

- 1 Department of Chemical and Geological Sciences, University of Cagliari, Cagliari, Italy
- 2 Istituto Officina dei Materiali del Consiglio Nazionale delle Ricerche, UOS SLACS, Cagliari, Italy
- 3 Department of Biomedical Sciences, University of Cagliari, Cagliari, Italy
Natural Antimicrobial Peptides: A Palette of Extraordinary Colors
Virtually all multicellular organisms must ward off pathogenic microbes in order to survive and thrive on this planet. To accomplish this, most metazoans rely on gene-encoded antimicrobial peptides (AMPs) as an essential part of their innate immune system. The role played by AMPs – and by the other umoral and cellular components of innate immunity – is particularly crucial in those organisms (the vast majority) that have not developed the more sophisticated adaptive immune system. Even in higher vertebrates as humans, AMPs like defensins and cathelicidins (e.g., LL-37) do not only have direct microbicidal activity, but they also serve as signals which initiate, mobilize, and amplify adaptive immune host defenses, thus functioning as immunomodulatory and immunostimulatory elements (Giuliani and Rinaldi, 2010).
Despite a bewildering variety in their primary sequences, AMPs generally share a cationic character, a length that usually does not exceeds 50 residues (a large proportion of which are generally hydrophobic), and a globally amphipathic fold, with clearly distinguishable hydrophilic and hydrophobic faces. These structural features reflect their mode of action, which is primarily directed to the interaction with and damage of the pathogen cell’s plasma membrane, although the evidence that many AMPs may hit other targets is rapidly increasing. These evolutionarily conserved molecules display a broad spectrum antimicrobial activity against bacteria, fungi, protozoans, and even enveloped viruses. AMPs represent key components especially at epithelial surfaces, where the initial contact with pathogens takes place, thus being deployed at the very front line of the defense system, where rapid action is required before the more slowly responding adaptive immune system (if any) can be brought into action.
With the prospects of an ever-increasing bacterial resistance to conventional antibiotics looming at the horizon, much attention has been devoted to AMPs as a potential source of new anti-infective drugs (Mangoni, 2011; Yeung et al., 2011). However, despite intense research aimed at spotting possible ways for harnessing the therapeutic potential of these intriguing natural compounds, little practical outcome has been generated as yet. Multiple hurdles on this way exist, indeed, related to the fact that naturally occurring AMPs present serious drawbacks that limit their development into clinically applicable antibiotics. These include (but are not limited to) high costs of manufacture, susceptibility to protease degradation, reduced activity in the presence of salts as those present in serum. In addition, given the recognized immunomodulatory and immunostimulatory effect of several AMPs – that is some cases is so evident that the term Host Defense Peptides has been proposed as more indicative of the real role played by these molecules in vivo – using AMPs systemically to treat infections should necessarily suppose an advanced knowledge of (and possibility to control) the possible responses these peptides can trigger. Furthermore, as mentioned before, AMPs have been crafted by evolution to be part of the network of interacting and self-reinforcing components of the innate immune systems, thus expecting that they could stand alone as the new “magic bullet” against resistant microbes would be simply overoptimistic.
Given the inherent limitations of naturally occurring AMPs that have so far prevented their transformation into therapeutics, two general approaches have emerged to overcome this major obstacle, i.e., the modification of existing peptide sequences or the de novo synthesis of peptides, and the development of synthetic molecules that mimic the properties and activities of AMPs (Giuliani et al., 2008; Brogden and Brogden, 2011; Fjell et al., 2011; Giuliani and Rinaldi, 2011). Here, a few recent, significant examples pertaining to these research avenues will be highlighted.
Designed Peptide Antibiotics: Rationally Banking on Nature’s Work
Several computational approaches have been proposed for the design of promising novel peptide antibiotics based on our current understanding of structure–activity relationships of the naturally occurring AMPs. One of the most interesting of such directions was reported by the group led by Davor Juretić at the University of Split, Croatia. By inspecting a self-assembled database of natural AMPs where primary and secondary structures have been linked to the peptides’ antimicrobial activity and selectivity (i.e., the ability to discriminate bacterial versus host cells as targets), these researchers have extracted selected physicochemical peptide properties then used to construct a “AMP-Designer” algorithm (Juretić et al., 2009). A peptide (23 residues, glycine-rich) suggested by the algorithm, was synthesized and its activity and selectivity tested. This peptide, adepantin 1 (from “Automatically DEsigned Peptide ANTIbiotic Number 1”), is less than 50% identical to any other AMP, and displayed a potent antibacterial activity against Escherichia coli coupled to a significantly greater selectivity for bacterial cells than the best AMP present in the reference database (Juretić et al., 2009).
