Abstract
T cell signaling begins with the ligation of the T cell antigen receptor (TCR) by a cognate peptide and the phosphorylation of the receptor’s immunoreceptor tyrosine-based activation motif domains by the kinase Lck. However, the canonical receptor model is insufficient to explain how the constitutively active kinase Lck can discriminate between non-ligated and ligated TCRs. Here, we discuss the factors that are thought to regulate the spatial distribution of the TCR and Lck, and therefore critically influence TCR signaling initiation.
INTRODUCTION
The family of Src kinases play key roles in the signal transduction of many cell surface receptors in a diverse range of cellular functions, such as cell growth, differentiation, migration, and survival (Bromann et al., 2004; Parsons and Parsons, 2004). In T cells, the lymphocyte-specific protein tyrosine kinase (Lck) is critical in the early propagation and modulation of T cell receptor (TCR) signaling. TCR signaling is triggered by the recognition and engagement of antigenic peptides bound to major histocompatibility complex (MHC) and relies on the phosphorylation by Lck of receptor complex at immunoreceptor tyrosine-based activation motif (ITAM) consensus sites. Phosphorylated ITAMs recruit zeta-chain-associated protein kinase 70 (Zap70), which requires additional phosphorylation by Lck to be activated (Wange and Samelson, 1996; Palacios and Weiss, 2004). Zap70 in turn phosphorylates other proteins in TCR signaling cascade, which eventually leads to T cell activation.
Lck is bound to the plasma membrane via myristoylated and palmitoylated residues at its N-terminus. It also contains a SH3 and a SH2 domains next to the membrane anchor followed by a catalytic tyrosine kinase domain and a short C-terminal tail. As an essential kinase in the propagation of TCR signaling, the regulation of Lck activity is itself tightly controlled by conformational changes mainly relying on phosphorylation and dephosphorylation on two regulatory tyrosine residues. When the C-terminal inhibitory tyrosine (Tyr505) is phosphorylated, it can interact with its own SH2 domain, which causes an intra-molecular arrangement and locks the kinase in an inactive or “closed” conformation (Figure 1). SH3 and SH2 domains intra molecular interactions also contribute to stabilize the closed conformation (Boggon and Eck, 2004). In contrast, phosphorylation of the activating tyrosine (Tyr394) stabilizes the activation loop in an active conformation (Yamaguchi and Hendrickson, 1996; Boggon and Eck, 2004). An intermediary form in which Lck is phosphorylated on both regulatory tyrosine residues also exists and is catalytically active. This double phosphorylated form accounts for ~20% of all Lck in T lymphocyte (D’Oro and Ashwell, 1999; Nika et al., 2010). The inhibitory Tyr505 residue is phosphorylated by C-terminal Src kinase (Csk) and dephosphorylated by the tyrosine phosphatase CD45 (Alexander, 2000; Palacios and Weiss, 2004). The activating Tyr394 residue is phosphorylated by Lck in a process of transphosphorylation. Tyr394 is also dephosphorylated by SH2 domain-containing phosphatase-1 (SHP-1; Stefanová et al., 2003) and CD45, which can hence positively and negatively regulate Lck activity (Alexander, 2000; Saunders and Johnson, 2010). Currently the consensus is that CD45 acts predominately negatively on Lck activity (D’Oro and Ashwell, 1999; Wong et al., 2008) because high levels of CD45 are required to dephosphorylate Y394 while low levels are sufficient for Y505 dephosphorylation and Lck activation (McNeill et al., 2007; Zikherman et al., 2010). This suggests that Lck is kept inactive by high level of CD45 in resting cells, whereas partial segregation of the two molecules upon TCR activation reduces the levels of CD45 and favors dephosphorylation at Y505 and hence Lck activation. All in all, the various combinations of kinases and phosphatase accounts for the complex regulatory mechanism of Lck (Figure 1).
