 Xavier Blanchet1*
Xavier Blanchet1* Marcella Langer1*
Marcella Langer1* Christian Weber1,2,3
Christian Weber1,2,3 Rory R. Koenen1,2
Rory R. Koenen1,2 Philipp von Hundelshausen1
Philipp von Hundelshausen1
- 1 Institute for Cardiovascular Prevention, Ludwig-Maximilians-University of Munich, Munich, Germany
- 2 Cardiovascular Research Institute Maastricht, Maastricht University, Maastricht, Netherlands
- 3 Munich Heart Alliance, Munich, Germany
Chemoattractant cytokines or chemokines constitute a family of structurally related proteins found in vertebrates, bacteria, or viruses. So far, 48 chemokine genes have been identified in humans, which bind to around 20 chemokine receptors. These receptors belong to the seven transmembrane G-protein-coupled receptor family. Chemokines and their receptors were originally studied for their role in cellular trafficking of leukocytes during inflammation and immune surveillance. It is now known that they exert different functions under physiological conditions such as homeostasis, development, tissue repair, and angiogenesis but also under pathological disorders including tumorigenesis, cancer metastasis, inflammatory, and autoimmune diseases. Physicochemical properties of chemokines and chemokine receptors confer the ability to homo- and hetero-oligomerize. Many efforts are currently performed in establishing new therapeutically compounds able to target the chemokine/chemokine receptor system. In this review, we are interested in the role of chemokines in inflammatory disease and leukocyte trafficking with a focus on vascular inflammatory diseases, the operating synergism, and the emerging therapeutic approaches of chemokines.
Introduction
With the exceptions of CX3CL1/fractalkine and CXCL16/SR-PSOX, chemoattractant cytokines or chemokines constitute a family of small soluble signaling molecules of approximately 70 amino acid residues with a molecular weight of 7–12 kDa. In addition to their monomeric form, these proteins are able to associate, forming dimers, tetramers, or multimers (i.e., to oligomerize). Chemokines have crucial roles in both homeostasis and disease. Their homeostatic roles include leukocyte maturation and trafficking, development, tissue repair, and angiogenesis (Ransohoff, 2009). As disease modulators, chemokines have roles in a wide variety of inflammatory and immune responses through the chemoattraction of innate and adaptive immune cells. To date, around 50 chemokines have been identified in humans, which have been grouped into one of four families, CXC, CC, CX3C, and XC, based on the arrangement of cysteine residues involved in the formation of disulfide bonds (Table 1). In the CXC and CX3C chemokine family, one or three amino acid residues are inserted between the first two of four cysteine residues, respectively. The first and third cysteine residues are absent in the XC subfamily that possesses only one disulfide bond. In the CC subfamily, the first two cysteines are juxtaposed. Another family has been recently described in the zebrafish genome, namely the CX family, which lacks one of the four cysteine residues highly conserved amongst chemokines (Nomiyama et al., 2008). All chemokines arose from a single ancestral gene, originating approximately 650 million years ago (Nomiyama et al., 2010). Amongst vertebrates, the zebrafish genome has the highest number of chemokine genes with more than 100 genes while both pufferfish Tetraodon and Fugu genomes contain less than 20 chemokine genes each. The human genome encompasses more than 50 different chemokine genes and pseudo-genes. These genes have undergone a rapid evolution in both their sequences and their family gene size. The conventional name is still often used, which may lead to some confusion while the International Union of Immunological Societies/World Health Organization Subcommittee on Chemokine Nomenclature has assigned a name to each chemokine and chemokine receptor (Bacon et al., 2001). A large number of human chemokine genes are known to be clustered on specific chromosomal regions. There are two major gene clusters comprising exclusively either CXC or CC genes on chromosome 4q13.3-q21.1 and 17q12, respectively (Table 1). These major clusters can be subdivided into two regions. For the CXC gene cluster, the regions are named GRO and IP10 while the regions of the CC gene cluster are called MCP and MIP (Nomiyama et al., 2010). The GRO region contains the CXCL1–CXCL8 genes and the IP10 region the CXCL9–CXCL13 genes, respectively. In the CC major cluster, the MCP and MIP regions comprise 6 and 12 genes, respectively (CCL2, CCL7, CCL11, CCL8, CCL13, CCL1 versus CCL5, CCL16, CCL14, CCL15, CCL23, CCL18, CCL3, CCL4, CCL3L3, CCL4L1, CCL3L1, CCL4L2). In addition to the two major clusters, a CC “mini”-cluster is found on chromosome 7 (comprising the CCL26 and CCL24 genes), on chromosome 9 (CCL27, CCL19, CCL21), and on chromosome 16 (CCL22, CX3CL1, and CCL17), respectively. Both XCL1 and XCL2 are also found in a “mini”-cluster on chromosome 1.
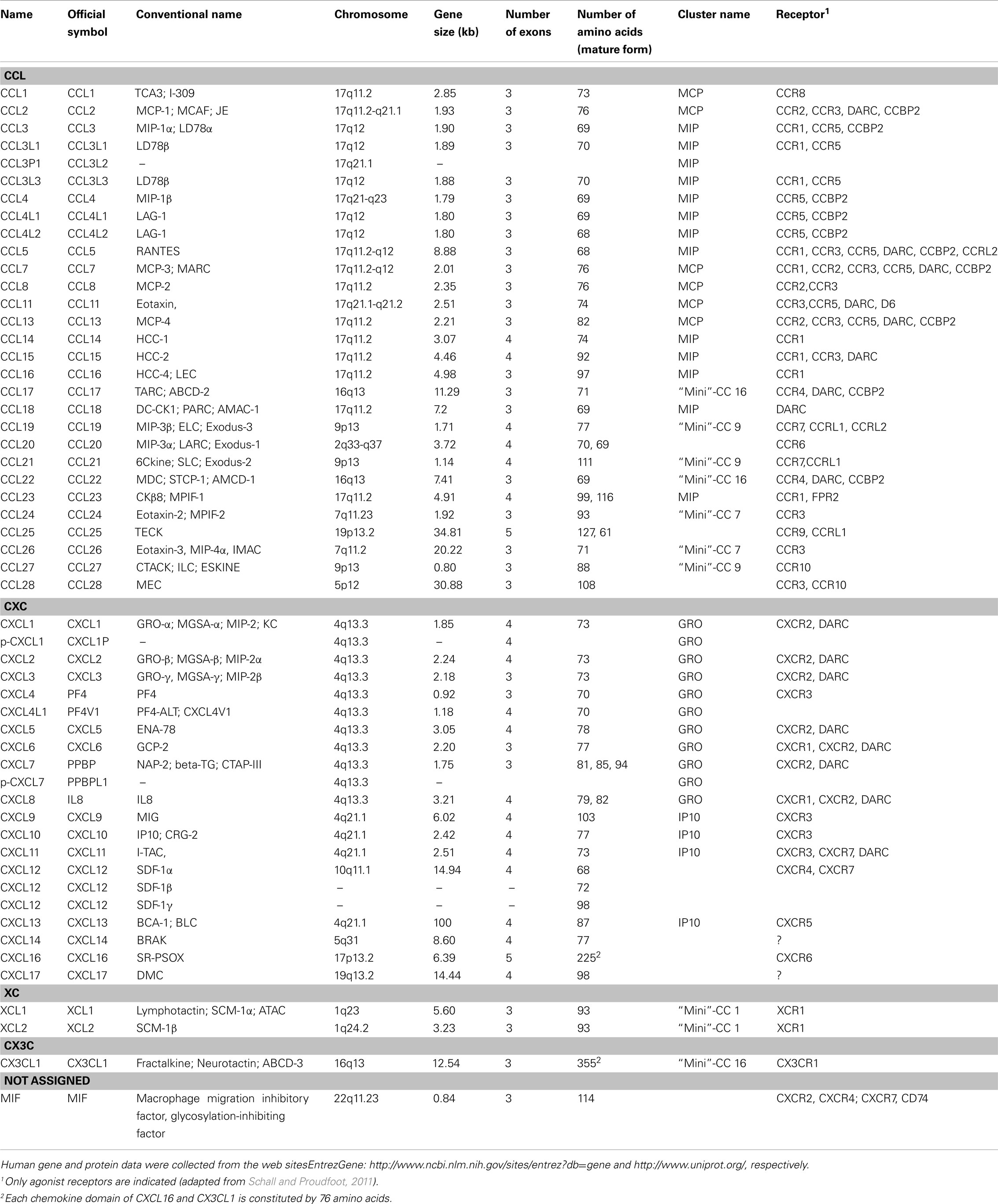
Table 1. Human chemokine genes.
