 Giuseppe A. Ramirez
Giuseppe A. Ramirez Stefano Franchini
Stefano Franchini Patrizia Rovere-Querini Maria Grazia Sabbadini
Patrizia Rovere-Querini Maria Grazia Sabbadini Angelo A. Manfredi* and Norma Maugeri
Angelo A. Manfredi* and Norma Maugeri
- Department of Immunology, San Raffaele Scientific Institute, Università Vita-Salute San Raffaele, Milano, Italy
Systemic sclerosis (SSc) is an inflammatory disease of unknown etiology characterized by widespread organ dysfunction due to fibrosis and ischemia. Its nebulous pathogenic background and the consequent absence of an etiological therapy prevent the adoption of satisfying treatment strategies, able to improve patients’ quality of life and survival and stimulate researchers to identify a unifying pathogenic target. Platelets show a unique biological behavior, lying at the crossroads between vascular function, innate and adaptive immunity, and regulation of cell proliferation. Consequently they are also emerging players in the pathogenesis of many inflammatory diseases, including SSc. In the setting of SSc platelets are detectable in a persistent activated state, which is intimately linked to the concomitant presence of an injured endothelium and to the widespread activation of the innate and adaptive immune system. As a consistent circulating source of bioactive compounds platelets contribute to the development of many characteristic phenomena of SSc, such as fibrosis and impaired vascular tone.
Platelets, Immunity, and Vessel Integrity
Platelets are anucleate cellular fragments emitted by a larger nucleate precursor, the megakaryocyte. Differently from other myeloid or lymphoid precursors this latter progenitor usually resides in the bone marrow, but can also be detected in the lung (Kosaki, 2005). During the evolution of the hemopoietic system, platelets progressively acquired a functional specialization in the control of vessel integrity and repair (Semple et al., 2011). However, as probably happened for other circulating innate mediators such as pentraxins (Manfredi et al., 2008; Martinez de la Torre et al., 2010), an ancestral link between vessel repair and patrolling (i.e., between hemostasis and immunity) still seems to characterize platelet biology in humans and “upper” species (Semple et al., 2011). In the setting of injured vessels, a tripartite network between the endothelium, platelets, and rolling neutrophils develops (Maugeri et al., 2012). Subsequently extensive physical and biochemical cross-talks between these cellular species take place: selectins provide a dual anchorage system to rolling neutrophils, since they are both expressed by the endothelium and by platelets (Maugeri et al., 2012); coagulation factors recruited at sites of vessel injury also constitute a common binding platform for leukocytes and platelets (Maugeri et al., 2012); finally a large array of membrane-bound and soluble alarmins and immune mediators (expressed by the endothelium, platelets, and neutrophils) cause the reciprocal activation of neutrophils and platelets (Maugeri et al., 2009). Notably, while a platelet–leukocyte network has long been recognized within the injured vessels, a growing number of studies is currently elucidating the role of circulating heterotypic aggregates in vessel biology and inflammation (Manfredi et al., 2010). After engagement of membrane-bound functional Toll-like receptors, platelets contribute to host defense by favoring neutrophil activation and NETting (Clark et al., 2007) and shuttle blood-borne pathogens to immune outposts (such as the spleen), thus promoting their recognition and clearance by immune cells (Verschoor et al., 2011). Moreover, although devoid of MHC class II molecules, platelets express large amounts of membrane-bound and soluble stimulatory molecules, such as CD154 (Semple et al., 2011) and in vivo and in vitro studies proved the efficacy of platelets in promoting T cell-mediated responses (Iannacone et al., 2005). Intravital microscopy studies also demonstrated the ability of activated platelets to restore lymphocyte rolling on high endothelial venules of peripheral lymph nodes in L-selectin deficient mice (Diacovo et al., 1996), thus reconstituting their susceptibility to a cutaneous delayed-type hypersensitivity reaction, elicited by 2,4-dinitrofluorobenzene (Diacovo et al., 1998; Ludwig et al., 2010). This platelet-assisted interaction between lymphocytes and vessel walls seems to involve both B and T lymphocytes, including memory T cells, and can thus play a role in the maintenance of sustained inflammation (Diacovo et al., 1996). Moreover a selective ability of T-helper 1 lymphocytes (Th1) to interact with P-selectin has been described in the T cell subset (Austrup et al., 1997), suggesting that platelets could actively regulate the characteristics of the immune infiltrate during chronic inflammation. Circulating platelets are also armed with a large array of other immune signaling quanta [e.g., the alarmin High Mobility Group 1 protein (HMGB1), IL1β, and many chemokines], as well as defensive mediators (the so-called thrombocidins), which exert direct killing actions against invading bacteria (Yeaman, 2010; Semple et al., 2011). Furthermore platelets store large amounts of growth factors like PDGF, VEGF, and TGFβ and, when recruited “at sites of inflammation and vessel injury through hemostatic mechanisms” (Nurden, 2011), platelets are able to release them and affect local tissue tropism (Nurden, 2011). The sprouting of new-vessels and the activation of fibroblasts within injured tissues or vessels are part of a physiological response, which has the “aim” of preventing hemorrhage and excessive exposure of the body to the hostile outside environment (Maugeri et al., 2009). However, when these remodeling responses occur constitutively in the absence of an injuring stimulus, as in the case of atherosclerosis (Mullenix et al., 2005) or large vessel vasculitides (Maugeri et al., 2009), the integrity and the conductive performances of vessel are impaired (eventually leading to ischemia or vessel disruption and hemorrhage; Maugeri et al., 2009). Similarly substitution of functional tissues with amorphous fibrosis (as happens in post-ischemic conditions during myocardial infarction and stroke) eventually impairs the whole organ function.
