
- 1 Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden
- 2 Molecular Dermatology, Department of Medicine Solna, Karolinska Institutet, Stockholm, Sweden
Cutaneous leishmaniasis (CL) is associated with chronic inflammation and ulceration of the skin. Tissue macrophages serve as host cells and immune activation is necessary for parasite clearance. The balance between immune-mediated tissue destruction and successful clearance of infection is delicate and ulceration has been proposed to be a result of infiltration of activated immune cells into the skin. FasL and TRAIL play a dual role in skin homeostasis through induction of apoptosis as well as proinflammatory signaling. During leishmaniasis, dysregulation of both FasL and TRAIL has been described by us and others but the resulting pathogenic effects in the skin during human leishmaniasis are not fully elucidated. Targeting disease specific immune deviations has proven to be a promising new approach for the therapy of autoimmune diseases. Potentially, targeting FasL or TRAIL in combination with microcidals could offer a future treatment strategy to reduce the disfiguring immunopathology associated with CL. In this mini review we will discuss how FasL and TRAIL-induced signaling may influence on the extent of tissue inflammation and the efficacy of parasite clearance in leishmaniasis.
Leishmania Infection of the Skin Cause Chronic Tissue Inflammation and Ulceration
Human leishmaniasis of epithelial surfaces presents with a plethora of clinical manifestations – from asymptomatic infection to chronic skin ulcers or devastating ulceration of mucous surfaces. Cutaneous leishmaniasis (CL) in the Old World is caused by Leishmania major, L. tropica, and L. aethiopica and careful histological studies have elucidated the patterns of inflammation in these lesions (Gaafar et al., 1995, 1999; Connor et al., 1997). Different subspecies of Leishmania are associated with heterogeneous cutaneous manifestations but large clinical variations are also noted in skin pathology caused by the same species of Leishmania. This is exemplified by L. aethiopica infection that induces localized CL (LCL) or diffuse CL (DCL; Figure 1). LCL is characterized by erosive ulcers and a strong T cell-mediated response (Akuffo et al., 1987) which typically results in spontaneous healing within a year, scar formation, and solid protection against re-infection (Lemma et al., 1969). In contrast, DCL is linked to non-ulcerative chronic nodular disease with abundant parasitic infiltration of the dermal compartment of the skin and antigen-specific T cell unresponsiveness (Nilsen and Mshana, 1987). In the Americas, L. braziliensis cause devastating mucocutaneous lesions as well as diffuse and ulcerating CL. L. braziliensis infection can also lead to disseminated CL with a high number of ulcerated lesions spread over the body at large devoid of detectable parasites (Costa et al., 1986; Carvalho et al., 1994; Turetz et al., 2002). Patients affected by disseminated cutaneous L. braziliensis infection show effective antileishmanial T cell responses in circulating lymphocytes and histological studies proposed pronounced T cell infiltration into the affected skin (Leopoldo et al., 2006; Machado et al., 2011). Ulceration and tissue destruction in CL seems to be mainly a consequence of immune activation evoked by the infection rather than a direct effect of the infectious burden of parasites although definite proofs of this hypothesis has not been shown to date. Nevertheless, a low infectious burden in combination with an efficient antileishmanial T cell response is detected in disseminating or localized L. braziliensis-induced CL or L. aethiopica-induced LCL and is linked to ulcerative disease. On the other hand, L. aethiopica-induced DCL, with a high infectious burden and inefficient T cell responses, is linked to non-ulcerative disease. One obvious future treatment strategy would be to try to modulate the immune response to Leishmania so that the antileishmanial effects are maximized but tissue destruction minimized.
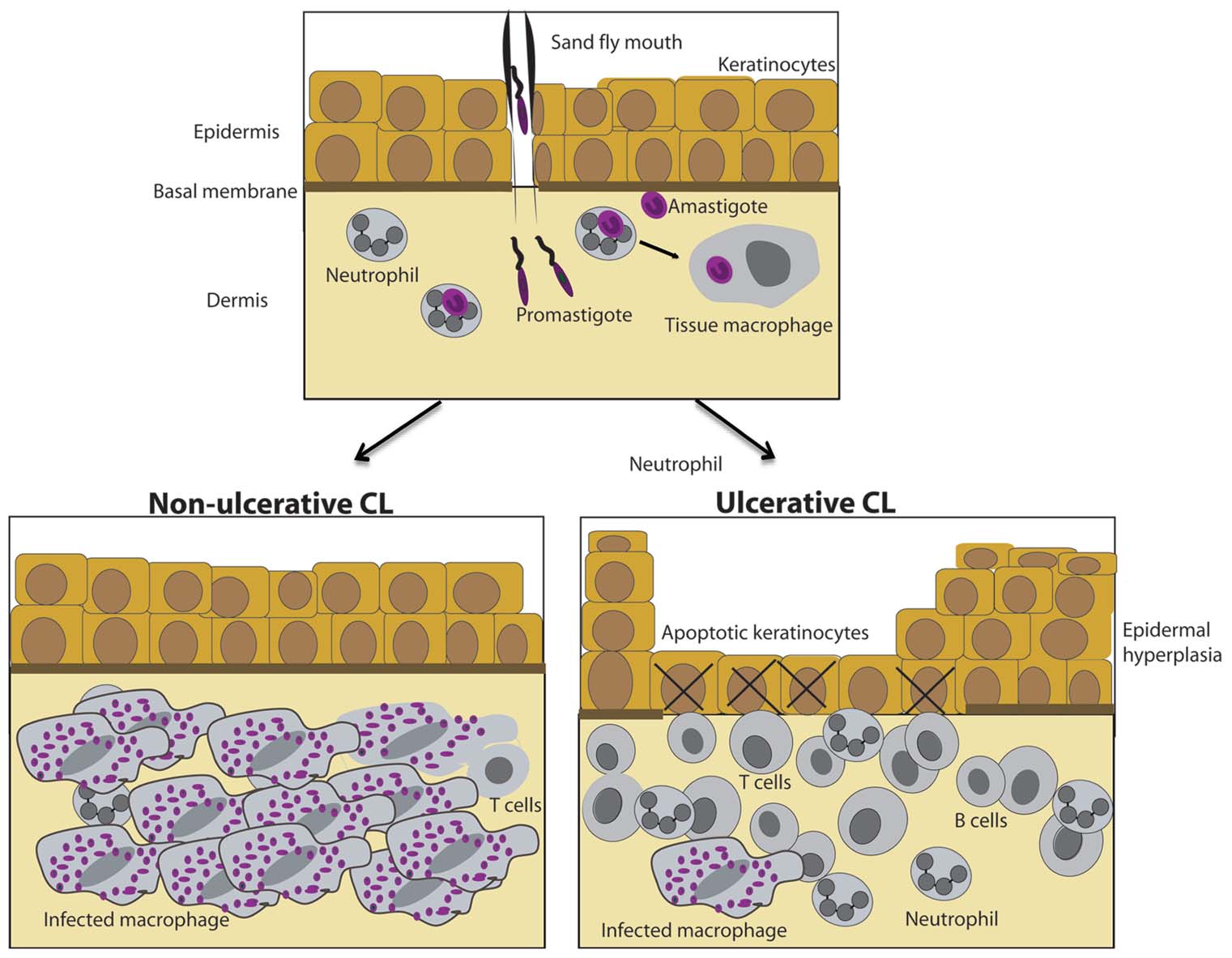
FIGURE 1. Histological hallmarks of diffuse and localized cutaneous leishmaniasis caused by L. aethiopica. (Top) Neutrophils, dendritic cells, and tissue macrophages are targets for infective Leishmania promastigotes and are present in the healthy skin. (Bottom) L. aethiopica cause non-ulcerative diffuse cutaneous leishmaniasis (DCL) or ulcerative localized cutaneous leishmaniasis (LCL). In non-ulcerative DCL (left), epidermis is intact and a large number of tissue macrophages packed with Leishmania amastigotes are present in dermis and the lymphocyte infiltrate is sparse as compared to ulcerative LCL (right). In ulcerative disease, apoptotic keratinocytes are present in the ulcerated epidermis. Epidermal hyperplasia and proliferating keratinocytes surround the ulcer and a dense inflammatory infiltrate is present in the dermal compartment. The number of infected tissue macrophages is low.