Besides their structural classification, another organization of chemokines has been proposed based on their expression and their functional activity. This classification groups chemokines into three “families”: pro-inflammatory, homeostatic, and mixed function (Mantovani et al., 2006). Pro-inflammatory chemokines are up-regulated under inflammatory conditions and are involved in the leukocyte recruitment to inflamed sites. Homeostatic chemokines are expressed constitutively at non-inflamed sites and are involved in homeostatic migration and homing of cells in physiological conditions such as lymphocyte homing. Some chemokines have both properties, and are thus called mixed-function chemokines.
Chemokines act by binding specialized receptors on the target cell surface. These chemokine receptors are also grouped into four families, CXCR, CCR, XCR, and CX3R, based on the chemokine family they bind (Nomiyama et al., 2011). The entire group of chemokine receptors belongs to the seven transmembrane domain G-protein-coupled receptors that usually combine the receptor to the Gαi subunit of heterotrimeric G proteins. So far, around 25 human chemokine receptor genes have been identified (Table 2). Interestingly, 12 of these receptors are found on human chromosome 13 and stretched around 13.5 megabases. In addition, several decoy receptors have been reported to bind chemokine ligands without eliciting signal transduction. These comprise CXCR7, CCBP2, Duffy antigen receptor for chemokines (DARC), CCRL1, and CCRL2. The first chemokine receptor genes appeared in the most primitive vertebrate, agnathan lamprey (hagfish), around 480 million years ago (Nomiyama et al., 2011).
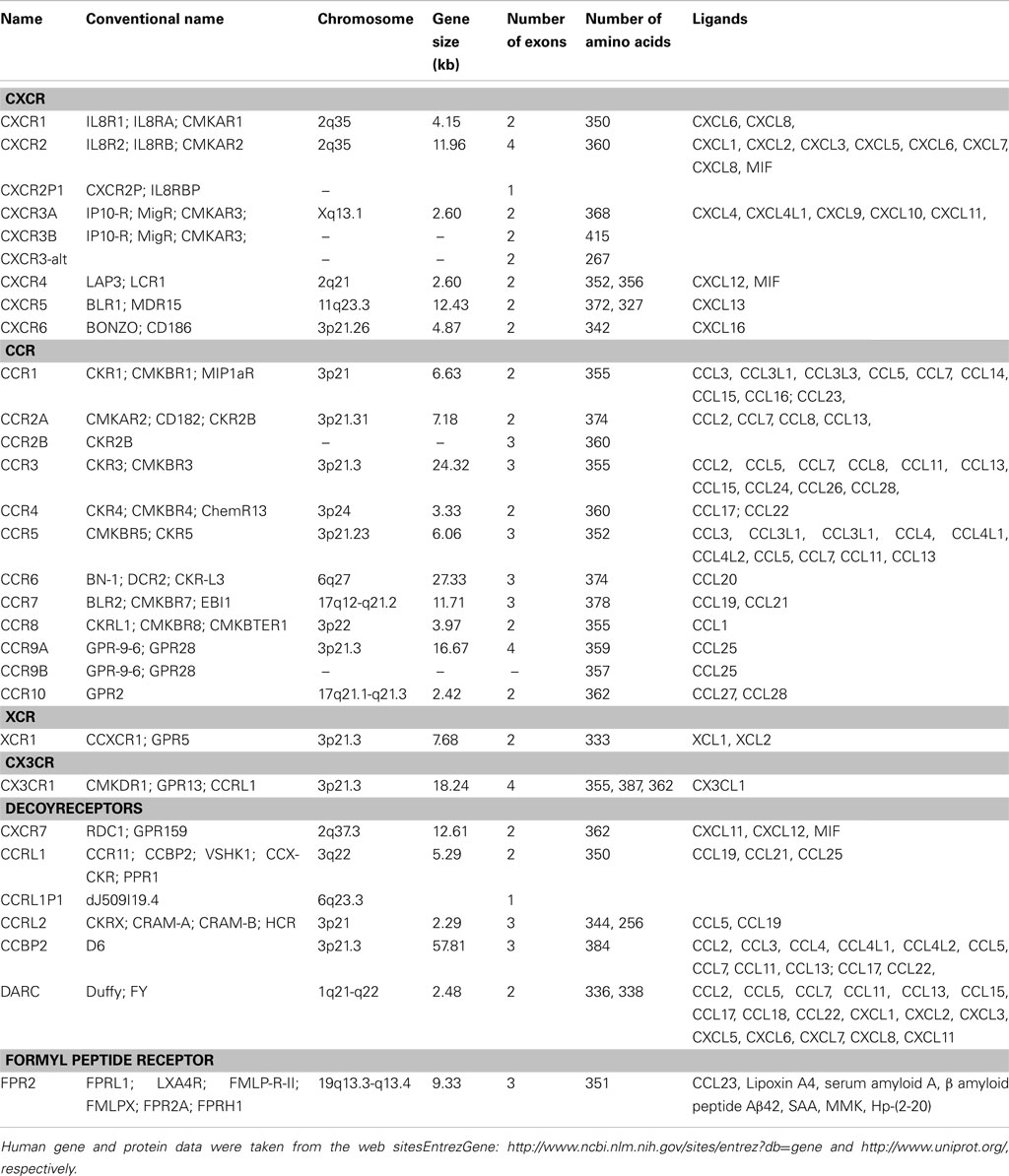
Table 2. Human chemokine receptor genes.
The chemokine/chemokine receptor system can be considered as a “puzzle” since many receptors have different chemokines as ligands and vice versa. However, thanks to the multiple combinations allowed, this system offers robustness. Indeed, even if one chemokine or receptor does not function, another one can replace it.
Chemokines in Leukocyte Trafficking and Inflammatory Diseases
Leukocyte recruitment represents a fundamental episode during infection, in inflammatory disorders, such as atherosclerosis, as well as in autoimmune diseases, such as in psoriasis, rheumatoid arthritis, and chronic lung disease (Luster et al., 2005). Initially, leukocyte extravasation was described as a three-step process namely rolling, activation, and arrest. Recently, new insights have allowed defining a more complex process by adding several steps to the three original, including tethering (or capture), slow rolling, adhesion strengthening, spreading, intravascular crawling, and finally paracellular and transcellular transmigration.
Whereas capture and slow rolling are mediated by reversible and transient interactions between E-, L-, or P-selectin and ligands such as P-selectin glycoprotein-1 (PSGL-1), the adhesion of leukocytes to endothelial cells is mediated by the interaction of VCAM1 and ICAM-1, receptor for advanced glycation endproducts (RAGE), or mucosal vascular cell-adhesion molecule 1 (MADCAM1) with leukocyte integrins. The common structure of integrins is a non-covalently associated α and β subunit. So far, 16 α subunits and 8 β subunits have been identified, and various combinations form at least 22 heterodimers. The principal neutrophil β2-integrins are CD11a/CD18 (LFA-1) and CD11b/CD18 (Mac-1, CR3; Luo et al., 2007) although neutrophils can express p150,95/αxβ2 (CD11c/CD18) and at low level very late antigen (VLA) 4/α4β1 (CD49d/CD18; Zarbock et al., 2012). LFA-1 and Mac-1 have been shown to mediate neutrophil adhesion by interacting with ICAM-1 while αxβ2 is able to bind the N-terminal part of the alpha chain of fibrinogen (Loike et al., 1991; Diamond and Springer, 1993; Lum et al., 2002). During neutrophil adhesion, LFA-1 and Mac-1 appear to have sequential roles binding ICAM-1 under shear conditions (Neelamegham et al., 1998; Hentzen et al., 2000). In a two-step process neutrophils adhere first to ICAM-1 by interacting with LFA-1 and then Mac-1 acts as a stabilizer of the LFA-1/ICAM-1 bond.