Multimeric peptides have proved to be another intriguing variation on the AMPs theme. Recently, several examples of peptide dendrimers endowed with antimicrobial properties were reported. These molecules were designed following the so-called multiple antigenic peptide (MAP) system, introduced many years ago, where multiple peptide sequences can be added using standard solid phase chemistry to a inner core of radially branched lysine residues. Starting from a linear AMP sequence originally identified by selecting a random phage library against whole E. coli cells, several cycles of rational modification and optimization led to the tetra-branched peptide known as SB041, found to be especially active against Gram-negative strains and able to strongly bind E. coli and Pseudomonas aeruginosa lipopolysaccharide (LPS) in vitro (Bruschi et al., 2010). More recently, a novel AMP with a dimeric dendrimer scaffold, SB056 (Figure 1A), was synthesized and showed high activity against Gram-negative bacteria and some limited activity against Gram-positive bacteria (Scorciapino et al., 2012). Its potency against Gram-negative strains was comparable (on a molar basis) to that of colistin and polymyxin B, with an even broader spectrum of activity than numerous other reference compounds. Biophysical characterization through circular dichroism, NMR, molecular dynamics simulation, and membrane affinity assayed through lipid monolayer surface pressure experiments, revealed that the peptide is membrane-active, and tends to folds into a β-type conformation in a lipid environment (Scorciapino et al., 2012). In general, dendrimeric peptides display increased activity compared to their monomeric counterparts – a fact probably attributable to the higher local concentration of bioactive units for multimeric peptides – as well as greater stability to peptidases and proteases, possibly due to the steric hindrance of the branching core that would limit the cleavage rates of plasma peptidases, thus increasing the peptides’ pharmacokinetics properties (Giuliani and Rinaldi, 2011).
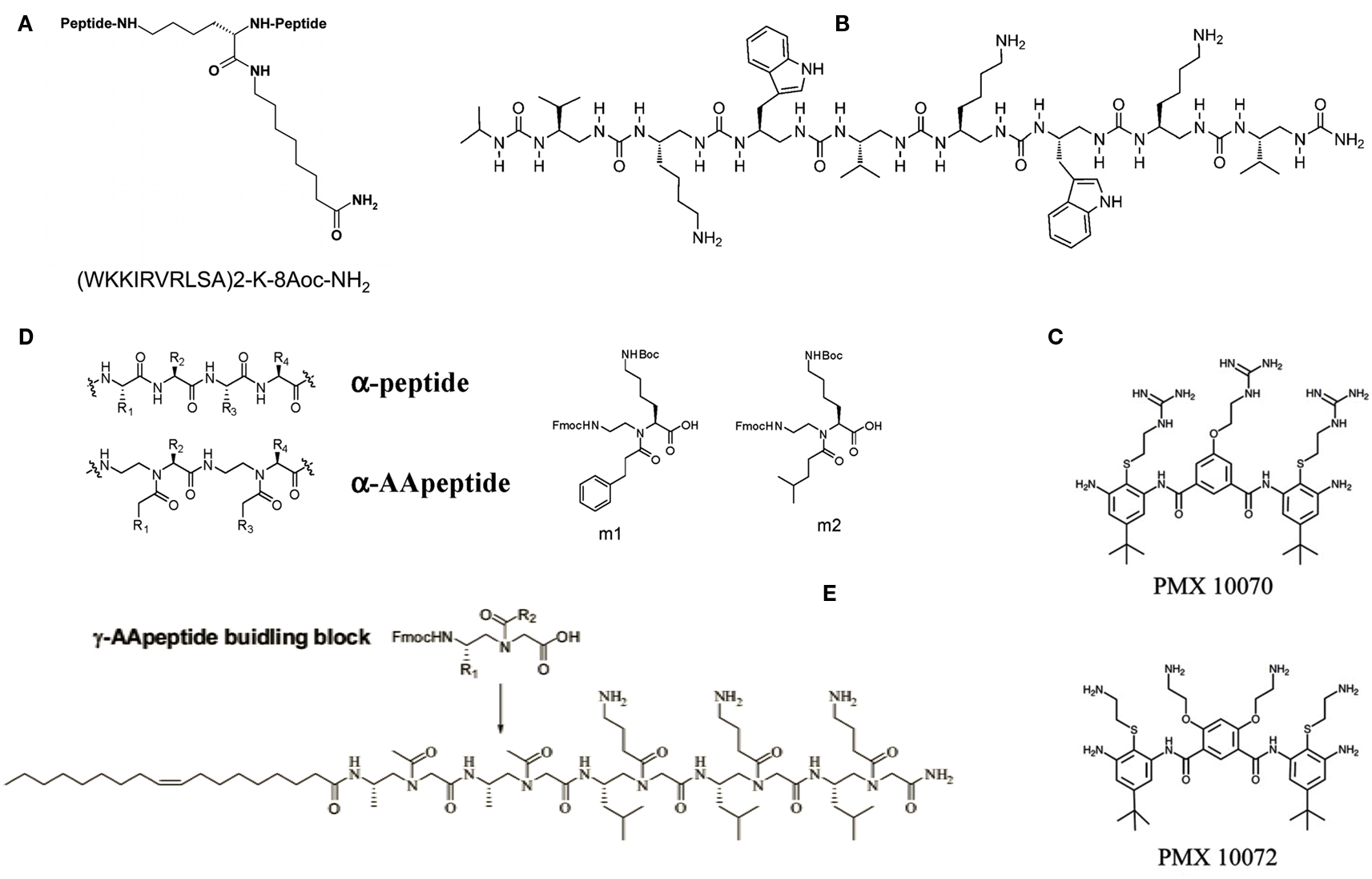
Figure 1. Examples of designed antimicrobial peptides and mimics. (A) The dendrimeric peptide SB056. 8-Aoc is 8-aminooctanoic acid, which is amidated in the peptide construct; (B) antimicrobial oligourea; (C) arylamide compounds; (D) structures of an α-peptide and a corresponding AApeptide, and α-AApeptide building blocks (m1 and m2) used for the preparation of antimicrobial α-AApeptides; (E) lipo-γ-AApeptide. See text for more details and original studies.