FIGURE 1
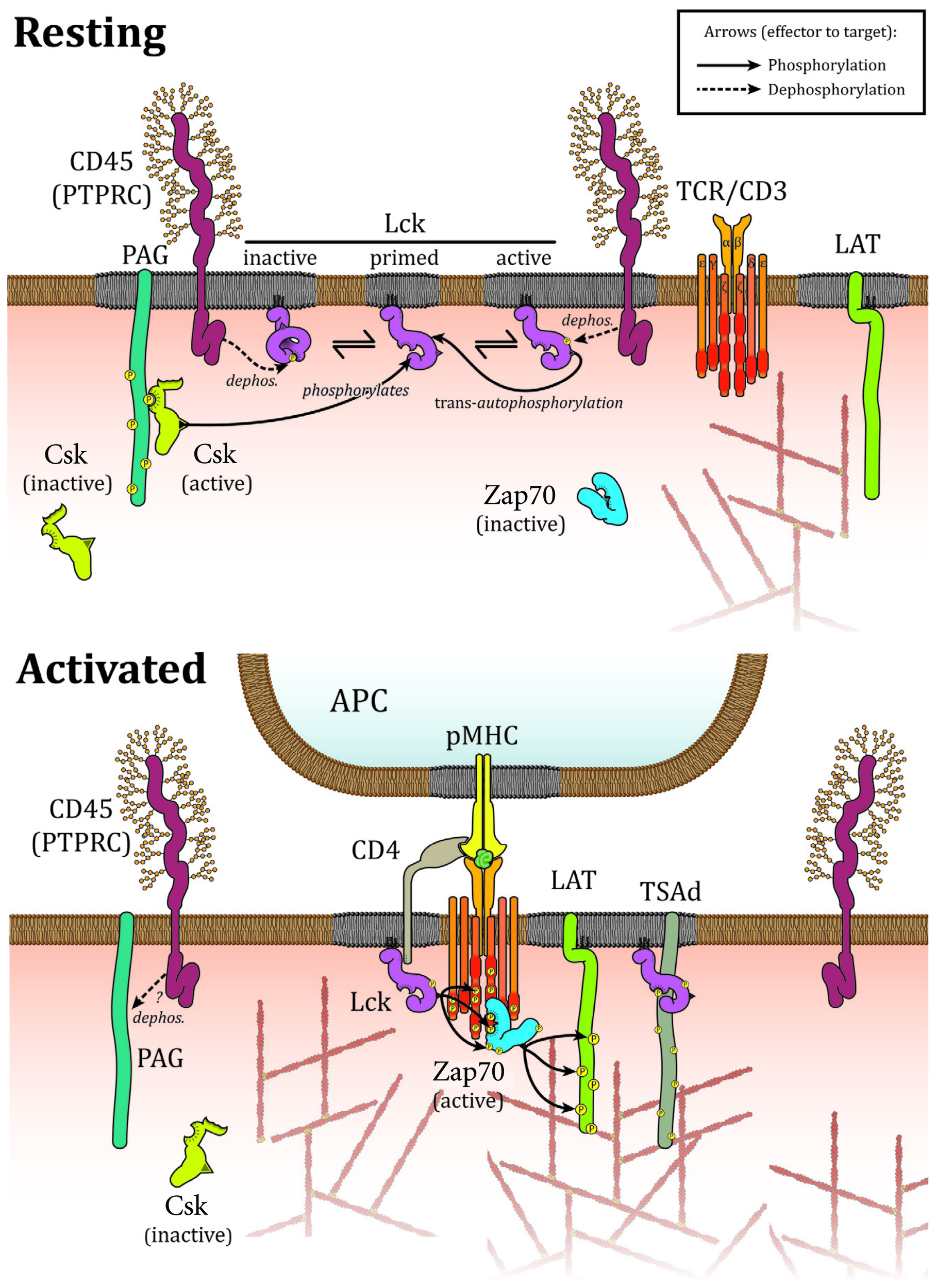
Interactions controlling Lck localization upon TCR activation. In resting cells, Lck is located in microdomains where it interacts directly with CD45 that acts both on activating Tyr394 and inhibiting Tyr505. Csk is targeted to membrane microdomains by PAG and aids to limit Lck activity via Tyr505 phosphorylation. Association with membrane domains and protein–protein interactions prevent constitutively active Lck to phosphorylate TCR or Zap70 in resting cells. Upon activation, CD45 is excluded from membrane microdomains and Csk detaches from the plasma membrane as a consequence of PAG dephosphorylation by CD45. Lck oligomerizes, binds to and phosphorylates TCR and Zap70 to mediate TCR signaling. Interactions with TSAd and CD4/8 contribute to enhance TCR binding and signaling.
In resting T lymphocytes, a large percentage of Lck is constitutively active. More surprisingly, however, TCR activation has no impact on Lck activity with the proportion of the different phosphorylation and conformational states of Lck remaining relatively unchanged (Secrist et al., 1993; Paster et al., 2009; Nika et al., 2010). This implies that a pool of constitutively active Lck is responsible for the propagation of TCR signaling. Lck activity is thought to be held in check under resting conditions by phosphatases and Csk. Consequently, suppression of phosphatases activity by pervanadate treatment markedly enhances phosphorylation of Y394 and promotes T cell activation (Secrist et al., 1993). Similarly, inhibiting Csk activity triggers phosphorylation of Lck at Tyr394 and TCR signaling (Schoenborn et al., 2011). Taken together, it seems that Lck kinase activity and TCR phosphorylation are critically regulated by the interaction probabilities of kinases, phosphatases, and their substrates.
SPATIAL PATTERNING OF THE IMMUNOLOGICAL SYNAPSE
Since the binding of TCR to antigenic peptides does not increase the proportion of active Lck, the mechanisms that allow the kinase to initiate signaling only when the receptor is peptide-bound are likely to depend on interaction probabilities and hence spatial distributions within the plasma membrane of Lck relative to its modulators and targets. In this model, ITAM phosphorylation is balanced by the access of constitutively active Lck to TCRs relative to the action of phosphatases. To characterize the spatial patterns of signaling proteins at T cell activation sites and immunological synapses and identify the mechanisms of the lateral membrane organization has therefore been a long-standing goal of many researchers.