Characteristic Features of Systemic Sclerosis
Systemic sclerosis (SSc) is an autoimmune disease, in which excessive connective tissue deposition, inflammation and autoimmunity and vascular dysfunction (the three main pathophysiological hallmarks of the disease), rise from a largely unknown pathogenic background. The clinical fallout of such complex pathophysiological phenomena consists in a large spectrum of clinical manifestations, characterized by failure of multiple organs (including lung, kidney, heart, gastrointestinal tract, and skin), due to ischemia or to the impairment of motility or substance exchange with the environment after substitution of functional tissues with amorphous fibrotic tissue. Due to the scarcity of effective therapies and to the frequent involvement of major organs, SSc significantly impacts on the patients quality of life and is associated with higher overall mortality (Pattanaik et al., 2011).
Signs of systemic inflammation and autoimmunity are detectable in almost all patients with SSc and comprise the activation of cellular and humoral as well as innate and adaptive immune responses (Kahaleh and LeRoy, 1999; Postlethwaite and Chiang, 2007; Yoshizaki et al., 2009; Pattanaik et al., 2011). A characteristic antiviral-like interferon-mediated response has been recognized in patients with SSc (Lafyatis and York, 2009) and constitutes a shared pathogenic hallmark of many connective tissue diseases (Banchereau and Pascual, 2006). Marked signs of humoral innate immunity activation are also detectable in SSc both in circulating blood (Yoshizaki et al., 2009) and affected tissues (Luchetti et al., 2004; see also below), and have been shown to correlate with disease activity (Yoshizaki et al., 2009). On the other hand extensive antibody and cellular autoimmunity against vessel and connective tissue constituents is thought to directly contribute to tissue damage, stimulate the development of aberrant misrepair responses (Pattanaik et al., 2011) and enhance the activation of innate immune players (including platelets; Postlethwaite and Chiang, 2007).
Vascular impairment is one of the main and early appearing hallmarks of SSc and develops as a consequence of the concurrent action of autoimmune reactions against vessel walls (Holt et al., 1989; Kahaleh and Fan, 1997), endothelial dysfunction (with prominent imbalance between vasoconstrictors and vasodilators; Pattanaik et al., 2011), abnormalities in neural control of the vascular tone (Freedman et al., 1999) and possibly alterations in the hemostatic function. The development of ischemia, which ultimately follows vascular impairment in SSc, leads to the release of large amounts of active mitogens. These latter mediators of cell proliferation, instead of initiating a physiological formation of neo-vessels, give rise to aberrant and unfunctional neoangiogenic responses (see below) and further stimulate fibrogenesis (Trojanowska, 2008; Pattanaik et al., 2011).
Fibrosis arises as a consequence of uncontrolled proliferation and secretive activity of fibroblasts, due to intense stimulation by immune cells and ischemic tissue on a likely already abnormal genetic background (Gardner et al., 2006). In tissue affected by SSc fibroblasts proliferate from resident precursors, trans-differentiating cells or bone marrow-derived precursors (Frid et al., 2002). Beside the functional and structural damage due to the substitution of normal tissues with collagen and other glycoproteins, contraction of the fibrotic tissue due to the activity of a particular subtype of fibroblasts (i.e., the myofibroblasts) ultimately worsen tissue ischemia and organ dysfunction.
Platelet Activation and Pathogenesis of SSc
Activation of Platelets in SSc
Enhanced activation of platelets and increased tendency to aggregation have long been observed in SSc patients (Kahaleh et al., 1982; Goodfield et al., 1993) and are generally attributed to the concomitant dysfunction of the endothelium (Pattanaik et al., 2011). Moreover the elevation of markers of platelet activation like the HMGB1 or P-selectin has been shown to correlate with disease activity in SSc (Agache et al., 2007; Yoshizaki et al., 2009), as well as in other diseases characterized by prominent vascular inflammation (Harris et al., 2012).