Researchers at the University of Zurich and Polyphor Ltd.1 have used a different approach to come out with a Pseudomonas specific antibiotic with a novel mode of action. The lead, termed POL7080, was derived from the so-called PEM (protein epitope mimetic) technology platform, which takes advantage of the fact that molecular recognition involving proteins is often mediated by surface-exposed secondary structure motifs such as β-hairpins and α-helices (Obrecht et al., 2012). Following this path, researchers have developed epitope mimetics of protegrin I – an AMP first discovered in porcine leukocytes – that specifically target P. aeruginosa in the nanomolar range via a mechanism of action that is distinct from the membrane-disrupting activity of the parent peptide. Studies have shown that POL7080 rather targets an essential bacterial β-barrel outer membrane protein called LptD, which functions in outer membrane biogenesis (Srinivas et al., 2010). The β-hairpin mimetic POL7080 is currently in a Phase I clinical trial in Europe, and could represent an important drive in the treatment of chronic lung infections in patients with cystic fibrosis where P. aeruginosa plays a crucial role.
The Raise of Peptide Mimetics
Along the years, a number of mimics of AMPs, both (unconventionally) peptidic and non-peptidic in nature, have been explored as for their capacity of working as antimicrobial substances with potential therapeutic interest. These different classes of compounds include oligoacyllysines, ceragenins, peptoids, phenylene-ethynylene oligomers, and others. Some further examples are detailed below. The main idea behind the design of these mimics is to build foldamers that could maintain the amphiphilic character of naturally occurring AMPs while circumventing the major drawbacks that have prevented peptide-based antimicrobials from having significant success in practical terms.
Among the AMP-mimicking synthetic oligomers built with unnatural building blocks, foldamers such as aromatic and aliphatic N,N′-linked oligoureas (Figure 1B) have received considerable attention (Violette et al., 2006; Claudon et al., 2010). High resistant to proteolytic degradation oligoureas have been shown to adopt a remarkably stable helical fold stabilized by 12- and 14-membered H-bonded rings, with helical propensity being enhanced in the presence of phospholipid vesicles, and to possess significant antibacterial activity against Gram-negative and Gram-positive bacteria (Violette et al., 2006; Claudon et al., 2010). Recently, a detailed solid-state NMR structural investigation was performed on aliphatic oligoureas synthesized with 15N at selected positions (Aisenbrey et al., 2012). This permitted to acquire information on the oligomers’ conformation, dynamics, and interactions with model membranes. The acquired data were indicative of an alignment of the oligourea helix parallel to the surface of the phospholipid bilayers, in agreement with the amphipathic character of the foldamer and consistent with previous models explaining the modes of action of AMPs (Aisenbrey et al., 2012).