When stimulated with antigen presenting cells, TCR and signaling proteins redistribute to first form so-called TCR microclusters (Seminario and Bunnell, 2008) and then the bull-eye pattern of a mature immunological synapse (Grakoui et al., 1999; Dustin, 2009). The diffusion of Lck molecules within the plasma membrane is indeed affected by TCR activation: Lck molecules display heterogeneous diffusion in activated cells with an increased residency time in microdomains that contain TCR (Ike et al., 2003; Douglass and Vale, 2005). Lck distribution matches that of TCR in activated cells, and both molecules are recruited simultaneously to the immunological synapse upon activation (Ehrlich et al., 2002) where they co-localize in the center of the synapse (Monks et al., 1998; Freiberg et al., 2002). However, active Lck is found at the periphery of the immunological synapse, where it initiates TCR phosphorylation and signaling cascades (Campi et al., 2005). Interestingly, TCR clusters form before and independently of Lck (Campi et al., 2005) suggesting that Lck is somehow recruited to pre-formed TCR clusters. Importantly, CD45 and Lck have distinct and mutually exclusive localizations in activated T cells, consistent with the notion that upon TCR triggering, CD45 mainly limits Lck activity and TCR phosphorylation (D’Oro and Ashwell, 1999; McNeill et al., 2007; Wong et al., 2008). Several studies report CD45 exclusion from TCR-containing areas within the immunological synapse (Johnson, 2000; Leupin et al., 2000) and more precisely from TCR microclusters (Varma et al., 2006; Kaizuka et al., 2009). Additionally, CD45 is excluded from CD2 clusters that contain Lck and the adaptor linker for activation of T cells (LAT; Douglass and Vale, 2005). Similarly, Csk loses its membrane association upon T cell activation, segregating de facto from membrane-associated Lck (Brdika et al., 2000; Davidson et al., 2003). In summary, the distribution of Lck is modified by TCR engagement, resulting in the recruitment of Lck to TCR microclusters and segregation of Lck from CD45 and Csk. However, the molecular mechanisms of these redistributions are poorly understood. Below, we discuss the interactions that may regulate Lck localization and distribution.
INTERACTIONS GOVERNING Lck LOCALIZATION AND DISTRIBUTION
The first described mechanism that controls Lck localization is the binding of Lck to the coreceptors CD4 and CD8. Lck directly associate with CD4 and CD8 in T cells (Rudd et al., 1988; Veillette et al., 1988) – an interaction that is mediated by zinc, which allows the cytoplasmic tails of CD4/8 to interact with the N-terminal region of Lck and to form heterodimers (Lin et al., 1998; Kim et al., 2003). The major function of CD4/8 is to enhance the recruitment of Lck to the immunological synapse (Holdorf et al., 2002) and deliver Lck to the TCR–pMHC complex (Li et al., 2004; Van Laethem et al., 2007; Artyomov et al., 2010). However, the TCR complex was able to induce signaling even the absence of coreceptors (Locksley et al., 1993; Schilham et al., 1993; Van Laethem et al., 2007), emphasizing a role for CD4/8 mediated-delivery of Lck in enhancing specificity of TCR binding to pMHC rather than being a prerequisite for TCR signaling.
Protein–protein interactions play a dominant role in membrane distributions since such interactions regulate diffusion and often result in immobile clusters. The SH2 domain of Lck, which is essential for TCR signaling, is a mediator of such protein interactions (Table 1). In the inactive conformation, the SH2 domain of Lck binds to the inhibitory phosphotyrosine (pTyr505) of Lck, preventing interactions with other proteins (Eck et al., 1994; Xu et al., 1999). However, the affinity of the SH2 domain for pTyr505 is relatively weak (Nika et al., 2007) allowing competitive binding with other proteins such as TCR and Zap70 (Duplay et al., 1994; Straus et al., 1996; Yamasaki et al., 1996). Interestingly, it is the phosphotyrosine binding property of the SH2 domain that mediates the interactions with the phosphorylated ζ chain of TCR and phosphorylated Zap70 (Lewis et al., 1997). Thus, the SH2 domain can maintain a stable association of Lck with its main targets after they have been phosphorylated, effectively consolidating the protein network for sustained signaling and signal amplification (Pawson, 2004). After TCR engagement, oligomerization of Lck, driven by a “head-to-toe” SH2 and SH3 domains association (Lee-Fruman et al., 1996) likely contributes to the amplification of TCR signaling. However, to which extend specific SH2 interactions are essential or could substitute CD4/8 coreceptor engagement is currently not clear. The SH2 domains may also confer intrinsic adaptor properties to Lck, as a potential kinase independent function (Xu and Littman, 1993).