Several studies have revealed an increased responsiveness of SSc platelets to 5-hydroxytryptamine (5HT), adrenaline, ADP, and collagen (Friedhoff et al., 1984; Goodfield et al., 1988; Postlethwaite and Chiang, 2007). More recently SSc platelets were shown to upregulate a specific non-integrin receptor for type I collagen (Chiang et al., 2006). Type I collagen is in turn abundantly expressed in (injured) vessel walls and is one of the main constituent of fibrotic tissues in SSc. Overexpression of the non-integrin receptor for collagen I together with enhanced downstream signaling ultimately leads to further pro-coagulant activation of platelets, which undergo extensive cytoskeletal remodeling and mobilization of intracellular calcium (Postlethwaite and Chiang, 2007; Figure 1).
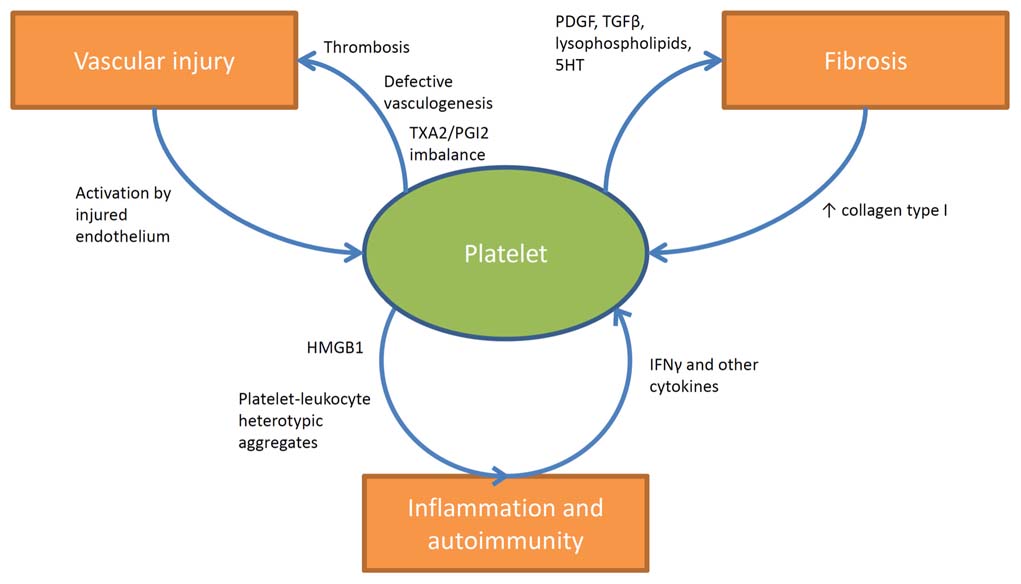
FIGURE 1. Involvement of platelets in the pathogenesis of SSc. Platelets are stimulated by the injured endothelium and contribute to vascular dysfunction and ischemia by participating in thrombotic events and by releasing vaso-active moieties (such as TXA2). Increased expression of type I collagen as a consequence of tissue fibrosis further enhances platelet activation. On the other hand platelets actively stimulate tissue fibrosis by releasing fibrogenic mediators such as PDGF, TGF-β, lysophospholipids, and serotonin. The interactions of platelets with immune cells in SSc are less clearly understood. Platelets are known sources of active pro-inflammatory mediators like HMGB1, and interestingly increased circulating levels of this prototypic alarmin have been shown to correlate with disease activity in SSc. Moreover evidence is growing about the prominent role of circulating heterotypic aggregates (between platelets and innate immune cells) in the pathogenesis of inflammatory and vascular diseases and recent studies are currently evaluating their impact in the pathogenesis of SSc. Furthermore other studies suggested the existence of a specific network between collagen-reactive T lymphocyte and platelets in SSc. In this latter setting platelet activation would be enhanced by the release of IFNγ and a wider array of cytokines from autoreactive T lymphocyte, which in turn would affect megakaryocyte maturation and platelet basal activation state.
Lungs, Platelets, and SSc-Pathogenesis
The involvement of the lung is the leading cause of mortality in SSc (Steen and Medsger, 2007). Pulmonary disease usually encompasses interstitial lung disease and pulmonary arterial hypertension and it is clinically evident as progressive respiratory insufficiency. Moreover histologic evidence of lung injury can be detected in up to 80% of patients with SSc (D’Angelo et al., 1969). In fact as the lung vasculature is the main portal system in the body, it is highly exposed to the activity of circulating inflammatory humoral and cellular mediators (including platelets) and thus it constitutes the ideal setting for the complete expression of some of the most characteristic pathogenic features of SSc, i.e., vascular dysfunction and subsequent aberrant vessel and tissue remodeling. Moreover, as suggested by the pathogenesis of transfusion-related acute lung injury (TRALI; Nurden, 2011), by clinical reports of non-SSc interstitial lung disease after the onset of severe idiopathic thrombocytopenic purpura (ITP; Fontana et al., 2007) and finally by detection of platelet activation markers in bronchoalveolar lavages of SSc patients with active pulmonary involvement (Kowal-Bielecka et al., 2005), lungs constitute a preferential target for platelet-induced inflammatory injury.