One of the most interesting approaches to chemically mimicking AMPs, is that of small arylamide foldamers, i.e., an arylamide backbone and various charged and hydrophobic groups yielding a topographically amphiphilic structure. The best known compound belonging to this class is PMX-30063, a “defensin-mimetic” lead compound being developed by PolyMedix for the broad treatment of Staphylococcus aureus infections. PMX-30063 has just completed a Phase 2 clinical trial to treat patients with acute bacterial skin and skin structure infections caused by S. aureus: the study objectives were met, demonstrating clinical efficacy and safety in all evaluated doses of PMX-300632. A recent investigation on the mechanism of action of two arylamides – namely PMX 10070, which has a 2-ethyl guanidinium charged substitution, and PMX 10072, which has a 2-ethyl amine substitution on the arylamide backbone (Figure 1C) – has shown that the arylamide compounds significantly disrupted the permeability of the outer membrane of E. coli cells, possibly by binding to the LPS component of cell wall (Mensa et al., 2011). Perturbation of the inner membrane was comparably less dramatic, but arylamide exposure nevertheless led to increased permeability of the inner membrane to small ions and to defects in protein translocation across the membrane, thus contributing to arylamides’ lethality, authors concluded. “Compared to several natural AMPs, the membrane permeabilization caused by arylamides is less extensive, which could minimize the risk of detrimental inflammatory responses to leakage of bacterial cellular contents and thus prove advantageous in a therapeutic setting,” they remarked (Mensa et al., 2011).
An entire new class of peptide mimetics, namely α-AApeptides, with broad spectrum activity against both Gram-negative and Gram-positive bacteria and fungi, was recently reported. AApeptides are oligomers of N-acylated-N-aminoethyl amino acids, designed on the principle of globally amphipathic structures as drivers for membrane-active antimicrobial compounds (Figure 1D). “Coupled with straightforward solid phase synthesis, virtually limitless structural possibilities, low cost of production, simple tunability and programmability, and resistance to protease hydrolysis, α-AApeptides may lead to a new class of antimicrobial peptidomimetics,” remarked authors (Padhee et al., 2011). In their hands, one of the synthesized oligomers displayed minimal inhibitory concentration (MIC) values – defined as the lowest inhibitor concentration that completely inhibits the growth of microbes during a 24 h incubation period at 37°C – as low as 2.1 μg/ml against the Gram-negative E. coli and 0.9 μg/ml against the Gram-positive Bacillus subtilis, respectively, while being non-hemolytic (Padhee et al., 2011). These levels of activities are equal to or better than those of most natural AMPs and peptidomimetics reported so far. The same group, led by Jianfeng Cai at the University of South Florida in Tampa, FL, USA, lately reported the design and synthesis of lipidated γ-AApeptides (Figure 1E) as antimicrobial agents (Niu et al., 2012). According to the authors, the introduction of an unsaturated lipid chain significantly decreased hemolytic activity, thereby enhancing target selectivity. One of the synthesized lipo-γ-AApeptides did not induce drug resistance in methicillin-resistant S. aureus, even after 17 passages (Niu et al., 2012).
Looking Back, Looking Ahead
Considering that AMPs, like cecropins and defensins, have been identified some three decades ago, the failure in transforming their potential into therapeutics against resistant microbial infections might be felt as scorchingly disappointing. However, great advances in our understanding of AMPs’ physicochemical properties and modes of action are fostering the development of a new wave of peptidomimetics, inspired by naturally occurring examples, that could remove the hurdles that have prevented the adoption of AMPs in clinical settings. The next few years will be crucial in showing whether this view is correct.
Footnotes
References
Aisenbrey, C., Pendem, N., Guichard, G., and Bechinger, B. (2012). Solid state NMR studies of oligourea foldamers: interaction of 15N-labelled amphiphilic helices with oriented lipid membranes. Org. Biomol. Chem. 10, 1440–1447.
Brogden, N. K., and Brogden, K. A. (2011). Will new generations of modified antimicrobial peptides improve their potential as pharmaceuticals? Int. J. Antimicrob. Agents 38, 217–225.
Bruschi, M., Pirri, G., Giuliani, A., Nicoletto, S. F., Baster, I., Scorciapino, M. A., Casu, M., and Rinaldi, A. C. (2010). Synthesis, characterization, antimicrobial activity and LPS-interaction properties of SB041, a novel dendrimeric peptide with antimicrobial properties. Peptides 31, 1459–1467.