Table 1
| Interacting with | Lck part involved | Type of interaction | Effect onTCR signaling | Reference |
|---|---|---|---|---|
| CD4/8 | N-terminus | Zinc mediated | Enhanced | Lin et al. (1998), Kim et al. (2003) |
| Zap70 | SH2 | SH2–pTyr | Enhanced | Duplay et al. (1994), Straus et al. (1996), Yamasaki et al. (1996) |
| TCRζ | SH2 | SH2–pTyr | Enhanced | Straus et al. (1996) |
| pZap70, pTCRζ | SH2 | SH2–pTyr | Enhanced | Lewis et al. (1997) |
| Lck | SH2, SH3 | SH2–SH3 | Enhanced | Lee-Fruman et al. (1996) |
| CD45 | SH2 | pTry independent SH2 | Decreased | Ng et al. (1996) |
| Csk | pTyr394 | SH2–pTyr | Decreased | Bougeret et al. (1996) |
| LIME | SH2 | SH2–pTyr | Decreased | Brdicková et al. (2003), Hur et al. (2003) |
| TSAd | SH2 | SH2–pTyr | Increased | Marti et al. (2006), Granum et al. (2008) |
| LAT | Unknown | Charges mediated | Decreased | Kabouridis et al. (2011) |
Lck molecular interactions.
SH2 domain interactions not only regulate Lck access to TCR and Zap70, but also take part in the control of Lck distribution relative to proteins that regulate its activity (Table 1). A phosphotyrosine-independent interaction mediates the binding of Lck’s SH2 domain to CD45 (Ng et al., 1996). Additionally, the SH2 domain of Csk binds to activating pTyr394 in Lck (Bougeret et al., 1996), probably preventing Lck hyperactivation. Lck and Csk interact indirectly as well, via SH2 mediated binding to the adaptor LIME (Brdicková et al., 2003; Hur et al., 2003). The SH2 domains of Lck also bind to the T cell-specific adapter protein (TSAd) that in turn positively regulates Lck activity upon TCR activation (Marti et al., 2006; Granum et al., 2008). Hence, the SH2 domain of Lck does not only contribute to the enhancement and propagation of TCR signaling, but also participate in regulating Lck activity by linking it to CD45 Csk and TSAd. Additionally, direct interaction of CD45 with CD3ζ ITAM domains may contribute to the differential localization and interaction of Lck and CD45 in resting and activated cells (Furukawa, 1994).
Other SH2 domain-independent protein–protein interactions also play a role in controlling Lck distribution. The diffusion and localization of membrane proteins including Lck can also be regulated by the actin cytoskeleton. Actin polymerization is essential in the formation of TCR microclusters, but is not required for their maintenance once they are established (Campi et al., 2005; Seminario and Bunnell, 2008; Kaizuka et al., 2009). Lck associates with actin upon TCR engagement, and is responsible for the SH2 domain-dependant anchoring of TCRζ to the actin cytoskeleton (Rozdzial et al., 1998). However, more evidence is required to understand how Lck distribution is regulated by the actin cytoskeleton. Other protein–protein interactions of Lck with effectors include binding to LAT, which is recruited and phosphorylated by Zap70 and interacts with the active form of Lck upon TCR stimulation. Lck interaction with LAT is electrostatic and impacts negatively on Lck activity (Kabouridis et al., 2011). Finally, extracellular protein–protein interactions can also act on Lck distribution: a lattice formed by galectin, a glycoprotein binding protein, can contribute to maintain Lck and CD45 in the common membrane microdomains in resting cells, favoring Lck inhibition by CD45 (Chen et al., 2007).
It has previously suggested that residency in membrane microdomains or lipid rafts controls Lck distribution relative to effectors and targets. The myristoylation and palmitoylation of Lck’s membrane binding domain target the kinase to cholesterol- and glycolipid-enriched microdomains (Shenoy-Scaria et al., 1994; Filipp et al., 2003; Rodgers et al., 2005). These membrane microdomains are thought to aid the translocation of Lck to the immunological synapse after activation by antigen presenting cells (Jordan and Rodgers, 2003). More specifically, active Lck translocates to lipid rafts upon TCR engagement (Filipp et al., 2003). Such membrane compartmentalization is especially important for the regulation of Lck distributions relative to CD45 and Csk, thereby governing Lck activation status (Figure 1). Indeed, replacing the membrane targeting motif of Lck with that of Fyn results in aberrant TCR signaling (Salmond et al., 2011) and hyper-phosphorylation of the inhibitory Tyr505 (Gervais and Veillette, 1995). However, even though TCR activation reduces CD45 association with lipid rafts (Edmonds and Ostergaard, 2002), the evidence whether CD45 and Lck localization in the same microdomains favors or prevents Lck activation is confusing (Rodgers and Rose, 1996; Chichili et al., 2010). PAG/Cbp is a raft-associated membrane protein that interacts with Csk and localizes the kinase to lipid rafts in resting cells (Brdika et al., 2000; Kawabuchi et al., 2000) where it contributes to Lck inhibition by phosphorylating the inhibitory Tyr505. Upon T cell activation, PAG is dephosphorylated and Csk is removed from the plasma membrane to allow Lck activation (Brdika et al., 2000; Davidson et al., 2003). Other evidence for the importance of lipid domains comes from coreceptors: CD28 induces the recruitment of Lck to lipid rafts (Tavano et al., 2006), where Lck associates with CD4, which in turn controls raft aggregation (Fragoso et al., 2003). However, the controversy about methodologies used lipid raft studies (Munro, 2003) and the questions about their very existence (Nichols, 2005) as well as the fact that CD45 can either activate and deactivate Lck (Saunders and Johnson, 2010) prevent a clear understanding of the lipid rafts role in the control of Lck distribution. In a landmark study, Douglass and Vale (2005) have shown that the membrane anchor of Lck that targets the proteins to lipid raft does not control Lck diffusion and microclusters localization. Although membrane lipid composition is likely to be involved in the control of Lck distribution, it could be via other mechanisms, such as the electrostatic effects of the charges-mediated binding of TCRζ chain basic residues to the plasma membrane, which controls TCR–Lck interactions and hence localizations (Zhang et al., 2011). It should also be noted that spatial control of Lck distribution can be achieved without proteins or lipids interactions as proposed by the kinetic-segregation model. This model postulates that close-contacts zones are created when engaged TCR shorten the distance between T cell and APC membranes. CD45 that bears a large ectodomain is excluded from these areas, generating a favorable environment for TCR and Zap70 phosphorylation by Lck (Davis and van der Merwe, 2006).