Lungs have long been recognized as secondary sites of thrombocytopoiesis (Kosaki, 2005). Differently from the bone marrow (which is mainly constituted by immature immune cells and is protected by the blood–marrow barrier), in the setting of the lung megakaryocytes are potentially exposed to powerful inflammatory stimuli, provided by resident or circulating mature immune cells, such as lymphocytes (Kosaki, 2005; Postlethwaite and Chiang, 2007; Nurden, 2011). A recent series of studies suggested that autoreactive T lymphocytes, directed against collagen I, could stimulate platelets through specific patterns of cytokines and induce them to increase their own response to collagen I and enhance their aggregation activity (Postlethwaite and Chiang, 2007). Although the precise way by which innate immune players (as platelets are) could elicit a selective activation against a single autoantigen in response to signals from adaptive immunity is still incompletely clear, this kind of evidence provides an interesting clue toward a better comprehension of the links between platelets abnormalities and autoimmunity.
Activated or destructed platelets are known to release large amounts of bioactive compounds in the bloodstream, most of which are actively metabolized by the lungs, which are thus chronically exposed to platelet-derived endogenous toxins. In particular the lungs are the main physiologic filters for circulating 5HT, a known marker of platelet activation and a key player in platelet aggregation (Nurden, 2011), whose aberrant activity in tissue and vessel remodeling is well-demonstrated by carcinoid heart disease (Palaniswamy et al., 2012). Excessive circulating 5HT and defective platelet content (i.e., increased release of 5HT by platelets) has been observed in patients with SSc (Klimiuk et al., 1989) and other connective tissue diseases (Biondi et al., 1988; maybe accounting for the presence of mild to severe pulmonary involvement in systemic lupus erythematosus or inflammatory myopathies). Moreover platelet-derived serotonin has recently been linked to the development of fibrosis (Dees et al., 2011; see below). Along with this line inhibition of platelet loading with 5HT by the administration of selective serotonin reuptake inhibitors (SSRI) has been proposed as a possible therapeutic option to face the presence of extensive fibrogenic activity and vasoconstriction as well as increased incidence of mood disorders in SSc (Coleiro et al., 2001; Garcia-Porrua et al., 2004).
An imbalance between endothelial prostacyclin (PGI2) and platelet-derived thromboxane (TXA2) has long been recognized as a key factor in determining the risk of ischemia by means of platelet aggregation and vessel vasoconstriction (and it constitutes the main rationale for the use of low-dose aspirin for anti-thrombotic prevention). Diffuse microvascular dysfunction with sustained vasoconstriction, due to defective synthesis of PGI2, is a characteristic feature of SSc lungs (Tuder et al., 1999) and accounts for the frequent development of pulmonary hypertension and the efficacy of prostaglandins analogs like iloprost (Herrick, 2011). Moreover this deficit in prostacyclin activity likely promotes platelets aggregation by acting synergistically with the known resistance of SSc platelets to PGI2 signaling (Belch et al., 1985) and the enhanced production of TXA2, due to collagen stimulation (Postlethwaite and Chiang, 2007; Figure 1). Accordingly the use of prostacyclin analogs as well as other vasodilators has been suggested to affect and improve also platelet function (Candela et al., 2001; Herrick, 2011).
Kidneys, Platelets, and Thrombotic Microangiopathy
Kidney disease in SSc manifests itself as the so-called “scleroderma renal crisis,” the leading cause of death by SSc before the introduction of angiotensin converting enzyme inhibitors (ACEi; Steen and Medsger, 2007). Although the hallmark of renal injury in SSc is the aberrant activation of the renin–angiotensin–aldosterone system, the initial sequence of vasoconstriction and ischemia (which gives rise to a vicious circle, broken by the administration of ACEi) appears to be primed by microangiopathic processes (Trostle et al., 1988) similar to those seen in other SSc-vessels (and characterized by deposition of platelet antigens within the vessel walls (Miller et al., 1980). These processes, which lead to the development of schistocytosis, thrombocytopenia, renal insufficiency, proteinuria and an active urinary sediment, also resemble the pathophysiological features of thrombotic thrombocytopenic purpura (TTP), from which scleroderma renal crisis is often hardly distinguishable. Platelet consumption due to defective von Willebrand factor (vWF) turnover is the hallmark of TTP, but notably the presence of “supranormal” multimers of vWF has also been recognized in sera from SSc patients (Mannucci et al., 1989).
Platelets and Abnormalities of Cell Proliferation and Secretion in SSc
Aberration in cell proliferation and secretion is a hallmark of SSc and expresses on the one hand as “frustrated angiogenesis,” a process ultimately leading to chronic ischemia, and on the other hand as excessive myofibroblast proliferation and deposition of connective tissue. Such anomalies in the control of the cell life-cycle are globally due to excessive release of mitogens like TGF-β, PDGF, VEGF, FGF, IGF1, endothelin 1, IL6, IL4, and IL13 in response to inflammation and/or ischemic injury (Mauviel, 2005; Trojanowska, 2008). The importance of these molecules in the pathogenesis of SSc has been confirmed recently by studies utilizing DNA microarray technologies (Whitfield et al., 2003; Gardner et al., 2006). However such kind of studies can only suggest the nature of the complex intercellular interactions underlying the development of such humoral alterations and may even fail to adequately evaluate subtle functional variations in the biological behavior of cytoplasts containing small amounts of RNA, as are platelets (Postlethwaite and Chiang, 2007).
