

- Division of Inflammation Biology, La Jolla Institute for Allergy and Immunology, La Jolla, CA, USA
Lymphocyte function-associated antigen-1 (LFA-1) is a heterodimeric integrin consisting of αL (gene name, Itgal) and β2 (gene name, Itgb2) subunits expressed in all leukocytes. LFA-1 is essential for neutrophil recruitment to inflamed tissue. Activation of LFA-1 by chemokines allows neutrophils and other leukocytes to undergo arrest, resulting in firm adhesion on endothelia expressing intercellular adhesion molecules (ICAMs). In mice, CXCR2 is the primary chemokine receptor involved in triggering neutrophil arrest, and it does so through “inside-out” activation of LFA-1. CXCR2 signaling induces changes in LFA-1 conformation that are coupled to affinity upregulation of the ligand-binding headpiece (extended with open I domain). Unlike naïve lymphocytes, engagement of P-selectin glycoprotein ligand-1 (PSGL-1) on neutrophils stimulates a slow rolling behavior that is mediated by LFA-1 in a distinct activation state (extended with closed I domain). How inside-out signaling cascades regulate the structure and function of LFA-1 is being studied using flow chambers, intravital microscopy, and flow cytometry for ligand and reporter antibody binding. Here, we review how LFA-1 activation is regulated by cellular signaling and ligand binding. Two FERM domain-containing proteins, talin-1 and Kindlin-3, are critical integrin co-activators and have distinct roles in the induction of LFA-1 conformational rearrangements. This review integrates these new results into existing models of LFA-1 activation.
LFA-1 Structure, Function, and Inside-Out Activation
Twenty-four different integrins are expressed in humans, each composed of non-covalently associated α and β chains (Hynes, 2002). Integrins have large extracellular domains, single-pass transmembrane segments, and short intracellular tails. Lacking enzymatic activity, the short integrin cytoplasmic domains serve as scaffolds for signaling and structural proteins that allow integrins to be a conduit of bidirectional communication between the cytoplasm and extracellular ligands (Legate and Fassler, 2009; Moser et al., 2009b) [Box 1]. The integrin ligand recognition site spans the β subunit inserted-like (βI) domain and α subunit β-propeller domain or, for about half of the integrin family, resides entirely within the α subunit I (αI) domain (Lu et al., 2001; Luo et al., 2007). All β2 integrins, including LFA-1 (αLβ2 or CD11a/CD18) [Box 2], contain this extra αI domain within the headpiece. The structural features of the various domains of LFA-1 have been reviewed in great detail elsewhere (Luo et al., 2007).
Box 1. “Inside-out” and “outside-in”signaling.
Inside-out signaling refers to a process by which a cellular stimulus, for example, a chemokine binding to its receptor, leads to integrin activation by intracellular signaling pathways. Inside-out signaling leads to vast conformational changes in integrins, but not directly to clustering (redistribution of integrin heterodimers in the plane of the plasma membrane). By contrast, outside-in activation refers to integrin activation induced by ligand binding. Although the terminology is often confused, outside-in signaling, distinct from outside-in activation, refers to a process whereby the cell receives signals from the extracellular environment through integrins that have already been activated and are now bound to immobilized ligands on another cell or in the extracellular matrix. Adhesion strengthening, or stabilization, is a process following integrin activation and initial ligand binding. Adhesion strengthening most likely involves integrin clustering and is required to keep arrested leukocytes from being washed away.
Box 2. β2 integrin family.
The β2 subfamily of integrins comprises four heterodimers. αLβ2 (LFA-1, CD11a/CD18) is expressed on all leukocytes; αMβ2 (Mac-1, CD11b/CD18) is expressed on granulocytes, monocytes, macrophages, and subsets of activated lymphocytes; αXβ2 (P150,95, CD11c/CD18) is expressed on dendritic cells, macrophages, and small subsets of blood monocytes; and αDβ2 (CD11d/CD18) is expressed on tissue-specific subsets of macrophages.
The macromolecular structure of integrins is coupled to the accessibility of the ligand-binding pocket and to its ligand-binding affinity. Therefore, the regulation of integrin conformation is critical for their adhesive and signaling function (Luo et al., 2007). Studies employing crystallography (Shimaoka et al., 2003b), nuclear magnetic resonance (Huth et al., 2000; Legge et al., 2000), electron microscopy (Nishida et al., 2006), and molecular dynamics simulation (Jin et al., 2004) have suggested that LFA-1 can assume at least three distinct conformational states (Figure 1). In its inactive state, the LFA-1 extracellular domain has a bent structure shaped like an inverted V (Nishida et al., 2006) with the low affinity headpiece closely approaching the plasma membrane (Larson et al., 2005; Nishida et al., 2006), similar to what has been shown for other integrins (Xiong et al., 2001; Chigaev et al., 2003; Zhu et al., 2008; Xie et al., 2010). The inactive state is also characterized by inter-domain contacts between the N-terminal headpiece and membrane-proximal lower legs, between the lower legs of the α and β chains, and between the α and β transmembrane domains (TMDs; Takagi et al., 2002; Li et al., 2005; Luo et al., 2005; Partridge et al., 2005; Nishida et al., 2006). Based primarily on mutational studies, it is thought that close association of TMDs stabilizes the inactive state and disruption of this association leads to integrin activation (Hughes et al., 1996; Lu and Springer, 1997; Vinogradova et al., 2002; Luo et al., 2005).
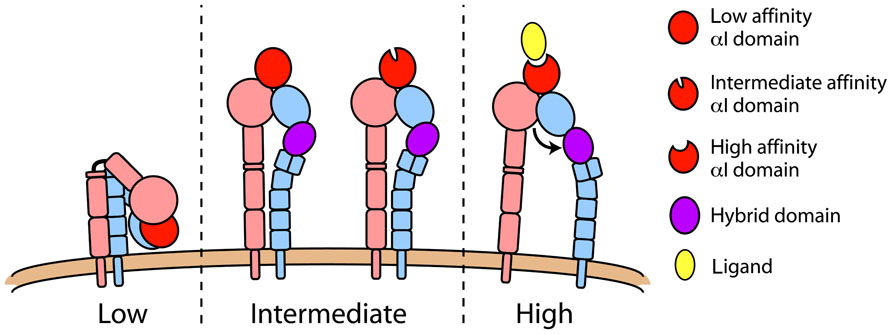
FIGURE 1. LFA-1 conformations. Integrin function is structurally regulated and three distinct conformations have been demonstrated. In its low affinity state (left), LFA-1 is compact with a sharp bend at the “genu” and headpiece closely approaching the plasma membrane. Extended conformations (middle) differ in the orientation of their hybrid domain (purple), with swingout of this domain representing a conversion from intermediate to high affinity state (right). Data suggest that the extended/closed conformation (middle) can have either a low or intermediate affinity αI domain.
In contrast to the compact structure of inactive LFA-1, active LFA-1 conformations exhibit an extended extracellular domain with the ligand-binding headpiece situated more than 20 nm above the membrane (Nishida et al., 2006). Extended structures of LFA-1 (Figure 1) differ in the conformation of the headpiece and αI domain, the angle between the βI domain and hybrid domain into which the βI is inserted, and the distance between the αL and β2 TMDs in the plane of the plasma membrane. The extended ectodomain with a closed headpiece has also been termed the “intermediate affinity state” integrin (Luo et al., 2007). It is thought that the spatial orientation of the headpiece alone in the extended intermediate affinity structure may account for enhanced recognition of ligands, as steric hinderance may prevent large, immobilized ligands from accessing the αI domain in the bent, inactive LFA-1 conformer. This idea is supported by data showing that the isolated LFA-1 αI domain alone, in its basal state, can mediate transient interactions with intercellular adhesion molecule-1 (ICAM-1) that result in a rolling phenotype (Knorr and Dustin, 1997; Eniola et al., 2005), whereas the full-length LFA-1 molecule does not support interaction under flow conditions (Salas et al., 2002). However, in a series of disulfide-stabilized crystal structures of the αL I domain, distinct conformations with low, intermediate, and high affinity were identified, where the transition from low to intermediate affinity was mainly driven by an increase in the bimolecular association rate (kon) and the transition from intermediate to high affinity was mainly driven by lower koff (slower release of bound ligand; Shimaoka et al., 2003b). Notably, the αI metal ion-dependent adhesion site (MIDAS) that recognizes ligand retains the same coordination and conformation in the putative αI low and intermediate affinity conformations. Therefore, in addition to the macromolecular integrin structures, it is apparent that the αI domain may exist in three distinct conformations regulated by the relative position of the α7 helix. There is ample evidence, as will be discussed below, that extended LFA-1 mediates neutrophil slow rolling by transiently binding to ICAM-1. During slow rolling, the αL I domain is most likely not in the high affinity conformation.