CONCLUSION
Since Lck is constitutively active and TCR engagement does not modulate Lck activity per se, it is thought that the spatial distributions of Lck relative to its substrates and other kinases and phosphatases minimize ITAM phosphorylation in resting T cells while fast and efficiently phosphorylating the receptor in activated cells. Evidence has been presented for various mechanisms that may regulate Lck localizations in the plasma membrane such as SH2 domain-mediated interactions with TCR signaling proteins and Lck activity-regulating proteins, SH2 domain-independent scaffolding functions, association with lipid rafts and redistribution caused by the close contact zone between cellular membranes. It is very likely that Lck localization is regulated by the same processes that allow the formation of compositionally distinct TCR microclusters and synapse patterns upon TCR triggering. New single-molecule fluorescence microscopy methods that measure protein distributions with nanometer precision may afford new insights into protein distributions during T cellactivation (Lillemeier et al., 2010; Williamson et al., 2011) that allow us to determine the interdependencies between Lck activity, conformational states and TCR phosphorylation.
Statements
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Alexander D. R. (2000). The CD45 tyrosine phosphatase: a positive and negative regulator of immune cell function.Semin. Immunol.12349–359.
2
Artyomov M. N. Lis M. Devadas S. Davis M. M. Chakraborty A. K. (2010). CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery.Proc. Natl. Acad. Sci. U.S.A.10716916–16921.
3
Boggon T. J. Eck M. J. (2004). Structure and regulation of Src family kinases.Oncogene237918–7927.
4
Bougeret C. Delaunay T. Romero F. Jullien P. Siegmund F. (1996). Detection of a physical and functional interaction between Csk and Lck which involves the SH2 domain of Csk and is mediated by autophosphorylation of Lck on tyrosine 394.J. Biol. Chem.2717465–7472.
5
Brdicková N. Brdicka T. Angelisová P. Horváth O. Spicka J. Hilgert I. Paces J. Simeoni L. Kliche S. Merten C. Schraven B Horejsí V. (2003). LIME: a new membrane Raft-associated adaptor protein involved in CD4 and CD8 coreceptor signaling.J. Exp. Med.1981453–1462.
6
Brdika T. Pavlistova D. Albrecht L. Bruyns E. Schraven B. (2000). Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase Csk and is involved in regulation of T cell activation.J. Exp. Med.1911591–1604.
7
Bromann P. A. Korkaya H. Courtneidge S. A. (2004). The interplay between Src family kinases and receptor tyrosine kinases.Oncogene237957–7968.
8
Campi G. Varma R. Dustin M. L. (2005). Actin and agonist MHC–peptide complex-dependent T cell receptor microclusters as scaffolds for signaling.J. Exp. Med.2021031–1036.
9
Chen I.-J. Chen H.-L. Demetriou M. (2007). Lateral compartmentalization of T cell receptor versus CD45 by galectin-N-glycan binding and microfilaments coordinate basal and activation signaling.J. Biol. Chem.28235361–35372.
10
Chichili G. R. Westmuckett a. D. Rodgers W. (2010). T cell signal regulation by the actin cytoskeleton.J. Biol. Chem.28514737–14746.
11
Davidson D. Bakinowski M. Thomas M. L. Horejsi V. Veillette A. (2003). Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor.Mol. Cell. Biol.232017–2028.
12
Davis S. J van der Merwe P. A. (2006). The kinetic-segregation model: TCR triggering and beyond.Nat. Immunol.7803–809.
13
D’Oro U. Ashwell J. D. (1999). Cutting edge: the CD45 tyrosine phosphatase is an inhibitor of Lck activity in thymocytes.J. Immunol.1621879–1883.