Several other studies have indeed recognized the fundamental role of platelets in determining tissue remodeling in SSc (Silveri et al., 2001) as in many other diseases related to vascular injury (May et al., 2002; Donners et al., 2008; Nurden, 2011; Maugeri et al., 2012):
(1) as the main constituents of blood clots, platelets are directly responsible for the development of ischemia by means of (micro)-vascular thrombosis;
(2) by their productive interactions with leukocytes (Maugeri et al., 2012), they actively drive the subsequent inflammatory and (mis)-repair response (Figure 1);
(3) in the setting of injured vessels, they also directly release active mitogens like TGF-β, PDGF, or lysophospholipids (Pattanaik et al., 2011) and stimulate fibroblasts sensitivity by releasing serotonin (Dees et al., 2011; Figure 1);
Defective vasculogenesis characterize SSc (Kuwana et al., 2004) and develops as a consequence of the overwhelming activity of anti-angiogenic factors such as the long pentraxin PTX3 (which is released by the endothelium and by activated myofibroblasts in SSc; Luchetti et al., 2004; Margheri et al., 2010) upon a yet enhanced VEGF/FGF-mediated angiogenic response (Giusti et al., 2006; Margheri et al., 2010), which shows the important contribution of circulating platelets (Solanilla et al., 2009). PTX3, an emerging marker and a central player in vascular/inflammatory injury (Bottazzi et al., 2010; Maugeri et al., 2012), besides its widespread regulatory functions in innate immunity (Manfredi et al., 2008), acts in the circulating blood as a key modulator of neutrophil-platelet cross-talk: after release by neutrophil secondary granules PTX3 inhibits platelet–leukocyte aggregation and prevents the binding of fibrinogen to activated platelets (Maugeri et al., 2012). Less is known about the role of tissue-derived PTX3 on platelets, especially in the setting of SSc.
Conclusion
Besides their ancient role in hemostasis, platelets exert a complex set of interactions with circulating and tissue residing cells, which connotes them as circulating regulators of vascular homeostasis, immune function, and tissue remodeling. Along with this line they are actively involved in the development of the main pathological phenomena of SSc, although variations in their biological behavior are probably underestimated markers of disease activity and treatment response to date. Further researches about the involvement of platelets in the initiation ad perpetuation of the pathogenic phenomena of SSc would possibly disclose new therapeutic perspectives for such a nebulous and still health-impacting disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge the support of the Ministero della Salute (to Angelo A. Manfredi, Patrizia Rovere-Querini, and Maria G. Sabbadini) and of the MIUR (to Angelo A. Manfredi and Maria G. Sabbadini).
References
Agache, I., Radoi, M., and Duca, L. (2007). Platelet activation in patients with systemic scleroderma – pattern and significance. Rom. J. Intern. Med. 45, 183–191.
Austrup, F., Vestweber, D., Borges, E., Lohning, M., Brauer, R., Herz, U., Renz, H., Hallmann, R., Scheffold, A., Radbruch, A., and Hamann, A. (1997). P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflammed tissues. Nature 385, 81–83.
Banchereau, J., and Pascual, V. (2006). Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25, 383–392.
Belch, J. J., O’Dowd, A., Forbes, C. D., and Sturrock, R. D. (1985). Platelet sensitivity to a prostacyclin analogue in systemic sclerosis. Br. J. Rheumatol. 24, 346–350.
Biondi, M. L., Marasini, B., Bianchi, E., and Agostoni, A. (1988). Plasma free and intraplatelet serotonin in patients with Raynaud’s phenomenon. Int. J. Cardiol. 19, 335–339.
Bottazzi, B., Doni, A., Garlanda, C., and Mantovani, A. (2010). An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu. Rev. Immunol. 28, 157–183.
Candela, M., Pansoni, A., Jannino, L., Menditto, V. G., Natalini, M., Ravaglia, F., Da Lio, L., Scorza, R., Gabrielli, A., and Danieli, G. (2001). Coagulative modifications in patients with systemic sclerosis treated with iloprost or nifedipine. Ann. Ital. Med. Int. 16, 170–174.
Chiang, T. M., Takayama, H., and Postlethwaite, A. E. (2006). Increase in platelet non-integrin type I collagen receptor in patients with systemic sclerosis. Thromb. Res. 117, 299–306.