FasL and TRAIL Signaling Lead to Apoptosis or Production of Proinflammatory Mediators
FasL and TRAIL (tumor necrosis factor-related apoptosis inducing ligand) are death receptor ligands signaling through Fas, and TRAIL R1-4, respectively (Figure 2A). Fas, as well as TRAIL-R1 and TRAIL-R2 contain a functional intracellular death domain and may induce apoptosis but also alternative signals via NF-κB and MAPK activation (Falschlehner et al., 2007; Peter et al., 2007). TRAIL-R3 lack an intracellular cytoplastic domain and TRAIL-R4 contains an incomplete intracellular death domain but may induce proinflammatory signaling through NF-κB (Falschlehner et al., 2007). Fas regulates immune homeostasis and tolerance via inducing apoptosis of activated T, B, and dendritic cells (DCs), as suggested by the lymphoproliferative diseases associated with Fas or FasL mutations and by studies using targeted Fas deletion on T, B, and DCs (Stranges et al., 2007; Hao et al., 2008; Mabrouk et al., 2008; Turbyville and Rao, 2010). By killing APCs, FasL might decrease bystander T cell activation and in addition Fas can serve as a molecular break for T cell responses by complementing the effects of Bim to eliminate activated T cells (Hughes et al., 2008). Death receptor-induced apoptosis thus regulates turnover of effector cells during immune responses and additionally, due to FasL and TRAIL being expressed by activated T, NKT, and NK cells, it can eliminate cells infected by various pathogens. Death receptor-induced apoptosis can contribute to bystander apoptosis in inflamed tissues thereby increasing disease pathogenesis. Such activity is well studied during HIV infection where FasL, TNF, and TRAIL-mediated apoptosis have all been implicated in the depletion of non-infected T lymphocytes (Gougeon, 2003). FasL and TRAIL have also been shown to contribute to memory B cell apoptosis, possibly contributing to the decreased serological memory (Moir and Fauci, 2009; van Grevenynghe et al., 2011).
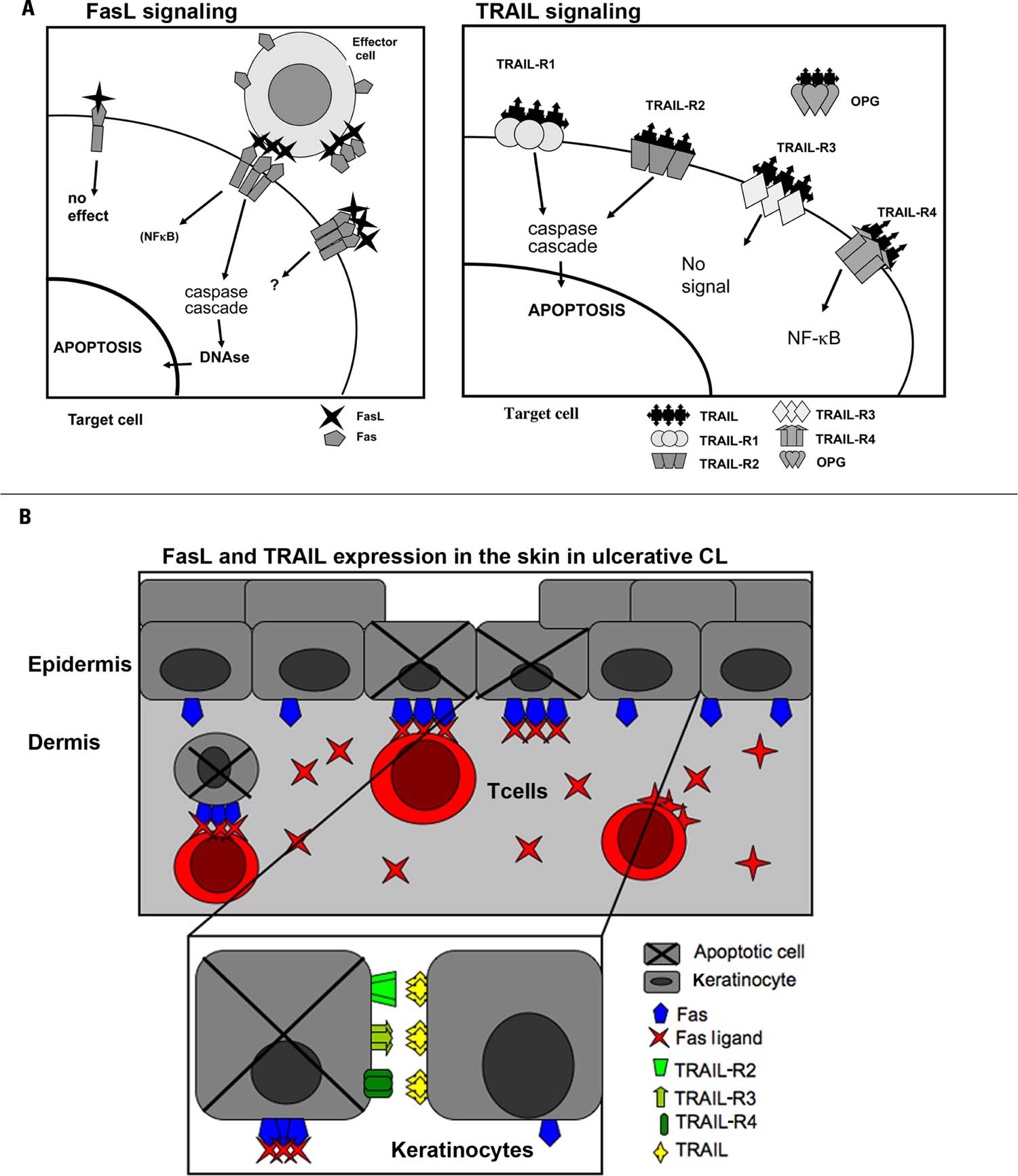
FIGURE 2. FasL and TRAIL signaling in ulcerative cutaneous leishmaniasis. (A) FasL and TRAIL signaling induce apoptosis or proinflammatory signaling depending on the receptor expression on the target cell. (Left) Fas is expressed as a homodimer on cell-surfaces. Upon FasL ligation to trimerized Fas caspase activation leading to apoptosis is initiated. Fas may also lead to NF-κb activation. sFas probably serves as a negative modulator of FasL. The function of sFasL is debated and it has been suggested that the concentration will determine if apoptosis can be triggered through Fas by sFasL. (Right) Apoptosis is induced through TRAIL activation of TRAIL-R1 and -R2. TRAIL-R3 is a decoy receptor and will not initiate intracellular signaling. Activation of TRAIL-R4 leads to NK-κB activation. OPG is a soluble receptor binding TRAIL and its function has not been elucidated. (B) A model for keratinocyte apoptosis in the ulcerative process of CL. Keratinocytes upregulate Fas, TRAIL-R2, -R4, and TRAIL as a result of the inflammatory reaction surrounding L. major-infected macrophages. FasL-expressing T cells and TRAIL-expressing inflammatory cells accumulate at the site of L. major infection. The Fas–FasL/Fas–sFasL and TRAIL/TRAIL-R interaction may lead to keratinocyte apoptosis and ulceration as well as to activation induced T cell apoptosis.