The transition of rolling to leukocyte arrest and activation is triggered by chemokines such as CXCL1/GRO-α while others like CCL2/MCP-1 per se are rather promoting transmigration. Arrest of rolling leukocytes is triggered by an increase in the affinity of integrins by chemokines (Ley et al., 2007; Chavakis et al., 2009).
Different cell types, such as mesenchymal stem cells, endothelial cells, and circulating blood cells including leukocytes or platelets produce and release a broad range of chemokines and other chemoattractants that facilitate and enhance the recruitment of leukocytes. Some of these pro-inflammatory mediators circulate in the plasma, others are only found in the inflamed tissue, and yet others are presented on endothelial cells. Furthermore, additional to direct endothelial deposition from the luminal side, chemokines are transported via caveolae through the endothelium and presented to the apical side of the cell instead of diffusing through endothelial cell junctions (Pruenster et al., 2009). This transcytosis requires the DARC (Middleton et al., 1997). Recently, a new mechanism has been highlighted introducing the concept of lymphocyte transendothelial migration by intraendothelial vesicle-stored chemokines beneath the apical membrane (Shulman et al., 2011).
Chemokines bind chemokine receptors expressed on leukocytes to induce activation. In addition, most chemokines are also able to bind extracellular matrix components, including glycosaminoglycans (GAGs), to get immobilized and be presented to leukocytes. This is essential in order to avoid to be swept away under flow conditions from the cell surface. This coimmobilization with adhesion molecules will promote leukocyte activation, adhesion, and migration.
The following section will give several examples of chemokine contribution in leukocyte trafficking and in inflammatory diseases with a particular focus on vascular inflammatory diseases.
Chemokines in Platelets
As outlined above, activated platelets are able to release chemokines as well as a battery of different mediators to modulate inflammation. Thus, platelets have been found to be involved in different diseases with an inflammatory component such as obesity, acute lung injury, or coronary artery disease where they interact with both endothelial cells and leukocytes leading to a diversity of effects (van Gils et al., 2009; von Hundelshausen et al., 2009). Platelets release chemotactic cytokines stored in α-granules upon activation. Inter alia, CXCL4/PF4, CCL5/RANTES, CXCL7/NAP-2, CXCL12/SDF-1, CXCL1/GRO-α, or CXCL5/ENA-78 are able to mediate the endothelial adhesion of different cells including monocytes, neutrophils, and progenitor cells (Lievens and von Hundelshausen, 2011).
Platelets secrete CXCL4 which is the first and most abundant chemokine identified in releasates from activated platelets and which is involved in a wide range of physiological processes such as proliferation and angiogenesis. This chemokine is also involved in numerous pathological processes. High levels of CXCL4 are positively correlated with Crohn’s disease activity index (Simi et al., 1987). In Heparin-induced thrombocytopenia (HIT), autoantibodies developed against high molecular complexes of CXCL4/heparin or CXCL4/GAG side chains. The presence of HIT antibodies can lead to platelet activation and depletion through platelet consumption in venous thrombosis (Greinacher, 2009). CXCL4 exerts chemotactic activities on different cells including neutrophils, monocytes (Deuel et al., 1981), and activated T-lymphocytes in a pertussis toxin-sensitive manner (Mueller et al., 2008). Recently, CXCL4 has also been shown to be able to induce a specific macrophage type with specific phenotypic and functional characteristics (Gleissner, 2012). Moreover, it promotes adhesion of neutrophils on endothelial cells (Petersen et al., 1999). Although CXCL4 has been reported to bind to and stimulate CXCR3 and a splice variant thereof (CXCR3B), the functional importance of these two receptors for the biological activity of CXCL4 is not clear. For instance, a recent report found CXCL4 to be involved in ligand driven monocyte down-regulation of chemokine receptors CCR1, CCR2, and CCR5 by releasing the respective ligands (CCL2-4) from CXCL4-activated monocytes in absence of CXCR3 highlighting the connection between platelets and monocytes (Schwartzkopff et al., 2012). CXCL4 can induce exocytosis and firm neutrophil adhesion to endothelium when incubated with the appropriate co-stimuli. A contribution of CXCL4 in cardiovascular diseases has been described in both human and mouse where CXCL4 has been found in the endothelium, neovasculature, macrophages, and calcified regions of atherosclerotic carotid arteries (Pitsilos et al., 2003). Moreover, a strong positive correlation between both luminal and neovascular CXCL4 staining and coronary artery disease and between CXCL4 in macrophages and the presence of symptomatic atherosclerotic disease has been found. In a murine model of atherosclerosis, the knock-out of CXCL4 has been shown to exert an atheroprotective effect reducing atherosclerotic lesion formation (Sachais et al., 2007). Activation of platelets results in a release of stored-P-selectin -CXCL4 and CCL5 from granules. CCL5 has been detected on the luminal surface of atherosclerotic murine and human carotid arteries or neointimal lesions after arterial injury and can be deposited on inflamed or atherosclerotic endothelium by activated platelets, thereby triggering monocyte recruitment under flow (von Hundelshausen et al., 2001; Schober et al., 2002). The deposition of platelet chemokines can be facilitated by platelet-derived microparticles (Mause et al., 2005). Injection of activated platelets into the tail vein of atherosclerosis prone mice results in exacerbated atherosclerotic lesions and increased endothelial deposition of CXCL4 and CCL5 dependent on the presence of P-selectin (Huo et al., 2003). Therefore, platelet adhesion molecules such as P-selectin are mediating transient interactions with endothelial cells enabling a local delivery of soluble chemokines. We have discovered that heterophilic interactions between CXCL4 and CCL5 (see below) are responsible for enhanced monocyte recruitment into the arterial wall which explains to a certain extent why activated platelets are strong promoters of atherosclerosis. Peptides inhibiting the association of CXCL4 and CCL5 decrease atherosclerosis and macrophage content of lesions (von Hundelshausen et al., 2005; Koenen et al., 2009).
A non-allelic variant form of CXCL4, called CXCL4L1 or PF4ALT which differs only in three amino acids in the C-terminal α-helix of the protein has been identified in different kind of cells including leukocytes, endothelial, or smooth muscle cells (Lasagni et al., 2007). CXCL4L1 is capable of inducing endothelial cell chemokinesis and has been characterized as a potent anti-angiogenic regulator similar to CXCL4. Important differences of CXCL4L1 and CXCL4 are a lower affinity for CCL5 (Sarabi et al., 2011) and GAGs, e.g., heparin (Dubrac et al., 2010). Substantiating the role of CCL5–CXCL4 heterodimers CXCL4L1 failed to increase CCL5-triggered monocyte adhesion (Sarabi et al., 2011). The decreased affinity of CXCL4L1 compared to CXCL4 may have critical implications for cell adhesion since CXCL4L1 will not be retained at its site of expression. The existence of heparan sulfate in the subendothelial extracellular matrix has been found to regulate the arrest function of CCL5 and CCL4/MIP-1β (Gilat et al., 1994). On the endothelial surface under flow conditions, both platelet-derived and recombinant CCL5 are able to bind to activated endothelium and to trigger the firm arrest and transmigration of monocytes (von Hundelshausen et al., 2001).
Moreover, the oligomerization of CCL5 is crucial for CCR1-mediated leukocyte arrest on inflamed endothelium but not for their transmigration via CCR5 (Baltus et al., 2003). In bronchial mucosa of patients with chronic obstructive pulmonary disease (COPD), CCL5, and to a lesser extent CXCL7, have been found to be the most abundant chemokine expressed in the bronchial epithelium and are associated with an increase of neutrophil activation (Di Stefano et al., 2009).
