
 Tobias Bopp2
Tobias Bopp2
- 1 Department of Dermatology, University Medical Center of the Johannes Gutenberg-University, Mainz, Germany
- 2 Institute for Immunology, University Medical Center of the Johannes Gutenberg-University, Mainz, Germany
Understanding tolerance mechanisms at the cellular and molecular level holds the promise to establish novel immune intervention therapies in patients with allergy or autoimmunity and to prevent transplant rejection. Administration of mAb against the CD4 molecule has been found to be exceptionally well suited for intentional tolerance induction in rodent and non-human primate models as well as in humanized mouse models. Recent evidence demonstrated that regulatory T cells (Treg) are directly activated by non-depleting CD4 ligands and suggests Treg activation as a central mechanism in anti-CD4-mediated tolerance induction. This review summarizes the current knowledge on the role of Treg in peripheral tolerance, addresses the putative mechanisms of Treg-mediated suppression and discusses the clinical potential of harnessing Treg suppressive activity through CD4 stimulation.
Regulatory T Cells in Maintenance of Peripheral Tolerance
The notion of peripheral immune regulation by T cells that shut off other immune cells has been around for many decades (Gershon and Kondo, 1971). Discovery of suitable surface markers (Sakaguchi et al., 1995; Takahashi et al., 1998) and a lineage-specific transcription factor (Hori et al., 2003; Walker et al., 2003) confirmed the existence of a distinct regulatory T cell (Treg) population. Originally identified by their aptitude to hold off autoimmune reactions (Sakaguchi et al., 1995; Takahashi et al., 1998; Wildin et al., 2002; Fontenot et al., 2005), Treg own far-ranging suppressive activity affecting the function, maturation, and survival of all types of immune cells (Thornton and Shevach, 1998; Jonuleit et al., 2001; Grossman et al., 2004; Kojima et al., 2005) in response to self and non-self antigens, including pathogens (Belkaid et al., 2002; Hasenkrug, 2003). Additionally, Treg have been shown to confer regulatory properties upon suppressed T cells implementing a second layer of regulation (Jonuleit et al., 2002; Stassen et al., 2004; Andersson et al., 2008).
Attempts to define the molecular basis of Treg suppression have lead to the description of numerous putative pathways and molecules (Tang and Bluestone, 2008; Shevach, 2009). A majority of studies agreed on cell contact-dependent suppression by Treg in vitro (Thornton and Shevach, 1998; Jonuleit et al., 2001) and the observation of persistent contacts between Treg and dendritic cells (DCs) during active suppression by intravital microscopy suggests that cell contact-dependent suppression might also play a role in vivo (Tang et al., 2006).
Analyzing the molecular mechanism of contact-dependent Treg suppression by comparison of gene expression in Treg and non-regulatory T cells, we found that Treg up-regulate cAMP in their cytosol upon activation and consign cAMP to conventional CD4+ T cells and DCs (Bopp et al., 2007; Becker et al., 2009; Fassbender et al., 2010) by gap junction intercellular communication (GJIC; Oviedo-Orta et al., 2000). Upon transfer cAMP inhibits the proliferation and differentiation of responder cells, most probably through the induction of inducible cAMP early repressor (ICER) expression (Foulkes et al., 1991; Bodor et al., 1996, 2007). Continuative work revealed that cAMP transmission is an essential component of Treg-mediated suppression in vivo (Bopp et al., 2007; Becker et al., 2009). Concurrent with stable and persistent Treg–DC interaction (Tang and Bluestone, 2006), transfer of Treg-derived cAMP into conventional T cells in vivo was inevitably dependent on the presence of antigen presenting cells (APC) and restricted to the draining lymph node (Bopp et al., 2007). Correspondingly, repression of cAMP accumulation in Treg either by adenylyl cyclase inhibition, application of a cAMP-specific antagonist or phosphodiesterase (PDE) overexpression abrogated Treg suppression (Bopp et al., 2007; Oberle et al., 2007; Becker et al., 2009; Klein et al., 2012; Martin et al., 2012). Inversely, blockade of cAMP degradation by PDE inhibition improved Treg-mediated suppression in a murine asthma model (Bopp et al., 2009).
Regarding cAMP regulation in Treg, Foxp3 has been shown to repress PDE3b expression (Gavin et al., 2006) thereby preventing cAMP degradation. More recently, Huang et al. (2009) showed that the high cAMP content in Treg and their suppressive property depend on Foxp3-mediated repression of the adenylyl cyclase 9 (AC9) regulating miRNA 142-3p. In line with these observations, Lahl et al. (2009) demonstrated that non-functional Treg in Foxp3 mutant scurfy mice harbor significantly reduced levels of cytosolic cAMP. Hence, the transcription factor Foxp3 participates in cAMP buildup by concomitantly regulating the expression of cAMP-generating and degrading enzymes. It is noteworthy that the transmission of cAMP is actually involved both in the suppression of other T cells (Bopp et al., 2007; Becker et al., 2009; Huang et al., 2009; Klein et al., 2012) and in suppression of DCs (Fassbender et al., 2010). Together these findings classify cAMP as a key component of Treg suppressive mechanism in vitro and in vivo and disclose cAMP-regulating enzymes as molecular targets for therapeutic intervention with Treg activity in pathological processes like allergy and autoimmunity.
Next to the transfer of cAMP through gap junctions, production of extracellular adenosine has been suggested as an alternative mechanism in cAMP-dependent suppression by Treg (Deaglio et al., 2007). Extracellular nucleotides are anti-inflammatory mediators produced by a variety of cell types including Treg (Deaglio et al., 2007; Mandapathil et al., 2009) and Th17 cells (Chalmin et al., 2012). Physiologically, extracellular nucleotide production represents a protective mechanism in response to tissue injury (Fredholm, 2007). In Treg suppression adenosine formation through the ectoenzymes CD39 and CD73, expressed by murine Treg and a subpopulation of human Treg (Mandapathil et al., 2009), has been assumed to induce cAMP production in conventional T cells or DCs upon binding to the A2A receptor (Deaglio et al., 2007; Ernst et al., 2010). However, the role of adenosine as a major suppressive mechanism employed specifically by Treg is questionable. Blockade of cAMP production in responder T cells by inhibition of adenylyl cyclases does not alter their susceptibility to Treg-mediated suppression (Klein et al., 2012). In addition, A2A receptor expression is detectable on T cells 4 days after stimulation (Deaglio et al., 2007) while T cells are susceptible to Treg suppression exclusively within the first 24 h after stimulation (Hagness et al., 2012). Finally, Blockage of ectonucleotidase activity only slightly abrogates suppression of human T cells by CD39 expressing Treg (Mandapathil et al., 2010). Thus, while nucleotides certainly affect numerous cellular functions – including de novo cAMP generation in Treg – their role in Treg suppression is most likely of an indirect nature.
Interestingly, cAMP up-regulation in Treg coincides with another cell contact-dependent mechanism of suppression: Treg constitutively express the two co-inhibitory membrane-bound molecules CTLA-4 and TIGIT (Read et al., 2000; Takahashi et al., 2000) which are believed to provide inhibitory signals. In mice CTLA-4 deficiency (Bachmann et al., 1999), CTLA-4 blockade (Takahashi et al., 2000), and Treg-specific ablation of CTLA-4 (Wing et al., 2008) resulted in spontaneous autoimmunity. Yet, CTLA-4 deficient Treg remain suppressive in vitro and in vivo (Tang et al., 2004; Read et al., 2006) suggesting additional mechanisms to be involved. Studies on human Treg in vitro revealed only a minor role of CTLA-4 in Treg suppression (Birebent et al., 2004) or firmly excluded CTLA-4 as a suppressive mechanism (Baecher-Allan et al., 2001; Jonuleit et al., 2001; Levings et al., 2001). However, discrepancies regarding the importance of CTLA-4 in Treg suppression might in part be due to the use of different target cells. While the role of CTLA-4 in suppression of T cells remains uncertain, it is unequivocally required in the suppression of APC. Suppression of DCs by Treg via CTLA-4 has been shown to induce the downregulation of CD80 and CD86 (Cederbom et al., 2000) preventing effector T cell activation by the APC in vitro (Oderup et al., 2006) and in vivo (Wing et al., 2008). Notably, elevated cAMP levels in T cells have been shown to increase CTLA-4 expression (Vendetti et al., 2006) and cAMP and CTLA-4 expression are simultaneously up-regulated in Treg upon activation (Becker et al., 2009).