In the “high affinity state” structure, swingout of the hybrid domain away from the α subunit by approximately 60° is coupled to opening of the headpiece through the downward movement of the αI domain α7 helix that connects to the βI domain as an internal ligand for the βI MIDAS (Alonso et al., 2002; Xiao et al., 2004; Luo et al., 2007). This shift in the α7 helix is coupled to rearrangement of the αI domain, enhancing its affinity for ligand (Alonso et al., 2002; Yang et al., 2004). It is the high affinity LFA-1 conformer that mediates arrest of leukocytes on the endothelium (Constantin et al., 2000; Giagulli et al., 2004), and in vitro studies demonstrate that the open headpiece of LFA-1 is necessary and sufficient to mediate cell arrest under flow conditions (Salas et al., 2002, 2004).
The structure and affinity of integrin receptors is thought to be regulated by cells in an “inside-out” manner [Box 1] through signaling cascades that impinge upon the integrin cytoplasmic tails (Dustin and Springer, 1989; O’Toole et al., 1990; Sims et al., 1991). Two intracellular protein families have been implicated in the final events of integrin activation. Talins and Kindlins both contain a band 4.1/ezrin/radixin/moesin (FERM) domain with four independently folded subdomains (F0–F3; Moser et al., 2009b). While the FERM domain comprises the talin head that is sufficient for activating integrins (Calderwood et al., 1999; Ye et al., 2010), talins also have a flexible rod domain that contains actin-binding sequences and mediates cytoskeletal association (Critchley, 2009). Although many of the structural and functional studies that have dissected the role of talin in integrin activation have employed experimental systems where the talin head domain alone is used, the full-length molecule exists in a basal autoinhibited state that is mediated by contact between the integrin-binding F3 subdomain and rod domain (Goksoy et al., 2008). Binding to phospholipids, such as phosphatidylinositol 4,5-bisphosphate (PIP2), relieves this autoinhibition and activates talin for integrin binding (Martel et al., 2001; Goksoy et al., 2008). The ability of talins and Kindlins to activate integrins depends on their binding to membrane phospholipids. For talin, this occurs through positively charged surfaces within the F1, F2, and F3 subdomains (Anthis et al., 2009; Goult et al., 2010). Kindlins have an analogous surface within the F1 subdomain (Bouaouina et al., 2012) as well as an additional pleckstrin homology domain that is inserted within its F2 subdomain (Tu et al., 2003; Qu et al., 2011), both of which bind to membrane phospholipids and are important for integrin activation.
Talins and Kindlins mediate integrin activation by binding directly to separate NXX(Y/F) motifs within the integrin β chain short cytoplasmic tail (Tadokoro et al., 2003; Harburger et al., 2009). Multiple lines of evidence demonstrate that talins contribute to the separation of the integrin TMDs (Kim et al., 2003; Wegener et al., 2007), but the mechanisms by which Kindlins are involved in regulating integrin conformation are largely unknown (Moser et al., 2009b; Shattil et al., 2010). Mutations of Kindlin-3, the Kindlin isoform expressed in hematopoietic cells, were found to underlie the leukocyte adhesion deficiency type III (LAD-III) pathology (Malinin et al., 2009; Moser et al., 2009a; Svensson et al., 2009) [Box 3]. Kindlin-3-deficient leukocytes are unable to arrest on inflamed endothelium, but their selectin-mediated rolling capacity is normal, indicating an important role for Kindlin-3 in LFA-1 activation (Moser et al., 2009a). The roles of talin-1 and Kindlin-3 in regulating LFA-1 in neutrophils are discussed in further detail below.
Box 3. Leukocyte adhesion deficiency type III.
In the human disease leukocyte adhesion deficiency type III (LAD-III), activation of β2, β3, and some β1 integrins is defective. LAD-III was discovered in 1997 and initially called LAD-I variant (Kuijpers et al., 1997). Kuijpers and colleagues recognized that expression of β2 integrins was normal but their activation was defective in these patients (Alon et al., 2003; Alon and Etzioni, 2003; Malinin et al., 2009; Manevich-Mendelson et al., 2009; Svensson et al., 2009; Jurk et al., 2010; McDowall et al., 2010; Robert et al., 2011). Alon and colleagues (Pasvolsky et al., 2007) reported a point mutation in RASGRP2, the gene encoding CalDAG-GEFI, and thought that defective CalDAG-GEFI caused LAD-III. This idea was supported by the leukocyte and platelet adhesion deficiencies exhibited by CalDAG-GEFI knockout mice (Bergmeier et al., 2007). However, it was later found that mutation of the FERMT3 gene encoding Kindlin-3 that causes the pathology of LAD-III (Kuijpers et al., 2009; Svensson et al., 2009), dominates the phenotype of the subset of LAD-III patients with mutations in both FERMT3 and RASGRP2. Humans deficient in CalDAG-GEFI, but not Kindlin-3, have not been described. There is now consensus that mutations in the FERMT3 gene encoding kindlin-3 cause all known LAD-III cases (Abram and Lowell, 2009).
Models of LFA-1 Activation
There are several models of integrin activation by inside-out signaling. The dynamic equilibrium that exists between the various integrin conformational states (Figure 1) appears to vary between the 24 different integrin subtypes and even amongst the four different β2 integrins (Nishida et al., 2006). Therefore, aside from the most basic structural commonalities, integrin activation mechanisms may exhibit significant variability.
The switchblade model of integrin activation stems from the delineation of the three distinct affinity states described above, and was formulated based on crystallography (Xiong et al., 2001; Xiao et al., 2004) and studies of isolated integrins by electron microscopy (Takagi et al., 2002; Nishida et al., 2006). The switchblade model postulates that only the closed, unliganded integrin headpiece can stably exist in the overall bent structure. The switchblade-like extension of the extracellular domain occurs as a result of disruption of headpiece-lower leg and α/β lower leg stabilizing contacts, either by separation of the TMDs or by ligand-induced hybrid domain swingout (Luo et al., 2007). The switchblade model received support from the recent finding that single, full-length αIIbβ3 integrin embedded in membrane has the same compact structure observed in isolated extracellular domains and can undergo ectodomain extension in the presence of talin head domain bound to the integrin cytoplasmic tail (Ye et al., 2010). Most of the activated structures observed in this study, however, resembled the intermediate rather than the high affinity conformation. As discussed below, an important unresolved question is whether inside-out signaling alone (that is, in the absence of ligand binding) induces the high affinity structure with an open headpiece.
The deadbolt model of integrin activation was proposed (Xiong et al., 2003) based on the observation that crystals of the unbound (Xiong et al., 2001) and ligand-occupied (Xiong et al., 2002) integrin αVβ3 exhibited the same compact, bent structure. The crystal structure of ligand-occupied, bent αVβ3 was further supported by electron microscopy studies (Adair et al., 2005). In addition, FRET studies have shown that small ligand binding to α4β1 integrin (VLA-4) on the living cell surface can occur in the absence of ectodomain extension (Chigaev et al., 2007). The deadbolt model posits that a loop within the β subunit extracellular membrane-proximal β-tail domain contacts the α7 helix of the βI domain that regulates its conformation and affinity (Xiong et al., 2003). During activation, movement of the TMDs would disrupt the deadbolt and allow headpiece opening in the bent structure (Xiong et al., 2003). However, elimination of the β-tail domain loop that was proposed to form the deadbolt did not enhance ligand binding (Zhu et al., 2007).
More recent work suggests that inside-out integrin activation is more complicated than the switchblade and deadbolt models imply. For example, in kinetic studies of VLA-4 on live cells, binding of antibodies that recognize epitopes selectively exposed in extended integrin structures was minimal after triggering two different physiologic inside-out activation pathways, but was robust in the presence of a small ligand even without cell stimulation (Chigaev et al., 2009). The same group recently performed similar experiments with a fluorescent ligand probe of LFA-1 and found that small ligand binding to LFA-1 in the absence of inside-out activation was very slow (Chigaev et al., 2011). These findings indicate a greater restraint on inactive LFA-1 compared to VLA-4 with respect to ligand binding. Inside-out stimulation via chemokine or formyl peptide receptors [both G-protein coupled receptors (GPCRs)] rapidly enhanced LFA-1 small ligand binding, suggesting affinity upregulation of the headpiece. These studies suggest that mechanisms of integrin conformational regulation, including inside-out and ligand-induced outside-in activation, have both shared and distinct components between integrin subtypes and activating stimuli.