CXCL12 or SDF-1α, which is the ligand for CXCR4 and CXCR7, has both proatherogenic and antiatherosclerotic properties (Weber et al., 2011). Blocking the CXCR4–CXCL12 axis leads to a release of different leukocyte subsets into the circulation. In this context, monocytosis and neutrophilia are conditions positively correlated with the development and severity of atherosclerosis. On the other hand has CXCL12 been demonstrated to be crucial for the healing of arterial lesions by the regenerative capacity of progenitor cells which are attracted to adhere be CXCL12 (Massberg et al., 2006). In addition to its production by platelets, CXCL12 is expressed in the bone marrow and in cells directly relevant to atherogenesis, including endothelial cells, smooth muscle cells, and leukocytes, which enables it to regulate the trafficking and localization of immature and maturing leukocytes, including bone marrow stem cells, neutrophils, T cells, and monocytic cells (Abi-Younes et al., 2000; Zeiffer et al., 2004; Stellos et al., 2009). Furthermore, CXCL12 has been thought to play a pro-inflammatory role in various autoimmune diseases, especially in rheumatoid arthritis and nephritis, in murine lupus erythematosus as well as in ongoing experimental autoimmune encephalomyelitis (Meiron et al., 2008; Karin, 2010). Recently, changes in CXCL12 signaling patterns have been found to be necessary for bone marrow neutrophil mobilization and are involved in polymicrobial sepsis, where its inhibition resulted in peritoneal cavity neutropenia (Delano et al., 2011). While both CCL5 and CCL7/MCP-3 are able to activate and to induce the chemotaxis of eosinophil and basophil granulocytes in allergy (Baggiolini and Dahinden, 1994), CCL11/Eotaxin has been found to be a powerful attractant for eosinophils and has also been identified in atherosclerotic lesions (Baggiolini et al., 1997; Haley et al., 2000).
The expression of CXCL7 is restricted to the platelet-lineage. Proteolytic cleavage of the carboxy-terminal part of pro-platelet basic protein (PPBP) and the proteolytic removal of the N-terminal part of PPBP produces two other chemokines namely connective tissue-activating peptide III (CTAP-III) and beta-thromboglobulin (beta-TG; Walz and Baggiolini, 1990; von Hundelshausen et al., 2007). Dependent on CXCR2, CTAP-III, and CXCL7 promote neutrophil and monocyte adhesion to human endothelial cells under flow conditions, respectively (Schenk et al., 2002; Baltus et al., 2005). The chemotactic potential of CXCL7 is also enhanced in COPD patients (Traves et al., 2004).
CXCL5/ENA-78 has been shown to act as a potent chemoattractant and activator of neutrophil function via CXCR2 (Ahuja and Murphy, 1996). CXCL5 has also been found to be strongly correlated with the number of neutrophils in patients with acute respiratory distress syndrome (Goodman et al., 1996).
During early atherosclerosis, CXCL1/GRO-α immobilized on the surface of endothelial cells via heparin proteoglycans induces the firm adhesion of rolling monocytes expressing CXCR2 (Schwartz et al., 1994; Huo et al., 2001; Boisvert et al., 2006). Moreover, a recent study has shown that in vivo, lysophosphatidic acid increased the progression of atherosclerosis and recruited leukocytes to the vessel wall during early atherogenesis via lysophosphatidic acid receptor-mediated release of endothelial CXCL1 (Zhou et al., 2011). A study conducted on elderly COPD patients has also indicated that CXCL1 might be a relevant candidate biomarker for this disease (Tsai et al., 2010).
Chemokines in Mast Cells
In addition to their role as sentinels in the recognition of pathogens, mast cells (like platelets) are able to communicate with immune cells facilitating the recruitment of leukocytes to sites of infection. Indeed, mast cells are able to produce different chemokines including CCL4, CXCL8, or CCL11 assisting in the recruitment of CD8+ T cells, eosinophils, and natural killer cells, respectively (Abraham and St John, 2010).
CCL3/MIP-1α and CCL4/MIP-1β can initiate diverse cellular responses that regulate both acute and chronic inflammation via their interaction with CCR1 and CCR5. In addition, proteoglycan-bound CCL4 is used to effectively activate and induce the adhesion of circulating lymphocytes for their extravasation through lymph node endothelium (Tanaka et al., 1993). The quaternary structures of CCL3 and CCL4 are decisive for their biological activity. Aggregation of CCL3 and CCL4 can be considered as polymerization processes of MIP-1 dimers, which constitute the basic unit of MIP-1 proteins. MIP-1 monomers form dimers of the CC-type by creating an anti-parallel β-sheet of the N-termini (Lodi et al., 1994; Czaplewski et al., 1999; Ren et al., 2010). MIP-1 dimers associate to polymers consisting up to 50 units forming a double helixed rod like structure. Polymerization of MIP-1 protects MIP-1 from proteolytic degradation while the positively charged region of MIP-1, which is crucial for the receptor binding, is buried. The continuous and slow release of monomers from the polymer leads to a shallow gradient with a long gradient and effective range for leukocyte recruitment.
CXCL8/interleukin-8/IL8 has been found in intracellular granules from skin mast cells and mast cell lines (Möller et al., 1993). Recently, Kim et al. (2010) have shown that CXCL8 synthesis is induced via the leukotriene B4/leukotriene B4 Receptor 2 pathway in response to IL-1β in human primary mast cells and mast cell line HMC-1. CXCL8 released by mast cells is implicated in the selective chemotaxis of CXCR1-expressing natural killer cells (Burke et al., 2008). CXCL8 also induces neutrophil migration and activation by binding to G-protein-coupled receptors on their surface, namely human CXCR1 and CXCR2 (Wuyts et al., 1998). During inflammation, CXCL8 is produced and presented to the endothelial surface in association with GAGs. In a recent study, using obligate monomeric and dimeric IL8 mutants, the oligomerization state of CXCL8 was shown to have an influence on the kinetics of the neutrophil extravasation. The dimeric form initiated a fast robust but short lived vascular efflux whereas the monomeric form resulted in a weaker but longer-lasting response (Das et al., 2010). Also, this chemokine is among the most important in the recruitment of inflammatory cells, mostly neutrophils, in COPD (Barnes, 2004).
Chemokines in Dendritic Cells: CCL17 as Example
CCL17/TARC (thymus and activation regulated chemokine) together with CCL22/MDC (macrophage-derived chemokine) are expressed in relevant amounts by mature dendritic cells but occur as well in other cell types such as fibroblasts. CCL17 is constitutively expressed in the thymus (Saeki and Tamaki, 2006). CCL17 is a ligand for CCR4, which is predominantly expressed on Th2 lymphocytes, basophils, and natural killer cells. Recently, dendritic cell-derived CCL17 has been found to be critical in atherosclerosis (Weber et al., 2011). Indeed, deficiency of CCL17 in Apoe−/− mice results in a reduction of the plaque formation in aortic root since CCL17 inhibits the expansion of atheroprotective Tregs and attracts CD4+ and CD3+ T cells.
Membrane-Bound Chemokines
In addition to different types of cells such as T cells, macrophages, cytokine-induced smooth muscle cells, and endothelial cells, CXCL16/SR-PSOX has been recently identified for the first time in platelets (Seizer et al., 2011). This protein constitutes an atypical chemokine because it is expressed as a cell surface bound molecule but is also found in a soluble form after shedding. CXCL16 has also been involved in different diseases. Thus, a low plasma concentration of CXCL16 has been associated with coronary artery disease and has been found in atherosclerotic lesions in human and mice (Wuttge et al., 2004; Sheikine and Hansson, 2006). In vivo and in vitro, homocysteine, a homolog of cysteine that can promote atherosclerosis (Harker et al., 1976), has been found to stimulate CXCL16 production and deposition on the surface of endothelial cells via both production of ROS and a PPARγ-dependant pathway, thereby increasing adhesion of lymphocytes to endothelial cells (Postea et al., 2008).