14
Douglass A. D. Vale R. D. (2005). Single-molecule microscopy reveals plasma membrane microdomains created by protein–protein networks that exclude or trap signaling molecules in T cells.Cell121937–950.
15
Duplay P. Thome M. Hervé F. Acuto O. (1994). p56lck interacts via its src homology 2 domain with the ZAP-70 kinase.J. Exp. Med.1791163–1172.
16
Dustin M. L. (2009). The cellular context of T cell signaling.Immunity30482–492.
17
Eck M. J. Atwell S. K. Shoelson S. E. Harrison S. C. (1994). Structure of the regulatory domains of the Src-family tyrosine kinase Lck.Nature368764–769.
18
Edmonds S. D. Ostergaard H. L. (2002). Dynamic association of CD45 with detergent-insoluble microdomains in T lymphocytes.J. Immunol.1695036–5042.
19
Ehrlich L. I. R. Ebert P. J. R. Krummel M. F. Weiss A. Davis M. M. (2002). Dynamics of p56lck translocation to the T cell immunological synapse following agonist and antagonist stimulation.Immunity17809–822.
20
Filipp D. Zhang J. Leung B. L. Shaw A. Levin S. D. Veillette A. Julius M. (2003). Regulation of Fyn through translocation of activated Lck into lipid rafts.J. Exp. Med.1971221–1227.
21
Fragoso R. Ren D. Zhang X. Su M. W.-C. Burakoff S. J. Jin Y.-J. (2003). Lipid raft distribution of CD4 depends on its palmitoylation and association with Lck, and evidence for CD4-induced lipid raft aggregation as an additional mechanism to enhance CD3 signaling.J. Immunol.170913–921.
22
Freiberg B. A. Kupfer H. Maslanik W. Delli J. Kappler J. Zaller D. M. Kupfer A. (2002). Staging and resetting T cell activation in SMACs.Nat. Immunol.3911–917.
23
Furukawa T. (1994). Specific interaction of the CD45 protein-tyrosine phosphatase with tyrosine-phosphorylated CD3ζ; chain.Proc. Natl. Acad. Sci.9110928–10932.
24
Gervais F. G. Veillette A. (1995). The unique amino-terminal domain of p56lck regulates interactions with tyrosine protein phosphatases in T lymphocytes.Mol. Cell. Biol.152393–2401.
25
Grakoui A. Bromley S. K. Sumen C. Davis M. M. Shaw A. S. Allen P. M. Dustin M. L. (1999). The immunological synapse: a molecular machine controlling T cell activation.Science285221–227.
26
Granum S. Andersen T. C. B. Sørlie M. Jørgensen M. Koll L. Berge T. Lea T. Fleckenstein B. Spurkland A. Sundvold-Gjerstad V. (2008). Modulation of Lck function through multisite docking to T cell-specific adapter protein.J. Biol. Chem.28321909–21919.
27
Holdorf A. D. Lee K.-H. Burack W. R. Allen P. M. Shaw A. S. (2002). Regulation of Lck activity by CD4 and CD28 in the immunological synapse.Nat. Immunol.3259–264.
28
Hur E. M. Son M. Lee O.-H. Choi Y. B. Park C. Lee H. Yun Y. (2003). LIME, a novel transmembrane adaptor protein, associates with p56lck and mediates T cell activation.J. Exp. Med.1981463–1473.
29
Ike H. Kosugi A. Kato A. Iino R. Hirano H. Fujiwara T. Ritchie K. Kusumi A. (2003). Mechanism of Lck recruitment to the T-cell receptor cluster as studied by single-molecule-fluorescence video imaging.Chemphyschem4620–626.
30
Johnson K. G. (2000). A supramolecular basis for CD45 tyrosine phosphatase regulation in sustained T cell activation.Proc. Nat. Acad. Sci. U.S.A.9710138–10143.
31
Jordan S. Rodgers W. (2003). T cell glycolipid-enriched membrane domains are constitutively assembled as membrane patches that translocate to immune synapses.J. Immunol.17178–87.
32
Kabouridis P. S. Isenberg D. A. Jury E. C. (2011). A negatively charged domain of LAT mediates its interaction with the active form of Lck.Mol. Membr. Biol.28487–494.
33
Kaizuka Y. Douglass A. D. Vardhana S. Dustin M. L. Vale R. D. (2009). The coreceptor CD2 uses plasma membrane microdomains to transduce signals in T cells.J. Cell Biol.185521–534.
34
Kawabuchi M. Satomi Y. Takao T. Shimonishi Y. Nada S. Nagai K. Tarakhovsky A. Okada M. (2000). Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases.Nature404999–1003.
35
Kim P. W. Sun Z.-Y. J. Blacklow S. C. Wagner G. Eck M. J. (2003). A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8.Science3011725–1728.
36
Lee-Fruman K. K. Collins T. L. Burakoff S. J. (1996). Role of the Lck Src homology 2 and 3 domains in protein tyrosine phosphorylation.J. Biol. Chem.27125003–25010.