It is clear that inside-out signal transduction pathways, such as those elicited by chemokine receptor engagement, stimulate an increase in the affinity of integrins. The ability of LFA-1 to bind ICAM-1 in its soluble, rather than immobilized, form has been utilized as a sensitive assay that specifically reports the high affinity state (Constantin et al., 2000; Shimaoka et al., 2003a,b). Further support for this assay as a means to discriminate between extended/closed and extended/open conformations of LFA-1 was provided by Laudanna and colleagues (Bolomini-Vittori et al., 2009) by showing that a signaling defect in headpiece opening due to silencing of phosphatidylinositol-4-phosphate 5-kinase type I gamma (PIP5KC) blocks chemokine-induced soluble ICAM-1 binding to T lymphocytes despite the ability of LFA-1 to become extended (as indicated by a reporter mAb) under the same conditions. Many studies have thus demonstrated that chemokines induce a high affinity state of LFA-1 that is competent for soluble ICAM-1 binding and that mediates T lymphocyte arrest (Constantin et al., 2000; Giagulli et al., 2004; Bolomini-Vittori et al., 2009).
Despite these extensive studies, it is not clear whether chemokine stimulation alone, in the absence of ligand, triggers opening of the LFA-1 headpiece. Chemokines induce the binding of epitope-specific reporter antibodies, such as mAb 24 (Dransfield et al., 1992) and 327C mAb (Beals et al., 2001), that specifically recognize the extended/open high affinity conformation of LFA-1 (Bolomini-Vittori et al., 2009). However, these reporter antibodies also recognize ICAM-1-bound LFA-1 in the absence of cellular stimulation (Beals et al., 2001). Therefore, it remains possible that chemokines enhance the affinity of LFA-1 for ICAM-1 and permit a transition to the high affinity state, but do not directly induce opening of the headpiece associated with the high affinity αI domain. Rather, ligand engagement and a force pulling on the engaged integrin may subsequently be needed for LFA-1 to achieve its high affinity state. Affinity measurements of LFA-1 on the cell surface show that inside-out signaling in response to stromal cell-derived factor-1 (SDF-1, also known as chemokine CXCL12) or PMA, an activator of protein kinase C, enhanced binding of soluble monovalent ICAM-1, indicating affinity upregulation (Schurpf and Springer, 2011). However, artificially stabilizing specific LFA-1 structures resulted in ICAM-1 binding affinities of the intermediate affinity state that were similar to those achieved by inside-out activation, and of the high affinity state that were much higher. In a novel assay in which antibodies that specifically recognize extended LFA-1 conformers (KIM127 mAb and NKI-L16 mAb) or the putative high affinity state of LFA-1 (327C mAb) were co-immobilized with the chemokine CXCL12, it was found that peripheral blood lymphocytes bound only KIM127 and NKI-L16, but not 327C (Shamri et al., 2005). These data all suggest that chemokines trigger LFA-1 extension, but cannot induce the full affinity of the ligand-binding headpiece.
If inside-out activation does not shift integrin affinity to its highest state, how then does ligand binding do so? The demonstration of a catch bond between LFA-1 and ICAM-1 provides insight into a possible mechanism and also suggests an important role for force in affinity maturation of the αI domain (Chen et al., 2010). Using a biomembrane probe to ligate single LFA-1 molecules and measure bond kinetics in the absence or presence of a pulling force, Zhu and colleagues (Chen et al., 2010) demonstrate that ICAM-1-occupied LFA-1 passes through three distinct bond lifetime regimes with increasing force. Pulling on the bond first enhanced LFA-1 affinity (catch bond) and then, for larger forces, resulted in a decrease in bond lifetime (slip bond). They propose that pulling on the αI domain results in its movement away from the βI domain, causing the anchored (to the βI MIDAS) α7 helix to experience a relative shift downward (as discussed above) and resulting in affinity upregulation of the αI domain (Chen et al., 2010). Thus, in the context of physiologic leukocyte–endothelial interactions in which LFA-1 bonds experience a force from blood flow acting on rolling or adherent cells, chemokines may stimulate LFA-1 extension while enhancing affinity of the headpiece only moderately. Upon ICAM-1 engagement, the bond would then undergo further affinity maturation and transition into the high affinity conformation supporting long-lived bonds. This model of force-mediated LFA-1 affinity maturation also suggests a role for cytoskeletal anchorage of the integrin tail (Alon and Dustin, 2007; Zhu et al., 2008). Talin represents the best candidate for mediating such actin linkage through sequences in its rod domain (Critchley, 2009), though a specific role for talin-mediated actin filament association in LFA-1 activation is yet to be described. A potential role for Kindlins in mediating indirect cytoskeletal anchorage is also discussed below in further detail.
Regulation of LFA-1 Activation in Neutrophils
Neutrophils play a central role in immunity as the first leukocyte subset to enter tissues in response to infection or injury. Inflammatory cues produced locally in the tissue are relayed to neutrophils through the expression of adhesion molecules, such as E-selectin and ICAM-1, on the luminal surface of endothelial cells. This induced expression of adhesion molecules initiates a cascade of progressive interactions between leukocytes and the vascular wall that precede transmigration across the endothelial barrier and into the affected tissue (Ley et al., 2007).
Members of the β2 integrin family [Box 2], whose expression is restricted to leukocytes and leukocyte-derived tissue cells, are involved at several steps of the leukocyte adhesion cascade. Initial capture and rolling of neutrophils is mediated primarily by endothelial E- and P-selectins binding to P-selectin glycoprotein ligand-1 (PSGL-1) and other selectin ligands on the surface of neutrophils. In addition to its function as an adhesion receptor, PSGL-1 transduces an intracellular signal that partially activates LFA-1 (Zarbock et al., 2007b; Miner et al., 2008; Kuwano et al., 2010; Figure 2). Although other leukocyte subsets, including some memory T lymphocytes, express functional PSGL-1, it seems that ligation of PSGL-1 triggers LFA-1 extension only in myeloid cells. It may be that the lymphoid homologs of one or more molecules downstream of PSGL-1 do not participate in this signaling pathway (Alon and Ley, 2008). The molecular details of the signal transduction pathway triggered by PSGL-1 engagement and leading to LFA-1 activation in neutrophils has been the subject of recent reviews (Zarbock et al., 2009, 2011), and new details on this signaling cascade continue to emerge (Stadtmann et al., 2011; Block et al., 2012; Lefort et al., 2012; Shao et al., 2012; Spertini et al., 2012). During neutrophil rolling interactions with P- and E-selectin, PSGL-1 signaling results in the conversion of LFA-1 from an inactive state to an extended conformation that then interacts with ICAM-1 to reduce neutrophil rolling velocity (Chesnutt et al., 2006; Zarbock et al., 2007b; Kuwano et al., 2010). Two main pieces of evidence demonstrate that the extended LFA-1 conformer with a closed headpiece mediates slow rolling in both murine and human neutrophils. First, an LFA-1 allosteric antagonist that binds to the βI domain and blocks structural communication with the αI domain, thus preventing headpiece opening, completely abrogates soluble ICAM-1 binding (Shimaoka et al., 2003a) and neutrophil arrest, but does not impair LFA-1-dependent slow rolling (Zarbock et al., 2007b). Second, the KIM127 mAb (Robinson et al., 1992) and NKI-L16 mAb (van Kooyk et al., 1991) epitopes that report extension of the β2 and αL legs, respectively, are exposed in human neutrophils rolling on E-selectin in flow chambers (Kuwano et al., 2010). In the same assay, the mAb 24 (Dransfield et al., 1992) recognition site that is only accessible in the high affinity state remains buried unless neutrophils are activated by Mn2+ (Kuwano et al., 2010). Thus, ligation of PSGL-1 induces LFA-1 extension, but not (full) opening of the headpiece.
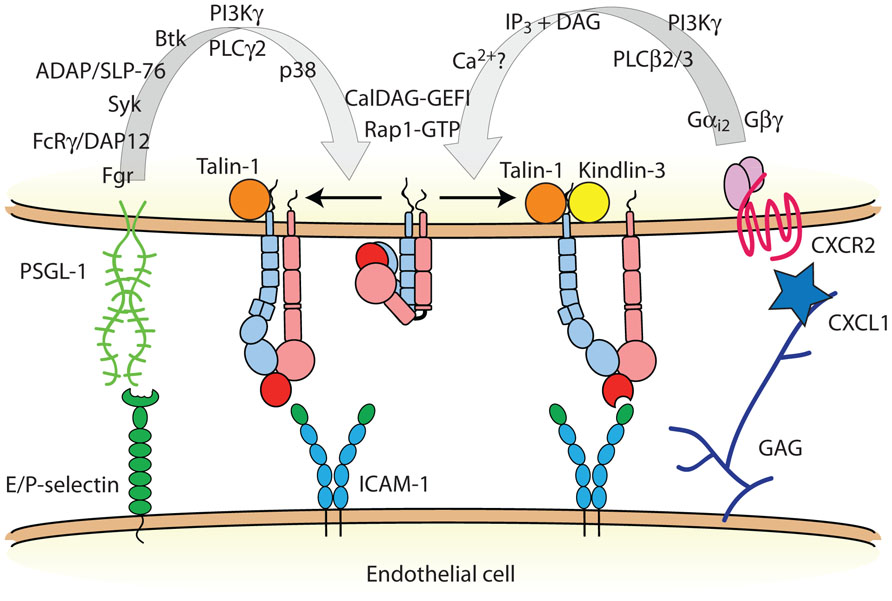
FIGURE 2. Neutrophil inside-out activation of LFA-1. Two distinct signal transduction pathways modulate LFA-1 activation in neutrophils: (1) selectin binding to PSGL-1, and (2) chemokine receptor engagement. PSGL-1 signals talin-1-dependent LFA-1 extension, whereas chemokine GPCRs signal LFA-1 activation to a high affinity state that requires both talin-1 and Kindlin-3. Some of these signaling molecules may exist in preformed modules to facilitate rapid integrin activation. Abbreviations: Fgr, feline Gardner–Rasheed sarcoma kinase; FcRγ, immunoglobulin gamma Fc receptor; DAP12, DNAX activating protein of 12 kDa; Syk, spleen tyrosine kinase; ADAP, adhesion and degranulation promoting adaptor protein; SLP-76, SH2 domain-containing leukocyte phosphoprotein of 76 kDa; Btk, Bruton tyrosine kinase; PI3K, phosphoinositide 3-kinase; PLC, phospholipase C; p38, p38 mitogen activated protein kinase; IP3, inositol trisphosphate; DAG, diacylglycerol; CalDAG-GEFI, calcium- and diacylglycerol-regulated guanine nucleotide exchange factor I; PSGL-1, P-selectin glycoprotein ligand-1; ICAM-1, intercellular adhesion molecule-1; GAG, glycosaminoglycan.
During inflammation, neutrophils encounter immobilized chemokines as they roll on the vascular wall. LFA-1 is the primary integrin receptor involved in leukocyte arrest on inflamed endothelium, with little or no detectable contribution of Mac-1 (Ding et al., 1999; Ley et al., 2007). SDF-1α (a ligand for CXCR4) and chemokine ligands for CXCR2 trigger soluble ICAM-1 binding and rapid lymphocyte arrest on immobilized ICAM-1 (Constantin et al., 2000; Giagulli et al., 2004; Shamri et al., 2005; Bolomini-Vittori et al., 2009). This has been interpreted to mean that high affinity LFA-1 is required to mediate leukocyte arrest. Intravital microscopy analysis of the mouse cremaster muscle, a thin tissue that envelops the testes, after intravenous injection of CXCL1 provides a sensitive arrest assay, as neutrophils rapidly (within 15 s) transition from rolling to arrest on the endothelium. This assay provides the ability to distinguish the arrest step from adhesion stabilization, as neutrophils lacking signaling molecules involved in the latter process, such as PI3Kγ (Smith et al., 2006) and PKCθ (Bertram et al., 2012), quickly detach and return to the bulk flow. Since chemokine signaling also contributes to the stabilization and strengthening of leukocyte adhesion following arrest by enhancing LFA-1 mobility and allowing clustering to occur (Constantin et al., 2000; Giagulli et al., 2004, 2006), the analysis of rapid arrest in vivo is critical for assessing the functional role of molecules in inside-out activation of LFA-1. CXCL1 (also known as keratinocyte-derived chemokine) is the primary chemokine involved in murine neutrophil arrest through CXCR2 and activation of the Gαi2 signaling cascade (Ley, 2003; Smith et al., 2004; Zarbock et al., 2007a). In the context of inflammation, both the PSGL-1 and CXCR2 signaling pathways that regulate LFA-1 activation contribute to neutrophil adhesion and recruitment (Smith et al., 2004; Zarbock et al., 2007b). PSGL-1 signaling seems to induce only the extended conformation of LFA-1, and even pulling on LFA-1 as it transiently engages with ICAM-1 in slow rolling is not sufficient to induce rapid neutrophil arrest mediated by the high affinity conformation. By contrast, in the presence of chemokines, neutrophil LFA-1 reaches its high affinity state and mediates arrest. Another potential difference between these two signaling pathways is that chemokine receptors trigger rapid and locally restricted LFA-1 activation (Shamri et al., 2005), whereas PSGL-1 signals to LFA-1 may not be as spatially confined (Alon and Ley, 2008; Kuwano et al., 2010). It has been proposed that the downstream signaling proteins involved in integrin activation may be pre-assembled into complexes, thus allowing for leukocyte arrest to occur on the timescales observed experimentally (Alon and Ley, 2008). Consistent with this idea, a signaling module containing SKAP55 (Src kinase-associated phosphoprotein of 55 kDa), ADAP (adhesion and degranulation promoting adaptor protein), RIAM (Rap1-interacting adaptor molecule), and Kindlin-3 exists constitutively in unstimulated human T lymphocytes, and inducibly associates with the common integrin activator Rap1 GTPase (Kliche et al., 2012).
New Mechanistic Insights into Inside-Out LFA-1 Activation
LFA-1 has been a model for studying integrin activation for more than 20 years. The role of inside-out signaling in regulating LFA-1 structure and affinity is a topic of great interest not only in the fields of immunology and leukocyte adhesion, but also among integrin biologists. Until recently, the roles of two families of common integrin co-activators, talins and Kindlins, in regulating LFA-1 conformational activation had not been studied. Using mixed chimeric mice with genetic deletion of either Fermt3 encoding Kindlin-3 or Tln1 encoding talin-1, we tested the individual roles of these two proteins in neutrophil slow rolling and arrest mediated by LFA-1 (Lefort et al., 2012). We reasoned that since different conformations of LFA-1 are induced by stimulating either the PSGL-1 or CXCR2 signaling cascades, it would be possible to separately test whether talin-1 and Kindlin-3 were involved in LFA-1 extension versus headpiece opening. We observed that LFA-1-dependent neutrophil slow rolling on E-selectin/ICAM-1 substrates in flow chambers and on inflamed endothelium in vivo was impaired in talin-1-deficient neutrophils, but was unaffected by knockout of Kindlin-3. In contrast, both talin-1 and Kindlin-3 were required for CXCL1-stimulated rapid neutrophil arrest and soluble ICAM-1 binding to LFA-1, indicating an important role for these co-activators in reaching the high affinity state of LFA-1. Our findings in neutrophils were corroborated by studies measuring LFA-1 affinity states using the reporter antibodies KIM127 (Robinson et al., 1992), NKI-L16 (van Kooyk et al., 1991), mAb 24 (Dransfield et al., 1992), and 2E8 (Carreno et al., 2010) in human HL-60 cells stimulated with an active Rap1a peptide. Together, these results show that talin-1 is needed for LFA-1 extension, while Kindlin-3 is involved in transition of the LFA-1 headpiece to its high affinity state. These data are the first to demonstrate distinct functions of talin-1 and Kindlin-3 in inducing specific conformers of LFA-1. It remains to be shown whether talin-1 plays an additional role in LFA-1 headpiece opening to reach the high affinity state, or whether LFA-1 extension is a prerequisite for headpiece opening in the context of chemokine-induced neutrophil arrest. Furthermore, previous studies of cultured effector T lymphocytes derived from LAD-III patients suggest that Kindlin-3 is needed for LFA-1 extension in the context of chemokine stimulation (Manevich-Mendelson et al., 2009). It is apparent that there are diverse mechanisms among the various modalities of integrin activation in leukocytes.
What do these new insights into talin-1 and Kindlin-3 regulation of LFA-1 structure tell us about the mechanisms of inside-out activation? It was not surprising to find that talin-1 is needed for LFA-1 extension. As shown for αIIbβ3 in a reconstituted system, the talin head domain (but not intact, autoinhibited talin) is sufficient to induce extension of the integrin extracellular domain (Ye et al., 2010). Several studies have shown that talin directly disrupts the basal association of integrin TMDs by contacting the salt bridge between the α and β chains close to the inner membrane leaflet (Wegener et al., 2007; Anthis et al., 2009). Talin is likely involved in further rearrangement of the TMDs, as mutations that disrupt the α/β salt bridge do not fully overcome the requirement for talin in integrin activation (Tadokoro et al., 2003; Wegener et al., 2007). That Kindlin-3 was not required for neutrophil slow rolling mediated by the extended/closed conformer of LFA-1 (Lefort et al., 2012) suggests that it may not be involved in initial disruption of the LFA-1 TMDs. However, structural studies of LFA-1 suggest that the TMDs and lower legs are separated even further in high affinity than in intermediate affinity LFA-1 (Nishida et al., 2006). Thus, Kindlin-3 may be involved in inducing swingout of the hybrid domain by causing a further separation of the TMDs. It is also possible that Kindlin-3 mediates the indirect linkage of the β2 integrin tail to the actin cytoskeleton through a scaffolding function, thus providing an anchor for force-induced conversion of ligated integrins to the high affinity state. It was recently found that LFA-1-mediated neutrophil slow rolling is not dependent on the linkage of LFA-1 to the actin cytoskeleton (Shao et al., 2012), but chemokine-stimulated arrest of lymphocytes (Shamri et al., 2005) and neutrophils (Shao et al., 2012) is impaired by reagents that disrupt actin microfilaments. These studies support the idea that LFA-1 extension and headpiece opening are distinctly regulated processes, and that Kindlin-3 may be involved in the transition to a high affinity state through an indirect interaction with the actin cytoskeleton. Kindlins have been shown to bind to several actin-binding proteins and complexes, including integrin-linked kinase (Mackinnon et al., 2002), migfilin (Tu et al., 2003), focal adhesion kinase, and α-actinin (Has et al., 2009). The expression and role of these adaptor molecules in LFA-1 activation will need to be tested in leukocyte arrest under flow conditions.
The distinct roles of talin-1 and Kindlin-3 in neutrophil slow rolling and arrest raise several questions about the two signaling pathways that stimulate these separate behaviors. Clearly, LFA-1 engages ICAM-1 during slow rolling interactions induced by PSGL-1 signaling. If chemokines directly stimulate only LFA-1 extension and not conversion to the high affinity state, why does CXCR2 engagement lead to neutrophil behavior qualitatively distinct from the PSGL-1 pathway that has also been shown to induce LFA-1 extension? Likewise, if force on the LFA-1/ICAM-1 bond results in affinity maturation to the long-lived state, how are neutrophils able to use LFA-1 for rolling interactions that require a transient bond with a fast off-rate? Structural communication between the βI and αI domains through the α7 helix is needed for headpiece opening (Shimaoka et al., 2003a). Perhaps Kindlin-3 provides a permissive signal or induces a structural rearrangement that allows coupling of the βI and αI domains, such as swingout of the hybrid domain. We speculate that Kindlin-3 may be actively excluded from binding to the β2 cytoplasmic tail after inside-out activation of LFA-1 by the PSGL-1 signaling pathway and this prevents the transition to the open headpiece and neutrophil arrest. This could occur by promoting the binding of a competing molecule to the Kindlin-3 binding NPKF site on the β2 tail, or by regulating phosphorylation of threonine residues that also contribute to Kindlin-3 binding (Ma et al., 2008). Indeed, T cell receptor signaling affects the phosphorylation of β2T758 and promotes binding of 14-3-3 proteins and LFA-1 activation (Fagerholm et al., 2005; Gronholm et al., 2011). Whether this is also true for chemokine signaling is unknown.
A potential alternative mechanism for LFA-1 extension resulting in distinct slow rolling and arrest behaviors after PSGL-1 and CXCR2 signaling, respectively, may be that chemokines also stimulate an increase in LFA-1 mobility in the membrane (Constantin et al., 2000) that could lead to ligand-driven LFA-1 clustering and subsequent firm adhesion. This mechanism would require that LFA-1 ligands like ICAM-1 are pre-clustered on endothelial cells (Barreiro et al., 2008), because integrin clustering is most likely a post-ligand binding event (Kim et al., 2004). In the case of slow rolling, if LFA-1 is immobile following PSGL-1 engagement then clustering may be disfavored and the force on individual LFA-1/ICAM-1 bonds could be sufficiently high so that LFA-1 resides in the slip bond regime with short enough lifetimes (high koff) to support rolling behavior. Clearly, much remains to be tested to uncover the mechanisms regulating LFA-1 structure and affinity on the surface of leukocytes.
Concluding Remarks
LFA-1 plays important roles in the trafficking of multiple leukocyte subsets during the immune response. At least two distinct signaling pathways impact the adhesive function of LFA-1 by altering its structure, and thus affinity for extracellular ligands. Post-ligand binding LFA-1 clustering alters its distribution on the plasma membrane. Our understanding of how cellular factors regulate LFA-1 conformation by binding to its short cytoplasmic tails has evolved as Kindlin-3 and talin-1 have been identified as major and distinct players. The concepts explaining how structural rearrangements are propagated through the TMDs and integrin legs to the ligand-binding headpiece are being refined. It has recently become clear that force plays an integral role in these processes, and future work will be aimed at determining the molecular requirements and mechanisms of LFA-1 affinity regulation by force in rolling and arresting leukocytes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abram, C. L., and Lowell, C. A. (2009). Leukocyte adhesion deficiency syndrome: a controversy solved. Immunol. Cell Biol. 87, 440–442.
Adair, B. D., Xiong, J. P., Maddock, C., Goodman, S. L., Arnaout, M. A., and Yeager, M. (2005). Three-dimensional EM structure of the ectodomain of integrin {alpha} V{beta}3 in a complex with fibronectin. J. Cell Biol. 168, 1109–1118.
Alon, R., Aker, M., Feigelson, S., Sokolovsky-Eisenberg, M., Staunton, D. E., Cinamon, G., Grabovsky, V., Shamri, R., and Etzioni, A. (2003). A novel genetic leukocyte adhesion deficiency in subsecond triggering of integrin avidity by endothelial chemokines results in impaired leukocyte arrest on vascular endothelium under shear flow. Blood 101, 4437–4445.
Alon, R., and Dustin, M. L. (2007). Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity 26, 17–27.
Alon, R., and Etzioni, A. (2003). LAD-III, a novel group of leukocyte integrin activation deficiencies. Trends Immunol. 24, 561–566.
Alon, R., and Ley, K. (2008). Cells on the run: shear-regulated integrin activation in leukocyte rolling and arrest on endothelial cells. Curr. Opin. Cell Biol. 20, 525–532.
Alonso, J. L., Essafi, M., Xiong, J. P., Stehle, T., and Arnaout, M. A. (2002). Does the integrin alphaA domain act as a ligand for its betaA domain? Curr. Biol. 12, R340–R342.
Anthis, N. J., Wegener, K. L., Ye, F., Kim, C., Goult, B. T., Lowe, E. D., Vakonakis, I., Bate, N., Critchley, D. R., Ginsberg, M. H., and Campbell, I. D. (2009). The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 28, 3623–3632.
Barreiro, O., Zamai, M., Yanez-Mo, M., Tejera, E., Lopez-Romero, P., Monk, P. N., Gratton, E., Caiolfa, V. R., and Sanchez-Madrid, F. (2008). Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J. Cell Biol. 183, 527–542.
Beals, C. R., Edwards, A. C., Gottschalk, R. J., Kuijpers, T. W., and Staunton, D. E. (2001). CD18 activation epitopes induced by leukocyte activation. J. Immunol. 167, 6113–6122.
Bergmeier, W., Goerge, T., Wang, H. W., Crittenden, J. R., Baldwin, A. C., Cifuni, S. M., Housman, D. E., Graybiel, A. M., and Wagner, D. D. (2007). Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J. Clin. Invest. 117, 1699–1707.
Bertram, A., Zhang, H., Von Vietinghoff, S., De Pablo, C., Haller, H., Shushakova, N., and Ley, K. (2012). Protein kinase C-theta is required for murine neutrophil recruitment and adhesion strengthening under flow. J. Immunol. 188, 4043–4051.
Block, H., Herter, J. M., Rossaint, J., Stadtmann, A., Kliche, S., Lowell, C. A., and Zarbock, A. (2012). Crucial role of SLP-76 and ADAP for neutrophil recruitment in mouse kidney ischemia-reperfusion injury. J. Exp. Med. 209, 407–421.
Bolomini-Vittori, M., Montresor, A., Giagulli, C., Staunton, D., Rossi, B., Martinello, M., Constantin, G., and Laudanna, C. (2009). Regulation of conformer-specific activation of the integrin LFA-1 by a chemokine-triggered Rho signaling module. Nat. Immunol. 10, 185–194.
Bouaouina, M., Goult, B. T., Huet-Calderwood, C., Bate, N., Brahme, N. N., Barsukov, I. L., Critchley, D. R., and Calderwood, D. A. (2012). A conserved lipid-binding loop in the kindlin FERM F1 domain is required for kindlin-mediated alphaIIbbeta3 integrin coactivation. J. Biol. Chem. 287, 6979–6990.
Calderwood, D. A., Zent, R., Grant, R., Rees, D. J., Hynes, R. O., and Ginsberg, M. H. (1999). The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274, 28071–28074.
Carreno, R., Brown, W. S., Li, D., Hernandez, J. A., Wang, Y., Kim, T. K., Craft, J. W. Jr., Komanduri, K. V., Radvanyi, L. G., Hwu, P., Molldrem, J. J., Legge, G. B., Mcintyre, B. W., and Ma, Q. (2010). 2E8 binds to the high affinity I-domain in a metal ion-dependent manner: a second generation monoclonal antibody selectively targeting activated LFA-1. J. Biol. Chem. 285, 32860–32868.
Chen, W., Lou, J., and Zhu, C. (2010). Forcing switch from short- to intermediate- and long-lived states of the alphaA domain generates LFA-1/ICAM-1 catch bonds. J. Biol. Chem. 285, 35967–35978.
Chesnutt, B. C., Smith, D. F., Raffler, N. A., Smith, M. L., White, E. J., and Ley, K. (2006). Induction of LFA-1-dependent neutrophil rolling on ICAM-1 by engagement of E-selectin. Microcirculation 13, 99–109.
Chigaev, A., Buranda, T., Dwyer, D. C., Prossnitz, E. R., and Sklar, L. A. (2003). FRET detection of cellular alpha4-integrin conformational activation. Biophys. J. 85, 3951–3962.
Chigaev, A., Smagley, Y., Zhang, Y., Waller, A., Haynes, M. K., Amit, O., Wang, W., Larson, R. S., and Sklar, L. A. (2011). Real-time analysis of the inside-out regulation of lymphocyte function-associated antigen-1 revealed similarities to and differences from very late antigen-4. J. Biol. Chem. 286, 20375–20386.
Chigaev, A., Waller, A., Amit, O., Halip, L., Bologa, C. G., and Sklar, L. A. (2009). Real-time analysis of conformation-sensitive antibody binding provides new insights into integrin conformational regulation. J. Biol. Chem. 284, 14337–14346.
Chigaev, A., Waller, A., Zwartz, G. J., Buranda, T., and Sklar, L. A. (2007). Regulation of cell adhesion by affinity and conformational unbending of alpha4beta1 integrin. J. Immunol. 178, 6828–6839.
Constantin, G., Majeed, M., Giagulli, C., Piccio, L., Kim, J. Y., Butcher, E. C., and Laudanna, C. (2000). Chemokines trigger immediate beta2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity 13, 759–769.
Critchley, D. R. (2009). Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annu. Rev. Biophys. 38, 235–254.
Ding, Z. M., Babensee, J. E., Simon, S. I., Lu, H., Perrard, J. L., Bullard, D. C., Dai, X. Y., Bromley, S. K., Dustin, M. L., Entman, M. L., Smith, C. W., and Ballantyne, C. M. (1999). Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 163, 5029–5038.
Dransfield, I., Cabanas, C., Craig, A., and Hogg, N. (1992). Divalent cation regulation of the function of the leukocyte integrin LFA-1. J. Cell Biol. 116, 219–226.
Dustin, M. L., and Springer, T. A. (1989). T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature 341, 619–624.
Eniola, A. O., Krasik, E. F., Smith, L. A., Song, G., and Hammer, D. A. (2005). I-domain of lymphocyte function-associated antigen-1 mediates rolling of polystyrene particles on ICAM-1 under flow. Biophys. J. 89, 3577–3588.
Fagerholm, S. C., Hilden, T. J., Nurmi, S. M., and Gahmberg, C. G. (2005). Specific integrin alpha and beta chain phosphorylations regulate LFA-1 activation through affinity-dependent and -independent mechanisms. J. Cell Biol. 171, 705–715.
Giagulli, C., Ottoboni, L., Caveggion, E., Rossi, B., Lowell, C., Constantin, G., Laudanna, C., and Berton, G. (2006). The Src family kinases Hck and Fgr are dispensable for inside-out, chemoattractant-induced signaling regulating beta 2 integrin affinity and valency in neutrophils, but are required for beta 2 integrin-mediated outside-in signaling involved in sustained adhesion. J. Immunol. 177, 604–611.
Giagulli, C., Scarpini, E., Ottoboni, L., Narumiya, S., Butcher, E. C., Constantin, G., and Laudanna, C. (2004). RhoA and zeta PKC control distinct modalities of LFA-1 activation by chemokines: critical role of LFA-1 affinity triggering in lymphocyte in vivo homing. Immunity 20, 25–35.
Goksoy, E., Ma, Y. Q., Wang, X., Kong, X., Perera, D., Plow, E. F., and Qin, J. (2008). Structural basis for the autoinhibition of talin in regulating integrin activation. Mol. Cell 31, 124–133.
Goult, B. T., Bouaouina, M., Elliott, P. R., Bate, N., Patel, B., Gingras, A. R., Grossmann, J. G., Roberts, G. C., Calderwood, D. A., Critchley, D. R., and Barsukov, I. L. (2010). Structure of a double ubiquitin-like domain in the talin head: a role in integrin activation. EMBO J. 29, 1069–1080.
Gronholm, M., Jahan, F., Marchesan, S., Karvonen, U., Aatonen, M., Narumanchi, S., and Gahmberg, C. G. (2011). TCR-induced activation of LFA-1 involves signaling through Tiam1. J. Immunol. 187, 3613–3619.
Harburger, D. S., Bouaouina, M., and Calderwood, D. A. (2009). Kindlin-1 and -2 directly bind the C-terminal region of beta integrin cytoplasmic tails and exert integrin-specific activation effects. J. Biol. Chem. 284, 11485–11497.
Has, C., Herz, C., Zimina, E., Qu, H. Y., He, Y., Zhang, Z. G., Wen, T. T., Gache, Y., Aumailley, M., and Bruckner-Tuderman, L. (2009). Kindlin-1 is required for RhoGTPase-mediated lamellipodia formation in keratinocytes. Am. J. Pathol. 175, 1442–1452.
Hughes, P. E., Diaz-Gonzalez, F., Leong, L., Wu, C., Mcdonald, J. A., Shattil, S. J., and Ginsberg, M. H. (1996). Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J. Biol. Chem. 271, 6571–6574.
Huth, J. R., Olejniczak, E. T., Mendoza, R., Liang, H., Harris, E. A., Lupher, M. L. Jr., Wilson, A. E., Fesik, S. W., and Staunton, D. E. (2000). NMR and mutagenesis evidence for an I domain allosteric site that regulates lymphocyte function-associated antigen 1 ligand binding. Proc. Natl. Acad. Sci. U.S.A. 97, 5231–5236.
Jin, M., Andricioaei, I., and Springer, T. A. (2004). Conversion between three conformational states of integrin I domains with a C-terminal pull spring studied with molecular dynamics. Structure 12, 2137–2147.
Jurk, K., Schulz, A. S., Kehrel, B. E., Rapple, D., Schulze, H., Mobest, D., Friedrich, W. W., Omran, H., Deak, E., Henschler, R., Scheele, J. S., and Zieger, B. (2010). Novel integrin-dependent platelet malfunction in siblings with leukocyte adhesion deficiency-III (LAD-III) caused by a point mutation in FERMT3. Thromb. Haemost. 103, 1053–1064.
Kim, M., Carman, C. V., and Springer, T. A. (2003). Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301, 1720–1725.
Kim, M., Carman, C. V., Yang, W., Salas, A., and Springer, T. A. (2004). The primacy of affinity over clustering in regulation of adhesiveness of the integrin {alpha}L{beta}2. J. Cell Biol. 167, 1241–1253.
Kliche, S., Worbs, T., Wang, X., Degen, J., Patzak, I., Meineke, B., Togni, M., Moser, M., Reinhold, A., Kiefer, F., Freund, C., Forster, R., and Schraven, B. (2012). CCR7-mediated LFA-1 functions in T cells are regulated by 2 independent ADAP/SKAP55 modules. Blood 119, 777–785.
Knorr, R., and Dustin, M. L. (1997). The lymphocyte function-associated antigen 1 I domain is a transient binding module for intercellular adhesion molecule (ICAM)-1 and ICAM-3 in hydrodynamic flow. J. Exp. Med. 186, 719–730.
Kuijpers, T. W., Van De Vijver, E., Weterman, M. A., De Boer, M., Tool, A. T., Van Den Berg, T. K., Moser, M., Jakobs, M. E., Seeger, K., Sanal, O., Unal, S., Cetin, M., Roos, D., Verhoeven, A. J., and Baas, F. (2009). LAD-1/variant syndrome is caused by mutations in FERMT3. Blood 113, 4740–4746.
Kuijpers, T. W., Van Lier, R. A., Hamann, D., De Boer, M., Thung, L. Y., Weening, R. S., Verhoeven, A. J., and Roos, D. (1997). Leukocyte adhesion deficiency type 1 (LAD-1)/variant. A novel immunodeficiency syndrome characterized by dysfunctional beta2 integrins. J. Clin. Invest. 100, 1725–1733.
Kuwano, Y., Spelten, O., Zhang, H., Ley, K., and Zarbock, A. (2010). Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood 116, 617–624.
Larson, R. S., Davis, T., Bologa, C., Semenuk, G., Vijayan, S., Li, Y., Oprea, T., Chigaev, A., Buranda, T., Wagner, C. R., and Sklar, L. A. (2005). Dissociation of I domain and global conformational changes in LFA-1: refinement of small molecule-I domain structure-activity relationships. Biochemistry 44, 4322–4331.
Lefort, C. T., Rossaint, J., Moser, M., Petrich, B. G., Zarbock, A., Monkley, S. J., Critchley, D. R., Ginsberg, M. H., Fassler, R., and Ley, K. (2012). Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 119, 4275–4282.
Legate, K. R., and Fassler, R. (2009). Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J. Cell Sci. 122, 187–198.
Legge, G. B., Kriwacki, R. W., Chung, J., Hommel, U., Ramage, P., Case, D. A., Dyson, H. J., and Wright, P. E. (2000). NMR solution structure of the inserted domain of human leukocyte function associated antigen-1. J. Mol. Biol. 295, 1251–1264.
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689.
Li, W., Metcalf, D. G., Gorelik, R., Li, R., Mitra, N., Nanda, V., Law, P. B., Lear, J. D., Degrado, W. F., and Bennett, J. S. (2005). A push-pull mechanism for regulating integrin function. Proc. Natl. Acad. Sci. U.S.A. 102, 1424–1429.
Lu, C., Shimaoka, M., Ferzly, M., Oxvig, C., Takagi, J., and Springer, T. A. (2001). An isolated, surface-expressed I domain of the integrin alphaLbeta2 is sufficient for strong adhesive function when locked in the open conformation with a disulfide bond. Proc. Natl. Acad. Sci. U.S.A. 98, 2387–2392.
Lu, C. F., and Springer, T. A. (1997). The alpha subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1. J. Immunol. 159, 268–278.
Luo, B. H., Carman, C. V., and Springer, T. A. (2007). Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647.
Luo, B. H., Carman, C. V., Takagi, J., and Springer, T. A. (2005). Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc. Natl. Acad. Sci. U.S.A. 102, 3679–3684.
Ma, Y. Q., Qin, J., Wu, C., and Plow, E. F. (2008). Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J. Cell Biol. 181, 439–446.
Mackinnon, A. C., Qadota, H., Norman, K. R., Moerman, D. G., and Williams, B. D. (2002). C. elegans PAT-4/ILK functions as an adaptor protein within integrin adhesion complexes. Curr. Biol. 12, 787–797.
Malinin, N. L., Zhang, L., Choi, J., Ciocea, A., Razorenova, O., Ma, Y. Q., Podrez, E. A., Tosi, M., Lennon, D. P., Caplan, A. I., Shurin, S. B., Plow, E. F., and Byzova, T. V. (2009). A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat. Med. 15, 313–318.
Manevich-Mendelson, E., Feigelson, S. W., Pasvolsky, R., Aker, M., Grabovsky, V., Shulman, Z., Kilic, S. S., Rosenthal-Allieri, M. A., Ben-Dor, S., Mory, A., Bernard, A., Moser, M., Etzioni, A., and Alon, R. (2009). Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood 114, 2344–2353.
Martel, V., Racaud-Sultan, C., Dupe, S., Marie, C., Paulhe, F., Galmiche, A., Block, M. R., and Albiges-Rizo, C. (2001). Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J. Biol. Chem. 276, 21217–21227.
McDowall, A., Svensson, L., Stanley, P., Patzak, I., Chakravarty, P., Howarth, K., Sabnis, H., Briones, M., and Hogg, N. (2010). Two mutations in the KINDLIN3 gene of a new leukocyte adhesion deficiency III patient reveal distinct effects on leukocyte function in vitro. Blood 115, 4834–4842.
Miner, J. J., Xia, L., Yago, T., Kappelmayer, J., Liu, Z., Klopocki, A. G., Shao, B., Mcdaniel, J. M., Setiadi, H., Schmidtke, D. W., and Mcever, R. P. (2008). Separable requirements for cytoplasmic domain of PSGL-1 in leukocyte rolling and signaling under flow. Blood 112, 2035–2045.
Moser, M., Bauer, M., Schmid, S., Ruppert, R., Schmidt, S., Sixt, M., Wang, H. V., Sperandio, M., and Fassler, R. (2009a). Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15, 300–305.
Moser, M., Legate, K. R., Zent, R., and Fassler, R. (2009b). The tail of integrins, talin, and kindlins. Science 324, 895–899.
Nishida, N., Xie, C., Shimaoka, M., Cheng, Y., Walz, T., and Springer, T. A. (2006). Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity 25, 583–594.
O’Toole, T. E., Loftus, J. C., Du, X. P., Glass, A. A., Ruggeri, Z. M., Shattil, S. J., Plow, E. F., and Ginsberg, M. H. (1990). Affinity modulation of the alpha IIb beta 3 integrin (platelet GPIIb-IIIa) is an intrinsic property of the receptor. Cell Regul. 1, 883–893.
Partridge, A. W., Liu, S., Kim, S., Bowie, J. U., and Ginsberg, M. H. (2005). Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J. Biol. Chem. 280, 7294–7300.
Pasvolsky, R., Feigelson, S. W., Kilic, S. S., Simon, A. J., Tal-Lapidot, G., Grabovsky, V., Crittenden, J. R., Amariglio, N., Safran, M., Graybiel, A. M., Rechavi, G., Ben-Dor, S., Etzioni, A., and Alon, R. (2007). A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. J. Exp. Med. 204, 1571–1582.
Qu, H., Tu, Y., Shi, X., Larjava, H., Saleem, M. A., Shattil, S. J., Fukuda, K., Qin, J., Kretzler, M., and Wu, C. (2011). Kindlin-2 regulates podocyte adhesion and fibronectin matrix deposition through interactions with phosphoinositides and integrins. J. Cell Sci. 124, 879–891.
Robert, P., Canault, M., Farnarier, C., Nurden, A., Grosdidier, C., Barlogis, V., Bongrand, P., Pierres, A., Chambost, H., and Alessi, M. C. (2011). A novel leukocyte adhesion deficiency III variant: kindlin-3 deficiency results in integrin- and nonintegrin-related defects in different steps of leukocyte adhesion. J. Immunol. 186, 5273–5283.
Robinson, M. K., Andrew, D., Rosen, H., Brown, D., Ortlepp, S., Stephens, P., and Butcher, E. C. (1992). Antibody against the Leu-CAM beta-chain (CD18) promotes both LFA-1- and CR3-dependent adhesion events. J. Immunol. 148, 1080–1085.
Salas, A., Shimaoka, M., Chen, S., Carman, C. V., and Springer, T. (2002). Transition from rolling to firm adhesion is regulated by the conformation of the I domain of the integrin lymphocyte function-associated antigen-1. J. Biol. Chem. 277, 50255–50262.
Salas, A., Shimaoka, M., Kogan, A. N., Harwood, C., Von Andrian, U. H., and Springer, T. A. (2004). Rolling adhesion through an extended conformation of integrin alphaLbeta2 and relation to alpha I and beta I-like domain interaction. Immunity 20, 393–406.
Schurpf, T., and Springer, T. A. (2011). Regulation of integrin affinity on cell surfaces. EMBO J. 30, 4712–4727.
Shamri, R., Grabovsky, V., Gauguet, J. M., Feigelson, S., Manevich, E., Kolanus, W., Robinson, M. K., Staunton, D. E., Von Andrian, U. H., and Alon, R. (2005). Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat. Immunol. 6, 497–506.
Shao, B., Yago, T., Coghill, P. A., Klopocki, A. G., Mehta-D’souza, P., Schmidtke, D. W., Rodgers, W., and Mcever, R. P. (2012). Signal-dependent slow leukocyte rolling does not require cytoskeletal anchorage of P-selectin glycoprotein ligand-1 (PSGL-1) or integrin alphaLbeta2. J. Biol. Chem. 287, 19585–19598.
Shattil, S. J., Kim, C., and Ginsberg, M. H. (2010). The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 11, 288–300.
Shimaoka, M., Salas, A., Yang, W., Weitz-Schmidt, G., and Springer, T. A. (2003a). Small molecule integrin antagonists that bind to the beta2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity 19, 391–402.
Shimaoka, M., Xiao, T., Liu, J. H., Yang, Y., Dong, Y., Jun, C. D., Mccormack, A., Zhang, R., Joachimiak, A., Takagi, J., Wang, J. H., and Springer, T. A. (2003b). Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 112, 99–111.
Sims, P. J., Ginsberg, M. H., Plow, E. F., and Shattil, S. J. (1991). Effect of platelet activation on the conformation of the plasma membrane glycoprotein IIb-IIIa complex. J. Biol. Chem. 266, 7345–7352.
Smith, D. F., Deem, T. L., Bruce, A. C., Reutershan, J., Wu, D., and Ley, K. (2006). Leukocyte phosphoinositide-3 kinase {gamma} is required for chemokine-induced, sustained adhesion under flow in vivo. J. Leukoc. Biol. 80, 1491–1499.
Smith, M. L., Olson, T. S., and Ley, K. (2004). CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J. Exp. Med. 200, 935–939.
Spertini, C., Baisse, B., and Spertini, O. (2012). Ezrin–radixin–moesin-binding sequence of PSGL-1 glycoprotein regulates leukocyte rolling on selectins and activation of extracellular signal-regulated kinases. J. Biol. Chem. 287, 10693–10702.
Stadtmann, A., Brinkhaus, L., Mueller, H., Rossaint, J., Bolomini-Vittori, M., Bergmeier, W., Van Aken, H., Wagner, D. D., Laudanna, C., Ley, K., and Zarbock, A. (2011). Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur. J. Immunol. 41, 2074–2085.
Svensson, L., Howarth, K., McDowall, A., Patzak, I., Evans, R., Ussar, S., Moser, M., Metin, A., Fried, M., Tomlinson, I., and Hogg, N. (2009). Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 15, 306–312.
Tadokoro, S., Shattil, S. J., Eto, K., Tai, V., Liddington, R. C., De Pereda, J. M., Ginsberg, M. H., and Calderwood, D. A. (2003). Talin binding to integrin beta tails: a final common step in integrin activation. Science 302, 103–106.
Takagi, J., Petre, B. M., Walz, T., and Springer, T. A. (2002). Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599–511.
Tu, Y., Wu, S., Shi, X., Chen, K., and Wu, C. (2003). Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 113, 37–47.
van Kooyk, Y., Weder, P., Hogervorst, F., Verhoeven, A. J., Van Seventer, G., Te Velde, A. A., Borst, J., Keizer, G. D., and Figdor, C. G. (1991). Activation of LFA-1 through a Ca2+ -dependent epitope stimulates lymphocyte adhesion. J. Cell Biol. 112, 345–354.
Vinogradova, O., Velyvis, A., Velyviene, A., Hu, B., Haas, T., Plow, E., and Qin, J. (2002). A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell 110, 587–597.
Wegener, K. L., Partridge, A. W., Han, J., Pickford, A. R., Liddington, R. C., Ginsberg, M. H., and Campbell, I. D. (2007). Structural basis of integrin activation by talin. Cell 128, 171–182.
Xiao, T., Takagi, J., Coller, B. S., Wang, J. H., and Springer, T. A. (2004). Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432, 59–67.
Xie, C., Zhu, J., Chen, X., Mi, L., Nishida, N., and Springer, T. A. (2010). Structure of an integrin with an alphaI domain, complement receptor type 4. EMBO J. 29, 666–679.
Xiong, J. P., Stehle, T., Diefenbach, B., Zhang, R., Dunker, R., Scott, D. L., Joachimiak, A., Goodman, S. L., and Arnaout, M. A. (2001). Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science 294, 339–345.
Xiong, J. P., Stehle, T., Goodman, S. L., and Arnaout, M. A. (2003). New insights into the structural basis of integrin activation. Blood 102, 1155–1159.
Xiong, J. P., Stehle, T., Zhang, R., Joachimiak, A., Frech, M., Goodman, S. L., and Arnaout, M. A. (2002). Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science 296, 151–155.
Yang, W., Shimaoka, M., Salas, A., Takagi, J., and Springer, T. A. (2004). Intersubunit signal transmission in integrins by a receptor-like interaction with a pull spring. Proc. Natl. Acad. Sci. U.S.A. 101, 2906–2911.
Ye, F., Hu, G., Taylor, D., Ratnikov, B., Bobkov, A. A., Mclean, M. A., Sligar, S. G., Taylor, K. A., and Ginsberg, M. H. (2010). Recreation of the terminal events in physiological integrin activation. J. Cell Biol. 188, 157–173.
Zarbock, A., Deem, T. L., Burcin, T. L., and Ley, K. (2007a). Galphai2 is required for chemokine-induced neutrophil arrest. Blood 110, 3773–3779.
Zarbock, A., Lowell, C. A., and Ley, K. (2007b). Spleen tyrosine kinase Syk is necessary for E-selectin-induced alpha(L)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity 26, 773–783.
Zarbock, A., Ley, K., Mcever, R.P., and Hidalgo, A. (2011). Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood 118, 6743–6751.
Zarbock, A., Muller, H., Kuwano, Y., and Ley, K. (2009). PSGL-1-dependent myeloid leukocyte activation. J. Leukoc. Biol. 86, 1119–1124.
Zhu, J., Boylan, B., Luo, B. H., Newman, P. J., and Springer, T. A. (2007). Tests of the extension and deadbolt models of integrin activation. J. Biol. Chem. 282, 11914–11920.
Keywords: neutrophil, chemokine, integrin, LFA-1, inflammation
Citation: Lefort CT and Ley K (2012) Neutrophil arrest by LFA-1 activation. Front. Immun. 3:157. doi: 10.3389/fimmu.2012.00157
Received: 07 April 2012; Paper pending published: 24 April 2012;
Accepted: 25 May 2012; Published online: 12 June 2012.
Edited by:
Ronen Alon, The Weizmann Institute of Science, IsraelReviewed by:
Ronen Alon, The Weizmann Institute of Science, IsraelTatsuo Kinashi, Kansai Medical University, Japan
Carlo Laudanna, University of Verona, Italy
Copyright: © 2012 Lefort and Ley. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Klaus Ley, La Jolla Institute for Allergy and Immunology, 9420 Athena Circle, La Jolla, CA 92037, USA. e-mail:a2xhdXNAbGlhaS5vcmc=