Non-apoptotic signaling activities of Fas stimulate T cell proliferation, induce proinflammatory responses in various immune cells, promote neuronal tissue remodeling, liver regeneration, and tumor progression (Peter et al., 2007). Apoptotic and non-apoptotic signals are probably occurring concomitantly in response to death receptor triggering, with the outcome determined by the activity of several other mechanisms influencing apoptosis sensitivity. Specifically, DISC localization at the cell membrane, high c-FLIP expression or the activity of Toso have been shown to increase stimulatory signals from death receptors and the threshold for apoptosis (Lee et al., 1998; Peter et al., 2007; Nguyen et al., 2011). In addition, the same cell types can respond differently to death receptor triggering depending on the stage of activation. Fas triggering can induce early apoptosis among activated T cells and enhanced proliferation of the surviving T cells (Maksimow et al., 2003). Triggering Fas on non-activated T cells of HIV-infected individuals can lead to apoptosis, whereas the same signal contributed to strong activation of T cells that received weak, otherwise ignorable TCR stimuli (Rethi et al., 2008). In addition to activation or death decided at the level of single cells, death receptor-induced apoptosis of one cell can modulate the activities of others thereby playing a complex regulatory role during immune responses. As will be discussed later, Fas-induced apoptosis of neutrophils during Leishmania infection can lead to macrophage activation or inactivation depending on the host’s genetic background, which strongly influence the immune response against the pathogen (Ribeiro-Gomes et al., 2004, 2005).
FasL and TRAIL Signaling in Skin Homeostasis and Inflammation
Epidermis is a stratified epithelium mainly composed of keratinocytes. It is separated from the underlying dermis by the basal membrane and basal keratinocytes are susceptible to FasL and TRAIL-induced apoptosis (Jansen et al., 2003; Vassina et al., 2005). IFNγ renders keratinocytes more susceptible to death receptor-induced apoptosis (Konur et al., 2005). TRAIL, as well as TRAIL-R1, -R2, and -R4 are expressed in keratinocytes in the healthy skin (Leverkus et al., 2000; Stander and Schwarz, 2005). TRAIL induces apoptosis in IFNγ stimulated keratinocytes and was shown to induce apoptosis in proliferating but not differentiating human keratinocytes (Jansen et al., 2003). To further show the complex role of TRAIL signaling in the skin, TRAIL was shown to induce caspase-dependent differentiation of primary keratinocytes (Wu et al., 2011).
FasL-mediated killing of IFNγ sensitized keratinocytes by T cells has been shown in the skin during atopic dermatitis (AD). In this setting, apoptotic keratinocytes were shown to release T cell chemoattractants. Thus, a loop of signaling between infiltrating, FasL-expressing T cells and activated, Fas-expressing keratinocytes was suggested which ultimately increased the number of inflammatory T cells in eczematous skin (Trautmann et al., 2000). FasL signaling has also been shown to induce production of several proinflammatory cytokines such as TNFα, IL1β, and IL-6 and activation of the epidermal growth factor receptor in keratinocytes in AD (Iordanov et al., 2005). FasL driven production of proinflammatory cytokines could in part explain the increased proliferation of keratinocytes which has been reported in many dermatoses including CL. TRAIL-expressing CD4+ and CD8+ T cells are present in dermis during AD (Vassina et al., 2005). TRAIL-expressing dermal DCs (Zaba et al., 2010) and upregulation of TRAIL-R1 and -R2 has been shown in the psoriatic skin (Goto and Lindoso, 2010). Nickel renders basal keratinocytes sensitive to TRAIL-mediated apoptosis through upregulation of TRAIL-R1 and -R2 in contact dermatitis toward nickel (Schmidt et al., 2010). Despite many descriptive studies showing the presence of TRAIL-expressing cells in the skin during inflammation, the consequences of such dysregulation are still at large unknown.
Alterations of Fas and FasL expression in keratinocytes (Viard et al., 1998) and in the effector T cell compartment (Abe et al., 2003) leading to keratinocyte death and ulceration has been described in toxic epidermal necrolysis (TEN). Recently, sTRAIL secreted by dermal DCs present in TEN lesions was shown to induce keratinocyte apoptosis (de Araujo et al., 2011). Thus, a synergistic killing of keratinocytes by FasL released from infiltrating T cells and TRAIL released by infiltrating DCs could occur in TEN. Intravenous immunoglobulins (IVIG) exhibits a broad spectrum of immunoregulatory activities and both activation and inhibition of Fas signaling (Prasad et al., 1998; Viard et al., 1998) has been ascribed IVIG and systemic treatment with IVIG-containing Fas-blocking antibodies limited the ulcerative process during TEN (Abe et al., 2003). To date, direct targeting of FasL signaling has not been tested in man during TEN.
Dysregulation of FasL and TRAIL and their Receptors During Human Leishmaniasis
Leishmania promastigotes are rapidly ingested by neutrophils and DCs present in the healthy skin (Laskay et al., 2008; Ng et al., 2008; Peters et al., 2008). In visceral leishmaniasis (VL), the parasites spreads from the skin to draining lymph nodes and further on throughout the reticuloendothelial organs in the body including the spleen, the liver, and the bone marrow. VL leads to a systemic and potentially lethal infection. Fas and FasL are highly expressed on spleen-derived leukocytes but not on peripheral blood mononuclear cells during VL, suggesting localized increase in Fas/FasL expression only at the site of infection (Eidsmo et al., 2002).
In CL, the infection is mainly confined to the skin and within a few weeks after infection tissue macrophages serves as the main host cells. Localized chronic tissue inflammation arises and Leishmania-laden macrophages need to be activated through signals from infiltrating leukocytes, primarily T cells but also B cells and neutrophils, to clear infection. Keratinocyte proliferation and activation with an upregulation of activation markers such as HLA DR is noted together with marked epidermal hyperplasia, necrosis, and apoptosis (ElHassan et al., 1995; Gaafar et al., 1999).
We and others have shown that Fas and FasL is upregulated in the skin in L. major and L. aethiopica-induced ulcerative CL (Mustafa et al., 2001; Eidsmo et al., 2005, 2007; Tasew et al., 2010; Table 1) and apoptotic keratinocytes surround ulcers (Eidsmo et al., 2005). In the case of L. aethiopica-induced CL, we have shown increased number of dermal FasL-expressing cells in ulcerating as compared to non-ulcerative lesions. In ulcerative CL, the number of apoptotic keratinocytes correlates with the number of FasL-expressing dermal cells (Eidsmo et al., 2005; Tasew et al., 2010). In agreement with our findings, increased numbers of FasL-expressing cells were detected in L. braziliensis-induced LCL and DCL (Carneiro et al., 2009) and the number of apoptotic cells correlated to the number of FasL-expressing cells in ulcerative LCL. In addition, increased expression of Fas and FasL was recently shown in oral mucosal leishmaniasis caused by L. braziliensis (Palmeiro et al., 2012). In ulcerative CL induced by L. major, TRAIL as well as the pro-apoptotic receptor TRAIL-R2 and the proinflammatory receptor TRAIL-4 were upregulated in infected skin as compared to healthy controls (Figure 2B; Eidsmo et al., 2007). In L. aethiopica-induced CL, higher levels of TRAIL and TRAIL-R2 were detected in patients as compared to healthy control and ulcerative LCL had higher levels of TRAIL and TRAIL-R2 as compared to non-ulcerative DCL (Eidsmo et al., 2007; Tasew et al., 2010) and (unpublished observations; Table 1 and Figure 2B).
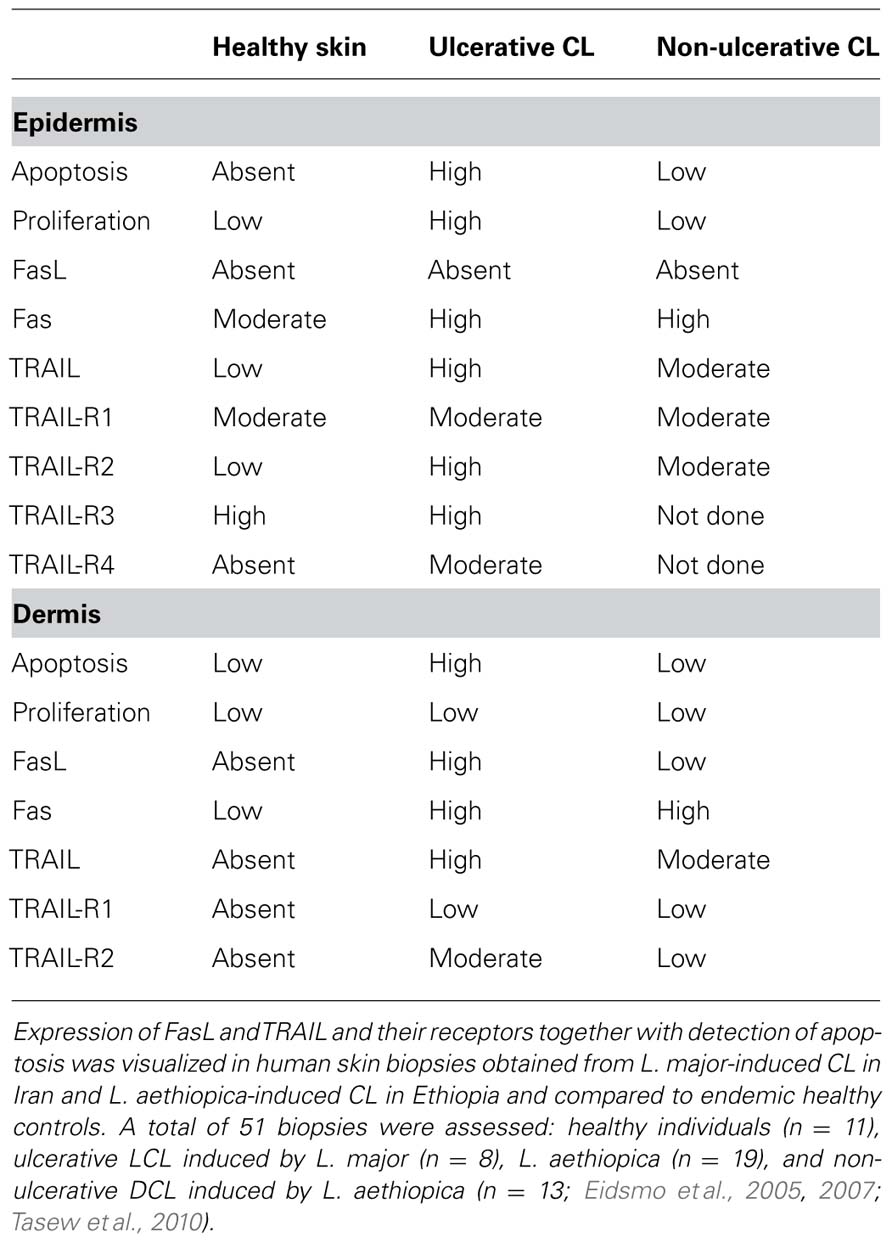
Table 1. Increased expression of FasL, TRAIL, TRAIL-R2, and -R4 at the site of infection during ulcerative leishmaniasis.
To test if the observed increased expression of FasL and TRAILwere indeed involved in ulceration during CL, we performed in vitro experiments using supernatants derived from L. major and L. aethiopica stimulated PBMCs that contained high levels of sFasL and sTRAIL. Primary keratinocytes and the keratinocyte cell line HaCaT exposed to sFasL- and sTRAIL-containing supernatants underwent apoptosis and this could be prevented by adding FasL and TRAIL neutralizing antibodies to the keratinocytes prior to exposure to supernatants. Interestingly, when strains of L. aethiopica isolated from patients with non-ulcerative DCL were used, keratinocyte apoptosis was not induced (Eidsmo et al., 2005, 2007; Tasew et al., 2010). To study the full consequences of FasL and TRAIL signaling during complex tissue infection and inflammation, we turned to an animal model of ulcerative CL. L. aethiopica inoculation in mice does not lead to productive infection or ulcerative disease. Murine CL can be obtained by L. major infection where in our hands the parasite strain Friedlin leads to ulcerative infection in BALB/c and the parasite strain LV39 leads to non-ulcerative infection. Thus, we have used L. major as a model organism to induce CL in mice as discussed below.
Regulation of Neutrophil Chemotaxis by Fas Activation in Leishmania Infection
During the chronic phase of CL, large numbers of neutrophils are present at the site of infection (Doerr and Seifert, 1995). Neutrophils age rapidly in the circulation and are constantly removed by macrophages in bone marrow, spleen, and liver (Furze and Rankin, 2008). Their activation leads to rapid apoptosis in which death receptors and Bcl-2 family member proteins play a role (Croker et al., 2011). Neutrophils serve as a first host during Leishmania infection and it has been shown in several settings that their spontaneous apoptosis is delayed upon infection and that infected, apoptotic neutrophils can serve as Trojan horses allowing the parasite a silent entry into tissue macrophages serving as the definite host (Aga et al., 2002; van Zandbergen et al., 2004).
FasL contributes to massive apoptosis of macrophages exposed to L. major in BALB/c mice and to the production of the chemokines CXCL1 and CCL3, both with neutrophil chemoattractant activity (Ribeiro-Gomes et al., 2005). Similarly to macrophages, Fas triggering on the surface of DCs, keratinocytes, and a subset of NKT cells induced the production of neutrophil chemoattractants (Giroux and Denis, 2005; Guo et al., 2005; Farley et al., 2006). The Fas/FasL molecule pair can thus serve as an accelerator of inflammation in addition to inducing apoptosis in various cell types. The FasL-induced neutrophil infiltration has several consequences. In addition to increasing pathogen clearance, the phagocytic cells might be important for the removal of apoptotic cells that accumulate as the result of the increased Fas activation. Apoptotic neutrophils can also initiate tissue repair mechanisms in macrophages, partly by inducing TGFβ secretion, which, on the other hand, facilitate Leishmania replication in macrophages (Afonso et al., 2008). In FasL-deficient gld mice, both macrophage apoptosis and chemokine production was reduced as well as the extravasation of neutrophils (Ribeiro-Gomes et al., 2005). Similarly, FasL neutralization using antibodies during ulcerative L. major (strain Friedlin V1) infection in BALB/c mice resulted in decreased neutrophil infiltration into infected skin (Tasew et al., 2010). The number of lesional neutrophils was approximately sixfold lower in a non-ulcerative model of L. major infection induced by the parasite-strain LV39. FasL neutralization in LV39-induced infection further reduced the number of lesional neutrophils (Tasew et al., 2010).
Modulation of Macrophage Response to Leishmania by Apoptotic Neutrophils
Neutrophils eliminate infecting pathogens via phagocytosis or by secreting reactive oxygen species, inflammatory cytokines, and chemokines and thereby increasing tissue inflammation. In addition, activated neutrophils can release a network formed of DNA and various granule proteins, called neutrophil extracellular traps, which can kill infecting pathogens including Leishmania (Guimaraes-Costa et al., 2009). Neutrophil responses are under tight regulation and the activation of these cells is rapidly followed by apoptosis, partly induced by Fas triggering. Apoptotic neutrophils can increase the activation of macrophages or DCs (Majai et al., 2010) and their phagocytozed material can be used as a source of pathogen-derived peptides for antigen presentation. It has been shown that apoptotic neutrophils regulate macrophage responses to Leishmania infection differently in BALB/c or C57BL/6 mice. In BALB/c mice, more sensitive to Leishmania infection, apoptotic neutrophils inhibit macrophage activation and facilitate pathogen replication in the cells via a PGE2 and TGF-β-dependent pathway (Ribeiro-Gomes et al., 2005). Interestingly, the absence of FasL fundamentally changed the role of neutrophils in the regulation of macrophage responses to Leishmania infection. The absence of FasL altered kinetics and probably the mechanism of apoptosis, and in this case neutrophils increased parasite clearance by macrophages via a NO-dependent mechanism (Ribeiro-Gomes et al., 2005).
In C57BL/6 mice, more resistant to Leishmania infection, apoptotic neutrophils increased parasite killing by macrophages through a neutrophil elastase-dependent induction of TNFα secretion (Ribeiro-Gomes et al., 2004). PGE2 and TGFβ, the neutrophil-induced factors in BALB/c mice that promote parasite growth in macrophages, were not induced during neutrophil–macrophage interactions in the C57BL/6 model. Thus, FasL turns neutrophils to suppressors of Leishmania clearance in BALB/c mice which was reflected by the finding that in vivo depletion of neutrophils decreased parasite load in wild-type (WT) but increased parasite load in gld mice (Ribeiro-Gomes et al., 2005). In C57BL/6 mice, in line with the macrophage stimulatory functions of neutrophils, granulocyte depletion led to increased parasite load (Ribeiro-Gomes et al., 2004).
Fas-Mediated Macrophage Apoptosis May Contribute to Parasite Clearance
In contrast to BALB mice, Fas or FasL deficiency on MLR and C57BL/6 background led to impaired parasite clearance when a high number of parasites was inoculated subcutaneously (Conceicao-Silva et al., 1998; Huang et al., 1998). Leishmania-infected macrophages upregulated Fas and apoptosis sensitivity in response to the Th1 cytokine IFNγ and to autocrine TNFα production. It was suggested that Fas-mediated elimination of Leishmania-infected macrophages play an important role in decreasing the parasite load (Conceicao-Silva et al., 1998). However, other studies have shown that gld mice on a C57BL/6 background control infection as efficient as WT control and that TNF, acting like another death receptor ligand, plays an essential role in pathogen clearance (Wilhelm et al., 2001, 2005). In these studies, the results varied depending on the Leishmania strain used for infection, indicating that the genetic background of both the host and the pathogen can influence the contribution of death receptors in the Leishmania-specific immune responses (Wilhelm et al., 2001; Ritter et al., 2004).
Short-Term Inhibition of FasL and TRAIL Signaling Decrease Ulceration in BALB/c Mice Infected with L. major
BALB/c serves as an ulcerative, non-healing model of CL and C57BL/6 mice serve as a non-ulcerative but healing model of CL. Thus, the characteristics of human CL are not completely mimicked by the models available. To study the effect of FasL or TRAIL neutralization during ulcerative CL, we utilized a model of injecting a low-infectious dose of metacyclics into dermis in BALB/c mice. Neutralization of both FasL and TRAIL inhibited ulcer development and no effect on the infectious load in the skin or draining lymph nodes was noted (Tasew et al., 2010). Interestingly, FasL neutralization reduced neutrophil infiltration into the skin during established infection, suggesting an additional proinflammatory role of FasL in addition to direct keratinocyte killing. FasL signaling resulting in recruitment of activated neutrophils into dermis may lead to destruction of the basal membrane and thus allow direct FasL-mediated killing of exposed keratinocytes in vivo.
A majority of patients affected by CL will request treatment despite the benign prognosis of the disease. The current treatment regimes are primarily aimed at parasite elimination and many will result in exacerbation of inflammation, tissue destruction, and scar formation (Mishra et al., 2007; Goto and Lindoso, 2010). Targeting specific immune mechanisms has proven to be a promising new approach for the therapy of cancer and autoimmune diseases and could potentially be used to improve treatment regimes of neglected tropical diseases including leishmaniasis. The current revolution in the development of immunotherapeutics could lead to novel effective treatments of neglected tropical diseases. Skin diseases serves as an excellent model system to test novel treatment regimes as the pathology and disease development can be followed by the naked eye. It has been suggested that TNFα-blocking antibodies could reduce tissue pathology during CL (Oliveira et al., 2011) and that inhibition of tyrosine kinases during VL could improve treatment efficacy (Dalton and Kaye, 2010; Dalton et al., 2010). We suggest that inhibition of reducing FasL and TRAIL signaling at the site of infection in combination with leishmanicidals could in theory reduce the chronic inflammation and the ulcerative process and subsequent scar formation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abe, R., Shimizu, T., Shibaki, A., Nakamura, H., Watanabe, H., and Shimizu, H. (2003). Toxic epidermal necrolysis and Stevens–Johnson syndrome are induced by soluble Fas ligand. Am. J. Pathol. 162, 1515–1520.
Afonso, L., Borges, V. M., Cruz, H., Ribeiro-Gomes, F. L., Dosreis, G. A., Dutra, A. N., Clarencio, J., De Oliveira, C. I., Barral, A., Barral-Netto, M., and Brodskyn, C. I. (2008). Interactions with apoptotic but not with necrotic neutrophils increase parasite burden in human macrophages infected with Leishmania amazonensis. J. Leukoc. Biol. 84, 389–396.
Aga, E., Katschinski, D. M., van Zandbergen, G., Laufs, H., Hansen, B., Muller, K., Solbach, W., and Laskay, T. (2002). Inhibition of the spontaneous apoptosis of neutrophil granulocytes by the intracellular parasite Leishmania major. J. Immunol. 169, 898–905.
Akuffo, H., Schurr, E., Andersson, G., Yamaneberhan, T., and Britton, S. (1987). Responsiveness in diffuse versus local cutaneous leishmaniasis is due to parasite differences. Scand. J. Immunol. 26, 717–721.
Carneiro, F. P., De Magalhaes, A. V., De Jesus Abreu Almeida Couto, M., Bocca, A. L., Muniz-Junqueira, M. I., and Ribeiro Sampaio, R. N. (2009). Foxp3 expression in lesions of the different clinical forms of American tegumentary leishmaniasis. Parasite Immunol. 31, 646–651.
Carvalho, E. M., Barral, A., Costa, J. M., Bittencourt, A., and Marsden, P. (1994). Clinical and immunopathological aspects of disseminated cutaneous leishmaniasis. Acta Trop. 56, 315–325.
Conceicao-Silva, F., Hahne, M., Schroter, M., Louis, J., and Tschopp, J. (1998). The resolution of lesions induced by Leishmania major in mice requires a functional Fas (APO-1, CD95) pathway of cytotoxicity. Eur. J. Immunol. 28, 237–245.
Connor, D., Chandler, F., Schwartz, D., Manz, H., and Lack, E. (1997). Pathology of Infectious Diseases. Stamford, CT: Appleton & Lange.
Costa, J. M., Marsden, P. D., Llanos-Cuentas, E. A., Netto, E. M., Carvalho, E. M., Barral, A., Rosa, A. C., Cuba, C. C., Magalhaes, A. V., and Barreto, A. C. (1986). Disseminated cutaneous leishmaniasis in a field clinic in Bahia, Brazil: a report of eight cases. J. Trop. Med. Hyg. 89, 319–323.
Croker, B. A., O’Donnell, J. A., Nowell, C. J., Metcalf, D., Dewson, G., Campbell, K. J., Rogers, K. L., Hu, Y., Smyth, G. K., Zhang, J. G., White, M., Lackovic, K., Cengia, L. H., O’Reilly, L. A., Bouillet, P., Cory, S., Strasser, A., and Roberts, A. W. (2011). Fas-mediated neutrophil apoptosis is accelerated by Bid, Bak, and Bax and inhibited by Bcl-2 and Mcl-1. Proc. Natl. Acad. Sci. U.S.A. 108, 13135–13140.
Dalton, J. E., and Kaye, P. M. (2010). Immunomodulators: use in combined therapy against leishmaniasis. Expert Rev. Anti Infect. Ther 8, 739–742.
Dalton, J. E., Maroof, A., Owens, B. M., Narang, P., Johnson, K., Brown, N., Rosenquist, L., Beattie, L., Coles, M., and Kaye, P. M. (2010). Inhibition of receptor tyrosine kinases restores immunocompetence and improves immune-dependent chemotherapy against experimental leishmaniasis in mice. J. Clin. Invest. 120, 1204–1216.
de Araujo, E., Dessirier, V., Lapree, G., Valeyrie-Allanore, L., Ortonne, N., Stathopoulos, E. N., Bagot, M., Bensussan, A., Mockenhaupt, M., Roujeau, J. C., and Tsapis, A. (2011). Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp. Dermatol. 20, 107–112.
Eidsmo, L., Fluur, C., Rethi, B., Eriksson Ygberg, S., Ruffin, N., De Milito, A., Akuffo, H., and Chiodi, F. (2007). FasL and TRAIL induce epidermal apoptosis and skin ulceration upon exposure to Leishmania major. Am. J. Pathol. 170, 227–239.
Eidsmo, L., Nylen, S., Khamesipour, A., Hedblad, M. A., Chiodi, F., and Akuffo, H. (2005). The contribution of the Fas/FasL apoptotic pathway in ulcer formation during Leishmania major-induced cutaneous Leishmaniasis. Am. J. Pathol. 166, 1099–1108.
Eidsmo, L., Wolday, D., Berhe, N., Sabri, F., Satti, I., El Hassan, A. M., Sundar, S., Chiodi, F., and Akuffo, H. (2002). Alteration of Fas and Fas ligand expression during human visceral leishmaniasis. Clin. Exp. Immunol. 130, 307–313.
ElHassan, A. M., Gaafar, A., and Theander, T. G. (1995). Antigen-presenting cells in human cutaneous leishmaniasis due to Leishmania major. Clin. Exp. Immunol. 99, 445–453.
Falschlehner, C., Emmerich, C. H., Gerlach, B., and Walczak, H. (2007). TRAIL signalling: decisions between life and death. Int. J. Biochem. Cell Biol. 39, 1462–1475.
Farley, S. M., Dotson, A. D., Purdy, D. E., Sundholm, A. J., Schneider, P., Magun, B. E., and Iordanov, M. S. (2006). Fas ligand elicits a caspase-independent proinflammatory response in human keratinocytes: implications for dermatitis. J. Invest. Dermatol. 126, 2438–2451.
Furze, R. C., and Rankin, S. M. (2008). The role of the bone marrow in neutrophil clearance under homeostatic conditions in the mouse. FASEB J. 22, 3111–3119.
Gaafar, A., El Kadaro, A. Y., Theander, T. G., Permin, H., Ismail, A., Kharazmi, A., and El Hassan, A. M. (1995). The pathology of cutaneous leishmaniasis due to Leishmania major in Sudan. Am. J. Trop. Med. Hyg. 52, 438–442.
Gaafar, A., Veress, B., Permin, H., Kharazmi, A., Theander, T. G., and El Hassan, A. M. (1999). Characterization of the local and systemic immune responses in patients with cutaneous leishmaniasis due to Leishmania major. Clin. Immunol. 91, 314–320.
Giroux, M., and Denis, F. (2005). CD1d-unrestricted human NKT cells release chemokines upon Fas engagement. Blood 105, 703–710.
Goto, H., and Lindoso, J. A. (2010). Current diagnosis and treatment of cutaneous and mucocutaneous leishmaniasis. Expert Rev. Anti Infect. Ther. 8, 419–433.
Gougeon, M. L. (2003). Apoptosis as an HIV strategy to escape immune attack. Nat. Rev. Immunol. 3, 392–404.
Guimaraes-Costa, A. B., Nascimento, M. T., Froment, G. S., Soares, R. P., Morgado, F. N., Conceicao-Silva, F., and Saraiva, E. M. (2009). Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. U.S.A. 106, 6748–6753.
Guo, Z., Zhang, M., Tang, H., and Cao, X. (2005). Fas signal links innate and adaptive immunity by promoting dendritic-cell secretion of CC and CXC chemokines. Blood 106, 2033–2041.
Hao, Z., Duncan, G. S., Seagal, J., Su, Y. W., Hong, C., Haight, J., Chen, N. J., Elia, A., Wakeham, A., Li, W. Y., Liepa, J., Wood, G. A., Casola, S., Rajewsky, K., and Mak, T. W. (2008). Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity 29, 615–627.
Huang, F. P., Xu, D., Esfandiari, E. O., Sands, W., Wei, X. Q., and Liew, F. Y. (1998). Mice defective in Fas are highly susceptible to Leishmania major infection despite elevated IL-12 synthesis, strong Th1 responses, and enhanced nitric oxide production. J. Immunol. 160, 4143–4147.
Hughes, P. D., Belz, G. T., Fortner, K. A., Budd, R. C., Strasser, A., and Bouillet, P. (2008). Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity 28, 197–205.
Iordanov, M. S., Sundholm, A. J., Simpson, E. L., Hanifin, J. M., Ryabinina, O. P., Choi, R. J., Korcheva, V. B., Schneider, P., and Magun, B. E. (2005). Cell death-induced activation of epidermal growth factor receptor in keratinocytes: implications for restricting epidermal damage in dermatitis. J. Invest. Dermatol. 125, 134–142.
Jansen, B. J., Van Ruissen, F., Cerneus, S., Cloin, W., Bergers, M., Van Erp, P. E., and Schalkwijk, J. (2003). Tumor necrosis factor related apoptosis inducing ligand triggers apoptosis in dividing but not in differentiating human epidermal keratinocytes. J. Invest. Dermatol. 121, 1433–1439.
Konur, A., Schulz, U., Eissner, G., Andreesen, R., and Holler, E. (2005). Interferon (IFN)-gamma is a main mediator of keratinocyte (HaCaT) apoptosis and contributes to autocrine IFN-gamma and tumour necrosis factor-alpha production. Br. J. Dermatol. 152, 1134–1142.
Laskay, T., van Zandbergen, G., and Solbach, W. (2008). Neutrophil granulocytes as host cells and transport vehicles for intracellular pathogens: apoptosis as infection-promoting factor. Immunobiology 213, 183–191.
Lee, S. H., Jang, J. J., Lee, J. Y., Kim, S. Y., Park, W. S., Shin, M. S., Dong, S. M., Na, E. Y., Kim, K. M., Kim, C. S., Kim, S. H., and Yoo, N. J. (1998). Fas ligand is expressed in normal skin and in some cutaneous malignancies. Br. J. Dermatol. 139, 186–191.
Lemma, A., Foster, W. A., Gemetchu, T., Preston, P. M., Bryceson, A., and Minter, D. M. (1969). Studies on leishmaniasis in Ethiopia. I. Preliminary investigations into the epidemiology of cutaneous leishmaniasis in the highlands. Ann. Trop. Med. Parasitol. 63, 455–472.
Leopoldo, P. T., Machado, P. R., Almeida, R. P., Schriefer, A., Giudice, A., De Jesus, A. R., Ho, J. L., Guimaraes, L. H., Bacellar, O., and Carvalho, E. M. (2006). Differential effects of antigens from L. braziliensis isolates from disseminated and cutaneous leishmaniasis on in vitro cytokine production. BMC Infect. Dis. 6, 75. doi: 10.1186/1471-2334-6-75
Leverkus, M., Neumann, M., Mengling, T., Rauch, C. T., Brocker, E. B., Krammer, P. H., and Walczak, H. (2000). Regulation of tumor necrosis factor-related apoptosis-inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res. 60, 553–559.
Mabrouk, I., Buart, S., Hasmim, M., Michiels, C., Connault, E., Opolon, P., Chiocchia, G., Levi-Strauss, M., Chouaib, S., and Karray, S. (2008). Prevention of autoimmunity and control of recall response to exogenous antigen by Fas death receptor ligand expression on T cells. Immunity 29, 922–933.
Machado, P. R., Rosa, M. E., Costa, D., Mignac, M., Silva, J. S., Schriefer, A., Teixeira, M. M., Bacellar, O., and Carvalho, E. M. (2011). Reappraisal of the immunopathogenesis of disseminated leishmaniasis: in situ and systemic immune response. Trans. R. Soc. Trop. Med. Hyg. 105, 438–444.
Majai, G., Gogolak, P., Ambrus, C., Vereb, G., Hodrea, J., Fesus, L., and Rajnavolgyi, E. (2010). PPARgamma modulated inflammatory response of human dendritic cell subsets to engulfed apoptotic neutrophils. J. Leukoc. Biol. 88, 981–991.
Maksimow, M., Santanen, M., Jalkanen, S., and Hanninen, A. (2003). Responding naive T cells differ in their sensitivity to Fas engagement: early death of many T cells is compensated by costimulation of surviving T cells. Blood 101, 4022–4028.
Mishra, J., Saxena, A., and Singh, S. (2007). Chemotherapy of leishmaniasis: past, present and future. Curr. Med. Chem. 14, 1153–1169.
Moir, S., and Fauci, A. S. (2009). B cells in HIV infection and disease. Nat. Rev. Immunol. 9, 235–245.
Mustafa, T., Bjune, T. G., Jonsson, R., Pando, R. H., and Nilsen, R. (2001). Increased expression of fas ligand in human tuberculosis and leprosy lesions: a potential novel mechanism of immune evasion in mycobacterial infection. Scand. J. Immunol. 54, 630–639.
Ng, L. G., Hsu, A., Mandell, M. A., Roediger, B., Hoeller, C., Mrass, P., Iparraguirre, A., Cavanagh, L. L., Triccas, J. A., Beverley, S. M., Scott, P., and Weninger, W. (2008). Migratory dermal dendritic cells act as rapid sensors of protozoan parasites. PLoS Pathog. 4, e1000222. doi: 10.1371/journal.ppat.1000222
Nguyen, X. H., Lang, P. A., Lang, K. S., Adam, D., Fattakhova, G., Foger, N., Kamal, M. A., Prilla, P., Mathieu, S., Wagner, C., Mak, T., Chan, A. C., and Lee, K. H. (2011). Toso regulates the balance between apoptotic and nonapoptotic death receptor signaling by facilitating RIP1 ubiquitination. Blood 118, 598–608.
Nilsen, R., and Mshana, R. N. (1987). In situ characterization of the cutaneous immune response in Ethiopian cutaneous leishmaniasis. Scand. J. Immunol. 26, 503–512.
Oliveira, F., Bafica, A., Rosato, A. B., Favali, C. B., Costa, J. M., Cafe, V., Barral-Netto, M., and Barral, A. (2011). Lesion size correlates with Leishmania antigen-stimulated TNF-levels in human cutaneous leishmaniasis. Am. J. Trop. Med. Hyg. 85, 70–73.
Palmeiro, M. R., Morgado, F. N., Valete-Rosalino, C. M., Martins, A. C., Moreira, J., Quintella, L. P., De Oliveira Schubach, A., and Conceicao-Silva, F. (2012). Comparative study of the in situ immune response in oral and nasal mucosal leishmaniasis. Parasite Immunol. 34, 23–31.
Peter, M. E., Budd, R. C., Desbarats, J., Hedrick, S. M., Hueber, A. O., Newell, M. K., Owen, L. B., Pope, R. M., Tschopp, J., Wajant, H., Wallach, D., Wiltrout, R. H., Zornig, M., and Lynch, D. H. (2007). The CD95 receptor: apoptosis revisited. Cell 129, 447–450.
Peters, N. C., Egen, J. G., Secundino, N., Debrabant, A., Kimblin, N., Kamhawi, S., Lawyer, P., Fay, M. P., Germain, R. N., and Sacks, D. (2008). In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 321, 970–974.
Prasad, N. K., Papoff, G., Zeuner, A., Bonnin, E., Kazatchkine, M. D., Ruberti, G., and Kaveri, S. V. (1998). Therapeutic preparations of normal polyspecific IgG (IVIg) induce apoptosis in human lymphocytes and monocytes: a novel mechanism of action of IVIg involving the Fas apoptotic pathway. J. Immunol. 161, 3781–3790.
Rethi, B., Vivar, N., Sammicheli, S., Fluur, C., Ruffin, N., Atlas, A., Rajnavolgyi, E., and Chiodi, F. (2008). Priming of T cells to Fas-mediated proliferative signals by interleukin-7. Blood 112, 1195–1204.
Ribeiro-Gomes, F. L., Moniz-De-Souza, M. C., Borges, V. M., Nunes, M. P., Mantuano-Barradas, M., D’Avila, H., Bozza, P. T., Calich, V. L., and Dosreis, G. A. (2005). Turnover of neutrophils mediated by Fas ligand drives Leishmania major infection. J. Infect. Dis. 192, 1127–1134.
Ribeiro-Gomes, F. L., Otero, A. C., Gomes, N. A., Moniz-De-Souza, M. C., Cysne-Finkelstein, L., Arnholdt, A. C., Calich, V. L., Coutinho, S. G., Lopes, M. F., and Dosreis, G. A. (2004). Macrophage interactions with neutrophils regulate Leishmania major infection. J. Immunol. 172, 4454–4462.
Ritter, U., Mattner, J., Rocha, J. S., Bogdan, C., and Korner, H. (2004). The control of Leishmania (Leishmania) major by TNF in vivo is dependent on the parasite strain. Microbes Infect. 6, 559–565.
Schmidt, M., Hupe, M., Endres, N., Raghavan, B., Kavuri, S., Geserick, P., Goebeler, M., and Leverkus, M. (2010). The contact allergen nickel sensitizes primary human endothelial cells and keratinocytes to TRAIL-mediated apoptosis. J. Cell. Mol. Med. 14, 1760–1776.
Stander, S., and Schwarz, T. (2005). Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is expressed in normal skin and cutaneous inflammatory diseases, but not in chronically UV-exposed skin and non-melanoma skin cancer. Am. J. Dermatopathol 27, 116–121.
Stranges, P. B., Watson, J., Cooper, C. J., Choisy-Rossi, C. M., Stonebraker, A. C., Beighton, R. A., Hartig, H., Sundberg, J. P., Servick, S., Kaufmann, G., Fink, P. J., and Chervonsky, A. V. (2007). Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity 26, 629–641.
Tasew, G., Nylen, S., Lieke, T., Lemu, B., Meless, H., Ruffin, N., Wolday, D., Asseffa, A., Yagita, H., Britton, S., Akuffo, H., Chiodi, F., and Eidsmo, L. (2010). Systemic FasL and TRAIL neutralisation reduce leishmaniasis induced skin ulceration. PLoS Negl. Trop. Dis. 4, e844. doi: 10.1371/journal.pntd.0000844
Trautmann, A., Akdis, M., Kleemann, D., Altznauer, F., Simon, H. U., Graeve, T., Noll, M., Brocker, E. B., Blaser, K., and Akdis, C. A. (2000). T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J. Clin. Invest. 106, 25–35.
Turbyville, J. C., and Rao, V. K. (2010). The autoimmune lymphoproliferative syndrome: a rare disorder providing clues about normal tolerance. Autoimmun. Rev. 9, 488–493.
Turetz, M. L., Machado, P. R., Ko, A. I., Alves, F., Bittencourt, A., Almeida, R. P., Mobashery, N., Johnson, W. D. Jr., and Carvalho, E. M. (2002). Disseminated leishmaniasis: a new and emerging form of leishmaniasis observed in northeastern Brazil. J. Infect. Dis. 186, 1829–1834.
van Grevenynghe, J., Cubas, R. A., Noto, A., Dafonseca, S., He, Z., Peretz, Y., Filali-Mouhim, A., Dupuy, F. P., Procopio, F. A., Chomont, N., Balderas, R. S., Said, E. A., Boulassel, M. R., Tremblay, C. L., Routy, J. P., Sekaly, R. P., and Haddad, E. K. (2011). Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J. Clin. Invest. 121, 3877–3888.
van Zandbergen, G., Klinger, M., Mueller, A., Dannenberg, S., Gebert, A., Solbach, W., and Laskay, T. (2004). Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J. Immunol. 173, 6521–6525.
Vassina, E., Leverkus, M., Yousefi, S., Braathen, L. R., Simon, H. U., and Simon, D. (2005). Increased expression and a potential anti-inflammatory role of TRAIL in atopic dermatitis. J. Invest. Dermatol. 125, 746–752.
Viard, I., Wehrli, P., Bullani, R., Schneider, P., Holler, N., Salomon, D., Hunziker, T., Saurat, J. H., Tschopp, J., and French, L. E. (1998). Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 282, 490–493.
Wilhelm, P., Ritter, U., Labbow, S., Donhauser, N., Rollinghoff, M., Bogdan, C., and Korner, H. (2001). Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking TNF. J. Immunol. 166, 4012–4019.
Wilhelm, P., Wiede, F., Meissner, A., Donhauser, N., Bogdan, C., and Korner, H. (2005). TNF but not Fas ligand provides protective anti-L. major immunity in C57BL/6 mice. Microbes Infect. 7, 1461–1468.
Wu, N. L., Lee, T. A., Tsai, T. L., and Lin, W. W. (2011). TRAIL-induced keratinocyte differentiation requires caspase activation and p63 expression. J. Invest. Dermatol. 131, 874–883.
Zaba, L. C., Fuentes-Duculan, J., Eungdamrong, N. J., Johnson-Huang, L. M., Nograles, K. E., White, T. R., Pierson, K. C., Lentini, T., Suarez-Farinas, M., Lowes, M. A., and Krueger, J. G. (2010). Identification of TNF-related apoptosis-inducing ligand and other molecules that distinguish inflammatory from resident dendritic cells in patients with psoriasis. J. Allergy Clin. Immunol. 125, 1261–1268 e1269.
Keywords: cutaneous leishmaniasis, FasL, TRAIL, ulceration, immunopathology, neutrophils, macrophages, keratinocytes
Citation: Rethi B and Eidsmo L (2012) FasL and TRAIL signaling in the skin during cutaneous leishmaniasis – implications for tissue immunopathology and infectious control. Front. Immun. 3:163. doi: 10.3389/fimmu.2012.00163
Received: 07 January 2012; Accepted: 31 May 2012;
Published online: 19 June 2012.
Edited by:
Nathan Peters, National Institute of Allergy and Infectious Diseases, USAReviewed by:
Adam Cunningham, University of Birmingham, UKGeorge DosReis, Federal University of Rio de Janeiro, Brazil
Copyright: © 2012 Rethi and Eidsmo. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Liv Eidsmo, Dermatology and Venereology Unit, CMM L8: 02, Karolinska University Hospital Solna, 171 77 Stockholm, Sweden. e-mail:bGl2LmVpZHNtb0BraS5zZQ==