37
Leupin O. Zaru R. Laroche T. Müller S. Valitutti S. (2000). Exclusion of CD45 from the T-cell receptor signaling area in antigen-stimulated T lymphocytes.Curr. Biol.10277–280.
38
Lewis L. Chung C. Chen J. Parnes J. Moran M. Patel V. Miceli M. (1997). The Lck SH2 phosphotyrosine binding site is critical for efficient TCR-induced processive tyrosine phosphorylation of the zeta-chain and IL-2 production.J. Immunol.1592292–2300.
39
Li Q.-J. Dinner A. R. Qi S. Irvine D. J. Huppa J. B. Davis M. M. Chakraborty A. K. (2004). CD4 enhances T cell sensitivity to antigen by coordinating Lck accumulation at the immunological synapse.Nat. Immunol.5791–799.
40
Lillemeier B. F. Mörtelmaier M. A. Forstner M. B. Huppa J. B. Groves J. T. Davis M. M. (2010). TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation.Nat. Immunol.1190–96.
41
Lin R. S. Rodriguez C. Veillette A. Lodish H. (1998). Zinc is essential for binding of p56lck to CD4 and CD8alpha.J. Biol. Chem.27332878–32882.
42
Locksley R. M. Reiner S. L. Hatam F. Littman D. R. Killeen N. (1993). Helper T cells without CD4: control of leishmaniasis in CD4-deficient mice.Science2611448–1451.
43
Marti F. Garcia G. G. Lapinski P. E. MacGregor J. N. King P. D. (2006). Essential role of the T cell-specific adapter protein in the activation of LCK in peripheral T cells.J. Exp. Med.203281–287.
44
McNeill L. Salmond R. J. Cooper J. C. Carret C. K. Cassady-Cain R. L. Roche-Molina M. Tandon P. Holmes N. Alexander D. R. (2007). The differential regulation of Lck kinase phosphorylation sites by CD45 is critical for T cell receptor signaling responses.Immunity27425–437.
45
Monks C. R. Freiberg B. A. Kupfer H. Sciaky N. Kupfer A. (1998). Three-dimensional segregation of supramolecular activation clusters in T cells.Nature39582–86.
46
Munro S. (2003). Lipid rafts: elusive or illusive?Cell115377–388.
47
Ng D. H. Watts J. D. Aebersold R. Johnson P. (1996). Demonstration of a direct interaction between p56[IMAGE] and the cytoplasmic domain of CD45 in vitro.J. Biol. Chem.2711295–1300.
48
Nichols B. (2005). Cell biology: without a raft.Nature436638–639.
49
Nika K. Soldani C. Salek M. Paster W. Gray A. Etzensperger R. Fugger L. Polzella P. Cerundolo V. Dushek O. (2010). Constitutively active Lck kinase in T cells drives antigen receptor signal transduction.Immunity32766–777.
50
Nika K. Tautz L. Arimura Y. Vang T. Williams S. Mustelin T. (2007). A weak Lck tail bite is necessary for Lck function in T cell antigen receptor signaling.J. Biol. Chem.28236000–36009.
51
Palacios E. H. Weiss A. (2004). Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation.Oncogene237990–8000.
52
Parsons S. J. Parsons J. T. (2004). Src family kinases, key regulators of signal transduction.Oncogene237906–7909.
53
Paster W. Paar C. Eckerstorfer P. Jakober A. Drbal K. Schütz G. J. Sonnleitner A. Stockinger H. (2009). Genetically encoded Förster resonance energy transfer sensors for the conformation of the Src family kinase Lck.J. Immunol.1822160–2167.
54
Pawson T. (2004). Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems.Cell116191–203.
55
Rodgers W. Farris D. Mishra S. (2005). Merging complexes: properties of membrane raft assembly during lymphocyte signaling.Trends Immunol.2697–103.
56
Rodgers W. Rose J. K. (1996). Exclusion of CD45 inhibits activity of p56lck associated with glycolipid-enriched membrane domains.J. Cell Biol.1351515–1523.
57
Rozdzial M. M. Pleiman C. M. Cambier J. C. Finkel T. H. (1998). pp56Lck mediates TCR zeta-chain binding to the microfilament cytoskeleton.J. Immunol.1615491–5499.
58
Rudd C. E. Trevillyan J. M. Dasgupta J. D. Wong L. L. Schlossman S. F. (1988). The CD4 receptor is complexed in detergent lysates to a protein-tyrosine kinase (pp58) from human T lymphocytes.Proc. Natl. Acad. Sci. U.S.A.855190–5194.
59
Salmond R. J. Filby A. Pirinen N. Magee A. I. Zamoyska R. (2011). Mislocalization of Lck impairs thymocyte differentiation and can promote development of thymomas.Blood117108–117.
60
Saunders A. E. Johnson P. (2010). Modulation of immune cell signaling by the leukocyte common tyrosine phosphatase, CD45.Cell. Signal.22339–348.
61
Schilham M. W. Fung-Leung W. P. Rahemtulla A. Kuendig T. Zhang L. Potter J. Miller R. G. Hengartner H. Mak T. W. (1993). Alloreactive cytotoxic T cells can develop and function in mice lacking both CD4 and CD8.Eur. J. Immunol.231299–1304.