Like CXCL16, CX3CL1 is an atypical multimodular chemokine that exists both in a membrane-tethered or soluble form. The immobilized form consists of a chemokine domain anchored to the plasma membrane through an extended mucin-like stalk, a transmembrane helix, and an intracellular domain. Besides, CX3CL1 has an anti-apoptotic and a proliferative effect on smooth muscle cells (White et al., 2010). Previous data have shown that CX3CL1 could serve as an adhesion molecule (Fong et al., 1998; Goda et al., 2000). However, more recent data indicated that, although CX3CL1 might mediate leukocyte adhesion, this phenomenon occurred only under low shear force and not under physiological conditions (Kerfoot et al., 2003). Regarding endothelial cells, CX3CL1 is expressed on the surface of IFN-γ/TNF-α-activated HUVEC and promotes leukocyte adhesion to atherosclerotic mouse arteries in vivo and under arterial flow in vitro. More precisely, CX3CL1 expressed by inflamed endothelial cells is recognized by CX3CR1 on activated platelets. Ligation of platelet CX3CR1 results in platelet activation and subsequent exposure of P-selectin on the surface of adherent platelets (Schulz et al., 2007). The inflamed CX3CL1-expressing endothelial cells can also recruit the non-classical subset of monocytes which highly express CXC3CR1 (Geissmann et al., 2010). Under homeostatic conditions, the disruption of the CX3CL1–CX3CR1 axis leads to a specific reduction of circulating non-classical monocytes in mice (Landsman et al., 2009). Addition of full-length recombinant soluble CX3CL1 to human monocytes has also shown to decrease apoptosis triggered by serum deprivation or treatment with 7-β-hydroxycholesterol. This reduction of apoptosis occurred in both CX3CR1-expressing CD14++CD16− and CD14+CD16+ monocyte subsets. However, the precise mechanism is still unclear. A recent study shows that platelets over-expressing CX3CR1 on their surface are recruited alone or in association with monocytes to the site of inflammation. This phenomenon might contribute to an acceleration of atherosclerotic lesions (Postea et al., 2012).
Chemorepulsion
Another aspect to take into consideration in the involvement of chemokines in leukocyte trafficking is the fact that chemokines could favor a “flight” of leukocytes from a tissue to reach the blood circulation or another tissue. In this case, leukocytes might run away from a chemokine gradient. This reverse migration from a peak concentration of chemokine is named chemorepulsion or fugetaxis. However, chemorepulsion refers more to a mediator that, depending on its concentration, can either repel or recruit cells using the same receptor. This phenomenon has been comprehensively studied in the context of T-cell trafficking during the process of thymic emigration and for which an extensive review has been recently published (Bunting et al., 2011). It has been suggested that chemorepulsion could participate in the thymic egress of human thymocytes. Thus, high concentration of CXCL12 has been shown to repulse human single positive thymocytes in vitro and this “run away” could be abolished using a neutralizing CXCL12 antibody (Poznansky et al., 2000). Moreover, this chemokine has also been shown to be a chemorepulsive agent of firm adhesion to activated pancreatic islet microvascular endothelium for both diabetogenic CD4 and CD8 T cells from NOD/LtJ mice. This repulsion results in a decrease of T-cell integrin activation in a CXCR4-independent manner (Sharp et al., 2008). Using a modified flow chamber containing a transwell insert on which HUVECs are cultured, Lee et al. (2009) have shown that T cells that have extravasated in response to subendothelial CCL5 may intravasate after exposure to subendothelial CXCL12 under flow conditions. High concentration of a chemokine, as already observable in the typically bell shaped response upon increasing chemokine concentration, is an important factor. However the exact molecular mechanisms of chemokine-induced chemorepulsion are still ill defined. Using a CXCL12 model, Zlatopolskiy and Laurence (2001) postulated that chemokine-mediated repulsion would be triggered by an excess of free ligand in the vicinity of the cell that would lead to a dimerization of the receptor, followed by an internalization of the ligand/receptor complexes. Internalization, digestion of the ligand, and recycling of the receptors would be realized under the same way than during the chemoattraction process. The difference would take place through the localization of the recycled receptors. The reappearance of the internalized receptors may occur not on the apical side of the cell but on the basal side resulting in a reverse movement. Summarizing, the gradient dependent direction of a chemokine triggered movement is concentration dependent. Thus, at least two different signaling pathways have to exist at the beginning, converging later again to reorganize the cytoskeleton for cell polarization and movement. Possible explanations for the chemorepulsion at high concentrations and chemoattraction at low concentrations are the chemokine dimerization at high concentrations, high- and low-affinity binding sites for chemokines on their cognate receptor, rapid recycling of GPCRs, apical rearrangements of recycled GPCRs, the oligomerization or homodimerization of GPCR with receptor and non-receptor proteins, and allosteric mechanisms.
Genetic Variations in Chemokine Genes
Different studies have been carried out in order to evaluate the relationship between chemokine/chemokine receptor genes and inflammatory diseases including cardiovascular diseases. Table 3 provides several examples illustrating the association of chemokine/chemokine receptor polymorphisms with cardiovascular diseases.

Table 3. Examples of chemokine/chemokine receptor single nucleotide polymorphisms (SNP) associated with cardiovascular diseases.
In order to identify genes involved in cardiovascular diseases and before the emergence of genome-wide association studies (GWAS), many efforts have been undertaken with gene candidate studies. In these studies several chemokine or chemokine receptor gene candidates have been found to be associated with cardiovascular diseases. For instance, a polymorphism in the promoter region of the CCL5 gene called rs2107538 has been found associated with coronary artery disease (Simeoni et al., 2004). However, an extensive analysis based on the MONICA/KORA Augsburg Case-Cohort, Athero-Express, and CARDIoGRAM Studies has been recently carried out. Though an association between high CCL5 levels and an unstable plaque phenotype has been found, no associations of either CCL5 serum levels or its content in carotid plaques or its different genotypes with CAD or other coronary events has been established (Herder et al., 2011). The result of this study suggests that CCL5 protein levels and its gene variants might not be considered as biomarkers for the risk of coronary events in humans. As discussed by Altshuler et al. (2008), studies of candidate genes are performed on specific variants that have a small a priori probability of being disease-causing. Those studies are also able to generate false positives due to the lack of knowledge of the genetic background of cases and controls. This could explain the low reproducibility in candidate gene studies and lack of recovery between GWAS and candidate gene studies.
Amongst the different GWAS for cardiovascular disease performed during the last years, chemokine CXCL12 gene polymorphisms have been associated with CAD (e.g., Samani et al., 2007; Kathiresan et al., 2009; Franceschini et al., 2011; Schunkert et al., 2011). In addition, the study conducted by Mehta et al. (2011) found the CAD risk locus 10q11 to regulate the level of CXCL12 transcripts.
Chemokine Synergism by Heteromerization
The regulation of chemokine activity during initiation and development of inflammatory diseases is crucial to reach a fast and directed response. There is evidence that the activity of chemokines can be modulated by posttranslational processing (reviewed by Proost et al., 2003) and synergistic cytokines, e.g., IFN-γ (Mortier et al., 2011). Especially at the early phase of inflammation the concentration of a specific chemokine might not be high enough for a sufficient cell response. Hence synergism would aid to speed up the chemokine-induced response of leukocyte migration and to increase combinatorial specificity (Gouwy et al., 2005; Paoletti et al., 2005). A mixture of low concentrated individual synergizing chemokines behaves like the receptor agonist at an adequate concentration. Although the synergism of some single chemokines has been explored, so far a complete overview how many chemokines are involved is still elusive. It was previously shown that chemokine receptor induced chemotaxis may be enhanced by addition of chemokines which have per se no effect are not cognate ligands of the respective receptor and are called synergy-induced chemokines (Paoletti et al., 2005). Currently it is still unclear how this effect may be mediated in detail. Several underlying mechanisms are conceivable and can depend mainly on the respective chemokine partners and their receptors. It is possible that homeostatic and inflammatory chemokines that exhibit a different functional activity can form heteromers and act together in a synergistic way. Furthermore the signaling of GPCR-agonists can be enhanced by non-ligand CXC- and/or CC-type chemokines. Additionally, the GPCR-agonist mono and dimer equilibrium may regulate the signaling of the specific GPCR, which results in a different recruitment pattern of the target cells (Drury et al., 2011). It is of strong interest how the chemokine–chemokine interactions occur in vivo but it is difficult to find feasible approaches for a direct observation of the processes in living organisms. Some examples for a chemokine–chemokine synergism are given in the next parts.