While a majority of studies firmly excluded soluble factors in Treg suppression in vitro, there is growing evidence that cytokines substantially add to the immune regulatory function of Treg in vivo. In particular, transforming growth factor-β (TGF-β) and IL-10 seem to be indispensable for sustained tolerance induction by Treg. A role for TGF-β in maintenance of peripheral tolerance was initially suggested by its importance in infectious tolerance (Chen et al., 2003) particularly its long-lasting production by CD4+ T cells from tolerant mice in long-term acceptance of allografts (Daley et al., 2007). However, in order to exert its biological functions, TGF-β needs to be converted from its latent (bound to latency associated peptide, LAP) into its active conformation by proteolytic cleavage (Khalil, 1999). Yet, there are multiple mechanisms of activating TGF-β from its latency (Lawrence, 2001; Annes et al., 2003) and it is unclear how TGF-β is activated in vivo.
Although repeatedly observed in disease models (Nakamura et al., 2001) a direct contribution of TGF-β in Treg suppression remained controversial because anti-TGF-β antibodies and soluble TGF-RII failed to affect the suppressive function of Treg (Andersson et al., 2008). Recently, “glycoprotein A repetitions predominant” (GARP) expressed on the surface of Treg upon activation (Wang et al., 2008, 2009; D’Alise et al., 2011) has been shown to act as a receptor for the TGF-β/LAP complex (Stockis et al., 2009). Reminiscent of infectious Treg suppression (Jonuleit et al., 2002; Stassen et al., 2004) latent TGF-β bound to GARP on the surface of activated Treg has been demonstrated to convert responder T cells into induced Treg (Andersson et al., 2008). Thus, apart from acting as a soluble modulator of immune cells, TGF-β supposedly helps Treg to execute their contact-dependent suppressive activity by binding to GARP (Battaglia and Roncarolo, 2009).
IL-10 has been unequivocally shown to form another important mediator in Treg suppression in vivo (Kearley et al., 2005; Collison et al., 2007) particularly in suppression of pathogenic Th17 cells (Chaudhry et al., 2011; Huber et al., 2011). Correspondingly, Treg-specific ablation of IL-10 leads to inflammation (Rubtsov et al., 2008). In contrast to general Treg deficiency, however, Treg-specific IL-10 paucity leads to mucosal but not systemic autoimmunity, suggesting mucosal restriction of IL-10-mediated Treg tolerance induction. This view is supported by our previous observation that human Treg expressing gut-homing β7 integrin preferentially induce IL-10 production in converted secondary T helper suppressor cells (Stassen et al., 2004).
Due to their far-ranging tolerizing capability Treg have become key targets in the development of tolerance-inducing therapies (Wing and Sakaguchi, 2010). Like other T cells, Treg require activation for their function. Attempts to exploit Treg for therapeutic purposes therefore depend on Treg activation, either by antigen or polyclonal stimulation (Jordan et al., 2001). Current efforts to increase the frequency and potency of Treg in vivo include the use of cytokines (Tawara et al., 2010), antigen targeting to immature DC (Mahnke et al., 2003), and monoclonal antibodies (mAb) against surface molecules (Belghith et al., 2003). As a whole population Treg are biased toward recognition of self-antigens (Hsieh et al., 2004), however, because antigenic specificities of Treg in diseases have not been elucidated, potential clinical applications have mainly focused on polyclonal Treg activation methods (Horwitz et al., 2004).
Coreceptor Engagement and Peripheral Tolerance
T cell surface molecules that participate in T cell receptor-mediated stimulation have a significant influence on T cell function. mAb against coreceptors have been successfully shown to allow intentional tolerance induction in rodent and non-human primate models (Krieger et al., 1996). One particularly well-established regimen of tolerance induction is the administration of anti-CD4 mAb (Waldmann and Cobbold, 1998). Although the mechanisms underlying tolerization by anti-CD4 mAb are not yet fully understood, the activation of Treg has been recognized as the entering wedge to successful tolerance induction (Becker et al., 2009; Kendal et al., 2011; Martin et al., 2012).
CD4, a 55-kDa glycoprotein with four extracellular domains (Littman, 1987), recruits the protein kinase p56lck (Rudd et al., 1988; Veillette et al., 1988) to the TCR complex (Holdorf et al., 2002; Kim et al., 2003; Nika et al., 2010) and strengthens the contact between T cells and APCs through its interaction with non-polymorphic regions of MHC class II molecules (Greenstein et al., 1984; Doyle and Strominger, 1987; Konig et al., 1992, 1995). CD4 molecules on T cell surface have been shown to preferentially form disulfide-linked dimers and tetramers (110 and 220 kDa; Li et al., 1998; Moldovan et al., 2002) and mutations disabling dimerization completely abrogate its coreceptor function (Vignali and Vignali, 1999). CD4 expression on mature T cells is uniform with the exception of polarized T helper 2 cells (Itoh et al., 2005) and Treg (Bryl et al., 2001) which both show decreased CD4 expression supposedly entailing altered proximal TCR signaling (Hannier et al., 2002; Itoh et al., 2005; Tsang et al., 2006).
Through its interaction with tyrosine kinase p56lck, CD4 engagement alone can induce TCR-independent signaling events in T cells (Zhou and Konig, 2003). Selective engagement of the CD4 coreceptor by certain mAb raises intracellular calcium and IL-2 production (Carrel et al., 1991), whereas other anti-CD4 mAb prime T helper cells to activation-dependent cell death triggered by subsequent TCR/CD3-mediated signals (Newell et al., 1990; Tamma et al., 1997). Comparing mAb against different CD4 epitopes, Baldari and colleagues suggested that the gene-activating and proapoptotic potential of different anti-CD4 mAb may be associated with different epitopes (Baldari et al., 1995; Di Somma et al., 1995; Milia et al., 1997). However, a similar range of divergent responses can be induced through a single CD4 epitope as demonstrated for the CD4-binding (Lasky et al., 1987) human immunodeficiency virus-1 (HIV-1) envelope protein gp120 (Liegler and Stites, 1994; Westendorp et al., 1995; Masci et al., 1999). It is therefore tempting to speculate that the functional outcome of CD4-stimulation might mainly depend on the functional state of the T cell addressed rather than on a specific CD4 epitope. However, the functional state is believed to affect the formation of CD4 oligomers, which, in turn, regulate the activation of the CD4 cytoplasmic tail-associated tyrosine kinase p56lck, by trans-phosphorylation (Veillette et al., 1989).
Even before the role of the CD4 molecule in T cell activation had been fully recognized, three groups reported that short courses of anti-CD4 mAb application induce long-term tolerance to foreign proteins (Benjamin and Waldmann, 1986; Benjamin et al., 1986; Goronzy et al., 1986; Gutstein et al., 1986). Subsequent studies revealed that anti-CD4-mediated tolerance induction was not based on T cell depletion but rather an activation of regulatory mechanisms (Benjamin et al., 1988; Carteron et al., 1988, 1989; Qin et al., 1990). Further, tolerance could not only be induced to foreign proteins but also to various transplanted allografts (Shizuru et al., 1987; Qin et al., 1989; Davies et al., 1996), demonstrating that the tolerizing potential of anti-CD4 mAb is not restricted to a particular type of antigen. Immunoregulatory mechanisms initially suggested to operate in anti-CD4 induced tolerance include a predisposure of developing T cells to selective deletion, or anergy in the thymus (Arima et al., 1997); immune deviation (Scully et al., 1997); receptor blockade (Fehervari et al., 2002; Harding et al., 2002); modulation of CD4 expression (Portoles et al., 1999); and transmission of negative signals (Chirmule et al., 1999). However, none of these – not mutually exclusive – processes could reasonably explain the “infectious tolerance” phenomenon (Qin et al., 1993). Rather than being submissive, anti-CD4 induced tolerance relied on dominant immune suppression by T cells activated in presence of the antibody. In regard to the dominant suppressive T cell type in charge several functionally and phenotypically different anti-CD4 mAb-induced tolerogenic CD4+ T cell populations have been proposed (Bushell et al., 2003; Chen et al., 2003; Cobbold et al., 2004; Karim et al., 2005). However, whether these had been directly or indirectly induced by anti-CD4 treatment remained undefined at first. The impressive capacity of Treg and their ability to confer regulatory properties upon suppressed T cells (Jonuleit et al., 2002; Stassen et al., 2004; Andersson et al., 2008) in particular, strongly suggested a role of Treg in anti-CD4-mediated “infectious tolerance” induction. In support of this assumption administration of non-depleting anti-CD4 mAb into mice had been shown to result in pre-activation of Treg in vivo (Karim et al., 2005; Yang et al., 2007). Eventually, using B6. Foxp3(hCD2) mice to ablate Treg with an anti-hCD2 mAb Kendal et al. (2011) formally demonstrated that Treg are crucial for infectious tolerance induced by non-ablative anti-T cell mAb.