Clark, S. R., Ma, A. C., Tavener, S. A., McDonald, B., Goodarzi, Z., Kelly, M. M., Patel, K. D., Chakrabarti, S., McAvoy, E., Sinclair, G. D., Keys, E. M., Allen-Vercoe, E., Devinney, R., Doig, C. J., Green, F. H., and Kubes, P. (2007). Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13, 463–469.
Coleiro, B., Marshall, S. E., Denton, C. P., Howell, K., Blann, A., Welsh, K. I., and Black, C. M. (2001). Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford) 40, 1038–1043.
D’Angelo, W. A., Fries, J. F., Masi, A. T., and Shulman, L. E. (1969). Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am. J. Med. 46, 428–440.
Dees, C., Akhmetshina, A., Zerr, P., Reich, N., Palumbo, K., Horn, A., Jungel, A., Beyer, C., Kronke, G., Zwerina, J., Reiter, R., Alenina, N., Maroteaux, L., Gay, S., Schett, G., Distler, O., and Distler, J. H. (2011). Platelet-derived serotonin links vascular disease and tissue fibrosis. J. Exp. Med. 208, 961–972.
Diacovo, T. G., Catalina, M. D., Siegelman, M. H., and von Andrian, U. H. (1998). Circulating activated platelets reconstitute lymphocyte homing and immunity in L-selectin-deficient mice. J. Exp. Med. 187, 197–204.
Diacovo, T. G., Puri, K. D., Warnock, R. A., Springer, T. A., and von Andrian, U. H. (1996). Platelet-mediated lymphocyte delivery to high endothelial venules. Science 273, 252–255.
Donners, M. M., Beckers, L., Lievens, D., Munnix, I., Heemskerk, J., Janssen, B. J., Wijnands, E., Cleutjens, J., Zernecke, A., Weber, C., Ahonen, C. L., Benbow, U., Newby, A. C., Noelle, R. J., Daemen, M. J., and Lutgens, E. (2008). The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood 111, 4596–4604.
Fontana, V., Horstman, L. L., Donna, E., Dudkiewicz, P., Ahn, E. R., and Ahn, Y. S. (2007). Interstitial lung disease (ILD) and severe ITP. Hematology 12, 75–80.
Freedman, R. R., Girgis, R., and Mayes, M. D. (1999). Endothelial and adrenergic dysfunction in Raynaud’s phenomenon and scleroderma. J. Rheumatol. 26, 2386–2388.
Frid, M. G., Kale, V. A., and Stenmark, K. R. (2002). Mature vascular endothelium can give rise to smooth muscle cells via endothelial–mesenchymal transdifferentiation: in vitro analysis. Circ. Res. 90, 1189–1196.
Friedhoff, L. T., Seibold, J. R., Kim, H. C., and Simester, K. S. (1984). Serotonin induced platelet aggregation in systemic sclerosis. Clin. Exp. Rheumatol. 2, 119–123.
Garcia-Porrua, C., Margarinos, C. C., and Gonzalez-Gay, M. A. (2004). Raynaud’s phenomenon and serotonin reuptake inhibitors. J. Rheumatol. 31, 2090; author reply 2090–2091.
Gardner, H., Shearstone, J. R., Bandaru, R., Crowell, T., Lynes, M., Trojanowska, M., Pannu, J., Smith, E., Jablonska, S., Blaszczyk, M., Tan, F. K., and Mayes, M. D. (2006). Gene profiling of scleroderma skin reveals robust signatures of disease that are imperfectly reflected in the transcript profiles of explanted fibroblasts. Arthritis Rheum. 54, 1961–1973.
Giusti, B., Fibbi, G., Margheri, F., Serrati, S., Rossi, L., Poggi, F., Lapini, I., Magi, A., Del Rosso, A., Cinelli, M., Guiducci, S., Kahaleh, B., Bazzichi, L., Bombardieri, S., Matucci-Cerinic, M., Gensini, G. F., Del Rosso, M., and Abbate, R. (2006). A model of anti-angiogenesis: differential transcriptosome profiling of microvascular endothelial cells from diffuse systemic sclerosis patients. Arthritis Res. Ther. 8, R115.
Goodfield, M. J., Orchard, M. A., and Rowell, N. R. (1988). Increased platelet sensitivity to collagen-induced aggregation in whole blood patients with systemic sclerosis. Clin. Exp. Rheumatol. 6, 285–288.
Goodfield, M. J., Orchard, M. A., and Rowell, N. R. (1993). Whole blood platelet aggregation and coagulation factors in patients with systemic sclerosis. Br. J. Haematol. 84, 675–680.
Harris, H. E., Andersson, U., and Pisetsky, D. S. (2012). HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 8, 195–202.
Herrick, A. L. (2011). Contemporary management of Raynaud’s phenomenon and digital ischaemic complications. Curr. Opin. Rheumatol. 23, 555–561.
Holt, C. M., Lindsey, N., Moult, J., Malia, R. G., Greaves, M., Hume, A., Rowell, N. R., and Hughes, P. (1989). Antibody-dependent cellular cytotoxicity of vascular endothelium: characterization and pathogenic associations in systemic sclerosis. Clin. Exp. Immunol. 78, 359–365.