Chemokine Heteromerization
Interaction between receptor agonist and non-ligand chemokines influences the activity of the chemokine receptor. All chemokines exhibit a typical tertiary structure homology which consists of a disordered N-terminus followed by three anti-parallel β-strands and the C-terminal α-helix. The quaternary structures of CC and CXC chemokines are different. Whereas the CXC-type forms dimers with a central β-sheet, the CC-type dimerizes through the interaction of both N-termini. In case of CC- and- CXC-type heteromers it is difficult to predict the proper structure. Our workgroup previously showed the synergistic interaction of CXCL4 and CCL5 to accelerate atherosclerosis by triggering monocyte arrest on endothelium (von Hundelshausen et al., 2005; Koenen et al., 2009). The synergistic effect is based on the heteromerization of these two chemokines, since peptides disrupting the heteromers abolish this synergism. Interestingly, the quaternary structure of the CCL5–CXCL4 complex features a CC-type heteromer, which exhibits paired N-termini, yet results in better receptor activation.
The response to CCR4 in skin-homing T-lymphocytes is enhanced by co-expressed chemokines in the inflamed skin. For example the CXCR3-agonist CXCL10 enhances the chemotaxis of CCR4-transfected preB-cells and T cells due to interaction with the CCR4-agonist CCL22 (Sebastiani et al., 2005). Further enhancement of the CCR4 activity evolves from the direct interaction of CCL22 with the CCR7-agonist CCL19. In addition, CCL22 was also shown to interact with the CCR3-agonist CCL7. In this last case, it was shown that a sequence of five amino acids of the first β-strand from CCL7, which contains two positively charged arginine residues, is needed to synergize with CCL22 and hence increases the CCR4 activation. In the same study a CCL4–CCL7 chimera lost the synergetic activity, being generated by substituting the first β-strand of CCL7 with that of the non-synergizing CCL4, lacking the positively charged amino acids. Thus the first β-strand of a chemokine, containing positive and hydrophilic amino acids, seems to have a crucial role in synergism and heteromer formation.
Furthermore monocyte recruitment is enhanced by the homeostatic chemokines CCL19 and CCL21 which are both CCR7-agonists. They synergize with CCL7 and CCL2 that result in an augmented CCR2 response to recruit monocytes (Kuscher et al., 2009). Interestingly the induced monocyte recruitment by CCL7 is enhanced 100 times by CCL19 and CCL21 whereas CCL2 showed less synergistic activity. By comparing a specific motif, comprising five amino acids in the first β-strand of all four chemokines, it has further been shown that CCL7 and CCL21 exhibit more positively charged amino acids which correlates with a higher synergistic effect confirming the importance of the first β-strand. Synergism of chemokines by heteromerization was also shown for other chemokines (Paoletti et al., 2005; Allen et al., 2007; Koenen et al., 2009). The authors suggest that heteromers of synergistically acting chemokines lead to a high affinity conformation of the respective receptor. Another study (Venetz et al., 2010) showed that heteromerization of CXCL12 with the inflammatory CXCR3-agonist CXCL9 results in a higher response of CXCR4-expressing T cells and malignant B cells on tumor vasculature.
Antagonism by Chemokine Dimers
Even if the neutrophil migration toward CXCL8 is enhanced by different CXC- and also CC-chemokines, i.e., CCL2 and CXCL12, the dimerization of CXCL8 decreases its binding to CXCR1 (Fernando et al., 2004; Weber and Koenen, 2006). This effect might not be due to structural change but rather to a loss of conformational flexibility which leads to a low-affinity configuration. Thus the dimer is not competent enough to bind the receptor N-domain. Moreover, heteromerization of CXCL8 with CXCL4 reduces the chemotactic propensity of CXCL8 (Dudek et al., 2003). These heterodimers enhance the anti-proliferative effect of CXCL4 on endothelial cells in culture, and the CXCL8-induced migration of CXCR2 transfected Baf3 cells as well (Nesmelova et al., 2005; Weber and Koenen, 2006). Inhibition of CXCL8-induced monocyte arrest is evoked by CXCL4. This effect might also be due to a less flexible CXCL8 molecule that has a lower affinity for its receptor. However, the availability of the monomer-dimer equilibrium of CXCL8 is crucial to regulate tissue-specific neutrophil recruitment given that the recruitment profile differs due to altered GAG-binding interaction (Gangavarapu et al., 2012).
Recently, it could be shown that a monomeric or dimeric state of CXCL12 plays a crucial role for the CXCR4 activation and its mode of signaling (Ray et al., 2012). The dimeric CXCL12 activates recruitment of β-arrestin 2 to CXCR4 and chemotaxis of CXCR4-expressing breast cancer cells, whereas the monomeric CXCL12 promotes the CXCR4 signaling through Gαi and Akt. Furthermore, another recent study (Drury et al., 2011) demonstrated that monomeric CXCL12 compared with the dimeric variant exhibits more contact sites for CXCR4 and thus results in different receptor signaling. To our knowledge it has not been tested, but supposedly a different or even inverse activity, e.g., the chemorepellent activity of higher concentrated CXCL12, may well be dependent on the preponderance of CXCL12 dimers.
Not only chemokine heteromerization may influence receptor driven signal transduction but as well the homooligomeric state changes the biological activity by either buried receptor binding sites, e.g., in polymeric MIP-1 or the different kinetics of monomeric versus dimeric CXCL8. These insights will be helpful to develop specific drugs interfering with oligomerization motifs thereby suppressing or enhancing desired chemokine effects.
Binding to GPCRs
In order that chemokine–chemokine partners unfold synergism it is suggested that first chemokine heteromers form and subsequently receptor binding follows. Besides the formation of a heteromeric chemokine complex, the binding to a receptor is required to mediate the synergistic effects. GAGs, as co-receptors of GPCRs, can also induce heteromerization of chemokines (Crown et al., 2006). In addition, it is speculated that instead of heteromerization, as mentioned above, different receptor binding sites for CCL2 and CCL7 are responsible for the synergistic activity as it was previously shown for CXCR3-agonists (Colvin et al., 2004).
Homeostatic chemokines like CCL21, CCL19, CXCL12, and CXCL13 are synergizing to promote a regulated lymphocyte trafficking across the lumen or basal lamina of high endothelial venules (HEVs) in lymph nodes. For example, CXCL12 augments through its receptor CXCR4 the CCR7-induced chemotaxis of T cells and therefore helps to transfer them across the HEVs without direct interactions with the CCR7-ligands CCL19 and CCL21 (Bai et al., 2009). Here the signaling through CXCR4 has a major impact because in T cells, deficient in CXCR4, no cooperative effect was observed. The synergistic effect is merely evident at suboptimal concentrations of the CCR7-ligands CCL19 and CCL21. In summary, CXCL12–CXCR4 signaling has the ability to cause a maximal T-cell response by a suboptimal CCR7-ligand concentration. A similar observation was also previously shown for CXCL13 (Paoletti et al., 2005; Bai et al., 2009). However, it is remarkable that the heteromerization of CXCL13 with CCR7-ligands is thought to be the responsible mechanistic reason for synergism, whereas synergy of CXCL12 with CCR7-ligands is independent of direct chemokine–chemokine interaction. In fact, it is assumed that CXCL12 increases the CCR7-signaling by ERK phosphorylation and actin polymerization in T cells (Bai et al., 2009). A similar conclusion was provided by van Damme’s group who showed that the synergism of CXCL8 or CXCL12 with CCL2 is mediated through CXCR1/2 (CXCL8) and CXCR4 (CXCL12; Gouwy et al., 2008, 2009). When the concentration of CCL2 is low CXCL8 helps to chemoattract monocytes. This requires binding of CXCL8 to CXCR1 and CXCR2. A further example is the synergism of CXCL12 with CCL2 where correct binding and signaling to CXCR4 and CCR2 is essential for synergistic interactions. Additionally a recent study has shown that CCR1-agonists like CCL5 and CCL3 are enhancing CXCR4-induced ERK phosphorylation and chemotaxis of mononuclear cells and it was further observed that this cooperative effect is inhibited by blocking CCR1 with specific antibodies and AMD3100 (Gouwy et al., 2011).