Motivated by the description of activated Treg in murine anti-CD4 tolerance models we previously analyzed the effect of CD4 binding agents on human Treg. Comparing numerous anti-CD4 mAb we found that certain anti-CD4 mAb have the potential to induce the suppressive function of isolated human Treg in a supposedly T cell receptor-independent manner (Becker et al., 2007). In addition, we and others observed that the CD4-binding HIV-1 surface protein gp120 activates the suppressive function of Treg (Nilsson et al., 2006; Kinter et al., 2007) in vitro and in two humanized mouse models in vivo (Becker et al., 2009; Ji and Cloyd, 2009) signifying that stimulation via the CD4 receptor represents an efficient Treg activating pathway with potential to induce immunological tolerance in humans.
Difference between anti-CD4 mAb to trigger Treg suppressive activity could not be related to a particular CD4 epitopes. However, comparing the Treg activating potential of different anti-CD4 mAb and CD4 binding virus envelopes we observed that one crucial event that separates Treg activating and non-activating CD4 ligands consists in up-regulation of the second messenger cAMP (Becker et al., 2009 and unpublished results). Moreover, the binding affinity of CD4 ligands seems to play a role as suggested by the fact that weak CD4 binding viral envelopes from HIV-2 (gp105) and SIV (gp130) did not activate human Treg in vitro and in vivo.
However, apart from these general observations the signaling events initiated by separate ligation of CD4 on Treg so far remain unexplored. In particular, it is unclear whether CD4 stimulation of Treg is truly independent of TCR signals, whether and how both pathways resemble or differ from another, and, most important, whether CD4-mediated signals are differently or similarly handled in Treg and conventional CD4+ T effector cells. The latter question is of particular interest since Treg are believed to maintain an activated phenotype through constant stimulation by self antigens, yet, require additional stimulation to become suppressive. Future insights into how TCR and CD4 signaling pathways drive the suppressive activity of Treg will undoubtedly help to understand Treg biology and discover alternative intervention points for functional manipulation of Treg suppressive activity.
As summarized in Figure 1 at least three different immune mechanisms can be distinguished that help to explain the tolerizing effect of CD4-specific agents: First, a general Treg-independent mechanism that consist in interference with proper CD4 coreceptor function resulting in induction of T cell anergy or T cell depletion (Figure 1A). This effect seems to depend either on CD4/MHC class II binding blockade or additional TCR-independent signaling. Second, by modulating antigenic stimulation, individual CD4 mAb induce differentiation of naive T cells into adaptive Tregs (Oliveira et al., 2008), which are suggested to control pathogenic effectors through TGF-β (Oliveira et al., 2011) or IL-10 release (Figure 1B). Finally, and crucially important for tolerance induction, CD4-specific mAb activate the suppressive function of Treg (Becker et al., 2007; Kendal et al., 2011), which, upon activation, exert control on pathogenic T cells by direct and linked suppression (Figure 1C). These different effects of CD4 stimulation are intrinsic functions of individual anti-CD4 mAb.
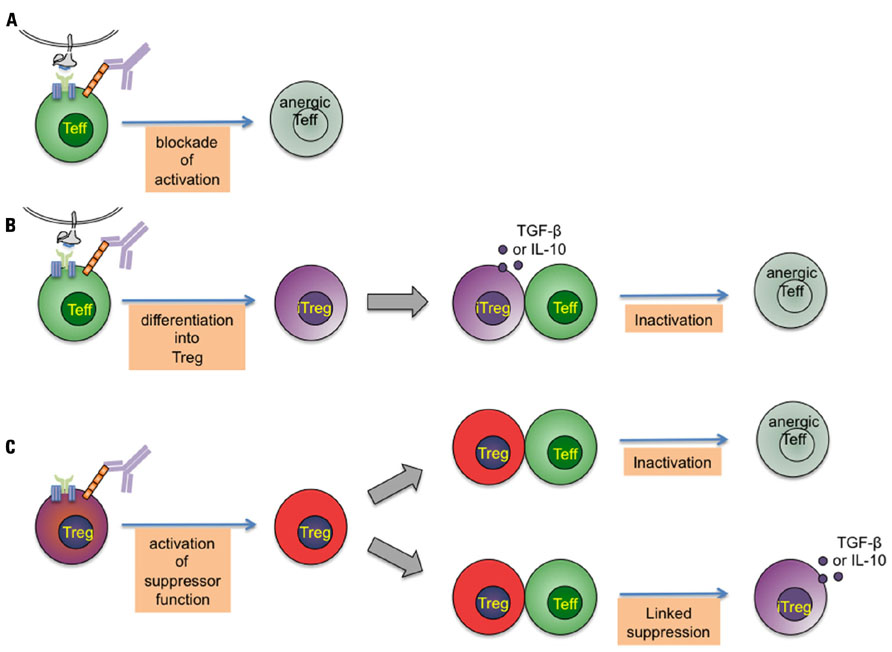
FIGURE 1. Potential modes of tolerance induction by CD4-specific monoclonal antibodies.A short-term treatment with non-depleting CD4-specific mAb induces dominant tolerance to foreign proteins and transplanted allografts. This figure represents the different immune mechanisms that have been proposed to explain the tolerizing effect of CD4-specific mAb. Three intervening points can be distinguished: (A) CD4 binding by non-depleting CD4-specific mAb modulates antigenic stimulation through the T-cell receptor complex resulting in induction of T cell anergy. (B) By modulating antigenic stimulation, CD4-specific mAb induce differentiation of naive T cells into adaptive regulatory T cells, which control pathogenic effectors through transforming growth factor-β (TGF-β) and IL-10 release. (C) Crucially important for tolerance induction, CD4-specific mAb activate the (cAMP-dependent) suppressive function of Treg, which, upon activation, exert control on pathogenic T cells by direct and linked suppression.
Clinical Approaches to Treg-Mediated Tolerance Induction
Current immunosuppressive therapies are efficient in preventing acute transplant rejection and dampening inflammation in autoimmune diseases such as rheumatoid arthritis or lupus. Nevertheless, immune suppression remains inadequate, as it comprises significant side effects such as organ toxicity and hypersuppression disabling protective immune responses against pathogens and enhancing the risk of chronic infections. Hence, there is a clinical need for novel immunotherapeutic drugs with the ability to rebalance the immunologic tolerance network without persistently affecting immune function. In contrast to pharmacological immune suppression, re-induction of tolerance through the exploitation of evolutionarily established tolerance mechanisms is expected to offer a parentally operative cure. Among mechanisms operative in self-tolerance, the immune-suppressive activity of Treg appears to be exceptionally well suited for therapeutic exploitation for several reasons: First, activated Treg dampen the function of a wide range of immune cells including T cells (Pandiyan et al., 2007), B cells (Lim et al., 2005), DC (Misra et al., 2004; Larmonier et al., 2007), and monocytes (Taams et al., 2005) and affect a broad range of immune contexts including cardiovascular disease (Ait-Oufella et al., 2006) and obesity-induced insulin resistance (Feuerer et al., 2009). Second, the activation of Treg is antigen-specific defined by the selected T cell receptor repertoire in the thymus. However, once activated the suppressive mechanisms of Treg operate in an antigen-non-specific manner, sidestepping the need to identify disease-specific antigens to affect a particular Treg population. Prime examples of the Treg immune dampening potential are experiments demonstrating that Treg can be expanded and re-infused to limit immune responses (Hoffmann et al., 2002) preventing GvHD induction without causing toxicity. While persistent polyclonal Treg activation would lead to general immune hyporesponsiveness, a short-term Treg activation – as established for tolerance induction with non-depleting anti-CD4 mAb in mice – is expected to induce (or re-induce) antigen-specific regulatory networks that maintain antigen-specific tolerance when Treg activity has returned to normal levels.
Based on the evidence for Treg activation by CD4 ligands as outlined above, anti-CD4 mAb seem to represent ideal compounds for Treg-mediated tolerance induction. However, although animal studies have provided a compelling basis for clinical application of anti-CD4-mediated tolerance induction, this approach has been remarkably unsuccessful when transferred to the clinic. Although short interventions with particular mAb have been shown to offer quick symptomatic relief, improvements supposedly caused by inactivation and depletion of CD4+ T cells (Kon et al., 2001; Choy et al., 2002) remained transient. Failure to establish an anti-CD4-based tolerogenic therapy in humans is most likely due to difficulties in translating the timing and dosage used in animal models for human application. Importantly, in contrast to animal models, mAb are administrated at late disease stages in clinical studies. Whereas the immature immune system seems to dependably allow tolerance induction with anti-CD4 mAb, it seems more difficult to tolerize the experienced immune system in patients, in part due to the presence of effector and memory T cells resistant to the suppressive action of Treg (Yang et al., 2007). In fact, Treg-based therapies have been found to be generally less effective in models of autoimmune diseases. Wehrens et al. (2011) for example observed that functionally active Treg failed to control hyperactivated T effector cells in rheumatoid arthritis patients with ongoing inflammation but prevented autoaggressive immune responses in non-inflammatory arthritis. Impaired Treg suppression under inflammatory conditions has been mainly ascribed to the influence of TNF-α, IL-1, and IL-6, which turn effector T cells resistant to Treg-mediated suppression (Walker, 2009; Goodman et al., 2011). Certainly, resistance to Treg-mediated suppression can be overcome by blockade of IL-6 (Chen et al., 2009) and supposedly, the beneficial effects of anti-TNF-α treatment include a similar effect too (Ehrenstein et al., 2004; Valencia et al., 2006). Thus, provided Treg can be sufficiently activated in the host, their suppressive efficiency might depend on the disease stage, which strongly argues for a combination of Treg enhancing strategies with biologicals that reverse Treg resistance in autoaggressive T effector cells. As exemplified with anti-CD3 mAb already in the clinic evacuation of T effectors cells and concomitant enhancement of Treg activity can form a very effective treatment (Chatenoud and Bluestone, 2007).
With regard to anti-CD4-mediated tolerance induction in humans, it is important to emphasize again that anti-CD4 mAb vary in their capacity to activate Treg (Becker et al., 2007) and antibodies used in clinical trials so far have not been analyzed with regard to their Treg activating potential. However, clinical trials with Treg enhancing agents such as the anti-CD4 mAb Tregalizumab in rheumatoid arthritis have been initiated to investigate the efficacy of Treg-based anti-CD4-mediated tolerance induction in patients with autoimmune diseases.
Concluding Remarks
In summary, polyclonal activation of Treg through their surface molecules by biologicals that enhance their intracellular cAMP level are effective to induce the suppressive function of Treg for re-induction of tolerance in small animal models and in humanized mice. It is therefore expected that polyclonal Treg activation forms a rational for tolerance induction in humans. However, both the exact conditions, efficiency in different stages of disease and cooperation with additional treatment regiments to diminish T effector cells need to be thoroughly explored. Moreover differential signals in Treg versus T effector cells are far from being clear. In addition to deepening our understanding of Treg biology investigation of the latter holds the key to define alternative entry points for therapeutic manipulation of Treg function.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) BE 3685/1-1 (to Christian Becker), Collaborative research center projects grants A1 (to Tobias Bopp), A2 (to Helmut Jonuleit), the GRK 1043, and the International Graduate School of Immunotherapy (to Tobias Bopp).
References
Abbracchio, M. P., Burnstock, G., Boeynaems, J. M., Barnard, E. A., Boyer, J. L., Kennedy, C., Knight, G. E., Fumagalli, M., Gachet, C., Jacobson, K. A., and Weisman, G. A. (2006). International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 58, 281–341.
Ait-Oufella, H., Salomon, B. L., Potteaux, S., Robertson, A. K., Gourdy, P., Zoll, J., Merval, R., Esposito, B., Cohen, J. L., Fisson, S., Flavell, R. A., Hansson, G. K., Klatzmann, D., Tedgui, A., and Mallat, Z. (2006). Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 12, 178–180.
Andersson, J., Tran, D. Q., Pesu, M., Davidson, T. S., Ramsey, H., O’Shea, J. J., and Shevach, E. M. (2008). CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J. Exp. Med. 205, 1975–1981.
Annes, J. P., Munger, J. S., and Rifkin, D. B. (2003). Making sense of latent TGFbeta activation. J. Cell Sci. 116, 217–224.
Arima, T., Lehmann, M., and Flye, M. W. (1997). Induction of donor specific transplantation tolerance to cardiac allografts following treatment with nondepleting (RIB 5/2) or depleting (OX-38) anti-CD4 mAb plus intrathymic or intravenous donor alloantigen. Transplantation 63, 284–292.
Bachmann, M. F., Kohler, G., Ecabert, B., Mak, T. W., and Kopf, M. (1999). Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J. Immunol. 163, 1128–1131.
Baecher-Allan, C., Brown, J. A., Freeman, G. J., and Hafler, D. A. (2001). CD4+CD25high regulatory cells in human peripheral blood. J. Immunol. 167, 1245–1253.
Baldari, C. T., Milia, E., Di Somma, M. M., Baldoni, F., Valitutti, S., and Telford, J. L. (1995). Distinct signaling properties identify functionally different CD4 epitopes. Eur. J. Immunol. 25, 1843–1850.
Battaglia, M., and Roncarolo, M. G. (2009). The Tregs’ world according to GARP. Eur. J. Immunol. 39, 3296–3300.
Becker, C., Kubach, J., Wijdenes, J., Knop, J., and Jonuleit, H. (2007). CD4-mediated functional activation of human CD4+CD25+ regulatory T cells. Eur. J. Immunol. 37, 1217–1223.
Becker, C., Taube, C., Bopp, T., Becker, C., Michel, K., Kubach, J., Reuter, S., Dehzad, N., Neurath, M. F., Reifenberg, K., Schneider, F. J., Schmitt, E., and Jonuleit, H. (2009). Protection from graft-versus-host disease by HIV-1 envelope protein gp120-mediated activation of human CD4+CD25+ regulatory T cells. Blood 114, 1263–1269.
Belghith, M., Bluestone, J. A., Barriot, S., Megret, J., Bach, J. F., and Chatenoud, L. (2003). TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat. Med. 9, 1202–1208.
Belkaid, Y., Piccirillo, C. A., Mendez, S., Shevach, E. M., and Sacks, D. L. (2002). CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420, 502–507.
Benjamin, R. J., Cobbold, S. P., Clark, M. R., and Waldmann, H. (1986). Tolerance to rat monoclonal antibodies. Implications for serotherapy. J. Exp. Med. 163, 1539–1552.
Benjamin, R. J., Qin, S. X., Wise, M. P., Cobbold, S. P., and Waldmann, H. (1988). Mechanisms of monoclonal antibody-facilitated tolerance induction: a possible role for the CD4 (L3T4) and CD11a (LFA-1) molecules in self-non-self discrimination. Eur. J. Immunol. 18, 1079–1088.
Benjamin, R. J., and Waldmann, H. (1986). Induction of tolerance by monoclonal antibody therapy. Nature 320, 449–451.
Birebent, B., Lorho, R., Lechartier, H., de Guibert, S., Alizadeh, M., Vu, N., Beauplet, A., Robillard, N., and Semana, G. (2004). Suppressive properties of human CD4+CD25+ regulatory T cells are dependent on CTLA-4 expression. Eur. J. Immunol. 34, 3485–3496.
Bodor, J., Fehervari, Z., Diamond, B., and Sakaguchi, S. (2007). Regulatory T cell-mediated suppression: potential role of ICER. J. Leukoc. Biol. 81, 161–167.
Bodor, J., Spetz, A. L., Strominger, J. L., and Habener, J. F. (1996). cAMP inducibility of transcriptional repressor ICER in developing and mature human T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 93, 3536–3541.
Bopp, T., Becker, C., Klein, M., Klein-Hessling, S., Palmetshofer, A., Serfling, E., Heib, V., Becker, M., Kubach, J., Schmitt, S., Stoll, S., Schild, H., Staege, M. S., Stassen, M., Jonuleit, H., and Schmitt, E. (2007). Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J. Exp. Med. 204, 1303–1310.
Bopp, T., Dehzad, N., Reuter, S., Klein, M., Ullrich, N., Stassen, M., Schild, H., Buhl, R., Schmitt, E., and Taube, C. (2009). Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J. Immunol. 182, 4017–4024.
Bryl, E., Gazda, M., Foerster, J., and Witkowski, J. M. (2001). Age-related increase of frequency of a new, phenotypically distinct subpopulation of human peripheral blood T cells expressing lowered levels of CD4. Blood 98, 1100.
Bushell, A., Karim, M., Kingsley, C. I., and Wood, K. J. (2003). Pretransplant blood transfusion without additional immunotherapy generates CD25+CD4+ regulatory T cells: a potential explanation for the blood-transfusion effect. Transplantation 76, 449–455.
Carrel, S., Salvi, S., Gallay, P., Rapin, C., and Sekaly, R. P. (1991). Positive signal transduction via surface CD4 molecules does not need coexpression of the CD3/TcR complex. Res. Immunol. 142, 97–108.
Carteron, N. L., Schimenti, C. L., and Wofsy, D. (1989). Treatment of murine lupus with F(ab’)2 fragments of monoclonal antibody to L3T4. Suppression of autoimmunity does not depend on T helper cell depletion. J. Immunol. 142, 1470–1475.
Carteron, N. L., Wofsy, D., and Seaman, W. E. (1988). Induction of immune tolerance during administration of monoclonal antibody to L3T4 does not depend on depletion of L3T4+ cells. J. Immunol. 140, 713–716.
Cederbom, L., Hall, H., and Ivars, F. (2000). CD4+CD25+ regulatory T cells downregulate co-stimulatory molecules on antigen-presenting cells. Eur. J. Immunol. 30, 1538–1543.
Chalmin, F., Mignot, G., Bruchard, M., Chevriaux, A., Végran, F., Hichami, A., Ladoire, S., Derangère, V., Vincent, J., Masson, D., Robson, S. C., Eberl, G., Pallandre, J. R., Borg, C., Ryffel, B., Apetoh, L., Rébé, C., and Ghiringhelli, F. (2012). Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 36, 362–373.
Chatenoud, L., and Bluestone, J. A. (2007). CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat. Rev. Immunol. 7, 622–632.
Chaudhry, A., Samstein, R. M., Treuting, P., Liang, Y., Pils, M. C., Heinrich, J. M., Jack, R. S., Wunderlich, F. T., Bruning, J. C., Muller, W., and Rudensky, A. Y. (2011). Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566–578.
Chen, W., Jin, W., Hardegen, N., Lei, K. J., Li, L., Marinos, N., McGrady, G., and Wahl, S. M. (2003). Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886.
Chen, X., Das, R., Komorowski, R., Beres, A., Hessner, M. J., Mihara, M., and Drobyski, W. R. (2009). Blockade of interleukin-6 signaling augments regulatory T-cell reconstitution and attenuates the severity of graft-versus-host disease. Blood 114, 891–900.
Chirmule, N., Avots, A., LakshmiTamma, S. M., Pahwa, S., and Serfling, E. (1999). CD4-mediated signals induce T cell dysfunction in vivo. J. Immunol. 163, 644–649.
Choy, E. H., Panayi, G. S., Emery, P., Madden, S., Breedveld, F. C., Kraan, M. C., Kalden, J. R., Rascu, A., Brown, J. C., Rapson, N., and Johnston, J. M. (2002). Repeat-cycle study of high-dose intravenous 4162W94 anti-CD4 humanized monoclonal antibody in rheumatoid arthritis. A randomized placebo-controlled trial. Rheumatology (Oxford) 41, 1142–1148.
Cobbold, S. P., Castejon, R., Adams, E., Zelenika, D., Graca, L., Humm, S., and Waldmann, H. (2004). Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J. Immunol. 172, 6003–6010.
Collison, L. W., Workman, C. J., Kuo, T. T., Boyd, K., Wang, Y., Vignali, K. M., Cross, R., Sehy, D., Blumberg, R. S., and Vignali, D. A. (2007). The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450, 566–569.
D’Alise, A. M., Ergun, A., Hill, J. A., Mathis, D., and Benoist, C. (2011). A cluster of coregulated genes determines TGF-beta-induced regulatory T-cell (Treg) dysfunction in NOD mice. Proc. Natl. Acad. Sci. U.S.A. 108, 8737–8742.
Daley, S. R., Ma, J., Adams, E., Cobbold, S. P., and Waldmann, H. (2007). A key role for TGF-beta signaling to T cells in the long-term acceptance of allografts. J. Immunol. 179, 3648–3654.
Davies, J. D., Leong, L. Y., Mellor, A., Cobbold, S. P., and Waldmann, H. (1996). T cell suppression in transplantation tolerance through linked recognition. J. Immunol. 156, 3602–3607.
Davis, S. J., Ikemizu, S., Evans, E. J., Fugger, L., Bakker, T. R., and van der Merwe, P. A. (2003). The nature of molecular recognition by T cells. Nat. Immunol. 4, 217–224.
Deaglio, S., Dwyer, K. M., Gao, W., Friedman, D., Usheva, A., Erat, A., Chen, J. F., Enjyoji, K., Linden, J., Oukka, M., Kuchroo, V. K., Strom, T. B., and Robson, S. C. (2007). Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265.
Di Somma, M. M., Nuti, S., Telford, J. L., and Baldari, C. T. (1995). p56lck plays a key role in transducing apoptotic signals in T cells. FEBS Lett. 363, 101–104.
Doyle, C., and Strominger, J. L. (1987). Interaction between CD4 and class II MHC molecules mediates cell adhesion. Nature 330, 256–259.
Ehrenstein, M. R., Evans, J. G., Singh, A., Moore, S., Warnes, G., Isenberg, D. A., and Mauri, C. (2004). Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J. Exp. Med. 200, 277–285.
Ernst, P. B., Garrison, J. C., and Thompson, L. F. (2010). Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J. Immunol. 185, 1993–1998.
Fassbender, M., Gerlitzki, B., Ullrich, N., Lupp, C., Klein, M., Radsak, M. P., Schmitt, E., Bopp, T., and Schild, H. (2010). Cyclic adenosine monophosphate and IL-10 coordinately contribute to nTreg cell-mediated suppression of dendritic cell activation. Cell. Immunol. 265, 91–96.
Fehervari, Z., Cooke, A., Brett, S., and Turner, J. (2002). Perturbation of naive TCR transgenic T cell functional responses and upstream activation events by anti-CD4 monoclonal antibodies. Eur. J. Immunol. 32, 333–340.
Feuerer, M., Herrero, L., Cipolletta, D., Naaz, A., Wong, J., Nayer, A., Lee, J., Goldfine, A. B., Benoist, C., Shoelson, S., and Mathis, D. (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 15, 930–939.
Fontenot, J. D., Dooley, J. L., Farr, A. G., and Rudensky, A. Y. (2005). Developmental regulation of Foxp3 expression during ontogeny. J. Exp. Med. 202, 901–906.
Foulkes, N. S., Laoide, B. M., Schlotter, F., and Sassone-Corsi, P. (1991). Transcriptional antagonist cAMP-responsive element modulator (CREM) down-regulates c-fos cAMP-induced expression. Proc. Natl. Acad. Sci. U.S.A. 88, 5448–5452.
Fredholm, B. B. (2007). Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 14, 1315–1323.
Gavin, M. A., Torgerson, T. R., Houston, E., DeRoos, P., Ho, W. Y., Stray-Pedersen, A., Ocheltree, E. L., Greenberg, P. D., Ochs, H. D., and Rudensky, A. Y. (2006). Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc. Natl. Acad. Sci. U.S.A. 103, 6659–6664.
Goodman, W. A., Young, A. B., McCormick, T. S., Cooper, K. D., and Levine, A. D. (2011). Stat3 phosphorylation mediates resistance of primary human T cells to regulatory T cell suppression. J. Immunol. 186, 3336–3345.
Goronzy, J., Weyand, C. M., and Fathman, C. G. (1986). Long-term humoral unresponsiveness in vivo, induced by treatment with monoclonal antibody against L3T4. J. Exp. Med. 164, 911–925.
Greenstein, J. L., Kappler, J., Marrack, P., and Burakoff, S. J. (1984). The role of L3T4 in recognition of Ia by a cytotoxic, H-2Dd-specific T cell hybridoma. J. Exp. Med. 159, 1213–1224.
Grossman, W. J., Verbsky, J. W., Barchet, W., Colonna, M., Atkinson, J. P., and Ley, T. J. (2004). Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21, 589–601.
Gutstein, N. L., Seaman, W. E., Scott, J. H., and Wofsy, D. (1986). Induction of immune tolerance by administration of monoclonal antibody to L3T4. J. Immunol. 137, 1127–1132.
Hagness, M., Henjum, K., Landskron, J., Brudvik, K. W., Bjørnbeth, B. A., Foss, A., Taskén, K., and Aandahl, E. M. (2012). Kinetics and activation requirements of contact-dependent immune suppression by human regulatory T cells. J. Immunol. 188, 5459–5466.
Hannier, S., Bitegye, C., and Demotz, S. (2002). Early events of TCR signaling are distinct in human Th1 and Th2 cells. J. Immunol. 169, 1904.
Harding, S., Lipp, P., and Alexander, D. R. (2002). A therapeutic CD4 monoclonal antibody inhibits TCR-zeta chain phosphorylation, zeta-associated protein of 70-kDa Tyr319 phosphorylation, and TCR internalization in primary human T cells. J. Immunol. 169, 230–238.
Hasenkrug, K. J. (2003). CD4+ regulatory T cells in chronic viral infection. Novartis Found. Symp. 252, 194–199; discussion 199–210.
Hoffmann, P., Ermann, J., Edinger, M., Fathman, C. G., and Strober, S. (2002). Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J. Exp. Med. 196, 389–399.
Holdorf, A. D., Lee, K. H., Burack, W. R., Allen, P. M., and Shaw, A. S. (2002). Regulation of Lck activity by CD4 and CD28 in the immunological synapse. Nat. Immunol. 3, 259–264.
Hori, S., Nomura, T., and Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061.
Horwitz, D. A., Zheng, S. G., Gray, J. D., Wang, J. H., Ohtsuka, K., and Yamagiwa, S. (2004). Regulatory T cells generated ex vivo as an approach for the therapy of autoimmune disease. Semin. Immunol. 16, 135–143.
Hsieh, C. S., Liang, Y., Tyznik, A. J., Self, S. G., Liggitt, D., and Rudensky, A. Y. (2004). Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity 21, 267–277.
Huang, B., Zhao, J., Lei, Z., Shen, S., Li, D., Shen, G. X., Zhang, G. M., and Feng, Z. H. (2009). miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep. 10, 180–185.
Huber, S., Gagliani, N., Esplugues, E., O’Connor, W. J., Huber, F. J., Chaudhry, A., Kamanaka, M., Kobayashi, Y., Booth, C. J., Rudensky, A. Y., Roncarolo, M. G., Battaglia, M., and Flavell, R. A. (2011). Th17 cells express interleukin-10 receptor and are controlled by Foxp3 and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565.
Hunsucker, S. A., Mitchell, B. S., and Spychala, J. (2005). The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol. Ther. 107, 1–30.
Itoh, Y., Wang, Z., Ishida, H., Eichelberg, K., Fujimoto, N., Makino, J., Ogasawara, K., and Germain, R. N. (2005). Decreased CD4 expression by polarized T helper 2 cells contributes to suboptimal TCR-induced phosphorylation and reduced Ca2 + signaling. Eur. J. Immunol. 35, 3187–3195.
Ji, J., and Cloyd, M. W. (2009). HIV-1 binding to CD4 on CD4+CD25+ regulatory T cells enhances their suppressive function and induces them to home to, and accumulate in, peripheral and mucosal lymphoid tissues: an additional mechanism of immunosuppression. Int. Immunol. 21, 283–294.
Jonuleit, H., Schmitt, E., Stassen, M., Tuettenberg, A., Knop, J., and Enk, A. H. (2001). Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J. Exp. Med. 193, 1285–1294.
Jonuleit, H., Schmitt, E., Kakirman, H., Stassen, M., Knop, J., and Enk, A. H. (2002). Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J. Exp. Med. 196, 255–260.
Jordan, M. S., Boesteanu, A., Reed, A. J., Petrone, A. L., Holenbeck, A. E., Lerman, M. A., Naji, A., and Caton, A. J. (2001). Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2, 301–306.
Karim, M., Feng, G., Wood, K. J., and Bushell, A. R. (2005). CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood 105, 4871–4877.
Kearley, J., Barker, J. E., Robinson, D. S., and Lloyd, C. M. (2005). Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J. Exp. Med. 202, 1539–1547.
Kendal, A. R., Chen, Y., Regateiro, F. S., Ma, J., Adams, E., Cobbold, S. P., Hori, S., and Waldmann, H. (2011). Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J. Exp. Med. 208, 2043–2053.
Kim, P. W., Sun, Z. Y., Blacklow, S. C., Wagner, G., and Eck, M. J. (2003). A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science 301, 1725–1728.
Kinter, A., McNally, J., Riggin, L., Jackson, R., Roby, G., and Fauci, A. S. (2007). Suppression of HIV-specific T cell activity by lymph node CD25+ regulatory T cells from HIV-infected individuals. Proc. Natl. Acad. Sci. U.S.A. 104, 3390–3395.
Klein, M., Vaeth, M., Scheel, T., Grabbe, S., Baumgrass, R., Berberich-Siebelt, F., Bopp, T., Schmitt, E., and Becker, C. (2012). Repression of cyclic adenosine monophosphate upregulation disarms and expands human regulatory T cells. J. Immunol. 188, 1091–1097.
Kojima, H., Kanno, Y., Hase, H., and Kobata, T. (2005). CD4+CD25+ regulatory T cells attenuate the phosphatidylinositol 3-kinase/Akt pathway in antigen-primed immature CD8+ CTLs during functional maturation. J. Immunol. 174, 5959–5967.
Kon, O. M., Sihra, B. S., Loh, L. C., Barkans, J., Compton, C. H., Barnes, N. C., Larche, M., and Kay, A. B. (2001). The effects of an anti-CD4 monoclonal antibody, keliximab, on peripheral blood CD4+ T-cells in asthma. Eur. Respir. J. 18, 45–52.
Konig, R., Huang, L. Y., and Germain, R. N. (1992). MHC class II interaction with CD4 mediated by a region analogous to the MHC class I binding site for CD8. Nature 356, 796–798.
Konig, R., Shen, X., and Germain, R. N. (1995). Involvement of both major histocompatibility complex class II alpha and beta chains in CD4 function indicates a role for ordered oligomerization in T cell activation. J. Exp. Med. 182, 779–787.
Krieger, N. R., Most, D., Bromberg, J. S., Holm, B., Huie, P., Sibley, R. K., Dafoe, D. C., and Alfrey, E. J. (1996). Coexistence of Th1- and Th2-type cytokine profiles in anti-CD2 monoclonal antibody-induced tolerance. Transplantation 62, 1285–1292.
Lahl, K., Mayer, C. T., Bopp, T., Huehn, J., Loddenkemper, C., Eberl, G., Wirnsberger, G., Dornmair, K., Geffers, R., Schmitt, E., Buer, J., and Sparwasser, T. (2009). Nonfunctional regulatory T cells and defective control of Th2 cytokine production in natural scurfy mutant mice. J. Immunol. 183, 5662–5672.
Larmonier, N., Marron, M., Zeng, Y., Cantrell, J., Romanoski, A., Sepassi, M., Thompson, S., Chen, X., Andreansky, S., and Katsanis, E. (2007). Tumor-derived CD4(+)CD25(+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol. Immunother. 56, 48–59.
Lasky, L. A., Nakamura, G., Smith, D. H., Fennie, C., Shimasaki, C., Patzer, E., Berman, P., Gregory, T., and Capon, D. J. (1987). Delineation of a region of the human immunodeficiency virus type 1 gp120 glycoprotein critical for interaction with the CD4 receptor. Cell 50, 975–985.
Levings, M. K., Sangregorio, R., and Roncarolo, M. G. (2001). Human CD25(+) CD4(+) T regulatory cells suppress naïve and memory T cell proliferation and can be expanded in vitro without loss of function. J. Exp. Med. 193, 1295–1302.
Li, S., Satoh, T., Korngold, R., and Huang, Z. (1998). CD4 dimerization and oligomerization: implications for T-cell function and structure-based drug design. Immunol. Today 19, 455–462.
Liegler, T. J., and Stites, D. P. (1994). HIV-1 gp120 and anti-gp120 induce reversible unresponsiveness in peripheral CD4 T lymphocytes. J. Acquir. Immune Defic. Syndr. 7, 340–348.
Lim, H. W., Hillsamer, P., Banham, A. H., and Kim, C. H. (2005). Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J. Immunol. 175, 4180–4183.
Mahnke, K., Qian, Y., Knop, J., and Enk, A. H. (2003). Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood 101, 4862–4869.
Mandapathil, M., Lang, S., Gorelik, E., and Whiteside, T. L. (2009). Isolation of functional human regulatory T cells (Treg) from the peripheral blood based on the CD39 expression. J. Immunol. Methods 346, 55–63.
Mandapathil, M., Hilldorfer, B., Szczepanski, M. J., Czystowska, M., Szajnik, M., Ren, J., Lang, S., Jackson, E. K., Gorelik, E., and Whiteside, T. L. (2010). Generation and accumulation of immunosuppressive adenosine by human CD4+ CD25high FOXP3+ regulatory T cells. J. Biol. Chem. 285, 7176–7186.
Martin, H., Reuter, S., Dehzad, N., Heinz, A., Bellinghausen, I., Saloga, J., Haasler, I., Korn, S., Jonuleit, H., Buhl, R., Becker, C., and Taube, C. (2012). CD4-mediated regulatory T-cell activation inhibits the development of disease in a humanized mouse model of allergic airway disease. J. Allergy Clin. Immunol. 129, 521–528, 528.e1–528.e7.
Masci, A. M., Paz, F. L., Borriello, A., Cassano, S., Della Pietra, V., Stoiber, H., Matarese, G., Della Ragione, F., Zappacosta, S., and Racioppi, L. (1999). Effects of human immunodeficiency virus type 1 on CD4 lymphocyte subset activation. Eur. J. Immunol. 29, 1879–1889.
Milia, E., Di Somma, M. M., Majolini, M. B., Ulivieri, C., Somma, F., Piccolella, E., Telford, J. L., and Baldari, C. T. (1997). Gene activating and proapoptotic potential are independent properties of different CD4 epitopes. Mol. Immunol. 34, 287–296.
Misra, N., Bayry, J., Lacroix-Desmazes, S., Kazatchkine, M. D., and Kaveri, S. V. (2004). Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J. Immunol. 172, 4676–4680.
Moldovan, M. C., Yachou, A., Levesque, K., Wu, H., Hendrickson, W. A., Cohen, E. A., and Sekaly, R. P. (2002). CD4 dimers constitute the functional component required for T cell activation. J. Immunol. 169, 6261–6268.
Nakamura, K., Kitani, A., and Strober, W. (2001). Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J. Exp. Med. 194, 629–644.
Newell, M. K., Haughn, L. J., Maroun, C. R., and Julius, M. H. (1990). Death of mature T cells by separate ligation of CD4 and the T-cell receptor for antigen. Nature 347, 286–289.
Nika, K., Soldani, C., Salek, M., Paster, W., Gray, A., Etzensperger, R., Fugger, L., Polzella, P., Cerundolo, V., Dushek, O., Hofer, T., Viola, A., and Acuto, O. (2010). Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 32, 766–777.
Nilsson, J., Boasso, A., Velilla, P. A., Zhang, R., Vaccari, M., Franchini, G., Shearer, G. M., Andersson, J., and Chougnet, C. (2006). HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood 108, 3808–3817.
Oberle, N., Eberhardt, N., Falk, C. S., Krammer, P. H., and Suri-Payer, E. (2007). Rapid suppression of cytokine transcription in human CD4+CD25 T cells by CD4+Foxp3+ regulatory T cells: independence of IL-2 consumption, TGF-beta, and various inhibitors of TCR signaling. J. Immunol. 179, 3578–3587.
Oderup, C., Cederbom, L., Makowska, A., Cilio, C. M., and Ivars, F. (2006). Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology 118, 240–249.
Oliveira, V., Sawitzki, B., Chapman, S., Appelt, C., Gebuhr, I., Wieckiewicz, J., Long, E., and Wood, K. J. (2008). Anti-CD4-mediated selection of Treg in vitro – in vitro suppression does not predict in vivo capacity to prevent graft rejection. Eur. J. Immunol. 38,1677–1688.
Oliveira, V. G., Caridade, M., Paiva, R. S., Demengeot, J., and Graca, L. (2011). Sub-optimal CD4+ T-cell activation triggers autonomous TGF-β-dependent conversion to Foxp3+ regulatory T cells. Eur. J. Immunol. 41,1249–1255.
Oviedo-Orta, E., Hoy, T., and Evans, W. H. (2000). Intercellular communication in the immune system: differential expression of connexin40 and 43, and perturbation of gap junction channel functions in peripheral blood and tonsil human lymphocyte subpopulations. Immunology 99, 578–590.
Pandiyan, P., Zheng, L., Ishihara, S., Reed, J., and Lenardo, M. J. (2007). CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 8, 1353–1362.
Portoles, P., de Ojeda, G., Criado, G., Fernandez-Centeno, E., and Rojo, J. M. (1999). Antibody-induced CD3-CD4 coligation inhibits TCR/CD3 activation in the absence of costimulatory signals in normal mouse CD4(+) T lymphocytes. Cell. Immunol. 195, 96–109.
Qin, S., Cobbold, S. P., Pope, H., Elliott, J., Kioussis, D., Davies, J., and Waldmann, H. (1993). “Infectious” transplantation tolerance. Science 259, 974–977.
Qin, S. X., Cobbold, S., Benjamin, R., and Waldmann, H. (1989). Induction of classical transplantation tolerance in the adult. J. Exp. Med. 169, 779–794.
Qin, S. X., Wise, M., Cobbold, S. P., Leong, L., Kong, Y. C., Parnes, J. R., and Waldmann, H. (1990). Induction of tolerance in peripheral T cells with monoclonal antibodies. Eur. J. Immunol. 20, 2737–2745.
Read, S., Malmstrom, V., and Powrie, F. (2000). Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 192, 295–302.
Read, S., Greenwald, R., Izcue, A., Robinson, N., Mandelbrot, D., Francisco, L., Sharpe, A. H., and Powrie, F. (2006). Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 177, 4376–4383.
Rubtsov, Y. P., Rasmussen, J. P., Chi, E. Y., Fontenot, J., Castelli, L., Ye, X., Treuting, P., Siewe, L., Roers, A., Henderson, W. R. J., Muller, W., and Rudensky, A. Y. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558.
Rudd, C. E., Trevillyan, J. M., Dasgupta, J. D., Wong, L. L., and Schlossman, S. F. (1988). The CD4 receptor is complexed in detergent lysates to a protein-tyrosine kinase (pp58) from human T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 85, 5190–5194.
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M., and Toda, M. (1995). Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164.
Scully, R., Cobbold, S. P., Mellor, A. L., Wissing, M., Arnold, B., and Waldmann, H. (1997). A role for Th2 cytokines in the suppression of CD8+ T cell-mediated graft rejection. Eur. J. Immunol. 27, 1663–1670.
Shevach, E. M. (2009). Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645.
Shizuru, J. A., Gregory, A. K., Chao, C. T., and Fathman, C. G. (1987). Islet allograft survival after a single course of treatment of recipient with antibody to L3T4. Science 237, 278–280.
Stassen, M., Fondel, S., Bopp, T., Richter, C., Muller, C., Kubach, J., Becker, C., Knop, J., Enk, A. H., Schmitt, S., Schmitt, E., and Jonuleit, H. (2004). Human CD25+ regulatory T cells: two subsets defined by the integrins alpha 4 beta 7 or alpha 4 beta 1 confer distinct suppressive properties upon CD4+ T helper cells. Eur. J. Immunol. 34, 1303–1311.
Stockis, J., Colau, D., Coulie, P. G., and Lucas, S. (2009). Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur. J. Immunol. 39, 3315–3322.
Taams, L. S., van Amelsfort, J. M., Tiemessen, M. M., Jacobs, K. M., de Jong, E. C., Akbar, A. N., Bijlsma, J. W., and Lafeber, F. P. (2005). Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum. Immunol. 66, 222–230.
Takahashi, T., Kuniyasu, Y., Toda, M., Sakaguchi, N., Itoh, M., Iwata, M., Shimizu, J., and Sakaguchi, S. (1998). Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10, 1969–1980.
Takahashi, T., Tagami, T., Yamazaki, S., Uede, T., Shimizu, J., Sakaguchi, N., Mak, T. W., and Sakaguchi, S. (2000). Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192, 303–310.
Tamma, S. M., Chirmule, N., McCloskey, T. W., Oyaizu, N., Kalyanaraman, V. S., and Pahwa, S. (1997). Signals transduced through the CD4 molecule interfere with TCR/CD3-mediated ras activation leading to T cell anergy/apoptosis. Clin. Immunol. Immunopathol. 85, 195–201.
Tang, Q., Boden, E. K., Henriksen, K. J., Bour-Jordan, H., Bi, M., and Bluestone, J. A. (2004). Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34, 2996–3005.
Tang, Q., Adams, J. Y., Tooley, A. J., Bi, M., Fife, B. T., Serra, P., Santamaria, P., Locksley, R. M., Krummel, M. F., and Bluestone, J. A. (2006). Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 7, 83–92.
Tang, Q., and Bluestone, J. A. (2006). Plasmacytoid DCs and T(reg) cells: casual acquaintance or monogamous relationship? Nat. Immunol. 7, 551–553.
Tang, Q., and Bluestone, J. A. (2008). The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat. Immunol. 9, 239–244.
Tawara, I., Shlomchik, W. D., Jones, A., Zou, W., Nieves, E., Liu, C., Toubai, T., Duran-Struuck, R., Sun, Y., Clouthier, S. G., Evers, R., Lowler, K. P., Levy, R. B., and Reddy, P. (2010). A crucial role for host APCs in the induction of donor CD4+CD25+ regulatory T cell-mediated suppression of experimental graft-versus-host disease. J. Immunol. 185, 3866–3872.
Thornton, A. M., and Shevach, E. M. (1998). CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188, 287–296.
Tsang, J. Y., Camara, N. O., Eren, E., Schneider, H., Rudd, C., Lombardi, G., and Lechler, R. (2006). Altered proximal T cell receptor (TCR) signaling in human CD4+CD25+ regulatory T cells. J. Leukoc. Biol. 80, 145–151.
Valencia, X., Stephens, G., Goldbach-Mansky, R., Wilson, M., Shevach, E. M., and Lipsky, P. E. (2006). TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood 108, 253-261.
Veillette, A., Bookman, M. A., Horak, E. M., and Bolen, J. B. (1988). The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell 55, 301–308.
Veillette, A., Bookman, M. A., Horak, E. M., Samelson, L. E., and Bolen, J. B. (1989). Signal transduction through the CD4 receptor involves the activation of the internal membrane tyrosine-protein kinase p56lck. Nature 338, 257–259.
Vendetti, S., Patrizio, M., Riccomi, A., and De Magistris, M. T. (2006). Human CD4+ T lymphocytes with increased intracellular cAMP levels exert regulatory functions by releasing extracellular cAMP. J. Leukoc. Biol. 80, 880–888.
Vignali, D. A., and Vignali, K. M. (1999). Profound enhancement of T cell activation mediated by the interaction between the TCR and the D3 domain of CD4. J. Immunol. 162, 1431–1439.
Waldmann, H., and Cobbold, S. (1998). How do monoclonal antibodies induce tolerance? A role for infectious tolerance? Annu. Rev. Immunol. 16, 619–644.
Walker, L. S. (2009). Regulatory T cells overturned: the effectors fight back. Immunology 126, 466–474.
Walker, M. R., Kasprowicz, D. J., Gersuk, V. H., Benard, A., Van Landeghen, M., Buckner, J. H., and Ziegler, S. F. (2003). Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J. Clin. Invest. 112, 1437–1443.
Wang, R., Kozhaya, L., Mercer, F., Khaitan, A., Fujii, H., and Unutmaz, D. (2009). Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 106, 13439–13444.
Wang, R., Wan, Q., Kozhaya, L., Fujii, H., and Unutmaz, D. (2008). Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLoS ONE, 3, e2705. doi: 10.1371/journal.pone.0002705
Wehrens, E. J., Mijnheer, G., Duurland, C. L., Klein, M., Meerding, J., van Loosdregt, J., de Jager, W., Sawitzki, B., Coffer, P. J., Vastert, B., Prakken, B. J., and van Wijk, F. (2011). Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood 118, 3538–3548.
Westendorp, M. O., Frank, R., Ochsenbauer, C., Stricker, K., Dhein, J., Walczak, H., Debatin, K. M., and Krammer, P. H. (1995). Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375, 497–500.
Wildin, R. S., Smyk-Pearson, S., and Filipovich, A. H. (2002). Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J. Med. Genet. 39, 537–545.
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., Nomura, T., and Sakaguchi, S. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275.
Wing, K., and Sakaguchi, S. (2010). Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 11, 7–13.
Yang, J., Brook, M. O., Carvalho-Gaspar, M., Zhang, J., Ramon, H. E., Sayegh, M. H., Wood, K. J., Turka, L. A., and Jones, N. D. (2007). Allograft rejection mediated by memory T cells is resistant to regulation. Proc. Natl. Acad. Sci. U.S.A. 104, 19954–19959.
Keywords: anti-CD4, cAMP, monoclonal antibody, regulatory T cells, tolerance
Citation: Becker C, Bopp T and Jonuleit H (2012) Boosting regulatory T cell function by CD4 stimulation enters the clinic. Front. Immun. 3:164. doi: 10.3389/fimmu.2012.00164
Received: 10 April 2012; Accepted: 31 May 2012;
Published online: 18 June 2012.
Edited by:
Stephen Paul Cobbold, University of Oxford, UKReviewed by:
Wayne Hancock, University of Pennsylvania School of Medicine, USAVigo Heissmeyer, Helmholtz Zentrum München, German Research Center for Environmental Health, Germany
Copyright: © 2012 Becker, Bopp and Jonuleit. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Christian Becker, Department of Dermatology, University Medical Center of the Johannes Gutenberg-University, Langenbeckstr. 1, D-55101 Mainz, Germany. e-mail:Y2hyaXN0aWFuLmJlY2tlckB1bmltZWRpemluLW1haW56LmRl