62
Schoenborn J. R. Tan Y. X. Zhang C. Shokat K. M. Weiss A. (2011). Feedback circuits monitor and adjust basal Lck-dependent events in T cell receptor signaling.Sci. Signal.4ra59.
63
Secrist J. P. Burns L. A. Karnitz L. Koretzky G. A. Abraham R. T. (1993). Stimulatory effects of the protein tyrosine phosphatase inhibitor, pervanadate, on T-cell activation events.J. Biol. Chem.2685886–5893.
64
Seminario M.-C. Bunnell S. C. (2008). Signal initiation in T-cell receptor microclusters.Immunol. Rev.22190–106.
65
Shenoy-Scaria A. M. Link D. C. Douglas M. L. (1994). Cysteine3 of Src family protein tyrosine kinase determines palmitoylation and localization in caveolae.J. Cell Biol.126353–363.
66
Stefanová I. Hemmer B. Vergelli M. Martin R. Biddison W. E. Germain R. N. (2003). TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways.Nat. Immunol.4248–254.
67
Straus D. B. Chan A. C. Weiss A. (1996). SH2 domain function is essential for the role of the Lck tyrosine kinase in T cell receptor signal transduction.J. Biol. Chem.2719976–9981.
68
Tavano R. Contento R. L. Baranda S. J. Soligo M. Tuosto L. Manes S. Viola A. (2006). CD28 interaction with filamin-A controls lipid raft accumulation at the T-cell immunological synapse.Nat. Cell Biol.81270–1276.
69
Varma R. Campi G. Yokosuka T. Saito T. Dustin M. L. (2006). T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster.Immunity25117–127.
70
Van Laethem F. Sarafova S. D. Park J.-H. Tai X. Pobezinsky L. Guinter T. I. Adoro S. Adams A. Sharrow S. O. Feigenbaum L. Singer A. (2007). Deletion of CD4 and CD8 coreceptors permits generation of alphabetaT cells that recognize antigens independently of the MHC.Immunity27735–750.
71
Veillette A. Bookman M. A. Horak E. M. Bolen J. B. (1988). The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck.Cell55301–308.
72
Wange R. L. Samelson L. E. (1996). Complex complexes: signaling at the TCR.Immunity5197–205.
73
Williamson D. J. Owen D. M. Rossy J. Magenau A. Wehrmann M. Gooding J. J. Gaus K. (2011). Pre-existing clusters of the adaptor Lat do not participate in early T cell signaling events.Nat. Immunol.12655–662.
74
Wong N. K. Y. Lai J. C. Y. Birkenhead D. Shaw A. S. Johnson P. (2008). CD45 down-regulates Lck-mediated CD44 signaling and modulates actin rearrangement in T cells.J. Immunol.1817033–7043.
75
Xu H. Littman D. R. (1993). A kinase-independent function of Lck in potentiating antigen-specific T cell activation.Cell74633–643.
76
Xu W. Doshi A. Lei M. Eck M. J. Harrison S. C. (1999). Crystal structures of c-Src reveal features of its autoinhibitory mechanism.Mol. Cell3629–638.
77
Yamaguchi H. Hendrickson W. A. (1996). Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation.Nature384484–489.
78
Yamasaki S. Takamatsu M. Iwashima M. (1996). The kinase, SH3, and SH2 domains of Lck play critical roles in T-cell activation after ZAP-70 membrane localization.Mol. Cell. Biol.167151–7160.
79
Zhang H. Cordoba S.-P. Dushek O Anton van der Merwe P. (2011). Basic residues in the T-cell receptor ζ cytoplasmic domain mediate membrane association and modulate signaling.Proc. Natl. Acad. Sci. U.S.A.10819323–19328.
80
Zikherman J. Jenne C. Watson S. Doan K. Raschke W. Goodnow C. C. Weiss A. (2010). CD45-Csk phosphatase-kinase titration uncouples basal and inducible T cell receptor signaling during thymic development.Immunity32342–54.
Summary
Keywords
membrane organization, T cell receptor, Lck kinase, signal transduction, protein distribution
Citation
Rossy J, Williamson DJ and Gaus K (2012) How does the kinase Lck phosphorylate the T cell receptor? Spatial organization as a regulatory mechanism. Front. Immun. 3:167. doi: 10.3389/fimmu.2012.00167
Received
25 January 2012
Accepted
04 June 2012
Published
19 June 2012
Volume
3 - 2012
Edited by
Michael Dustin, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, USA
Reviewed by
Michael Dustin, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, USA Christopher E. Rudd, University of Cambridge, UK
Copyright
© Rossy, Williamson and Gaus.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Katharina Gaus, Centre for Vascular Research, University of New South Wales, Sydney, NSW 2052, Australia. e-mail: k.gaus@unsw.edu.au
This article was submitted to Frontiers in T Cell Biology, a specialty of Frontiers in Immunology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.