In summary, synergism of chemokines crucially depends on increased activation of the GPCR by heteromerization of ligand and non-ligand chemokines or cooperative interactions after chemokine activation of distinct GPCRs. Heteromerization of receptors has been observed. However the mechanistic role of these complexes in respect of ligand binding is still unclear, it might be that chemokine heteromers can stabilize and change the functional activity of receptor heteromers (Thelen et al., 2010; Kramp et al., 2011).
Therapeutics
GPCRs as therapeutic targets have been reviewed extensively (Rek et al., 2009; Koenen and Weber, 2010b, 2011; Bennett et al., 2011; Schall and Proudfoot, 2011; Kanzler et al., 2012). A lot has already been done and it is still in progress to find appropriate therapeutic drugs, particularly for the prevention and treatment of HIV. Since chemokines and the subsequent receptor signaling are involved in many diseases, there is hope that good antagonists will increase the means to treat them.
The different interactions between chemokines, which result in a changed biological activity, can be used to find new targets against inflammatory diseases. There are several possibilities for therapeutically targeting chemokines involved in inflammation. In the next part, several examples are illustrating how to alter inflammatory properties by blocking heterophilic interactions, multiple chemokine axes, direct blocking of chemokine receptors as well as blocking of GAG-binding sites.
Modified Chemokines
Modifying the target chemokine is one option to create antagonists since changing the molecular structure leads to a different binding pattern and receptor response. Especially the N-terminal part is crucial for receptor signaling and thus a change in this domain can lead to alteration or loss of receptor activation. Variants of chemokines with an extended or modified N-terminal part are for example N-methylated CCL5 (Met-RANTES) or amino-oxypentane-RANTES which block the CCL5 receptors CCR1, CCR3, and CCR5 (Proudfoot et al., 1996; Elsner et al. 1997; Proudfoot et al. 1999; Veillard et al., 2004). In liver fibrosis and atherosclerosis it was shown that inhibiting CCL5 receptors through Met-RANTES was sufficient to reduce inflammation in mice (Veillard et al., 2004; Berres et al., 2010). This phenomenon was observed in vivo and in vitro. Another example is R6H-CXCL8, a variant of CXCL8 with substitutions on the conserved ELR-triad and CXC-motif which exclusively activates CXCR1 without effecting CXCR2. This is based on a distinct CXCL8 binding mechanism: the CXCR2 activation is mediated by the N-terminal ELR- and CXC-motif whereas the N-loop of CXCL8 is essential for CXCR1 activation (Sarmiento et al., 2011). Furthermore, the above mentioned CXCL8 variant displays anti-inflammatory properties since it activates CXCR1 by desensitization of the CXCR2 response in human neutrophils. In fact, this agonist could help to clarify the biological and physiological function, especially of CXCR1, in inflammatory diseases. Thus R6H-CXCL8 is a potential candidate as a therapeutic molecule.
Glycosaminoglycan Binding Affinity
Most chemokines have the ability to bind GAGs located on the cell surface. The enhancement or reduction of this property can diminish the GPCR signaling by an indirect blockade of chemokine binding to its receptor, since GAGs are co-factors for GPCR activation. It is assumed that chemokines first bind the GAG co-receptor followed by GPCR activation.
A variety of chemokines was previously designed with altered GAG-binding affinities resulting in a loss of GPCR activation (Proudfoot et al., 2008; Shahrara et al., 2008; Rek et al., 2009). The activity of the pro-inflammatory chemokine CCL5 depends on the binding to GAGs. The substitution of positively charged residues into alanine in the 40s loop ([44AANA47]-CCL5mutant) results in defective heparin binding and loss of the ability to recruit monocytes. The heteromerization of both CCL5 variants leads to non-functional heteromers with a lack of GAG-binding efficiency (Johnson et al., 2004; Koenen and Weber, 2010b). Another study (Braunersreuther et al., 2008) additionally confirms [44AANA47]-CCL5 as a potential therapeutic agent against atherosclerosis. But in contrast to Met-RANTES, [44AANA47]-CCL5 does not directly abolish GPCR activation. Thus variations of CCL5 mutants acting in different ways can lead to anti-inflammatory properties by a direct blockade of the GPCR or by indirect inhibition through prevention of chemokine binding to GAGs on the cell surface. Another way to block chemokine activity using the affinity for GAGs is to design dominant-negative mutants with a higher GAG-binding affinity compared to the wild type chemokine (Brandner et al., 2009). H23K-RANTES showed attenuation of autoimmune uveitis in rats based on displacement of wild type CCL5 from its proteoglycan-co-receptor. Mutants with increased GAG-binding potential were designed for CCL2, as well. The PA508 mutant of CCL2 exhibits no ability for CCR2 activation but a fourfold higher GAG affinity compared to the wild type CCL2 (Piccinini et al., 2010). In a recent study in mice, PA508-CCL2 showed prevention of neointima formation and reduction of tissue damage after myocardial infarction without notable side effects. Therefore, it could be a candidate as a therapeutic agent in reducing restenosis in stents (Liehn et al., 2010). Additionally, a mutant of CXCL12 with a deficiency in heparan sulfate binding can still transduce signals through CXCR4 but is not able to promote transendothelial migration in vitro. In vivo experiments could further show that this mutant efficiently down-regulates the CXCR4 expression and desensitizes the chemotactic response toward CXCL12. Hence, this modified chemokine might work in anti-inflammatory therapies (O’Boyle et al., 2009).
Small Molecules and Antibodies
The development of small molecules blocking GPCR activation is a powerful tool for the treatment of inflammatory diseases. Major efforts have been done to find drugs for blocking HIV infection. Maraviroc (Celsentri/Selzentry; Pfizer) was established as a functional anti-HIV drug by blocking CCR5 as important entry receptor. In inflammatory diseases, like atherosclerosis, TAK779 and nbI-74330 antagonists for CCR5 and CXCR9, respectively, represent suitable therapeutic agents (Koenen and Weber, 2010b). Antagonizing CXCR4 by TAK779 additionally blocks leukocyte trafficking induced by CXCL12 (Sohy et al., 2009). Recently, DF 2156A was introduced as a novel dual inhibitor of CXCL8 receptors CXCR1 and CXCR2 (Bertini et al., 2012). This dual function is based on a non-competitive inhibition resulting in a stabilized binding between DF 2156A and the two CXCL8 receptors due to formation of specific ionic bonds in the allosteric binding site (Bertini et al., 2012). Some CXCR2- and CCR2-specific antagonists (i.e., reparixin and MLN1202) have already been tested as therapeutic drugs in clinical trials, like MLN1202, which is a CCR2-blocking monoclonal antibody shown to reduce high-sensitivity CRP as surrogate parameter for atherosclerosis (Allegretti et al., 2008; Gilbert et al., 2011).
Chemokine Heteromerization
As mentioned before some chemokines inherently exhibit synergistic function toward other chemokines which mostly depends on heteromerization. Disruption or changing these critical heterophilic interactions might entail a decrease in the physiological response which has an impact on the degree of inflammation. A prominent example is the heterophilic interaction between CXCL4 and CCL5 which results in a synergistic enhancement of CCL5 induced signaling accompanied by increased monocyte recruitment to the inflamed endothelium (Koenen et al., 2009; Koenen and Weber, 2010a). Interruption of the chemokine heteromerization by cyclic peptides was shown to eliminate synergistic effect in vitro and in vivo. Recently it was shown (Grommes et al., 2012)that small peptide antagonists, disrupting CXCL4–CCL5 heteromer formation in mouse models of acute lung injury, result in improved lung edema, less neutrophil infiltration, and reduced tissue damage. Thus targeting heterophilic chemokine interactions can act as therapeutic approach by attenuating inflammatory disease in a mild way.