Claudon, P., Violette, A., Lamour, K., Decossas, M., Fournel, S., Heurtault, B., Godet, J., Mély, Y., Jamart-Grégoire, B., Averlant-Petit, M. C., Briand, J. P., Duportail, G., Monteil, H., and Guichard, G. (2010). Consequences of isostructural main-chain modifications for the design of antimicrobial foldamers: helical mimics of host-defense peptides based on a heterogeneous amide/urea backbone. Angew. Chem. Int. Ed. Engl. 49, 333–336.
Fjell, C. D., Hiss, J. A., Hancock, R. E. W., and Schneider, G. (2011). Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 16, 37–51.
Giuliani, A., Pirri, G., Bozzi, A., Di Giulio, A., Aschi, M., and Rinaldi, A. C. (2008). Antimicrobial peptides: natural templates for synthetic membrane-active compounds. Cell. Mol. Life Sci. 65, 2450–2460.
Giuliani, A., and Rinaldi, A. C. (2010). Antimicrobial peptides. Methods and Protocols: Methods in Molecular Biology, Vol. 618. New York: Humana Press.
Giuliani, A., and Rinaldi, A. C. (2011). Beyond natural antimicrobial peptides: multimeric peptides and other peptidomimetic approaches. Cell. Mol. Life Sci. 68, 2255–2266.
Juretić, D., Vukicević, D., Ilić, N., Antcheva, N., and Tossi, A. (2009). Computational design of highly selective antimicrobial peptides. J. Chem. Inf. Model. 49, 2873–2882.
Mangoni, M. L. (2011). Host-defense peptides: from biology to therapeutic strategies. Cell. Mol. Life Sci. 68, 2157–2159.
Mensa, B., Kim, Y. H., Choi, S., Scott, R., Caputo, G. A., and De Grado, W. F. (2011). Antibacterial mechanism of action of arylamide foldamers. Antimicrob. Agents Chemother. 55, 5043–5053.
Niu, Y., Padhee, S., Wu, H., Bai, G., Qiao, Q., Hu, Y., Harrington, L., Burda, W. N., Shaw, L. N., Cao, C., and Cai, J. (2012). Lipo-γ-AApeptides as a new class of potent and broad-spectrum antimicrobial agents. J. Med. Chem. 55, 4003–4009.
Obrecht, D., Chevalier, E., Moehle, K., and Robinson, J. A. (2012). β-Hairpin protein epitope mimetic technology in drug discovery. Drug Discov. Today 9, e63–e69.
Padhee, S., Hu, Y., Niu, Y., Bai, G., Wu, H., Costanza, F., West, L., Harrington, L., Shaw, L. N., Cao, C., and Cai, J. (2011). Non-hemolytic α-AApeptides as antimicrobial peptidomimetics. Chem. Commun. (Camb.) 47, 9729–9731.
Scorciapino, M. A., Pirri, G., Vargiu, A. V., Ruggerone, P., Giuliani, A., Casu, M., Bürck, J., Wadhwani, P., Ulrich, A. S., and Rinaldi, A. C. (2012). A novel dendrimeric peptide with antimicrobial properties: in vitro characterization of SB056. Biophys. J. 102, 1039–1048.
Srinivas, N., Jetter, P., Ueberbacher, B. J., Werneburg, M., Zerbe, K., Steinmann, J., Van der Meijden, B., Bernardini, F., Lederer, A., Dias, R. L. A., Misson, P. E., Henze, H., Zumbrunn, J., Gombert, F. O., Obrecht, D., Hunziker, P., Schauer, S., Ziegler, U., Kach, A., Eberl, L., Riedel, K., DeMarco, S. J., and Robinson, J. A. (2010). Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 327, 1010–1013.
Violette, A., Fournel, S., Lamour, K., Chaloin, O., Frisch, B., Briand, J.-P., Monteil, H., and Guichard, G. (2006). Mimicking helical antibacterial peptides with nonpeptidic folding oligomers. Chem. Biol. 13, 531–538.
Citation: Scorciapino MA and Rinaldi AC (2012) Antimicrobial peptidomimetics: reinterpreting nature to deliver innovative therapeutics. Front. Immun. 3:171. doi: 10.3389/fimmu.2012.00171
Received: 02 June 2012; Accepted: 05 June 2012;
Published online: 28 June 2012.
Edited by:
Mark W. Robinson, University of Technology, AustraliaReviewed by:
Mark W. Robinson, University of Technology, AustraliaCopyright: © 2012 Scorciapino and Rinaldi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence:cmluYWxkaUB1bmljYS5pdA==