Iannacone, M., Sitia, G., Isogawa, M., Marchese, P., Castro, M. G., Lowenstein, P. R., Chisari, F. V., Ruggeri, Z. M., and Guidotti, L. G. (2005). Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat. Med. 11, 1167–1169.
Kahaleh, M. B., and Fan, P. S. (1997). Mechanism of serum-mediated endothelial injury in scleroderma: identification of a granular enzyme in scleroderma skin and sera. Clin. Immunol. Immunopathol. 83, 32–40.
Kahaleh, M. B., and LeRoy, E. C. (1999). Autoimmunity and vascular involvement in systemic sclerosis (SSc). Autoimmunity 31, 195–214.
Kahaleh, M. B., Osborn, I., and Leroy, E. C. (1982). Elevated levels of circulating platelet aggregates and beta-thromboglobulin in scleroderma. Ann. Intern. Med. 96, 610–613.
Klimiuk, P. S., Grennan, A., Weinkove, C., and Jayson, M. I. (1989). Platelet serotonin in systemic sclerosis. Ann. Rheum. Dis. 48, 586–589.
Kosaki, G. (2005). In vivo platelet production from mature megakaryocytes: does platelet release occur via proplatelets? Int. J. Hematol. 81, 208–219.
Kowal-Bielecka, O., Kowal, K., Lewszuk, A., Bodzenta-Lukaszyk, A., Walecki, J., and Sierakowski, S. (2005). Beta thromboglobulin and platelet factor 4 in bronchoalveolar lavage fluid of patients with systemic sclerosis. Ann. Rheum. Dis. 64, 484–486.
Kuwana, M., Okazaki, Y., Yasuoka, H., Kawakami, Y., and Ikeda, Y. (2004). Defective vasculogenesis in systemic sclerosis. Lancet 364, 603–610.
Lafyatis, R., and York, M. (2009). Innate immunity and inflammation in systemic sclerosis. Curr. Opin. Rheumatol. 21, 617–622.
Luchetti, M. M., Sambo, P., Majlingova, P., Svegliati Baroni, S., Peri, G., Paroncini, P., Introna, M., Stoppacciaro, A., Mantovani, A., and Gabrielli, A. (2004). Scleroderma fibroblasts constitutively express the long pentraxin PTX3. Clin. Exp. Rheumatol. 22(Suppl. 33), S66–S72.
Ludwig, R. J., Bergmann, P., Garbaraviciene, J., von Stebut, E., Radeke, H. H., Gille, J., Diehl, S., Hardt, K., Henschler, R., Kaufmann, R., Pfeilschifter, J. M., and Boehncke, W. H. (2010). Platelet, not endothelial, P-selectin expression contributes to generation of immunity in cutaneous contact hypersensitivity. Am. J. Pathol. 176, 1339–1345.
Manfredi, A. A., Rovere-Querini, P., Bottazzi, B., Garlanda, C., and Mantovani, A. (2008). Pentraxins, humoral innate immunity and tissue injury. Curr. Opin. Immunol. 20, 538–544.
Manfredi, A. A., Rovere-Querini, P., and Maugeri, N. (2010). Dangerous connections: neutrophils and the phagocytic clearance of activated platelets. Curr. Opin. Hematol. 17, 3–8.
Mannucci, P. M., Lombardi, R., Lattuada, A., Perticucci, E., Valsecchi, R., and Remuzzi, G. (1989). Supranormal von Willebrand factor multimers in scleroderma. Blood 73, 1586–1591.
Margheri, F., Serrati, S., Lapucci, A., Chilla, A., Bazzichi, L., Bombardieri, S., Kahaleh, B., Calorini, L., Bianchini, F., Fibbi, G., and Del Rosso, M. (2010). Modulation of the angiogenic phenotype of normal and systemic sclerosis endothelial cells by gain-loss of function of pentraxin 3 and matrix metalloproteinase 12. Arthritis Rheum. 62, 2488–2498.
Martinez de la Torre, Y., Fabbri, M., Jaillon, S., Bastone, A., Nebuloni, M., Vecchi, A., Mantovani, A., and Garlanda, C. (2010). Evolution of the pentraxin family: the new entry PTX4. J. Immunol. 184, 5055–5064.
Maugeri, N., Baldini, M., Ramirez, G. A., Rovere-Querini, P., and Manfredi, A. A. (2012). Platelet–leukocyte deregulated interactions foster sterile inflammation and tissue damage in immune-mediated vessel diseases. Thromb. Res. 129, 267–273.
Maugeri, N., Rovere-Querini, P., Baldini, M., Sabbadini, M. G., and Manfredi, A. A. (2009). Translational mini-review series on immunology of vascular disease: mechanisms of vascular inflammation and remodelling in systemic vasculitis. Clin. Exp. Immunol. 156, 395–404.