Conclusion
It is still elusive which consequences the blockade of one chemokine has when entering clinical trials. For example, the dendritic cell-derived CCL17 could be identified as a catalyzer for atherosclerosis due to interference of Treg homeostasis in mice (Weber et al., 2011). Blocking CCL17 with an antibody abolished this pro-inflammatory effect. Nevertheless the blocking mechanism is unclear and which consequences the blocking has for other physiological signal cascades. For example the CXCL12–CXCR4 axis is crucial for the CXCL12-dependent recruitment of progenitor cells. Consequently a reduction of the CXCR4 level diminishes this important process in regeneration but inversely a decreased expression of CXCR4 was efficient to limit myocardial infarct size in mice (Liehn et al., 2011). Detailed knowledge and clarity of how a specific chemokine oligomerizes, binds to GAG and its GPCR as well as its interaction with other chemokines, with regard of the resulting signal cascade and immune response, are required.
Targeting GAG-binding sites of specific chemokines is a promising approach for developing drugs against chemokine driven diseases, given that the GPCR binding is not directly affected. Also the disruption of chemokine–chemokine interactions seems to become attractive, since synergistic effects can be prevented without reducing the function of the respective chemokine per se.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abi-Younes, S., Sauty, A., Mach, F., Sukhova, G. K., Libby, P., and Luster, A. D. (2000). The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ. Res. 86, 131–138.
Abraham, S. N., and St John, A. L. (2010). Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 10, 440–452.
Ahuja, S. K., and Murphy, P. M. (1996). The CXC chemokines growth-regulated oncogene (GRO) alpha, GRObeta, GROgamma, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J. Biol. Chem. 271, 20545–20550.
Allegretti, M., Bertini, R., Bizzarri, C., Beccari, A., Mantovani, A., and Locati, M. (2008). Allosteric inhibitors of chemoattractant receptors: opportunities and pitfalls. Trends Pharmacol. Sci. 29, 280–286.
Allen, S. J., Crown, S. E., and Handel, T. M. (2007). Chemokine: receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 25, 787–820.
Altshuler, D., Daly, M. J., and Lander, E. S. (2008). Genetic mapping in human disease. Science 322, 881–888.
Bacon, K., Baggiolini, M., Broxmeyer, H., Horuk, R., Lindley, I., Mantovani, A., Matsushima, K., Murphy, P., Nomiyama, H., Oppenheim, J., Rot, A., Schall, T., Tsang, M., Thorpe, R., Van Damme, J., Wadhwa, M., Yoshie, O., Zlotnik, A., and Zoon, K. (2001). Chemokine/chemokine receptor nomenclature. J. Leukoc. Biol. 70, 465–466.
Baggiolini, M., and Dahinden, C. A. (1994). CC chemokines in allergic inflammation. Immunol. Today 15, 127–133.
Baggiolini, M., Dewald, B., and Moser, B. (1997). Human chemokines: an update. Annu. Rev. Immunol. 15, 675–705.
Bai, Z., Hayasaka, H., Kobayashi, M., Li, W., Guo, Z., Jang, M. H., Kondo, A., Choi, B. I., Iwakura, Y., and Miyasaka, M. (2009). CXC chemokine ligand 12 promotes CCR7-dependent naive T cell trafficking to lymph nodes and Peyer’s patches. J. Immunol. 182, 1287–1295.
Baltus, T., Von Hundelshausen, P., Mause, S. F., Buhre, W., Rossaint, R., and Weber, C. (2005). Differential and additive effects of platelet-derived chemokines on monocyte arrest on inflamed endothelium under flow conditions. J. Leukoc. Biol. 78, 435–441.
Baltus, T., Weber, K. S., Johnson, Z., Proudfoot, A. E., and Weber, C. (2003). Oligomerization of RANTES is required for CCR1-mediated arrest but not CCR5-mediated transmigration of leukocytes on inflamed endothelium. Blood 102, 1985–1988.
Barnes, P. J. (2004). Mediators of chronic obstructive pulmonary disease. Pharmacol. Rev. 56, 515–548.
Bennett, L. D., Fox, J. M., and Signoret, N. (2011). Mechanisms regulating chemokine receptor activity. Immunology 134, 246–256.
Berres, M. L., Koenen, R. R., Rueland, A., Zaldivar, M. M., Heinrichs, D., Sahin, H., Schmitz, P., Streetz, K. L., Berg, T., Gassler, N., Weiskirchen, R., Proudfoot, A., Weber, C., Trautwein, C., and Wasmuth, H. E. (2010). Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J. Clin. Invest. 120, 4129–4140.
Bertini, R., Barcelos, L., Beccari, A., Cavalieri, B., Moriconi, A., Bizzarri, C., Di Benedetto, P., Di Giacinto, C., Gloaguen, I., Galliera, E., Corsi, M., Russo, R., Andrade, S., Cesta, M., Nano, G., Aramini, A., Cutrin, J., Locati, M., Allegretti, M., and Teixeira, M. (2012). Receptor binding mode and pharmacological characterization of a potent and selective dual CXCR1/CXCR2 non-competitive allosteric inhibitor. Br. J. Pharmacol. 165, 436–454.
Boisvert, W. A., Rose, D. M., Johnson, K. A., Fuentes, M. E., Lira, S. A., Curtiss, L. K., and Terkeltaub, R. A. (2006). Up-regulated expression of the CXCR2 ligand KC/GRO-alpha in atherosclerotic lesions plays a central role in macrophage accumulation and lesion progression. Am. J. Pathol. 168, 1385–1395.
Brandner, B., Rek, A., Diedrichs-Mohring, M., Wildner, G., and Kungl, A. J. (2009). Engineering the glycosaminoglycan-binding affinity, kinetics and oligomerization behavior of RANTES: a tool for generating chemokine-based glycosaminoglycan antagonists. Protein Eng. Des. Sel. 22, 367–373.
Braunersreuther, V., Steffens, S., Arnaud, C., Pelli, G., Burger, F., Proudfoot, A., and Mach, F. (2008). A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler. Thromb. Vasc. Biol. 28, 1090–1096.
Bunting, M. D., Comerford, I., and Mccoll, S. R. (2011). Finding their niche: chemokines directing cell migration in the thymus. Immunol. Cell Biol. 89, 185–196.
Burke, S. M., Issekutz, T. B., Mohan, K., Lee, P. W., Shmulevitz, M., and Marshall, J. S. (2008). Human mast cell activation with virus-associated stimuli leads to the selective chemotaxis of natural killer cells by a CXCL8-dependent mechanism. Blood 111, 5467–5476.
Chavakis, E., Choi, E. Y., and Chavakis, T. (2009). Novel aspects in the regulation of the leukocyte adhesion cascade. Thromb. Haemost. 102, 191–197.
Colvin, R. A., Campanella, G. S., Sun, J., and Luster, A. D. (2004). Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J. Biol. Chem. 279, 30219–30227.
Crown, S. E., Yu, Y., Sweeney, M. D., Leary, J. A., and Handel, T. M. (2006). Heterodimerization of CCR2 chemokines and regulation by glycosaminoglycan binding. J. Biol. Chem. 281, 25438–25446.
Czaplewski, L. G., Mckeating, J., Craven, C. J., Higgins, L. D., Appay, V., Brown, A., Dudgeon, T., Howard, L. A., Meyers, T., Owen, J., Palan, S. R., Tan, P., Wilson, G., Woods, N. R., Heyworth, C. M., Lord, B. I., Brotherton, D., Christison, R., Craig, S., Cribbes, S., Edwards, R. M., Evans, S. J., Gilbert, R., