

- 1 Department of Microbiology and Immunology, McGill University, Montreal, QC, Canada
- 2 Federation of Clinical Immunology Societies Center of Excellence, Research Institute of the McGill University Health Center, Montreal General Hospital, Montreal, QC, Canada
Peripheral immune tolerance requires a controlled balance between the maintenance of self-tolerance and the capacity to engage protective immune responses against pathogens. Dendritic cells (DCs) serve as sentinels of the immune system by sensing environmental and inflammatory signals, and play an essential role in the maintenance of immune tolerance. To achieve this, DC play a key role in dictating the outcome of immune responses by influencing the balance between inflammatory or Foxp3+ regulatory T (Treg) cell responses. At the heart of this immunological balance is a finely regulated DC and Treg cell crosstalk whereby Treg cells modulate DC phenotype and function, and DC drive the differentiation of Foxp3+ Treg cells in order to control immune responses. This review will focus on recent advances, which highlight the importance of this bidirectional DC and Treg cell crosstalk during the induction of tolerance and organ-specific autoimmunity. More specifically, we will discuss how Treg cells modulate DC function for the suppression of inflammatory responses and how DC subsets employ diverse mechanisms to drive differentiation of Treg cells. Finally, we will discuss the therapeutic potential of tolerogenic DCs for the induction of tolerance in autoimmune diseases.
Introduction
Immune tolerance consists of two main processes, namely central and peripheral tolerance. Central tolerance takes place in the thymus where most of the self-reactive T cells are deleted at an immature stage of their development (Bluestone, 2011). Despite negative selection, self-reactive T cells can escape thymic clonal deletion, and subsequently provoke autoimmune diseases such as type 1 diabetes (T1D), multiple sclerosis (MS), and inflammatory bowel disease (IBD) unless they are controlled by one of many peripheral mechanisms (Sakaguchi et al., 1995).
Regulatory T Cells are Major Mediators of Peripheral Self-Tolerance
In order to ensure tolerance induction of such auto-reactive T cells in the peripheral immune system, a number of mechanisms including a network of regulatory T (Treg) cells exist to achieve this function. Treg cells constitute 1–10% of thymic and peripheral CD4+ T cells in humans and mice, and arise during a thymic selection (Sakaguchi, 2000). They are characterized by the constitutive expression of the IL-2Rα chain (CD25) and expression of the forkhead winged helix transcriptional regulator Foxp3 (Hori et al., 2003). The importance of Foxp3 has been demonstrated by natural mutations of the foxp3 gene that result in a loss of Treg cell function and the development of severe autoimmune diseases, including T1D, in scurfy mice and IPEX patients (Bennett et al., 2001; Chang et al., 2006; d’Hennezel et al., 2009). Treg cell population can be divided into the naturally occurring Foxp3 Treg population (here defined as Treg), generated in the thymus and anyone of many inducible Treg cell populations (here defined as iTreg), that are derived in the periphery from CD4+Foxp3- precursors upon activation in presence of differentiating signals like TGF-β and IL-10 (e.g. Th3, Tr1 cells) (Haribhai et al., 2011; Pot et al., 2011).
Dendritic Cells Dictate the Balance Between Tolerance and Immunity
Numerous studies show that different cell populations of the innate immune system such as dendritic cells (DCs), macrophages, natural killer cells, and γδ T cells can regulate tolerance induction (Lehuen et al., 2010). DCs represent a heterogeneous population of bone marrow-derived cells and are the most potent antigen presenting cells (APCs) (Banchereau and Steinman, 1998; Shortman and Naik, 2007). DCs are derived from multiple lineages, have a distinct stage of development, activation, and maturation state (Banchereau and Steinman, 1998; Steinman et al., 2003). DCs exist as a distinct subset and differ in their ontogeny, surface molecule expression, and biological functions (Banchereau and Steinman, 1998; Steinman and Nussenzweig, 2002). These factors seems to determine the T cells polarizing signals and type T cells responses induced by DCs namely Th1, Th2, Th17, or Treg cells. Although all DCs are able to prime T cells, they differ in their in vivo niches, migration, function, and requirements from the environment for their generation and activation (Shortman and Naik, 2007). DCs are divided into conventional, myeloid, or plasmacytoid DC (pDC) subsets. In mice, three main subsets of conventional CD11chi DCs have been identified in the spleen and lymph nodes, namely the CD8+, CD4+CD8-, and CD4-CD8-DCs (Vremec et al., 2000). Lymph nodes contain two additional DCs subsets; skin-derived Langerhans cells and tissue interstitial DCs that arrive from the periphery through the lymphatic circulation (Vremec et al., 2000).
Dendritic cells receive the maturation signals through the pathogen associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs) receptors that detect certain microbial and tissue damage signals via activation of nuclear factor-κB (NF-κB) and interferon regulatory factors (IRF) families (Maldonado and von Andrian, 2010). Upon activation, DCs up-regulate a wide variety of gene products involved in antigen presentation and co-stimulation such as MHC II, CD86, CD80, OX40-L, inducible co-stimulator (ICOS) ligand as well cytokines involved in the modulation of effector function such as IL-1β, IL-2, IL-6, IL-8, IL-12, and IL-18 (Maldonado and von Andrian, 2010). These changes are required for DCs to initiate a three-step T cell activation process: MHC molecules displaying cognate peptide (signal 1), co-stimulatory signal expression (signal 2), and cytokine production by DCs (signal 3).
While potently capable to initiate inflammatory responses, DCs also play an important role in modulating tolerance induction. Tolerogenic DCs are characterized by high antigen uptake and processing capabilities in order to present antigen to antigen-specific T cells, but fail to deliver proper co-stimulatory signal for effector T (Teff) cells activation and proliferation (Steinman et al., 2003). This results in T cell death, T cell anergy, or induction and expansion of Treg cells subsets. As such, tolerogenic DCs have been shown to suppress experimental autoimmune disease and play an important role in alloimmunity (Morelli and Thomson, 2007).
Subsets of Tolerogenic DCs: Division of Labor in Tolerance Induction
Different DCs subsets are specialized in tolerance vs. inflammatory immune response decisions. Specific markers capable of discriminating tolerogenic from inflammatory DCs are still ill-defined. However, CD8+ DCs expressing CD95L and DEC205 often possess tolerogenic properties (Mahnke et al., 2002; Yamazaki et al., 2008). Expression of inhibitory immunoglobulin-like transcript (ILT) receptors is also frequently observed in some subsets of tolerogenic DCs, where ILT-3 and ILT-4 mediated signals on DCs inhibits expression of co-stimulatory molecules and induce a tolerogenic state (Manicassamy and Pulendran, 2011). In humans, ILT-3 and ILT-4 expressing tolerogenic DCs can promote antigen-specific unresponsiveness in CD4+ T cells and induce Treg cells. Recent studies have shown that Indoleamine 2,3-dioxygenase (IDO) and IL-10 can induce the expression of ILT-3 and ILT-4 on DCs and promote tolerogenic response (Manavalan et al., 2003). Activation of ILT-3 receptor on DCs leads to the recruitment of protein phosphate SHP-1 and SHIP-1 to the immunoreceptor tyrosine-based inhibitory motif (ITIM) resulting in the inhibition of NF-κB and p38MAPK pathways that are critical for inflammatory responses (Cella et al., 1997; Svajger et al., 2008). While immature DCs appear to be good indicators for DC tolerogenicity, mature DCs might not always induce immunity. Hence, other factors such as exposure to certain differentiation signals or cues from the local environment might condition DCs beyond their expression of co-stimulatory molecules. Therefore, tolerance is not only a consequence of T cells receiving insufficient signals 2 and 3, but additional tolerance inducing factors might be in play.
Generation of Tolerogenic DCs
Treg Cells, Through Various Mechanisms, Promote the Generation of Tolerogenic DCs
While several environmental factors and cytokines can promote the tolerogenic phenotype of DCs, there is accumulating evidence that Treg cells can also induce a tolerogenic phenotype in DCs by modulating their maturation and function. Several studies indicate that Treg cells can suppress the capacity of DCs to activate Teff cells by down-regulating CD80/CD86 expression on bone marrow-derived or splenic DCs in vitro (Cederbom et al., 2000). Depletion of Treg cells from asthma susceptible mice resulted in DCs with higher expression levels of MHC II, CD80, CD86 and displayed a increased T cell stimulatory activity (Mahnke et al., 2007). Although the mechanism by which Treg cells achieve this is unknown, this might be mediated through cell surface molecules or cytokines such as IL-10, CTLA-4, and TGF-β (Figure 1).
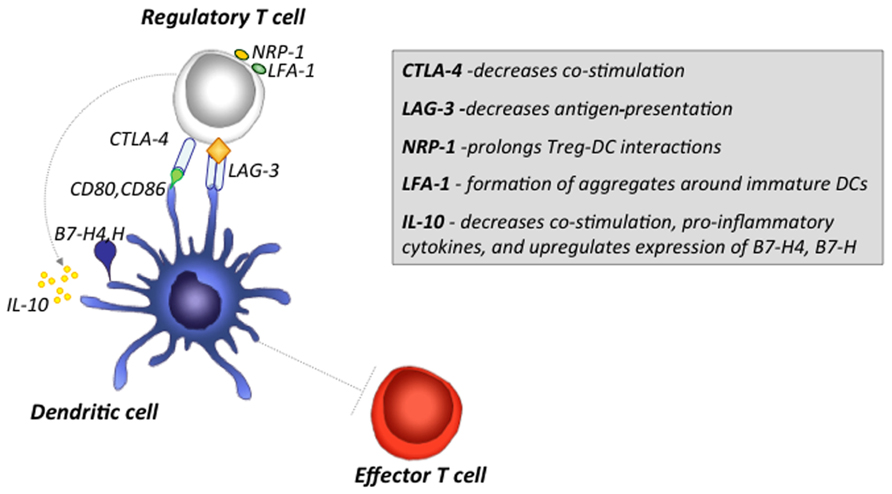
FIGURE 1. Major mechanisms by which Treg cells induce tolerogenic DCs and inhibit Teff cell activation. Treg cells can inhibit the function of DCs through various mechanisms. CTLA-4 expression on Treg cells down-regulates up-regulation of CD80 and CD86 co-stimulatory molecules on DCs. LAG3 binding to MHCII-expressing immature DCs results in inhibitory signals that suppress DCs maturation and co-stimulatory capacity. Nrp-1 expression on Treg cells promotes sustained interactions between Treg cells and DCs, and limits the access of Teff cells to DCs. Expression of LFA-1 on Treg cells, promotes aggregate formation around DCs, and inhibit the up-regulation of CD80 and CD86 on DCs. IL-10 inhibits up-regulation of MHCI/II and B7 co-stimulatory molecule expression, suppresses release of pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, and IL-12, and up-regulates of B7-H4 and B7-H inhibitory molecules.
Role of Treg Cell-Derived IL-10 in Induction of Tolerogenic DCs
The production and activity of the immunomodulatory cytokine IL-10 is often associated with tolerogenic responses. IL-10 inhibits multiple aspects of DC function including MHCI/II and CD80/CD86 co-stimulatory molecules expression and a release of pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, and IL-12 (Mahnke et al., 2002, 2007; Alexander, 2005). Interestingly, IL-10 mediated effects could be only observed when immature DCs were exposed to IL-10 (Read et al., 2000), as mature DCs were insensitive to IL-10 stimulation and displayed stable, mature phenotype (Read et al., 2000). In addition, DCs cultured in presence of Treg cells has been shown to secrete of IL-10, TGF-β, and IL-27 cytokines (Awasthi et al., 2007). IL-27 cytokine plays an important role in suppressing production of Th17 polarizing cytokines, such as IL-1β, IL-6, IL-23 derived from DCs and act on naïve T cells to induce Tr1 differentiation (Pot et al., 2009). IL-10 treated DCs have also been shown to promote tumor growth, and prevent transplant rejection and ameliorate MS (Mahnke et al., 2007).
CTLA-4 is a Potent Inducer of Tolerogenic DCs
CTLA-4 plays an important role in Treg cell-mediated suppression (Salomon et al., 2000). Treg cells constitutively express high levels of CTLA-4 and conditional deficiency of CTLA-4 in Treg cells results in lymphoproliferation and variety of autoimmune disease, an outcome analogous to global CTLA-4 deficient mice (Wing et al., 2008). Monoclonal antibody blockade of CTLA-4 exacerbates T1D in non-obese diabetic (NOD) mice and induce IBD (Read et al., 2000; Wing and Sakaguchi, 2010). CTLA-4 mediates suppression through down regulation of CD80 and CD86 expression on DCs. Treg cells from CTLA-4 deficient mice or administration of CTLA-4 blocking antibodies results in reduced down-modulation of B7 molecules and increased T cells proliferation (Oderup et al., 2006). IDO, a potent regulatory molecule that is know to induce the production of pro-apoptotic metabolites from the catabolism of tryptophan, results in the suppression of Teff cells through a mechanism dependent on interaction between CTLA-4 and CD80/CD86 (Fallarino et al., 2003; Baban et al., 2009). Baban et al. (2009) recently demonstrated that IDO is a critical molecular switch that stimulates potent Treg cells suppression while simultaneously block IL-6 driven reprogramming of Treg cells into Th17 cells and production pro-inflammatory cytokines such as IL-17, IFN-γ, TNF-α, and IL-2. Interestingly, it has also been demonstrated that Treg cells can modulate DCs to express IDO and more specifically CTLA-4 immunoglobulin fusion protein was found to induce IDO expression through ligation to CD80 and CD86 molecules in mouse and human DCs (Fallarino et al., 2003; Mellor and Munn, 2004; Mellor et al., 2004). In addition, IL-10 produced by Treg cells is critical for sustaining IDO expression in DCs (Manicassamy and Pulendran, 2011). Thus, IDO is one of the molecular switches controlling the balance between Treg and Teff cell responses.
B7-H4 and B7-H: B7 Family Members that Negatively Regulate T Cell Immunity
A number of other mechanisms have been proposed by which Treg cells can either abrogate the antigen presenting capacity of DCs or promote the secretion immunomodulatory cytokines. For example, Treg cells are able to induce the expression of B7-H and B7-H4, a ligands responsible for negative regulation of cell-mediated immunity in peripheral tissues (Mahnke et al., 2007). Recently, Treg cells have also been shown to trigger high levels of IL-10 production by APCs and in turn stimulate B7-H4 expression in an autocrine fashion and render these APCs immunosuppressive (Kryczek et al., 2006).
Lymphocyte Activation Gene 3 (LAG3)
Lymphocyte activation gene 3 is cell surface molecule expressed on Treg cells that can modulate DC phenotype and function (Huang et al., 2004; Workman and Vignali, 2005; Liang et al., 2008). LAG3 is a CD4 homolog that binds MHC II molecules with very high affinity, has a negative regulatory intrinsic function and is required for maximal Treg cells suppression (Huang et al., 2004; Workman and Vignali, 2005). Binding of LAG3 to MHC II molecules on immature DCs initiates a receptor tyrosine-based activation motif (ITAM) mediated inhibitor signaling pathway that suppress DC maturation (Liang et al., 2008). These findings provide a novel tolerogenic pathway that may endow Treg cells to enhance tolerance by inhibiting DCs functions.
Treg–DCs Cognate Interactions Mechanism
It has been shown that Treg cells are more motile that naïve T cells in vitro, in turn out-competing the latter for physical access to DCs (Tran et al., 2009). This Treg–DC cell aggregation process is antigen-dependent, and the selective advantage of Treg cells over Teff cells for physical interactions with DCs can be attributed, at least in part, to the expression of LFA-1 (Lymphocyte function-associated antigen 1) by Treg cells, as deficiency or blockade of LFA-1 abolishes this process. By forming aggregates, Treg cells inhibit the up-regulation of CD80, CD86 on immature DCs and also down-regulate the expression of CD80 and CD86 by mature DCs without affecting the expression of CD40 or MHC II molecules (Onishi et al., 2008). This modification of CD80 and CD86 is CTLA-4 dependent as Treg cells from CTLA-4 deficient mice fail to modify expression of CD80 and CD86 (Onishi et al., 2008). Therefore at least in vitro, Treg cell contact dependent suppression can be dissected by two consequent steps; initial LFA-1 dependent formation of aggregates around immature DCs and subsequent LFA-1 and CTLA-4 dependent active down-regulation of CD80 and CD86 expression (Onishi et al., 2008). Overall, this results in down-regulation of Teff cell activation and induction of immune suppression and tolerance. The significance of DCs and Treg cells interaction has been also demonstrated in studies using NOD mice. The Bluestone group, by means of intravital microscopy, demonstrated that direct interactions between Treg cells and DCs in vivo resulted in inhibition of Teff cells activation (Tadokoro et al., 2006; Tang et al., 2006) suggesting a potential feedback loop between Treg cells and DCs. In this model, Treg cells up-regulate LFA-1 expression upon activation by immature DCs, adhere to DCs, and suppress the expression of CD86 and CD80, in turn inhibiting the activation and expansion of Teff cells (Sakaguchi et al., 2009). While Foxp3+ Treg must be activated by antigen in order to exert their suppressive functions in vitro and in vivo (Takahashi et al., 1998), activated Foxp3+ Treg cells can mediate in vitro bystander suppression of Teff cells with different antigen specificities. However, it still remains to be determined whether such mechanism can also apply to other cell types like DCs, and to what extent such bystander suppression occurs in vivo.
Recently it has been shown that neuropilin (Nrp-1) also plays a critical role in mediating Treg–DC interactions (Sarris et al., 2008). Nrp-1 is a ligand binding receptor for a class of semaphorins, and is preferentially expressed by Treg cells. Nrp-1 expression can be induced by ectopic expression of Foxp3 in Foxp3- T cells, and antibody blockade of Nrp-1 reduces the frequency of Treg–DCs interactions. Furthermore, retroviral introduction of Nrp-1 endows T helper cells with ability to establish a long interactions with DCs (Sarris et al., 2008). Interestingly, it has been demonstrated that Nrp-1 can confer a high adhesive property of Treg cells in their interaction with DCs under steady conditions; however, this potential by Treg cells is lost under inflammatory conditions (Sarris et al., 2008).
The results described above demonstrate the capacity of Foxp3+ Treg cells, through the expression of LAG3 and Nrp-1 molecules, to influence immature but not mature DCs. Mature DCs, in contrast to immature DCs, produce pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1β which can down-regulate Foxp3 expression and abolish Foxp3+ Treg cell-mediated suppression (Rutella et al., 2006). Although IL-6 production induced by LPS stimulation only slightly reduced Foxp3+ Treg cells-mediated suppression, DC-derived IL-6 may render responder T cells resistant to suppression (Pasare and Medzhitov, 2003). Moreover, IL-6 might also facilitate the reprogramming of Foxp3+ Treg cells into Th17 lineage (Bettelli et al., 2007).
In other instances, it has been demonstrated that Treg cells, through the expression of LFA-1 and CTLA-4, can suppress TNF and LPS maturated DCs by selectively down-regulating CD80, CD86, PDL1, and PDL2, but not MHCII and CD40, expression on the DC surface. Overall, these findings demonstrate that Treg cells may employ specific mechanism(s) to suppress DCs functions in different microenvironments of antigen priming.
The Gut Environment Promotes Development of Tolerogenic DCs
The identification of tolerogenic DCs subsets in microenvironments such as gut, skin, and lungs suggest that local signals induce tolerogenic DCs in situ. Given the enormous amount of microbial stimuli in the gut, intestinal DCs represent a key regulatory mechanism to prevent excessive inflammation. Specifically, CD11c+ DCs expressing CD103 have been identified in the gut-associated lymphoid tissue (GALT) and mesenteric lymph node with the specialized function of inducing Foxp3+ Treg cells and maintaining gut immune homeostasis (Coombes et al., 2007; Sun et al., 2007). This CD11c+ DC subset expresses high levels of CD103, MHC II, CD80, and CD86. Interestingly, absence of CD103 on DCs led to the abrogation of Treg cell activity indicating a crucial role for CD103 in maintaining the balance between Treg and Teff cells (Annacker et al., 2005). While mesenteric lymph node derived CD103+ DCs are prone to imprint expression of CCR9 on T cells, an important homing receptor enabling homing to the gut, CD103- DCs promote the differentiation of CD4+ T cells producing IFN-γ (Jaensson et al., 2008). Collectively, CD103+ and CD103- DCs represent functional subsets and CD103 is critical to regulate Teff and Treg cells in the gut (Annacker et al., 2005).
Apoptotic DCs Favor Formation of Tolerogenic DCs
Recent studies show that immature DCs can uptake apoptotic and necrotic DCs without being recognized as an inflammatory event (Kushwah et al., 2010). This uptake results in conversion of mature DCs into tolerogenic DCs that remain resistant to LPS induced maturation and induces the production of TGF-β via the mTOR signaling pathway (Kushwah et al., 2010). TGF-β producing DCs subsequently interact with naïve T cells and drive the differentiation of iTreg cells (Kushwah et al., 2010; Kushwah and Hu, 2011). Moreover, recent studies showed that α-CD3 mAb treatment transiently depletes large numbers of T cells and induce long term immune tolerance (Perruche et al., 2008; Esplugues et al., 2011). The mechanism underlying this regulatory outcome is due to increased production of TGF-β by immature DCs after engulfment of apoptotic T cells (Perruche et al., 2008). A recent study demonstrated that α-CD3 mAb treatment resulted in elimination of the inflammatory Th17 cells from intestinal lumen or resulted in acquisition Th17 cells producing a IL-10 (Esplugues et al., 2011). However, it is not known which specific subset of gut-residing DCs is responsible for production of TGF-β and induction of IL-10 producing Th17 cells with regulatory capacity.
Immune Modulation by Tolerogenic DCs
DC Employs Various Mechanisms to Drive the Differentiation of Treg Cells
B7 Co-Stimulatory Molecules Promote Differentiation of Treg Cells
In the steady state, DCs are largely immature and present antigens to T cells in tolerogenic manner. Such DCs are characterized by low expression of CD80, CD86, and CD40 (Steinman and Nussenzweig, 2002; Steinman et al., 2003; Hubert et al., 2007). Moreover, targeting of antigens to immature DCs via the regulatory receptor DEC205 results in tolerance through deletion of antigen-specific T cells and preferential induction of Treg cells (Hawiger et al., 2001). Interestingly, antibodies bound to DEC205 are efficiently internalized and delivered to antigen processing compartments, however internalization of the antigen by DEC205 does not induce the maturation of the DCs (Mahnke et al., 2000). Immature DCs had been shown to induce Treg cells in vitro and anti-DEC205 targeting of antigens to immature DCs led to anergic T cells in vivo (Mahnke et al., 2003). These data show that that the DEC205 receptor is critical for DCs in the steady state to promote tolerance (Mahnke et al., 2003). Moreover, treatment with respective anti-DEC antigen conjugates results in significant improvement of autoimmune disorders such as T1D and MS (Hawiger et al., 2004; Bruder et al., 2005). DCs can also favor tolerance by utilizing ICOS, another CD28 family member, to specifically drive Tr1 cell differentiation (Akbari et al., 2002; Ito et al., 2007).
Despite the established role of immature DCs as inducers of Treg cells, recent studies have shown that mature DCs, expressing high levels of CD86, also have the potential to preferentially expand Treg cells in vitro, and prevent T1D development (Yamazaki et al., 2003; Tarbell et al., 2004; Sgouroudis et al., 2011). Interestingly, DCs isolated from Peyer’s patches, eye, or lungs display a mature phenotype, secrete IL-10, but not IL-12 and drive the development of Tr1 cells (Akbari et al., 2001). Thus, signals derived from the local tissue environment might play a role in conditioning tolerogenic DCs and drive the differentiation of Treg cells. All together, these findings suggest that the previously established paradigm whereby immature DCs leads to Treg cell differentiation and mature DCs drive Teff cell responses might be revisited (Figure 2).
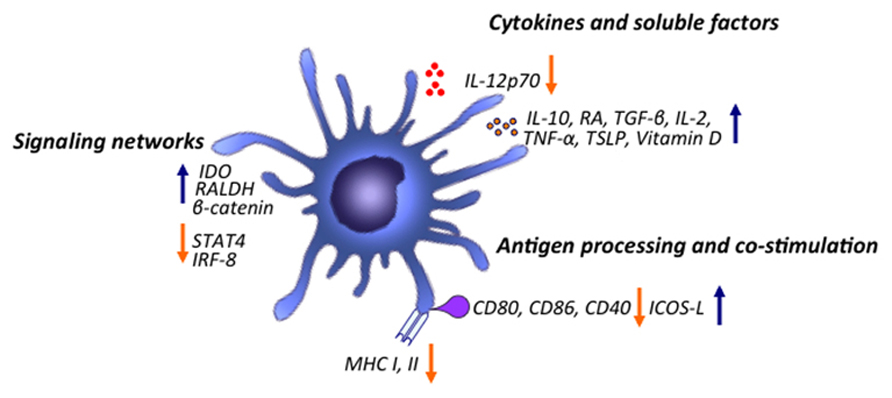
FIGURE 2. Major mechanisms of immune modulation by tolerogenic DCs. DCs modulate the immune system by promoting Treg cell generation through various mechanisms. Upon stimulation, DCs are able to increase production of various cytokines and inflammatory mediators such as IL-10, RA, vitamin D, TGF-β, IL-2, IL-10, TSLP, TNF-α and down-regulate expression of inflammatory cytokines such as IL-12p70. DCs, by down-regulating co-stimulation and antigen presentation, can favor induction of Treg cells. Various signaling pathways program DCs to induce tolerogenic responses. Activation of β-catenin signaling pathway promotes induction of anti-inflammatory factors such as vitamin A and is important for promoting Treg cell induction and limiting inflammatory responses. TSLPR signaling inhibits activation of STAT4 and IRF-8, factors critical for production of the Th1 polarizing factor IL-12. Triggering of DCs through TLR2-6 leads to Erk activation which mediates induction of RALDH and conversion of retinal to RA. TLR9 ligation can drive induction of IDO in DCs limiting IL-6 production and differentiation of Th17 cells while inducing differentiation of Treg cells.
DC-Derived Cytokines that Drives Differentiation of Treg Cells
IL-10
Dendritic cells can secrete high amounts of IL-10 upon stimulation and drive differentiation of naïve T cells into IL-10 secreting Tr1 cells (Kushwah and Hu, 2011). DCs of IL-10 transgenic mice display a particularly immature phenotype and Tr1 cells are significantly enriched in spleens of these mice (Wakkach et al., 2003). Langerhans cells in the skin also produce IL-10 and drive differentiation of Tr1 cells (Igyarto et al., 2009).
TNF-α
TNF-α can also induce tolerogenic state in DCs (Menges et al., 2002; Mahnke et al., 2007). Despite the fact that TNF-α treated DCs has a mature phenotype, they fail to secrete inflammatory cytokines such as IL-1β, IL-6, TNF-α, and IL-12 (Menges et al., 2002). Moreover, these tolerogenic DCs were able to reverse development of EAE where their suppressive effects were mediated by the induction of IL-10 producing Treg cells (Mahnke et al., 2007). However, there is also a controversial data demonstrating that TNF-α blockade prevented DC maturation and Treg cells were induced in the absence of TNF-α (Mahnke et al., 2007).
TGF-β
TGF-β-producing DCs might preferentially drive differentiation of Treg cells rather than Teff cells. Tumor-bearing mice contains a CD11c+ DC subset characterized by low expression of CD80 and CD86 co-stimulatory molecules and are endowed with the capacity to secrete TGF-β which promotes Treg cells differentiation and proliferation in vivo (Ghiringhelli et al., 2005).
IL-2
We and others have shown that a local deficiency in islets of IL-2, a critical cytokine for the homeostasis/fitness of Treg cells in vivo, compromises Treg cell function in islets, a defect readily corrected by low dose IL-2 therapy in NOD mice. NOD mice introgressed with a protective IL2 allelic variant from T1D-resistant C57BL6 mice (NOD.Idd3BL6) are resistant to autoimmune diabetes in contrast to wild-type NOD mice which are susceptible to the disease. Moreover, T1D-protective IL2 allelic variants in NOD mice impinge T1D development by bolstering IL-2 production by CD4+ Teff cells in turn, driving the expansion and homeostasis of CD4+Foxp3+ Treg cells in islets. Recently, we showed that CD11c+ DCs in NOD.Idd3BL6 have an increased maturation status relative to wild-type NOD CD11c+ DCs. We also showed that NOD.Idd3BL6 DCs are more potent activators of Treg cells functions in vitro and in vivo, and this increased capacity of congenic DCs to prime Treg cells is attributed to their ability to produce IL-2. Consistently, IL-2 blockade in vitro completely abolished the proliferative advantage conferred by Idd3BL6 DCs on Foxp3+ Treg cells (Sgouroudis et al., 2011). Interestingly, CD11c+ MHCII+ DCs isolated from pancreatic LN of NOD.Idd3BL6 mice promoted Foxp3+Treg cells expansion more efficiently that WT DCs isolated from similar sites, suggesting that Idd3BL6 DCs display tolerogenic phenotype specifically in the pancreatic sites to enhance Foxp3+ Treg cells functions (Sgouroudis et al., 2011). Thus, T1D-protective IL2 allelic variants impinge the development of β-islet autoimmunity by bolstering IL-2 mRNA expression and protein secretion by CD4+ Teff cells and DC and in turn, driving the functional homeostasis of CD4+Foxp3+ Treg cells in the target organ (Sgouroudis et al., 2011).
It has been previously shown that CD40/CD40L interaction regulates Treg cells homeostasis (Kumanogoh et al., 2001; Guiducci et al., 2005). Mature DCs lacking CD40L are impaired in sustaining Treg cells proliferation and survival. The underlying mechanism is mediated by fact that CD40L deficient DCs are not able to produce IL-2, to as similar extent as wild-type DCs. Administration of rhIL-2 in vivo restored Treg cells numbers in thymic and peripheral compartments of CD40L deficient mice, by increasing survival and homeostatic proliferation of Treg cells (Kumanogoh et al., 2001; Guiducci et al., 2005). Therefore, CD40 triggering by Treg cells contributes to induce IL-2 through CD40L on DCs needed for their maintenance. Therefore, several mechanisms have been established by DCs in order to maintain a peripheral pool of Treg cells through IL-2 production (Kumanogoh et al., 2001; Guiducci et al., 2005).
IDO Expression by DCs Drives Treg Cells Differentiation
Dendritic cells populations expressing IDO play a critical role in immune tolerance by promoting iTreg cells differentiation (Mellor and Munn, 2004; Matteoli et al., 2010). IDO expression by DCs appears to be dependent on Aryl hydrocarbon receptor (AHR) as DCs lacking AHR fail to up-regulate IDO and prime T cells responses rather than tolerance induction (Nguyen et al., 2010). Selective inhibition or genetic deletion of IDO affects the development of antigen-specific Treg cells while promotes Th1 and Th17 development and worsens T cells mediated and dextrin sulfate sodium (DSS) colitis in mice (Matteoli et al., 2010). It has been shown that TLR9 ligation can drive induction of IDO in pDCs that subsequently suppress IL-6 production and differentiation of Th17 cells while induce differentiation of Treg cells (Baban et al., 2009).
Site Specific Residing DCs Supports Treg Cells Differentiation
Thymic Residential DCs Play a Role in Induction of Foxp3+ Treg Cells
It has been shown that DCs and medullary thymic epithelial cells (mTECs) contribute to the selection of Foxp3+ Treg cells in the thymus. Epithelial cells in Hassall’s corpuscles in the thymus produce thymic stromal lymphopoietin (TSLP) which subsequently acts on thymic DCs by binding to TSLP receptor (TSLPR) and IL-7Rα complex and drives induction of CD80 and CD86 (Ziegler and Liu, 2006). These DCs subsequently drive differentiation of CD4+CD8+CD25- thymocytes into Foxp3+Treg cells, which highly depend from IL-2 and CD28 signaling (Ziegler and Liu, 2006). These data demonstrate that TSLP activated myeloid DCs are critical for selection of self-reactive thymocytes to develop into Foxp3+ Treg cells. In addition to the myeloid DCs, human thymic pDCs can also induce differentiation of Treg cells in a CD40L-dependant fashion (Hanabuchi et al., 2010; Martin-Gayo et al., 2010). Thymic pDCs express TSLPR along with IL-7Rα and are responsive to TSLP derived from thymic epithelial cells (Hanabuchi et al., 2010; Martin-Gayo et al., 2010). More evidence for the contribution of thymic DCs in Treg cell induction comes from studies done in mouse models, where targeting of neo-self antigens to thymic DCs instructed antigen-specific T cells to develop into Foxp3+ Treg cells (Proietto et al., 2008). Three thymic DCs subtypes, namely conventional DCs, thymus resident DCs, and pDCs were shown to be able of instructing Foxp3+ Treg cells differentiation in vitro, supporting the model where antigen recognition on thymic DCs can directly elicit Foxp3+ Treg cells differentiation (Wu and Shortman, 2005).
Gut-Residing DCs Maintain Treg Cell Stability via Production of Retinoic Acid and TGF-β
Dendritic cells from the lamina propria of the small intestine preferentially promote Treg cell induction relative to the DCs of lymphoid organs. This increased induction of Treg cells has been shown to be dependent on TGF-β and retinoic acid (RA), a vitamin A metabolite highly expressed in small intestine (Coombes et al., 2007; Sun et al., 2007; Belkaid and Oldenhove, 2008). DCs are the major subset of immune cells able of metabolizing vitamin A to RA. RA synthesis is a highly regulated process that involves several key enzymes: vitamin A is oxidized to retinaldehyde by alcohol dehydrogenase and then to RA by retinal dehydrogenase (RALDH). Manicassamy et al. (2009) and Manicassamy and Pulendran (2011) demonstrated that TLR2 and TLR6 signaling by zymosine promote induction of RALDH1 and RALDH2 in DCs via ERK dependent mechanism. This result in conversion of retinal to RA which then exerts an autocrine effect on DCs via RA receptor to induce SOCS3, this then suppress activation of p38MAPK and pro-inflammatory cytokines. Accumulating evidence shows that RA directly influences the development and function of various immune cells. For example, RA promotes the differentiation of Treg cells and suppress differentiation of Th1 and Th17 cells, and prevents IBD and EAE in mice (Kushwah and Hu, 2011). CD103+ DCs in the intestine lamina propria and mesenteric lymph nodes express high levels of RALDH and have ability to induce Foxp3+ Treg cells. It is well documented IL-6 is an essential cytokine driving Th17 cell differentiation, while down-regulate Foxp3 expression in Treg cells. Interestingly, Zhou et al. (2010) showed that RA maintains the stability of Foxp3+ Treg cells and sustains their suppressive potential in the presence of IL-6, through down-regulation of IL-6 receptor expression and signaling, therefore preventing Treg cells conversion into Th17 cells (Coombes and Powrie, 2008; Zhou et al., 2010). CD103+ DCs from the lamina propria of the small intestine and mesenteric lymph node have been shown to be significantly better than splenic DCs at mediating the conversion of naïve T cells into Foxp3+ T cells in the presence of exogenous TGF-β (Coombes et al., 2007). Moreover, expression of the αvβ8 integrin by DCs is important for TGF-β activation, accumulation of Treg cells in the small intestine and prevention of colitis development (Coombes and Powrie, 2008). Overall, gut-derived signals may condition DCs to produce TGF-β for Treg cell generation.
β Catenin: A Novel Regulator of Intestinal Homeostasis
Wnt-β-catenin signaling in intestinal DCs regulates the balance between inflammatory and regulatory responses. Recently, Manicassamy et al. (2009) performed a gene expression profile of lamina propria DCs and demonstrated that several Wnt family genes and β-catenin were constitutively expressed by intestinal DCs, but not by splenic DCs (Manicassamy et al., 2009). Furthermore, they showed that β-catenin signaling promotes the induction of Treg cells while suppress Th1 and Th17 cells in the gut, indicating that β-catenin expression by intestinal DCs is important for maintaining the balance between Treg and Teff cells in the gut (Manicassamy et al., 2010). In addition, intestinal DCs lacking β-catenin expression produced lower levels of Treg cell-promoting stimuli including RA metabolizing enzymes, IL-10, and TGF-β, but higher levels of Th17-promoting cytokines IL-23 and IL-6 (Manicassamy et al., 2010). Overall, β-catenin signaling is needed to maintain intestinal homeostasis through the induction of Treg cells and the suppression of pro-inflammatory factors.
The Gut Microenvironment Influences Intestinal DC Function
Many of the unique properties of intestinal DCs appear to be a result of environmental conditioning. Activation of NF-κB expression in intestinal epithelial cells, perhaps as a result of microflora signaling through PRRs, enhances TSLP production (Coombes and Powrie, 2008). Recently, important role for TSLP in dictating the quality of immune response has been suggested. TSLP and other epithelial cells factors limits the activation of STAT4 and IRF-8, essential factors for the production of Th1 polarizing cytokine IL-12-23p40 (Arima et al., 2010). In addition, TSLP signaling also induce the activation of STAT6, which programs DCs to secrete chemokines necessary for the recruitment of Th2 cells, and increase IL-10 and TGF-β production (Coombes and Powrie, 2008; Arima et al., 2010). TSLP has also been shown to confer human thymic DCs with ability to induce the differentiation of CD4+CD25- thymocytes into Foxp3+CD4+CD25+Treg cells (Watanabe et al., 2005). Thus, the increased ability of gut-residing CD103+ DCs to induce Foxp3 may be due to TSLP.
Overall, these findings illustrate that the intestinal immune system (i.e., GALT) is a preferential site for the induction of Treg cells, and provides a mechanism by which the thymus-derived Treg cell pool could be complemented by de novo generated Treg cells for the efficient control of inflammatory responses toward specific commensal bacteria and food antigens (Coombes and Powrie, 2008).
The Skin and Lung are Naturally Tolerogenic Environments
Like the intestine, the skin and lungs are also constantly exposed to various microbes and environmental antigens such as allergens. The interaction between vitamin D and RANK–RANKL signaling pathway in the skin plays a role in DC-mediated iTreg cells induction (Loser et al., 2006; Kushwah and Hu, 2011). This process occurs via the activated metabolite of vitamin D, VD3, which exerts its actions through its nuclear receptor, VDR. VDR is expressed on DCs and vitamin D treatment inhibits DCs maturation and their ability to prime alloreactive T cells responses (Loser et al., 2006). Similarly to the intestine, lung CD103+ DCs promote the induction of Treg cells, while the CD103- DCs represent the main producers of pro-inflammatory cytokines in response to airborne allergens and TLR ligands. In contrast to lung and mucosal sites, CD103-CD11b+ migratory dermal DCs, compared to CD103+CD11b+ DCs, are much more potent in promoting Treg cells induction (Guilliams et al., 2010; Pulendran et al., 2010).
Therapeutic Applications of Tolerogenic DCs
The possibility to generate or expand tolerogenic DCs in vitro provides significant opportunities for therapeutic interventions. DCs are generated in vitro from bone marrow precursors in rodents or blood monocytes in humans and can be rendered tolerogenic by modulating their culture conditions through exposure to cytokines, growth factors or pharmacologic mediators, or genetic engineering (Morelli and Thomson, 2007). In vitro generation of tolerogenic DC can be achieved through exposure to various anti-inflammatory agents such as vitamin D, as well as clinically approved suppressive drugs such as corticosteroids, cyclosporin, and rapamycin (Morelli and Thomson, 2007). Rapamycin acts as an inhibitor of the Akt–mTOR pathway, increases the number of Foxp3+ Treg cells, and promotes their resistance to apoptosis (Strauss et al., 2007; Basu et al., 2008). Moreover, rapamycin-conditioned myeloid DCs fail to produce IL-12p70 and TNF-α and are resistant to maturation induced by TLR ligands or by CD40 signaling (Turnquist et al., 2007). Advances in gene transfer technology offers the possibility to generate tolerogenic DCs by genetically inducing the expression of immunosuppressive molecules like IL-10, TGF-β, or CTLA-4, or blocking the expression of co-stimulatory molecules (Morelli and Thomson, 2007). Conversely, it is possible to generate tolerogenic DCs in vitro that drive Treg cells differentiation in a tissue specific manner in order to inhibit inflammation in a particular site (Rescigno, 2010). For example, intestinal epithelial cells release factors that drive the development of mucosal like tolerogenic DCs (Iliev et al., 2007). Incubation of DCs with intestinal, but not mammary or epithelial cell-derived supernatants induces the expression of CD103 on DCs while inhibiting the secretion of inflammatory cytokines. These in vitro generated CD103+ DCs can drive the induction of Treg cells and expression of the gut-associated homing receptor α4β7 (Iliev et al., 2009). Moreover, only intestinal epithelial cells conditioned DCs are able to protect against colitis development (Iliev et al., 2009). Therefore, DCs conditioned at local environment are important for generation of Treg cells that are able to suppress and home to specific sites.
Conclusion
Our understanding of the functional and phenotypic plasticity of DCs, as well the capacity to modulate DCs development and maturation in vitro and in vivo gives opportunity to use these cells for therapeutic purpose in autoimmunity and cancer. Tolerogenic DCs has a dual role, for example in cancer, a profound defect in DCs function are associated with accumulation of immature DCs in tumors where DCs are unable to initiate anti-tumor immune responses while contributes to the recruitment, expansion and function of Treg cells. While this have negative outcome in cancer settings, similar scenario would be helpful in the case of autoimmunity. Several studies in mice suggest that DCs might be used in the treatment of autoimmunity through their ability to induce Treg cells. For example, repetitive injections of semi-mature DCs results in protection from EAE and thyroiditis (Ueno et al., 2007). In the NOD mice, DC-mediated in vitro generation of Treg cells can inhibit spontaneous T1D development in these mice (Ueno et al., 2007). In recent years, several mechanisms have been identified to explain how tolerogenic DCs mediate their function. For example, IDO expression by intestinal DCs is important for regulating intestinal immune homeostasis by keeping the balance between Treg cells and Th17, Th1 cells. Deregulations of IDO activity results in increased intestinal inflammation, suggesting that IDO could be regarded as a new target for IBD. Therefore, pharmacological targeting of IDO during the chronic inflammation like IBD would be beneficial in dampening the inflammatory process and tissue damage in the gut (Matteoli et al., 2010). Moreover, as IL-6 is often present in inflammatory infiltrates in autoimmune settings, recent findings about ability of RA to stabilize Treg cells in the presence of IL-6 offers the possibility that RA treated Treg cells can be used in many autoimmune settings where achieve such a balance is essential.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential con-flict of interest.
References
Akbari, O., DeKruyff, R. H., and Umetsu, D. T. (2001). Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat. Immunol. 2, 725–731.
Akbari, O., Freeman, G. J., Meyer, E. H., Greenfield, E. A., Chang, T. T., Sharpe, A. H., Berry, G., DeKruyff, R. H., and Umetsu, D. T. (2002). Antigen-specific regulatory T cells develop via the ICOS–ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat. Med. 8, 1024–1032.
Annacker, O., Coombes, J. L., Malmstrom, V., Uhlig, H. H., Bourne, T., Johansson-Lindbom, B., Agace, W. W., Parker, C. M., and Powrie, F. (2005). Essential role for CD103 in the T cell mediated regulation of experimental colitis. J. Exp. Med. 202, 1051–1061.
Arima, K., Watanabe, N., Hanabuchi, S., Chang, M., Sun, S.-C., and Liu, Y.-J. (2010). Distinct signal codes generate dendritic cell functional plasticity. Sci. Signal. 3, ra4.
Awasthi, A., Carrier, Y., Peron, J. P. S., Bettelli, E., Kamanaka, M., Flavell, R. A., Kuchroo, V. K., Oukka, M., and Weiner, H. L. (2007). A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat. Immunol. 8, 1380–1389.
Baban, B., Chandler, P. R., Sharma, M. D., Pihkala, J., Koni, P. A., Munn, D. H., and Mellor, A. L. (2009). IDO activates regulatory T cells and blocks their conversion into Th17-Like T cells. J. Immunol. 183, 2475–2483.
Banchereau, J., and Steinman, R. M. (1998). Dendritic cells and the control of immunity. Nature 392, 245–252.
Basu, S., Golovina, T., Mikheeva, T., June, C. H., and Riley, J. L. (2008). Cutting edge: Foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J. Immunol. 180, 5794–5798.
Belkaid, Y., and Oldenhove, G. (2008). Tuning microenvironments: induction of regulatory T cells by dendritic cells. Immunity 29, 362–371.
Bennett, C. L., Christie, J., Ramsdell, F., Brunkow, M. E., Ferguson, P. J., Whitesell, L., Kelly, T. E., Saulsbury, F. T., Chance, P. F., and Ochs, H. D. (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21.
Bettelli, E., Oukka, M., and Kuchroo, V. K. (2007). TH-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 8, 345–350.
Bruder, D., Westendorf, A. M., Hansen, W., Prettin, S., Gruber, A. D., Qian, Y., von Boehmer, H., Mahnke, K., and Buer, J. (2005). On the edge of autoimmunity. Diabetes 54, 3395–3401.
Cederbom, L., Hall, H., and Ivars, F. (2000). CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur. J. Immunol. 30, 1538–1543.
Cella, M., Dohring, C., Samaridis, J., Dessing, M., Brockhaus, M., Lanzavecchia, A., and Colonna, M. (1997). A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J. Exp. Med. 185, 1743–1751.
Chang, X., Zheng, P., and Liu, Y. (2006). FoxP3: a genetic link between immunodeficiency and autoimmune diseases. Autoimmun. Rev. 5, 399–402.
Coombes, J. L., and Powrie, F. (2008). Dendritic cells in intestinal immune regulation. Nat. Rev. Immunol. 8, 435–446.
Coombes, J. L., Siddiqui, K. R. R., Arancibia-Cárcamo, C. V., Hall, J., Sun, C.-M., Belkaid, Y., and Powrie, F. (2007). A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764.
d’Hennezel, E., Ben-Shoshan, M., Ochs, H. D., Torgerson, T. R., Russell, L. J., Lejtenyi, C., Noya, F. J., Jabado, N., Mazer, B., and Piccirillo, C. A. (2009). FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. N. Engl. J. Med. 361, 1710–1713.
Esplugues, E., Huber, S., Gagliani, N., Hauser, A. E., Town, T., Wan, Y. Y., O’Connor, W., Rongvaux, A., Van Rooijen, N., Haberman, A. M., Iwakura, Y., Kuchroo, V. K., Kolls, J. K., Bluestone, J. A., Herold, K. C., and Flavell, R. A. (2011). Control of TH17 cells occurs in the small intestine. Nature 475, 514–518.
Fallarino, F., Grohmann, U., Hwang, K. W., Orabona, C., Vacca, C., Bianchi, R., Belladonna, M. L., Fioretti, M. C., Alegre, M.-L., and Puccetti, P. (2003). Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 4, 1206–1212.
Ghiringhelli, F. O., Puig, P. E., Roux, S., Parcellier, A., Schmitt, E., Solary, E., Kroemer, G., Martin, F. O., Chauffert, B., and Zitvogel, L. (2005). Tumor cells convert immature myeloid dendritic cells into TGF-β secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 202, 919–929.
Guiducci, C., Valzasina, B., Dislich, H., and Colombo, M. P. (2005). CD40/CD40L interaction regulates CD4+CD25+ T reg homeostasis through dendritic cell-produced IL-2. Eur. J. Immunol. 35, 557–567.
Guilliams, M., Crozat, K., Henri, S., Tamoutounour, S., Grenot, P., Devilard, E., de Bovis, B. A., Alexopoulou, L., Dalod, M., and Malissen, B. (2010). Skin-draining lymph nodes contain dermis-derived CD103 dendritic cells that constitutively produce retinoic acid and induce Foxp3+ regulatory T cells. Blood 115, 1958–1968.
Hanabuchi, S., Ito, T., Park, W.-R., Watanabe, N., Shaw, J. L., Roman, E., Arima, K., Wang, Y.-H., Voo, K. S., Cao, W., and Liu, Y.-J. (2010). Thymic stromal lymphopoietin-activated plasmacytoid dendritic cells induce the generation of FOXP3+ regulatory T cells in human thymus. J. Immunol. 184, 2999–3007.
Haribhai, D., Williams, J. B., Jia, S., Nickerson, D., Schmitt, E. G., Edwards, B., Ziegelbauer, J., Yassai, M., Li, S.-H., Relland, L. M., Wise, P. M., Chen, A., Zheng, Y.-Q., Simpson, P. M., Gorski, J., Salzman, N. H., Hessner, M. J., Chatila, T. A., and Williams, C. B. (2011). A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 35, 109–122.
Hawiger, D., Inaba, K., Dorsett, Y., Guo, M., Mahnke, K., Rivera, M., Ravetch, J. V., Steinman, R. M., and Nussenzweig, M. C. (2001). Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194, 769–780.
Hawiger, D., Masilamani, R. F., Bettelli, E., Kuchroo, V. K., and Nussenzweig, M. C. (2004). Immunological unresponsiveness characterized by increased expression of CD5 on peripheral T cells induced by dendritic cells in vivo. Immunity 20, 695–705.
Hori, S., Nomura, T., and Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 14, 1057–1061.
Huang, C.-T., Workman, C. J., Flies, D., Pan, X., Marson, A. L., Zhou, G., Hipkiss, E. L., Ravi, S., Kowalski, J., Levitsky, H. I., Powell, J. D., Pardoll, D. M., Drake, C. G., and Vignali, D. A. A. (2004). Role of LAG-3 in regulatory T cells. Immunity 21, 503–513.
Hubert, P., Jacobs, N., Caberg, J.-H., Boniver, J., and Delvenne, P. (2007). The cross-talk between dendritic and regulatory T cells: good or evil? J. Leukoc. Biol. 82, 781–794.
Igyarto, B. Z., Jenison, M. C., Dudda, J. C., Roers, A., Müller, W., Koni, P. A., Campbell, D. J., Shlomchik, M. J., and Kaplan, D. H. (2009). Langerhans cells suppress contact hypersensitivity responses via cognate CD4 interaction and Langerhans cell-derived IL-10. J. Immunol. 183, 5085–5093.
Iliev, I. D., Matteoli, G., and Rescigno, M. (2007). The yin and yang of intestinal epithelial cells in controlling dendritic cell function. J. Exp. Med. 204, 2253–2257.
Iliev, I. D., Mileti, E., Matteoli, G., Chieppa, M., and Rescigno, M. (2009). Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunol. 2, 340–350.
Ito, T., Yang, M., Wang, Y.-H., Lande, R., Gregorio, J., Perng, O. A., Qin, X.-F., Liu, Y.-J., and Gilliet, M. (2007). Plasmacytoid dendritic cells prime IL-10 producing T regulatory cells by inducible costimulator ligand. J. Exp. Med. 204, 105–115.
Jaensson, E., Uronen-Hansson, H., Pabst, O., Eksteen, B., Tian, J., Coombes, J. L., Berg, P.-L., Davidsson, T., Powrie, F., Johansson-Lindbom, B., and Agace, W. W. (2008). Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J. Exp. Med. 205, 2139–2149.
Kryczek, I., Wei, S., Zou, L., Zhu, G., Mottram, P., Xu, H., Chen, L., and Zou, W. (2006). Cutting edge: induction of B7-H4 on APCs through IL-10: novel suppressive mode for regulatory T cells. J. Immunol. 177, 40–44.
Kumanogoh, A., Wang, X., Lee, I., Watanabe, C., Kamanaka, M., Shi, W., Yoshida, K., Sato, T., Habu, S., Itoh, M., Sakaguchi, N., Sakaguchi, S., and Kikutani, H. (2001). Increased T cell autoreactivity in the absence of CD40–CD40 ligand interactions: a role of CD40 in regulatory T cell development. J. Immunol. 166, 353–360.
Kushwah, R., and Hu, J. (2011). Role of dendritic cells in the induction of regulatory T cells. Cell Biosci. 1, 20.
Kushwah, R., Wu, J., Oliver, J. R., Jiang, G., Zhang, J., Siminovitch, K. A., and Hu, J. (2010). Uptake of apoptotic DC converts immature DC into tolerogenic DC that induce differentiation of Foxp3+ Treg. Eur. J. Immunol. 40, 1022–1035.
Lehuen, A., Diana, J., Zaccone, P., and Cooke, A. (2010). Immune cell crosstalk in type 1 diabetes. Nat. Rev. Immunol. 10, 501–513.
Liang, B., Workman, C., Lee, J., Chew, C., Dale, B. M., Colonna, L., Flores, M., Li, N., Schweighoffer, E., Greenberg, S., Tybulewicz, V., Vignali, D., and Clynes, R. (2008). Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J. Immunol. 180, 5916–5926.
Loser, K., Mehling, A., Loeser, S., Apelt, J., Kuhn, A., Grabbe, S., Schwarz, T., Penninger, J. M., and Beissert, S. (2006). Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat. Med. 12, 1372–1379.
Mahnke, K., Guo, M., Lee, S., Sepulveda, H., Swain, S. L., Nussenzweig, M., and Steinman, R. M. (2000). The dendritic cell receptor for endocytosis, Dec-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 151, 673–684.
Mahnke, K., Johnson, T. S., Ring, S., and Enk, A. H. (2007). Tolerogenic dendritic cells and regulatory T cells: a two-way relationship. J. Dermatol. Sci. 46, 159–167.
Mahnke, K., Qian, Y., Knop, J., and Enk, A. H. (2003). Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood 101, 4862–4869.
Mahnke, K., Schmitt, E., Bonifaz, L., Enk, A. H., and Jonuleit, H. (2002). Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol. Cell Biol. 80, 477–483.
Maldonado, R. A., and von Andrian, U. H. (2010). How tolerogenic dendritic cells induce regulatory T cells. Adv. Immunol. 108, 111–165.
Manavalan, J. S., Rossi, P. C., Vlad, G., Piazza, F., Yarilina, A., Cortesini, R., Mancini, D., and Suciu-Foca, N. (2003). High expression of ILT3 and ILT4 is a general feature of tolerogenic dendritic cells. Transpl. Immunol. 11, 245–258.
Manicassamy, S., and Pulendran, B. (2011). Dendritic cell control of tolerogenic responses. Immunol. Rev. 241, 206–227.
Manicassamy, S., Ravindran, R., Deng, J., Oluoch, H., Denning, T. L., Kasturi, S. P., Rosenthal, K. M., Evavold, B. D., and Pulendran, B. (2009). Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat. Med. 15, 401–409.
Manicassamy, S., Reizis, B., Ravindran, R., Nakaya, H., Salazar-Gonzalez, R. M., Wang, Y.-C., and Pulendran, B. (2010). Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 329, 849–853.
Martin-Gayo, E., Sierra-Filardi, E., Corbí, A. L., and Toribio, M. L. (2010). Plasmacytoid dendritic cells resident in human thymus drive natural Treg cell development. Blood 115, 5366–5375.
Matteoli, G., Mazzini, E., Iliev, I. D., Mileti, E., Fallarino, F., Puccetti, P., Chieppa, M., and Rescigno, M. (2010). Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut 59, 595–604.
Mellor, A. L., Chandler, P., Baban, B., Hansen, A. M., Marshall, B., Pihkala, J., Waldmann, H., Cobbold, S., Adams, E., and Munn, D. H. (2004). Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int. Immunol. 16, 1391–1401.
Mellor, A. L., and Munn, D. H. (2004). Ido expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol. 4, 762–774.
Menges, M., Robner, S., Voigtlander, C., Schindler, H., Kukutsch, N. A., Bogdan, C., Erb, K., Schuler, G., and Lutz, M. B. (2002). Repetitive injections of dendritic cells matured with tumor necrosis factor induce antigen-specific protection of mice from autoimmunity. J. Exp. Med. 195, 15–22.
Morelli, A. E., and Thomson, A. W. (2007). Tolerogenic dendritic cells and the quest for transplant tolerance. Nat. Rev. Immunol. 7, 610–621.
Nguyen, N. T., Kimura, A., Nakahama, T., Chinen, I., Masuda, K., Nohara, K., Fujii-Kuriyama, Y., and Kishimoto, T. (2010). Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 19961–19966.
Oderup, C., Cederbom, L., Makowska, A., Cilio, C. M., and Ivars, F. (2006). Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+CD25+ regulatory T-cell-mediated suppression. Immunology 118, 240–249.
Onishi, Y., Fehervari, Z., Yamaguchi, T., and Sakaguchi, S. (2008). Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. U.S.A. 105, 10113–10118.
Pasare, C., and Medzhitov, R. (2003). Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036.
Perruche, S., Zhang, P., Liu, Y., Saas, P., Bluestone, J. A., and Chen, W. (2008). CD3-specific antibody-induced immune tolerance involves transforming growth factor-[beta] from phagocytes digesting apoptotic T cells. Nat. Med. 14, 528–535.
Pot, C., Apetoh, L., and Kuchroo, V. K. (2011). Type 1 regulatory T cells (Tr1) in autoimmunity. Semin. Immunol. 23, 202–208.
Pot, C., Jin, H., Awasthi, A., Liu, S. M., Lai, C.-Y., Madan, R., Sharpe, A. H., Karp, C. L., Miaw, S.-C., Ho, I.-C., and Kuchroo, V. K. (2009). Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol. 183, 797–801.
Proietto, A. I., van Dommelen, S., Zhou, P., Rizzitelli, A., D’Amico, A., Steptoe, R. J., Naik, S. H., Lahoud, M. H., Liu, Y., Zheng, P., Shortman, K., and Wu, L. (2008). Dendritic cells in the thymus contribute to T-regulatory cell induction. Proc. Natl. Acad. Sci. U.S.A. 105, 19869–19874.
Pulendran, B., Tang, H., and Manicassamy, S. (2010). Programming dendritic cells to induce TH2 and tolerogenic responses. Nat. Immunol. 11, 647–655.
Read, S., Malmstrom, V., and Powrie, F. (2000). Cytotoxic T lymphocyte associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 192, 295–302.
Rescigno, M. (2010). Dendritic cells in tolerance induction for the treatment of autoimmune diseases. Eur. J. Immunol. 40, 2119–2123.
Rutella, S., Danese, S., and Leone, G. (2006). Tolerogenic dendritic cells: cytokine modulation comes of age. Blood 108, 1435–1440.
Sakaguchi, S. (2000). Regulatory T cells: key controllers of immunologic self-tolerance. Cell 101, 455–458.
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M., and Toda, M. (1995). Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164.
Sakaguchi, S., Wing, K., Onishi, Y., Prieto-Martin, P., and Yamaguchi, T. (2009). Regulatory T cells: how do they suppress immune responses? Int. Immunol. 21, 1105–1111.
Salomon, B. T., Lenschow, D. J., Rhee, L., Ashourian, N., Singh, B., Sharpe, A., and Bluestone, J. A. (2000). B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12, 431–440.
Sarris, M., Andersen, K. G., Randow, F., Mayr, L., and Betz, A. G. (2008). Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity 28, 402–413.
Sgouroudis, E., Kornete, M., and Piccirillo, C. A. (2011). IL-2 production by dendritic cells promotes Foxp3+ regulatory T-cell expansion in autoimmune-resistant NOD congenic mice. Autoimmunity 44, 406–414.
Shortman, K., and Naik, S. H. (2007). Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7, 19–30.
Steinman, R. M., Hawiger, D., and Nussenzweig, M. C. (2003). Tolerogenic dendritic cells. Annu. Rev. Immunol. 21, 685–711.
Steinman, R. M., and Nussenzweig, M. C. (2002). Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. U.S.A. 99, 351–358.
Strauss, L., Whiteside, T. L., Knights, A., Bergmann, C., Knuth, A., and Zippelius, A. (2007). Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J. Immunol. 178, 320–329.
Sun, C.-M., Hall, J. A., Blank, R. B., Bouladoux, N., Oukka, M., Mora, J. R., and Belkaid, Y. (2007). Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204, 1775–1785.
Svajger, U., Vidmar, A., and Jeras, M. (2008). Niflumic acid renders dendritic cells tolerogenic and up-regulates inhibitory molecules ILT3 and ILT4. Int. Immunopharmacol. 8, 997–1005.
Tadokoro, C. E., Shakhar, G., Shen, S., Ding, Y., Lino, A. C., Maraver, A., Lafaille, J. J., and Dustin, M. L. (2006). Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J. Exp. Med. 203, 505–511.
Takahashi, T., Kuniyasu, Y., Toda, M., Sakaguchi, N., Itoh, M., Iwata, M., Shimizu, J., and Sakaguchi, S. (1998). Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10, 1969–1980.
Tang, Q., Adams, J. Y., Tooley, A. J., Bi, M., Fife, B. T., Serra, P., Santamaria, P., Locksley, R. M., Krummel, M. F., and Bluestone, J. A. (2006). Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 7, 83–92.
Tarbell, K. V., Yamazaki, S., Olson, K., Toy, P., and Steinman, R. M. (2004). CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J. Exp. Med. 199, 1467–1477.
Tran, D. Q., Glass, D. D., Uzel, G., Darnell, D. A., Spalding, C., Holland, S. M., and Shevach, E. M. (2009). Analysis of adhesion molecules, target cells, and role of IL-2 in human FOXP3+ regulatory T cell suppressor function. J. Immunol. 182, 2929–2938.
Turnquist, H. R., Raimondi, G., Zahorchak, A. F., Fischer, R. T., Wang, Z., and Thomson, A. W. (2007). Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J. Immunol. 178, 7018–7031.
Ueno, H., Klechevsky, E., Morita, R., Aspord, C., Cao, T., Matsui, T., Di Pucchio, T., Connolly, J., Fay, J. W., Pascual, V., Palucka, A. K., and Banchereau, J. (2007). Dendritic cell subsets in health and disease. Immunol. Rev. 219, 118–142.
Vremec, D., Pooley, J., Hochrein, H., Wu, L., and Shortman, K. (2000). CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164, 2978–2986.
Wakkach, A., Fournier, N., Brun, V. R., Breittmayer, J.-P., Cottrez, F. O., and Groux, H. (2003). Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity 18, 605–617.
Watanabe, N., Wang, Y.-H., Lee, H. K., Ito, T., Wang, Y.-H., Cao, W., and Liu, Y.-J. (2005). Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature 436, 1181–1185.
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., Nomura, T., and Sakaguchi, S. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275.
Wing, K., and Sakaguchi, S. (2010). Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 11, 7–13.
Workman, C. J., and Vignali, D. A. A. (2005). Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J. Immunol. 174, 688–695.
Wu, L., and Shortman, K. (2005). Heterogeneity of thymic dendritic cells. Semin. Immunol. 17, 304–312.
Yamazaki, S., Dudziak, D., Heidkamp, G. F., Fiorese, C., Bonito, A. J., Inaba, K., Nussenzweig, M. C., and Steinman, R. M. (2008). CD8+CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J. Immunol. 181, 6923–6933.
Yamazaki, S., Iyoda, T., Tarbell, K., Olson, K., Velinzon, K., Inaba, K., and Steinman, R. M. (2003). Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J. Exp. Med. 198, 235–247.
Zhou, X., Kong, N., Wang, J., Fan, H., Zou, H., Horwitz, D., Brand, D., Liu, Z., and Zheng, S. G. (2010). Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J. Immunol. 185, 2675–2679.
Keywords: Foxp3, immunity, suppression, tolerance, tolerogenic DC
Citation: Kornete M and Piccirillo CA (2012) Functional crosstalk between dendritic cells and Foxp3+ regulatory T cells in the maintenance of immune tolerance.Front. Immun. 3:165. doi: 10.3389/fimmu.2012.00165
Received: 05 April 2012; Paper pending published: 02 May 2012;
Accepted: 01 June 2012; Published online: 22 June 2012.
Edited by:
Francesca Granucci, University of Milano-Bicocca, ItalyReviewed by:
Francesca Granucci, University of Milano-Bicocca, ItalyIvan Zanoni, University of Milano-Bicocca, Italy
Copyright: © 2012 Kornete and Piccirillo. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ciriaco A. Piccirillo, Federation of Clinical Immunology Societies Center of Excellence, Research Institute of the McGill University Health Center, Montreal General Hospital, 1650 Cedar Avenue, Room L11.132, Montreal, QC, Canada H3G 1A4. e-mail:Y2lyby5waWNjaXJpbGxvQG1jZ2lsbC5jYQ==