Mauviel, A. (2005). Transforming growth factor-beta: a key mediator of fibrosis. Methods Mol. Med. 117, 69–80.
May, A. E., Kalsch, T., Massberg, S., Herouy, Y., Schmidt, R., and Gawaz, M. (2002). Engagement of glycoprotein IIb/IIIa (alpha(IIb)beta3) on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells. Circulation 106, 2111–2117.
Miller, K., Dresner, I. G., and Michael, A. F. (1980). Localization of platelet antigens in human kidney disease. Kidney Int. 18, 472–479.
Mullenix, P. S., Andersen, C. A., and Starnes, B. W. (2005). Atherosclerosis as inflammation. Ann. Vasc. Surg. 19, 130–138.
Nurden, A. T. (2011). Platelets, inflammation and tissue regeneration. Thromb. Haemost. 105(Suppl.1), S13–S33.
Pattanaik, D., Brown, M., and Postlethwaite, A. E. (2011). Vascular involvement in systemic sclerosis (scleroderma). J. Inflamm. Res. 4, 105–125.
Postlethwaite, A. E., and Chiang, T. M. (2007). Platelet contributions to the pathogenesis of systemic sclerosis. Curr. Opin. Rheumatol. 19, 574–579.
Semple, J. W., Italiano, J. E. Jr., and Freedman, J. (2011). Platelets and the immune continuum. Nat. Rev. Immunol. 11, 264–274.
Silveri, F., De Angelis, R., Poggi, A., Muti, S., Bonapace, G., Argentati, F., and Cervini, C. (2001). Relative roles of endothelial cell damage and platelet activation in primary Raynaud’s phenomenon (RP) and RP secondary to systemic sclerosis. Scand. J. Rheumatol. 30, 290–296.
Solanilla, A., Villeneuve, J., Auguste, P., Hugues, M., Alioum, A., Lepreux, S., Ducroix, J. P., Duhaut, P., Conri, C., Viallard, J. F., Nurden, A. T., Constans, J., and Ripoche, J. (2009). The transport of high amounts of vascular endothelial growth factor by blood platelets underlines their potential contribution in systemic sclerosis angiogenesis. Rheumatology (Oxford) 48, 1036–1044.
Steen, V. D., and Medsger, T. A. (2007). Changes in causes of death in systemic sclerosis, 1972–2002. Ann. Rheum. Dis. 66, 940–944.
Trojanowska, M. (2008). Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology (Oxford) 47(Suppl. 5), v2–v4.
Trostle, D. C., Bedetti, C. D., Steen, V. D., Al-Sabbagh, M. R., Zee, B., and Medsger, T. A. Jr. (1988). Renal vascular histology and morphometry in systemic sclerosis. A case–control autopsy study. Arthritis Rheum. 31, 393–400.
Tuder, R. M., Cool, C. D., Geraci, M. W., Wang, J., Abman, S. H., Wright, L., Badesch, D., and Voelkel, N. F. (1999). Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care. Med. 159, 1925–1932.
Verschoor, A., Neuenhahn, M., Navarini, A. A., Graef, P., Plaumann, A., Seidlmeier, A., Nieswandt, B., Massberg, S., Zinkernagel, R. M., Hengartner, H., and Busch, D. H. (2011). A platelet-mediated system for shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat. Immunol. 12, 1194–1201.
Whitfield, M. L., Finlay, D. R., Murray, J. I., Troyanskaya, O. G., Chi, J. T., Pergamenschikov, A., McCalmont, T. H., Brown, P. O., Botstein, D., and Connolly, M. K. (2003). Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc. Natl. Acad. Sci. U.S.A. 100, 12319–12324.
Yeaman, M. R. (2010). Platelets in defense against bacterial pathogens. Cell. Mol. Life Sci. 67, 525–544.
Keywords: platelets, systemic sclerosis, inflammation, autoimmunity, vascular injury, HMGB1, serotonin, collagen
Citation: Ramirez GA, Franchini S, Rovere-Querini P, Sabbadini MG, Manfredi AA and Maugeri N (2012) The role of platelets in the pathogenesis of systemic sclerosis. Front. Immun. 3:160. doi:10.3389/fimmu.2012.00160
Received: 03 May 2012; Accepted: 29 May 2012;
Published online: 18 June 2012.
Edited by:
Akihiko Yoshimura, Keio University, JapanReviewed by:
Ralf J. Ludwig, University of Lübeck, GermanyMartin Herrmann, Universitätsklinikum Erlangen, Germany
Copyright: © 2012 Ramirez, Franchini, Rovere-Querini, Sabbadini, Manfredi and Maugeri. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Angelo A. Manfredi, Department of Immunology, San Raffaele Scientific Institute, Università Vita-Salute San Raffaele, DIBIT 3A1, Via Olgettina 58, 20132 Milan, Italy. e-mail:bWFuZnJlZGkuYW5nZWxvQGhzci5pdA==