greger, S., Narayan, R., Chang, P.-L., Tran, T. A., Hickerson, S. M., Beverley, S. M., Wilson, M. E., Bruhn, K. W., and Craft, N. (2012). Killed but metabolically active Leishmania as a novel whole-cell vaccine for visceral leishmaniasis. Clin. Vaccine Immunol. 19, 490–498.
Brazolot Millan, C. L., Weeratna, R., Krieg, A. M., Siegrist, C. A., and Davis, H. L. (1998). CpG DNA can induce strong Th1 humoral and cell-mediated immune responses against hepatitis B surface antigen in young mice. Proc. Natl. Acad. Sci. U.S.A. 95, 15553–15558.
Brewer, J. M., Conacher, M., Hunter, C. A., Mohrs, M., Brombacher, F., and Alexander, J. (1999). Aluminium hydroxide adjuvant initiates strong antigen-specific Th2 responses in the absence of IL-4- or IL-13-mediated signaling. J. Immunol. 163, 6448–6454.
Buates, S., and Matlashewski, G. (1999). Treatment of experimental leishmaniasis with the immunomodulators imiquimod and S-28463: efficacy and mode of action. J. Infect. Dis. 179, 1485–1494.
Buates, S., and Matlashewski, G. (2001). Identification of genes induced by a macrophage activator, S-28463, using gene expression array analysis. Antimicrob. Agents Chemother. 45, 1137–1142.
Cabrera, M., Blackwell, J. M., Castes, M., Trujillo, D., Convit, J., and Shaw, M. A. (2000). Immunotherapy with live BCG plus heat killed Leishmania induces a T helper 1-like response in American cutaneous leishmaniasis patients. Parasite Immunol. 22, 73–79.
Campos-Neto, A., Webb, J. R., Greeson, K., Coler, R. N., Skeiky, Y. A., and Reed, S. G. (2002). Vaccination with plasmid DNA encoding TSA/LmSTI1 leishmanial fusion proteins confers protection against Leishmania major infection in susceptible BALB/c mice. Infect. Immun. 70, 2828–2836.
Carneiro, L. A., Travassos, L. H., and Girardin, S. E. (2007). Nod-like receptors in innate immunity and inflammatory diseases. Ann. Med. 39, 581–593.
Castes, M., Moros, Z., Martinez, A., Trujillo, D., Castellanos, P. L., Rondon, A. J., and Convit, J. (1989). Cell-mediated immunity in localized cutaneous leishmaniasis patients before and after treatment with immunotherapy or chemotherapy. Parasite Immunol. 11, 211–222.
Central Drugs Standard Control Organization. (2010). Vaccine Registration Certificate Data from Jan 2009 to July 2010. Available at: http://www.cdsco.nic.in/html/vaccinemain.html
Chakravarty, J., Kumar, S., Trivedi, S., Rai, V. K., Singh, A., Ashman, J. A., Laughlin, E. M., Coler, R. N., Kahn, S. J., Beckmann, A. M., Cowgill, K. D., Reed, S. G., Sundar, S., and Piazza, F. M. (2011). A clinical trial to evaluate the safety and immunogenicity of the LEISH-F1 + MPL-SE vaccine for use in the prevention of visceral leishmaniasis. Vaccine 29, 3531–3537.
Chan, S. H., Perussia, B., Gupta, J. W., Kobayashi, M., Pospísil, M., Young, H. A., Wolf, S. F., Young, D., Clark, S. C., and Trinchieri, G. (1991). Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J. Exp. Med. 173, 869–879.
Clements, D. E., Coller, B. A., Lieberman, M. M., Ogata, S., Wang, G., Harada, K. E., Putnak, J. R., Ivy, J. M., McDonell, M., Bignami, G. S., Peters, I. D., Leung, J., Weeks-Levy, C., Nakano, E. T., and Humphreys, T. (2010). Development of a recombinant tetravalent dengue virus vaccine: immunogenicity and efficacy studies in mice and monkeys. Vaccine 28, 2705–2715.
Coler, R. N., Bertholet, S., Moutaftsi, M., Guderian, J. A., Windish, H. P., Baldwin, S. L., Laughlin, E. M., Duthie, M. S., Fox, C. B., Carter, D., Friede, M., Vedvick, T. S., and Reed, S. G. (2011). Development and characterization of synthetic glucopyranosyl lipid adjuvant system as a vaccine adjuvant. PLoS ONE 6, e16333. doi: 10.1371/journal.pone.0016333
Coler, R. N., Goto, Y., Bogatzki, L., Raman, V., and Reed, S. G. (2007). Leish-111f, a recombinant polyprotein vaccine that protects against visceral leishmaniasis by elicitation of CD4+ T cells. Infect. Immun. 75, 4648–4654.
Coler, R. N., and Reed, S. G. (2005). Second-generation vaccines against leishmaniasis. Trends Parasitol. 21, 244–249.
Coler, R. N., Skeiky, Y. A., Bernards, K., Greeson, K., Carter, D., Cornellison, C. D., Modabber, F., Campos-Neto, A., and Reed, S. G. (2002). Immunization with a polyprotein vaccine consisting of the T-Cell antigens thiol-specific antioxidant, Leishmania major stress-inducible protein 1, and Leishmania elongation initiation factor protects against leishmaniasis. Infect. Immun. 70, 4215–4225.
Cooper, C. L., Davis, H. L., Morris, M. L., Efler, S. M., Adhami, M. A., Krieg, A. M., Cameron, D. W., and Heathcote, J. (2004a). CPG 7909, an immunostimulatory TLR9 agonist oligodeoxynucleotide, as adjuvant to Engerix-B HBV vaccine in healthy adults: a double-blind phase I/II study. J. Clin. Immunol. 24, 693–701.
Cooper, C. L., Davis, H. L., Morris, M. L., Efler, S. M., Krieg, A. M., Li, Y., Laframboise, C., Al Adhami, M. J., Khaliq, Y., Seguin, I., and Cameron, D. W. (2004b). Safety and immunogenicity of CPG 7909 injection as an adjuvant to Fluarix influenza vaccine. Vaccine 22, 3136–3143.
Crompton, P. D., Mircetic, M., Weiss, G., Baughman, A., Huang, C. Y., Topham, D. J., Treanor, J. J., Sanz, I., Lee, F. E., Durbin, A. P., Miura, K., Narum, D. L., Ellis, R. D., Malkin, E., Mullen, G. E., Miller, L. H., Martin, L. B., and Pierce, S. K. (2009). The TLR9 ligand CpG promotes the acquisition of Plasmodium falciparum-specific memory B cells in malaria-naive individuals. J. Immunol. 182, 3318–3326.
Daneshvar, H., Molaei, M. M., Kamiabi, H., Burchmore, R., Hagan, P., and Stephen Phillips, R. (2010). Gentamicin-attenuated Leishmania infantum: cellular immunity production and protection of dogs against experimental canine leishmaniasis. Parasite Immunol. 32, 722–730.
Darrah, P. A., Hegde, S. T., Patel, D. T., Lindsay, R. W., Chen, L., Roederer, M., and Seder, R. A. (2010). IL-10 production differentially influences the magnitude, quality, and protective capacity of Th1 responses depending on the vaccine platform. J. Exp. Med. 207, 1421–1433.
Darrah, P. A., Patel, D. T., De Luca, P. M., Lindsay, R. W., Davey, D. F., Flynn, B. J., Hoff, S. T., Andersen, P., Reed, S. G., Morris, S. L., Roederer, M., and Seder, R. A. (2007). Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat. Med. 13, 843–850.
Davis, H. L., Weeratna, R., Waldschmidt, T. J., Tygrett, L., Schorr, J., and Krieg, A. M. (1998). CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J. Immunol. 160, 870–876.
Davoudi, N., Tate, C. A., Warburton, C., Murray, A., Mahboudi, F., and McMaster, W. R. (2005). Development of a recombinant Leishmania major strain sensitive to ganciclovir and 5-fluorocytosine for use as a live vaccine challenge in clinical trials. Vaccine 23, 1170–1177.
Desjeux, P. (2001). The increase in risk factors for leishmaniasis worldwide. Trans. R. Soc. Trop. Med. Hyg. 95, 239–243.
Didierlaurent, A. M., Morel, S., Lockman, L., Giannini, S. L., Bisteau, M., Carlsen, H., Kielland, A., Vosters, O., Vanderheyde, N., Schiavetti, F., Larocque, D., Van Mechelen, M., and Garçon, N. (2009). AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. 183, 6186–6197.
Duthie, M. S., Raman, V. S., Piazza, F. M., and Reed, S. G. (2012). The development and clinical evaluation of second-generation leishmaniasis vaccines. Vaccine 30, 134–141.
Duthie, M. S., Windish, H. P., Fox, C. B., and Reed, S. G. (2011). Use of defined TLR ligands as adjuvants within human vaccines. Immunol. Rev. 239, 178–196.
Dynavax Technologies. (2010). Data from the Phase 1b Study of SD-101 in Treatment-naïve, Genotype 1 HCV Patients. Available at: http://www.dynavax.com/hepatitis_ct.html
Eisenbarth, S. C., Colegio, O. R., O’Connor, W., Sutterwala, F. S., and Flavell, R. A. (2008). Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453, 1122–1126.
Fernandez-Botran, R., Chilton, P. M., Hondowicz, B. D., Vetvickova, J., Yan, J., Jones, W. III, and Scott, P. (1999). Regulation of the production of soluble IL-4 receptors in murine cutaneous leishmaniasis. The roles of IL-12 and IL-4. J. Leukoc. Biol. 66, 481–488.
Firooz, A., Khamesipour, A., Ghoorchi, M. H., Nassiri-Kashani, M., Eskandari, S. E., Khatami, A., Hooshmand, B., Gorouhi, F., Rashighi-Firoozabadi, M., and Dowlati, Y. (2006). Imiquimod in combination with meglumine antimoniate for cutaneous leishmaniasis: a randomized assessor-blind controlled trial. Arch. Dermatol. 142, 1575–1579.
Flynn, B., Wang, V., Sacks, D. L., Seder, R. A., and Verthelyi, D. (2005). Prevention and treatment of cutaneous leishmaniasis in primates by using synthetic type D/A oligodeoxynucleotides expressing CpG motifs. Infect. Immun. 73, 4948–4954.
Fox, C. B., Dutill, T. S., Chesko, J., Anderson, R. C., Reed, S. G., and Vedvick, T. S. (2010a). Characterization of aluminum hydroxide gel and oil-in-water emulsion formulations containing CpG ODNs. Biopharm. Int. October, S14–S19.
Fox, C. B., Friede, M., Reed, S. G., and Ireton, G. C. (2010b). Synthetic and natural TLR4 agonists as safe and effective vaccine adjuvants. Subcell. Biochem. 53, 303–321.
Frazer, I. H., and Levin, M. J. (2011). Paradigm shifting vaccines: prophylactic vaccines against latent varicella-zoster virus infection and against HPV-associated cancer. Curr. Opin. Virol. 1, 268–279.
Fukao, T., Tanabe, M., Terauchi, Y., Ota, T., Matsuda, S., Asano, T., Kadowaki, T., Takeuchi, T., and Koyasu, S. (2002). PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 3, 875–881.
Garidel, P., Rappolt, M., Schromm, A. B., Howe, J., Lohner, K., Andrä, J., Koch, M. H., and Brandenburg, K. (2005). Divalent cations affect chain mobility and aggregate structure of lipopolysaccharide from Salmonella minnesota reflected in a decrease of its biological activity. Biochim. Biophys. Acta 1715, 122–131.
Gicheru, M. M., Olobo, J. O., Anjili, C. O., Orago, A. S., Modabber, F., and Scott, P. (2001). Vervet monkeys vaccinated with killed Leishmania major parasites and interleukin-12 develop a type 1 immune response but are not protected against challenge infection. Infect. Immun. 69, 245–251.
Goto, Y., Bogatzki, L. Y., Bertholet, S., Coler, R. N., and Reed, S. G. (2007). Protective immunization against visceral leishmaniasis using Leishmania sterol 24-c-methyltransferase formulated in adjuvant. Vaccine 25, 7450–7458.
Greenblatt, C. L. (1980). The present and future of vaccination for cutaneous leishmaniasis. Prog. Clin. Biol. Res. 47, 259–285.
Gupta, R. K., Chang, A. C., Griffin, P., Rivera, R., and Siber, G. R. (1996). In vivo distribution of radioactivity in mice after injection of biodegradable polymer microspheres containing 14C-labeled tetanus toxoid. Vaccine 14, 1412–1416.
Gurunathan, S., Prussin, C., Sacks, D. L., and Seder, R. A. (1998). Vaccine requirements for sustained cellular immunity to an intracellular parasitic infection. Nat. Med. 4, 1409–1415.
Gurunathan, S., Sacks, D. L., Brown, D. R., Reiner, S. L., Charest, H., Glaichenhaus, N., and Seder, R. A. (1997). Vaccination with DNA encoding the immunodominant LACK parasite antigen confers protective immunity to mice infected with Leishmania major. J. Exp. Med. 186, 1137–1147.
Handman, E. (2001). Leishmaniasis: current status of vaccine development. Clin. Microbiol. Rev. 14, 229–243.
Handman, E., Symons, F. M., Baldwin, T. M., Curtis, J. M., and Scheerlinck, J. P. (1995). Protective vaccination with promastigote surface antigen 2 from Leishmania major is mediated by a TH1 type of immune response. Infect. Immun. 63, 4261–4267.
Heinzel, F. P., Rerko, R. M., and Hujer, A. M. (1998). Underproduction of interleukin-12 in susceptible mice during progressive leishmaniasis is due to decreased CD40 activity. Cell. Immunol. 184, 129–142.
Heinzel, F. P., Schoenhaut, D. S., Rerko, R. M., Rosser, L. E., and Gately, M. K. (1993). Recombinant interleukin 12 cures mice infected with Leishmania major. J. Exp. Med. 177, 1505–1509.
Hemmi, H., Kaisho, T., Takeuchi, O., Sato, S., Sanjo, H., Hoshino, K., Horiuchi, T., Tomizawa, H., Takeda, K., and Akira, S. (2002). Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 3, 196–200.
Hemmi, H., Takeuchi, O., Kawai, T., Kaisho, T., Sato, S., Sanjo, H., Matsumoto, M., Hoshino, K., Wagner, H., Takeda, K., and Akira, S. (2000). A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745.
Hennessy, E. J., Parker, A. E., and O’Neill, L. A. (2010). Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307.
Hervas, J. A., Martin-Santiago, A., Hervás, D., Rojo, E., Mena, A., Rocamora, V., and Dueñas J. (2011). Old World Leishmania infantum cutaneous leishmaniasis unresponsive to liposomal amphotericin B treated with topical imiquimod. Pediatr. Infect. Dis. J. 31, 97–100.
Hilleman, M. R. (1966). Critical appraisal of emulsified oil adjuvants applied to viral vaccines. Prog. Med. Virol. 8, 131–182.
Hilleman, M. R. (1999). Personal historical chronicle of six decades of basic and applied research in virology, immunology, and vaccinology. Immunol. Rev. 170, 7–27.
Hondowicz, B. D., Park, A. Y., Elloso, M. M., and Scott, P. (2000). Maintenance of IL-12-responsive CD4+ T cells during a Th2 response in Leishmania major-infected mice. Eur. J. Immunol. 30, 2007–2014.
Hondowicz, B. D., Scharton-Kersten, T. M., Jones, D. E., and Scott, P. (1997). Leishmania major-infected C3H mice treated with anti-IL-12 mAb develop but do not maintain a Th2 response. J. Immunol. 159, 5024–5031.
Hsieh, C. S., Macatonia, S. E., Tripp, C. S., Wolf, S. F., O’Garra, A., and Murphy, K. M. (1993). Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 260, 547–549.
Hui, G. S., and Hashimoto, C. N. (2008). Adjuvant formulations possess differing efficacy in the potentiation of antibody and cell mediated responses to a human malaria vaccine under selective immune genes knockout environment. Int. Immunopharmacol. 8, 1012–1022.
Hutchins, A. S., Artis, D., Hendrich, B. D., Bird, A. P., Scott, P., and Reiner, S. L. (2005). Cutting edge: a critical role for gene silencing in preventing excessive type 1 immunity. J. Immunol. 175, 5606–5610.
Ioannou, X. P., Gomis, S. M., Hecker, R., Babiuk, L. A., and van Drunen Littel-van den Hurk, S. (2003). Safety and efficacy of CpG-containing oligodeoxynucleotides as immunological adjuvants in rabbits. Vaccine 21, 4368–4372.
Janeway, C. A. Jr. (1989). Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 54(Pt 1), 1–13.
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216.
Jones, T. R., Obaldia, N. III, Gramzinski, R. A., Charoenvit, Y., Kolodny, N., Kitov, S., Davis, H. L., Krieg, A. M., and Hoffman, S. L. (1999). Synthetic oligodeoxynucleotides containing CpG motifs enhance immunogenicity of a peptide malaria vaccine in Aotus monkeys. Vaccine 17, 3065–3071.
Kahl, L. P., Scott, C. A., Lelchuk, R., Gregoriadis, G., and Liew, F. Y. (1989). Vaccination against murine cutaneous leishmaniasis by using Leishmania major antigen/liposomes. Optimization and assessment of the requirement for intravenous immunization. J. Immunol. 142, 4441–4449.
Kamil, A. A., Khalil, E. A., Musa, A. M., Modabber, F., Mukhtar, M. M., Ibrahim, M. E., Zijlstra, E. E., Sacks, D., Smith, P. G., Zicker, F., and El-Hassan, A. M. (2003). Alum-precipitated autoclaved Leishmania major plus bacille Calmette-Guerrin, a candidate vaccine for visceral leishmaniasis: safety, skin-delayed type hypersensitivity response and dose finding in healthy volunteers. Trans. R. Soc. Trop. Med. Hyg. 97, 365–368.
Kane, M. M., and Mosser, D. M. (2001). The role of IL-10 in promoting disease progression in leishmaniasis. J. Immunol. 166, 1141–1147.
Kasturi, S. P., Skountzou, I., Albrecht, R. A., Koutsonanos, D., Hua, T., Nakaya, H. I., Ravindran, R., Stewart, S., Alam, M., Kwissa, M., Villinger, F., Murthy, N., Steel, J., Jacob, J., Hogan, R. J., García-Sastre, A., Compans, R., and Pulendran, B. (2011). Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470, 543–547.
Kedzierski, L., Curtis, J. M., Doherty, P. C., Handman, E., and Kedzierska, K. (2008). Decreased IL-10 and IL-13 production and increased CD44hi T cell recruitment contribute to Leishmania major immunity induced by non-persistent parasites. Eur. J. Immunol. 38, 3090–3100.
Kenney, R. T., Sacks, D. L., Sypek, J. P., Vilela, L., Gam, A. A., and Evans-Davis, K. (1999). Protective immunity using recombinant human IL-12 and alum as adjuvants in a primate model of cutaneous leishmaniasis. J. Immunol. 163, 4481–4488.
Klinman, D. M., Kamstrup, S., Verthelyi, D., Gursel, I., Ishii, K. J., Takeshita, F., and Gursel, M. (2000). Activation of the innate immune system by CpG oligodeoxynucleotides: immunoprotective activity and safety. Springer Semin. Immunopathol. 22, 173–183.
Klinman, D. M., Yamshchikov, G., and Ishigatsubo, Y. (1997). Contribution of CpG motifs to the immunogenicity of DNA vaccines. J. Immunol. 158, 3635–3639.
Klinman, D. M., Yi, A. K., Beaucage, S. L., Conover, J., and Krieg, A. M. (1996). CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc. Natl. Acad. Sci. U.S.A. 93, 2879–2883.
Kobayashi, M., Fitz, L., Ryan, M., Hewick, R. M., Clark, S. C., Chan, S., Loudon, R., Sherman, F., Perussia, B., and Trinchieri, G. (1989). Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J. Exp. Med. 170, 827–845.
Kool, M., Soullie, T., van Nimwegen, M., Willart, M. A., Muskens, F., Jung, S., Hoogsteden, H. C., Hammad, H., and Lambrecht, B. N. (2008). Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J. Exp. Med. 205, 869–882.
Krieg, A. M. (2008). Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 27, 161–167.
Krieg, A. M., Yi, A. K., Matson, S., Waldschmidt, T. J., Bishop, G. A., Teasdale, R., Koretzky, G. A., and Klinman, D. M. (1995). CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 374, 546–549.
Krug, A., Rothenfusser, S., Hornung, V., Jahrsdörfer, B., Blackwell, S., Ballas, Z. K., Endres, S., Krieg, A. M., and Hartmann, G. (2001). Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur. J. Immunol. 31, 2154–2163.
Kundi, M. (2007). New hepatitis B vaccine formulated with an improved adjuvant system. Expert Rev. Vaccines 6, 133–140.
Kuroda, E., Ishii, K. J., Uematsu, S., Ohata, K., Coban, C., Akira, S., Aritake, K., Urade, Y., and Morimoto, Y. (2011). Silica crystals and aluminum salts regulate the production of prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity 34, 514–526.
Laabs, E. M., Wu, W., and Mendez, S. (2009). Vaccination with live Leishmania major and CpG DNA promotes interleukin-2 production by dermal dendritic cells and NK cell activation. Clin. Vaccine Immunol. 16, 1601–1606.
Li, H., Nookala, S., and Re, F. (2007). Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. J. Immunol. 178, 5271–5276.
Li, H., Willingham, S. B., Ting, J. P., and Re, F. (2008). Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J. Immunol. 181, 17–21.
Li, Y., Ishii, K., Hisaeda, H., Hamano, S., Zhang, M., Nakanishi, K., Yoshimoto, T., Hemmi, H., Takeda, K., Akira, S., Iwakura, Y., and Himeno, K. (2004). IL-18 gene therapy develops Th1-type immune responses in Leishmania major-infected BALB/c mice: is the effect mediated by the CpG signaling TLR9? Gene Ther. 11, 941–948.
Liew, F. Y., Li, Y., and Millott, S. (1990a). Tumor necrosis factor-alpha synergizes with IFN-gamma in mediating killing of Leishmania major through the induction of nitric oxide. J. Immunol. 145, 4306–4310.
Liew, F. Y., Millott, S., Parkinson, C., Palmer, R. M., and Moncada, S. (1990b). Macrophage killing of Leishmania parasite in vivo is mediated by nitric oxide from L-arginine. J. Immunol. 144, 4794–4797.
Linghua, Z., Xingshan, T., and Fengzhen, Z. (2006). The efficacy of CpG oligodinucleotides, in combination with conventional adjuvants, as immunological adjuvants to swine streptococcic septicemia vaccine in piglets in vivo. Int. Immunopharmacol. 6, 1267–1276.
Lipford, G. B., Bauer, M., Blank, C., Reiter, R., Wagner, H., and Heeg, K. (1997a). CpG-containing synthetic oligonucleotides promote B and cytotoxic T cell responses to protein antigen: a new class of vaccine adjuvants. Eur. J. Immunol. 27, 2340–2344.
Lipford, G. B., Sparwasser, T., Bauer, M., Zimmermann, S., Koch, E. S., Heeg, K., and Wagner, H. (1997b). Immunostimulatory DNA: sequence-dependent production of potentially harmful or useful cytokines. Eur. J. Immunol. 27, 3420–3426.
Llanos-Cuentas, A., Calderon, W., Cruz, M., Ashman, J. A., Alves, F. P., Coler, R. N., Bogatzki, L. Y., Bertholet, S., Laughlin, E. M., Kahn, S. J., Beckmann, A. M., Cowgill, K. D., Reed, S. G., and Piazza, F. M. (2010). A clinical trial to evaluate the safety and immunogenicity of the LEISH-F1 + MPL-SE vaccine when used in combination with sodium stibogluconate for the treatment of mucosal leishmaniasis. Vaccine 28, 7427–7435.
Locksley, R. M., and Scott, P. (1991). Helper T-cell subsets in mouse leishmaniasis: induction, expansion and effector function. Immunol. Today 12, A58–A61.
Lousada-Dietrich, S., Jogdand, P. S., Jepsen, S., Pinto, V. V., Ditlev, S. B., Christiansen, M., Larsen, S. O., Fox, C. B., Raman, V. S., Howard, R. F., Vedvick, T. S., Ireton, G., Carter, D., Reed, S. G., and Theisen, M. (2011). A synthetic TLR4 agonist formulated in an emulsion enhances humoral and Type 1 cellular immune responses against GMZ2 – a GLURP-MSP3 fusion protein malaria vaccine candidate. Vaccine 29, 3284–3292.
Manetti, R., Parronchi, P., Giudizi, M. G., Piccinni, M. P., Maggi, E., Trinchieri, G., and Romagnani, S. (1993). Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J. Exp. Med. 177, 1199–1204.
Marichal, T., Ohata, K., Bedoret, D., Mesnil, C., Sabatel, C., Kobiyama, K., Lekeux, P., Coban, C., Akira, S., Ishii, K. J., Bureau, F., and Desmet, C. J. (2011). DNA released from dying host cells mediates aluminum adjuvant activity. Nat. Med. 17, 996–1002.
Marrack, P., McKee, A. S., and Munks, M. W. (2009). Towards an understanding of the adjuvant action of aluminium. Nat. Rev. Immunol. 9, 287–293.
Masihi, K. N., Lange, W., Brehmer, W., and Ribi E. (1986). Immunobiological activities of nontoxic lipid A: enhancement of nonspecific resistance in combination with trehalose dimycolate against viral infection and adjuvant effects. Int. J. Immunopharmacol. 8, 339–345.
Mayrink, W., da Costa, C. A., Magalhães, P. A., Melo, M. N., Dias, M., Lima, A. O., Michalick, M. S., and Williams, P. (1979). A field trial of a vaccine against American dermal leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 73, 385–387.
Mazumdar, T., Anam, K., and Ali, N. (2005). Influence of phospholipid composition on the adjuvanticity and protective efficacy of liposome-encapsulated Leishmania donovani antigens. J. Parasitol. 91, 269–274.
Mazumder, S., Ravindran, R., Banerjee, A., and Ali, N. (2007). Non-coding pDNA bearing immunostimulatory sequences co-entrapped with leishmanial antigens in cationic liposomes elicits almost complete protection against experimental visceral leishmaniasis in BALB/c mice. Vaccine 25, 8771–8781.
McGreal, E. P., Miller, J. L., and Gordon, S. (2005). Ligand recognition by antigen-presenting cell C-type lectin receptors. Curr. Opin. Immunol. 17, 18–24.
McKenzie, A., Watt, M., and Gittleson, C. (2010). ISCOMATRIX() vaccines: safety in human clinical studies. Hum. Vaccines 6, 237–246.
Medzhitov, R., and Janeway, C. A. Jr. (2002). Decoding the patterns of self and nonself by the innate immune system. Science 296, 298–300.
Mendez, S., Gurunathan, S., Kamhawi, S., Belkaid, Y., Moga, M. A., Skeiky, Y. A., Campos-Neto, A., Reed, S., Seder, R. A., and Sacks, D. (2001). The potency and durability of DNA- and protein-based vaccines against Leishmania major evaluated using low-dose, intradermal challenge. J. Immunol. 166, 5122–5128.
Mendez, S., Reckling, S. K., Piccirillo, C. A., Sacks, D., and Belkaid, Y. (2004). Role for CD4(+) CD25(+) regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J. Exp. Med. 200, 201–210.
Mendez, S., Tabbara, K., Belkaid, Y., Bertholet, S., Verthelyi, D., Klinman, D., Seder, R. A., and Sacks, D. L. (2003). Coinjection with CpG-containing immunostimulatory oligodeoxynucleotides reduces the pathogenicity of a live vaccine against cutaneous leishmaniasis but maintains its potency and durability. Infect. Immun. 71, 5121–5129.
Metcalfe, I. C., and Gluck, R. (2006). ”Virosomes for vaccine delivery,” Immunopotentiators in Modern Vaccines, eds V. E. Schijns and D. O’Hagan (Burlington, MA: Elsevier Academic Press), 179–189.
Metzger, D. W. (2010). Interleukin-12 as an adjuvant for induction of protective antibody responses. Cytokine 52, 102–107.
Middleton, D., Rockman, S., Pearse, M., Barr, I., Lowther, S., Klippel, J., Ryan, D., and Brown, L. (2009). Evaluation of vaccines for H5N1 influenza virus in ferrets reveals the potential for protective single-shot immunization. J. Virol. 83, 7770–7778.
Miranda-Verastegui, C., Llanos-Cuentas, A., Arévalo, I., Ward, B. J., and Matlashewski, G. (2005). Randomized, double-blind clinical trial of topical imiquimod 5% with parenteral meglumine antimoniate in the treatment of cutaneous leishmaniasis in Peru. Clin. Infect. Dis. 40, 1395–1403.
Miranda-Verastegui, C., Tulliano, G., Gyorkos, T. W., Calderon, W., Rahme, E., Ward, B., Cruz, M., Llanos-Cuentas, A., and Matlashewski, G. (2009). First-line therapy for human cutaneous leishmaniasis in Peru using the TLR7 agonist imiquimod in combination with pentavalent antimony. PLoS Negl. Trop. Dis. 3, e491. doi: 10.1371/journal.pntd.0000491
Mohanan, D., Slutter, B., Henriksen-Lacey, M., Jiskoot, W., Bouwstra, J. A., Perrie, Y., Kündig, T. M., Gander, B., and Johansen, P. (2010). Administration routes affect the quality of immune responses: a cross-sectional evaluation of particulate antigen-delivery systems. J. Control. Release 147, 342–349.
Moldoveanu, Z., Love-Homan, L., Huang, W. Q., and Krieg, A. M. (1998). CpG DNA, a novel immune enhancer for systemic and mucosal immunization with influenza virus. Vaccine 16, 1216–1224.
Momeni, A. Z., Jalayer, T., Emamjomeh, M., Khamesipour, A., Zicker, F., Ghassemi, R. L., Dowlati, Y., Sharifi, I., Aminjavaheri, M., Shafiei, A., Alimohammadian, M. H., Hashemi-Fesharki, R., Nasseri, K., Godal, T., Smith, P. G., and Modabber, F. (1999). A randomised, double-blind, controlled trial of a killed L. major vaccine plus BCG against zoonotic cutaneous leishmaniasis in Iran. Vaccine 17, 466–472.
Morein, B., Hu, K., Lövgren, K., and D’Hondt, E. (2007). ”New ISCOMs meet unsettled vaccine demands,” in Vaccine Adjuvants and Delivery Systems, ed. M. Singh (Hoboken, NJ: John Wiley & Sons, Inc.), 191–222.
Mougneau, E., Altare, F., Wakil, A. E., Zheng, S., Coppola, T., Wang, Z. E., Waldmann, R., Locksley, R. M., and Glaichenhaus, N. (1995). Expression cloning of a protective Leishmania antigen. Science 268, 563–566.
Mueller, M., Lindner, B., Kusumoto, S., Fukase, K., Schromm, A. B., and Seydel, U. (2004). Aggregates are the biologically active units of endotoxin. J. Biol. Chem. 279, 26307–26313.
Mullen, G. E., Aebig, J. A., Dobrescu, G., Rausch, K., Lambert, L., Long, C. A., Miles, A. P., and Saul, A. (2007). Enhanced antibody production in mice to the malaria antigen AMA1 by CPG 7909 requires physical association of CpG and antigen. Vaccine 25, 5343–5347.
Mullen, G. E., Ellis, R. D., Miura, K., Malkin, E., Nolan, C., Hay, M., Fay, M. P., Saul, A., Zhu, D., Rausch, K., Moretz, S., Zhou, H., Long, C. A., Miller, L. H., and Treanor, J. (2008). Phase 1 trial of AMA1-C1/Alhydrogel plus CPG 7909: an asexual blood-stage vaccine for Plasmodium falciparum malaria. PLoS ONE 3, e2940. doi: 10.1371/journal.pone.0002940
Musa, A. M., Khalil, E. A., Mahgoub, F. A., Elgawi, S. H., Modabber, F., Elkadaru, A. E., Aboud, M. H., Noazin, S., Ghalib, H. W., El-Hassan, A. M., and Leishmaniasis Research Group/Sudan. (2008). Immunochemotherapy of persistent post-kala-azar dermal leishmaniasis: a novel approach to treatment. Trans. R. Soc. Trop. Med. Hyg. 102, 58–63.
Nadim, A., Javadian, E., Tahvildar-Bidruni, G., and Ghorbani, M. (1983). Effectiveness of leishmanization in the control of cutaneous leishmaniasis. Bull. Soc. Pathol. Exot. Filiales 76, 377–383.
National Advisory Committee on Immunization. (2008). ”Type and contents of vaccines currently approved for use in Canada,” in 2006 Canadian Immunization Guide, 7th Edn. (Updated as of March 2008) (Ottawa: Public Health Agency of Canada).
O’Neill, L. A., and Bowie, A. G. (2007). The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364.
Onomoto, K., Yoneyama, M., and Fujita, T. (2007). Regulation of antiviral innate immune responses by RIG-I family of RNA helicases. Curr. Top. Microbiol. Immunol. 316, 193–205.
Othoro, C., Johnston, D., Lee, R., Soverow, J., Bystryn, J. C., and Nardin, E. (2009). Enhanced immunogenicity of Plasmodium falciparum peptide vaccines using a topical adjuvant containing a potent synthetic Toll-like receptor 7 agonist, imiquimod. Infect. Immun. 77, 739–748.
Ott, G., Singh, M., Kazzaz, J., Briones, M., Soenawan, E., Ugozzoli, M., and O’Hagan, D. T. (2002). A cationic sub-micron emulsion (MF59/DOTAP) is an effective delivery system for DNA vaccines. J. Control. Release 79, 1–5.
Owusu-Agyei, S., Ansong, D., Asante, K., Kwarteng Owusu, S., Owusu, R., Wireko Brobby, N. A., Dosoo, D., Osei Akoto, A., Osei-Kwakye, K., Adjei, E. A., Boahen, K. O., Sylverken, J., Adjei, G., Sambian, D., Apanga, S., Kayan, K., Vekemans, J., Ofori-Anyinam, O., Leach, A., Lievens, M., Demoitie, M. A., Dubois, M. C., Cohen, J., Ballou, W. R., Savarese, B., Chandramohan, D., Gyapong, J. O., Milligan, P., Antwi, S., Agbenyega, T., Greenwood, B., and Evans, J. (2009). Randomized controlled trial of RTS,S/AS02D and RTS,S/AS01E malaria candidate vaccines given according to different schedules in Ghanaian children. PLoS ONE 4, e7302. doi: 10.1371/journal.pone.0007302
Papadopoulou, B., Roy, G., Breton, M., Kündig, C., Dumas, C., Fillion, I., Singh, A. K., Olivier, M., and Ouellette, M. (2002). Reduced infectivity of a Leishmania donovani biopterin transporter genetic mutant and its use as an attenuated strain for vaccination. Infect. Immun. 70, 62–68.
Papadopoulou, G., Karagouni, E., and Dotsika, E. (1998). ISCOMs vaccine against experimental leishmaniasis. Vaccine 16, 885–892.
Park, A. Y., Hondowicz, B. D., and Scott, P. (2000). IL-12 is required to maintain a Th1 response during Leishmania major infection. J. Immunol. 165, 896–902.
Pashine, A., Valiante, N. M., and Ulmer, J. B. (2005). Targeting the innate immune response with improved vaccine adjuvants. Nat. Med. 11(Suppl.), S63–S68.
Persing, D. H., Coler, R. N., Lacy, M. J., Johnson, D. A., Baldridge, J. R., Hershberg, R. M., and Reed, S. G. (2002). Taking toll: lipid A mimetics as adjuvants and immunomodulators. Trends Microbiol. 10(Suppl.), S32–S37.
Pinoy, L. M. (1916a). Application a l’homme des vaccines en emulsion dans les corps gras (lipo-vaccins). C. R. Soc. Biol. 79, 352–354.
Pinoy, L. M. (1916b). Les vaccins en emulsion dans les corps gras ou ‘lipo-vaccins’. C. R. Soc. Biol. 79, 201–203.
Poltorak, A., He, X., Smirnova, I., Liu, M. Y., Van Huffel, C., Du, X., Birdwell, D., Alejos, E., Silva, M., Galanos, C., Freudenberg, M., Ricciardi-Castagnoli, P., Layton, B., and Beutler B. (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088.
Probst, P., Skeiky, Y. A., Steeves, M., Gervassi, A., Grabstein, K. H., Reed, S. G. (1997). A Leishmania protein that modulates interleukin (IL)-12, IL-10 and tumor necrosis factor-alpha production and expression of B7-1 in human monocyte-derived antigen-presenting cells. Eur. J. Immunol. 27, 2634–2642.
Querec, T., Bennouna, S., Alkan, S., Laouar, Y., Gorden, K., Flavell, R., Akira, S., Ahmed, R., and Pulendran, B. (2006). Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 203, 413–424.
Raman, V. S., Bhatia, A., Picone, A., Whittle, J., Bailor, H. R., O’Donnell, J., Pattabhi, S., Guderian, J. A., Mohamath, R., Duthie, M. S., and Reed, S. G. (2010). Applying TLR synergy in immunotherapy: implications in cutaneous leishmaniasis. J. Immunol. 185, 1701–1710.
Reed, S. G., Bertholet, S., Coler, R. N., and Friede, M. (2009). New horizons in adjuvants for vaccine development. Trends Immunol. 30, 23–32.
Reed, S. G., and Scott, P. (1993). T-cell and cytokine responses in leishmaniasis. Curr. Opin. Immunol. 5, 524–531.
Rhee, E. G., Mendez, S., Shah, J. A., Wu, C. Y., Kirman, J. R., Turon, T. N., Davey, D. F., Davis, H., Klinman, D. M., Coler, R. N., Sacks, D. L., and Seder, R. A. (2002). Vaccination with heat-killed leishmania antigen or recombinant leishmanial protein and CpG oligodeoxynucleotides induces long-term memory CD4+ and CD8+ T cell responses and protection against Leishmania major infection. J. Exp. Med. 195, 1565–1573.
Ribi, E., Cantrell, J. L., Takayama, K., Qureshi, N., Peterson, J., and Ribi, H. O. (1984). Lipid A and immunotherapy. Rev. Infect. Dis. 6, 567–572.
Richards, R. L., Swartz, G. M. Jr., Schultz, C., Hayre, M. D., Ward, G. S., Ballou, W. R., Chulay, J. D., Hockmeyer, W. T., Berman, S. L., and Alving, C. R. (1989). Immunogenicity of liposomal malaria sporozoite antigen in monkeys: adjuvant effects of aluminium hydroxide and non-pyrogenic liposomal lipid A. Vaccine 7, 506–512.
Row, R. (1912). The curative value of Leishmania culture ”vaccine” in oriental sore. Br. Med. J. 1, 540–541.
Salem, A. K., and Weiner, G. J. (2009). CpG oligonucleotides as immunotherapeutic adjuvants: innovative applications and delivery strategies. Adv. Drug Deliv. Rev. 61, 193–194.
Scharton-Kersten, T., Afonso, L. C., Wysocka, M., Trinchieri, G., and Scott, P. (1995). IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J. Immunol. 154, 5320–5330.
Schijns, V. E. (2000). Immunological concepts of vaccine adjuvant activity. Curr. Opin. Immunol. 12, 456–463.
Scott, P. (1991). Host and parasite factors regulating the development of CD4+ T-cell subsets in experimental cutaneous leishmaniasis. Res. Immunol. 142, 32–36.
Scott, P. (1998). Differentiation, regulation, and death of T helper cell subsets during infection with Leishmania major. Immunol. Res. 17, 229–238.
Scott, P., and Trinchieri, G. (1997). IL-12 as an adjuvant for cell-mediated immunity. Semin. Immunol. 9, 285–291.
Seubert, A., Monaci, E., Pizza, M., O’Hagan, D. T., and Wack, A. (2008). The adjuvants aluminum hydroxide and MF59 induce monocyte and granulocyte chemoattractants and enhance monocyte differentiation toward dendritic cells. J. Immunol. 180, 5402–5412.
Sharifi, I., FeKri, A. R., Aflatonian, M. R., Khamesipour, A., Nadim, A., Mousavi, M. R., Momeni, A. Z., Dowlati, Y., Godal, T., Zicker, F., Smith, P. G., and Modabber, F. (1998). Randomised vaccine trial of single dose of killed Leishmania major plus BCG against anthroponotic cutaneous leishmaniasis in Bam, Iran. Lancet 351, 1540–1543.
Shimizu, Y., Takagi, H., Nakayama, T., Yamakami, K., Tadakuma, T., Yokoyama, N., and Kojima, N. (2007). Intraperitoneal immunization with oligomannose-coated liposome-entrapped soluble leishmanial antigen induces antigen-specific T-helper type immune response in BALB/c mice through uptake by peritoneal macrophages. Parasite Immunol. 29, 229–239.
Singh, M., Chesko, J., Kazzaz, J., Ugozolli, M., Malyala, P., and O’Hagan, D. T. (2007). ”Surface-charged poly(lactide-co-glycolide) microparticles as novel antigen delivery systems,” Vaccine Adjuvants and Delivery Systems, ed. M. Singh (Hoboken, NJ: John Wiley & Sons), 223–248.
Sjolander, A., Baldwin, T. M., Curtis, J. M., Bengtsson, K. L., and Handman, E. (1998a). Vaccination with recombinant Parasite Surface Antigen 2 from Leishmania major induces a Th1 type of immune response but does not protect against infection. Vaccine 16, 2077–2084.
Sjolander, A., Baldwin, T. M., Curtis, J. M., and Handman, E. (1998b). Induction of a Th1 immune response and simultaneous lack of activation of a Th2 response are required for generation of immunity to leishmaniasis. J. Immunol. 160, 3949–3957.
Skeiky, Y. A., Benson, D. R., Costa, J. L., Badaro, R., and Reed, S. G. (1997). Association of Leishmania heat shock protein 83 antigen and immunoglobulin G4 antibody titers in Brazilian patients with diffuse cutaneous leishmaniasis. Infect. Immun. 65, 5368–5370.
Skeiky, Y. A., Benson, D. R., Guderian, J. A., Whittle, J. A., Bacelar, O., Carvalho, E. M., and Reed, S. G. (1995a). Immune responses of leishmaniasis patients to heat shock proteins of Leishmania species and humans. Infect. Immun. 63, 4105–4114.
Skeiky, Y. A., Guderian, J. A., Benson, D. R., Bacelar, O., Carvalho, E. M., Kubin, M., Badaro, R., Trinchieri, G., and Reed, S. G. (1995b). A recombinant Leishmania antigen that stimulates human peripheral blood mononuclear cells to express a Th1-type cytokine profile and to produce interleukin 12. J. Exp. Med. 181, 1527–1537.
Skeiky, Y. A., Kennedy, M., Kaufman, D., Borges, M. M., Guderian, J. A., Scholler, J. K., Ovendale, P. J., Picha, K. S., Morrissey, P. J., Grabstein, K. H., Campos-Neto, A., and Reed, S. G. (1998). LeIF: a recombinant Leishmania protein that induces an IL-12-mediated Th1 cytokine profile. J. Immunol. 161, 6171–6179.
Skidmore, B. J., Chiller, J. M., Weigle, W. O., Riblet, R., and Watson, J. (1976). Immunologic properties of bacterial lipopolysaccharide (LPS). III. Genetic linkage between the in vitro mitogenic and in vivo adjuvant properties of LPS. J. Exp. Med. 143, 143–150.
Soudi, S., Hosseini, A. Z., and Hashemi, S. M. (2011). Co-administration of rectal BCG and autoclaved Leishmania major induce protection in susceptible BALB/c mice. Parasite Immunol. 33, 561–571.
Stacey, K. J., and Blackwell, J. M. (1999). Immunostimulatory DNA as an adjuvant in vaccination against Leishmania major. Infect. Immun. 67, 3719–3726.
Stenger, S., Thuring, H., Röllinghoff, M., and Bogdan, C. (1994). Tissue expression of inducible nitric oxide synthase is closely associated with resistance to Leishmania major. J. Exp. Med. 180, 783–793.
Stobie, L., Gurunathan, S., Prussin, C., Sacks, D. L., Glaichenhaus, N., Wu, C. Y., and Seder, R. A. (2000). The role of antigen and IL-12 in sustaining Th1 memory cells in vivo: IL-12 is required to maintain memory/effector Th1 cells sufficient to mediate protection to an infectious parasite challenge. Proc. Natl. Acad. Sci. U.S.A. 97, 8427–8432.
Sypek, J. P., Chung, C. L., Mayor, S. E., Subramanyam, J. M., Goldman, S. J., Sieburth, D. S., Wolf, S. F., and Schaub, R. G. (1993). Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J. Exp. Med. 177, 1797–1802.
Tafaghodi, M., Khamesipour, A., and Jaafari, M. R. (2011). Immunization against leishmaniasis by PLGA nanospheres encapsulated with autoclaved Leishmania major (ALM) and CpG-ODN. Parasitol. Res. 108, 1265–1273.
Takayama, K., Qureshi, N., Mascagni, P., Nashed, M. A., Anderson, L., and Raetz, C. R. (1983). Fatty acyl derivatives of glucosamine 1-phosphate in Escherichia coli and their relation to lipid A. Complete structure of A diacyl GlcN-1-P found in a phosphatidylglycerol-deficient mutant. J. Biol. Chem. 258, 7379–7385.
Testerman, T. L., Gerster, J. F., Imbertson, L. M., Reiter, M. J., Miller, R. L., Gibson, S. J., Wagner, T. L., and Tomai, M. A. (1995). Cytokine induction by the immunomodulators imiquimod and S-27609. J. Leukoc. Biol. 58, 365–372.
The European Medicines Agency. (2005). Guideline on Adjuvants in Vaccines for Human Use. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003809.pdf
The Working Group on Research Priorities for Development of Leishmaniasis Vaccines, Costa, C. H., Peters, N. C., Maruyama, S. R., de Brito, E. C. Jr., and Santos, I. K. (2011). Vaccines for the leishmaniases: proposals for a research agenda. PLoS Negl. Trop. Dis. 5, e943. doi: 10.1371/journal.pntd.0000943
Titus, R. G., Gueiros-Filho, F. J., de Freitas, L. A., and Beverley, S. M. (1995). Development of a safe live Leishmania vaccine line by gene replacement. Proc. Natl. Acad. Sci. U.S.A. 92, 10267–10271.
Tokunaga, T. (1980). [A review on mechanism of anti-tumor action of BCG (author’s transl)]. Kekkaku 55, 351–357.
Tokunaga, T., Yamamoto, T., and Yamamoto, S. (1999). How BCG led to the discovery of immunostimulatory DNA. Jpn. J. Infect. Dis. 52, 1–11.
Tonui, W. K., Mejia, J. S., Hochberg, L., Mbow, M. L., Ryan, J. R., Chan, A. S., Martin, S. K., and Titus, R. G. (2004). Immunization with Leishmania major exogenous antigens protects susceptible BALB/c mice against challenge infection with L. major. Infect. Immun. 72, 5654–5661.
Traore, B., Kone, Y., Doumbo, S., Doumtabé, D., Traoré, A., Crompton, P. D., Mircetic, M., Huang, C. Y., Kayentao, K., Dicko, A., Sagara, I., Ellis, R. D., Miura, K., Guindo, A., Miller, L. H., Doumbo, O. K., and Pierce, S. K. (2009). The TLR9 agonist CpG fails to enhance the acquisition of Plasmodium falciparum-specific memory B cells in semi-immune adults in Mali. Vaccine 27, 7299–7303.
U.S. Food and Drug Administration. (2011). Complete List of Vaccines Licensed for Immunization and Distribution in the US. Available at: http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/UCM093833
Uzonna, J. E., Spath, G. F., Beverley, S. M., and Scott P. (2004). Vaccination with phosphoglycan-deficient Leishmania major protects highly susceptible mice from virulent challenge without inducing a strong Th1 response. J. Immunol. 172, 3793–3797.
Velez, I. D., Gilchrist, K., Martínez, S., Ramírez-Pineda, J. R., Ashman, J. A., Alves, F. P., Coler, R. N., Bogatzki, L. Y., Kahn, S. J., Beckmann, A. M., Cowgill, K. D., Reed, S. G., and Piazza, F. M. (2009). Safety and immunogenicity of a defined vaccine for the prevention of cutaneous leishmaniasis. Vaccine 28, 329–337.
von Stebut, E., Belkaid, Y., Jakob, T., Sacks, D. L., and Udey, M. C. (1998). Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J. Exp. Med. 188, 1547–1552.
Wack, A., Baudner, B. C., Hilbert, A. K., Manini, I., Nuti, S., Tavarini, S., Scheffczik, H., Ugozzoli, M., Singh, M., Kazzaz, J., Montomoli, E., Del Giudice, G., Rappuoli, R., and O’Hagan, D. T. (2008). Combination adjuvants for the induction of potent, long-lasting antibody and T-cell responses to influenza vaccine in mice. Vaccine 26, 552–561.
Wagner, T. L., Ahonen, C. L., Couture, A. M., Gibson, S. J., Miller, R. L., Smith, R. M., Reiter, M. J., Vasilakos, J. P., and Tomai, M. A. (1999). Modulation of TH1 and TH2 cytokine production with the immune response modifiers, R-848 and imiquimod. Cell. Immunol. 191, 10–19.
Walker, P. S., Scharton-Kersten, T., Krieg, A. M., Love-Homan, L., Rowton, E. D., Udey, M. C., and Vogel, J. C. (1999). Immunostimulatory oligodeoxynucleotides promote protective immunity and provide systemic therapy for leishmaniasis via IL-12- and IFN-gamma-dependent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 96, 6970–6975.
Webb, J. R., Campos-Neto, A., Skeiky, Y. A., and Reed, S. G. (1997). Molecular characterization of the heat-inducible LmSTI1 protein of Leishmania major. Mol. Biochem. Parasitol. 89, 179–193.
Webb, J. R., Kaufmann, D., Campos-Neto, A., and Reed, S. G. (1996). Molecular cloning of a novel protein antigen of Leishmania major that elicits a potent immune response in experimental murine leishmaniasis. J. Immunol. 157, 5034–5041.
Wenyon, C. M. (1928). Kala-azar and oriental sore: the problem of transmission. Br. Med. J. 2, 558–562.
White, R. G., Coons, A. H., and Connolly, J. M. (1955). Studies on antibody production. III. The alum granuloma. J. Exp. Med. 102, 73–82.
Whitmore, E. R. (1919). Lipovaccines, with special reference to public health work. Am. J. Public Health 9, 504–507.
WHO. (2004). 13 Vaccines that are in Common Use in Most Developing Countries. Available at: http://www.who.int/vaccines-documents/DoxTrng/h4iip.htm.
WHO. (2010). ”Control of the leishmaniases,” in Report of a Meeting of the WHO Expert Committee on the Control of Leishmaniases, Geneva, 22–26 March 2010. WHO Technical Report Series 949.
WHO Department of Immunization, Vaccines and Biologicals and UNICEF Programme Division, Health Section. (2005). Global Immunization Vision and Strategy 2006-2015. Available at: http://whqlibdoc.who.int/hq/2005/WHO_IVB_05.05.pdf
Wooldridge, J. E., Ballas, Z., Krieg, A. M., and Weiner, G. J. (1997). Immunostimulatory oligodeoxynucleotides containing CpG motifs enhance the efficacy of monoclonal antibody therapy of lymphoma. Blood 89, 2994–2998.
Wright, A. K., Briles, D. E., Metzger, D. W., and Gordon, S. B. (2008). Prospects for use of interleukin-12 as a mucosal adjuvant for vaccination of humans to protect against respiratory pneumococcal infection. Vaccine 26, 4893–4903.
Wu, W., Huang, L., and Mendez, S. (2010). A live Leishmania major vaccine containing CpG motifs induces the de novo generation of Th17 cells in C57BL/6 mice. Eur. J. Immunol. 40, 2517–2527.
Wu, W., Weigand, L., Belkaid, Y., and Mendez, S. (2006). Immunomodulatory effects associated with a live vaccine against Leishmania major containing CpG oligodeoxynucleotides. Eur. J. Immunol. 36, 3238–3247.
Yang, Z., Zhang, X., Darrah, P. A., and Mosser, D. M. (2010). The regulation of Th1 responses by the p38 MAPK. J. Immunol. 185, 6205–6213.
Zhang, W. W., and Matlashewski, G. (2008). Immunization with a Toll-like receptor 7 and/or 8 agonist vaccine adjuvant increases protective immunity against Leishmania major in BALB/c mice. Infect. Immun. 76, 3777–3783.
Keywords: vaccine, adjuvants, leishmania
Citation: Raman VS, Duthie MS, Fox CB, Matlashewski G and Reed SG (2012) Adjuvants for Leishmania vaccines: from models to clinical application. Front. Immun. 3:144. doi: 10.3389/fimmu.2012.00144
Received: 07 February 2012; Paper pending published: 13 March 2012;
Accepted: 18 May 2012; Published online: 11 June 2012.
Edited by:
Nathan Peters, National Institute of Allergy and Infectious Diseases, USAReviewed by:
Huji Xu, The Second Military Medical University, ChinaJude Ezeh Uzonna, University of Manitoba, Canada
Copyright: © 2012 Raman, Duthie, Fox, Matlashewski and Reed. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Vanitha S. Raman and Steven G. Reed, Pre-clinical Biology, Infectious Disease Research Institute, 1124 Columbia Street, Suite 400, Seattle, WA 98104, USA. e-mail:dnJhbWFuQGlkcmkub3Jn;c3JlZWRAaWRyaS5vcmc=
Abbreviations: ALM, autoclaved Leishmania major; AS, Adjuvant Systems; BCG, Bacille Calmette-Guérin; CL, cutaneous leishmaniasis; CLR, C-type lectin-like receptors; DTH, delayed type hypersensitivity; GLA, glucopyranosyl lipid adjuvant; IFN, interferon; IL, interleukin; ISCOM, immunostimulatory complexes; LPS, lipopolysaccharide; MPL, monophosphoryl lipid A; Mtb, Mycobacterium tuberculosis; NO, nitric oxide; ODN, oligodeoxynucleotides; PAMP, pathogen associated molecular patterns; PKDL, post-kala-azar dermal leishmaniasis; PRR, pattern recognition receptor; rh, recombinant human; RIG, retinoic acid inducible gene based; RLR, RIG-I-like receptors; SE, stable emulsion; SSG, sodium stibogluconate; TLR, Toll-like receptor; TNF, tumor necrosis factor; VL, visceral leishmaniasis.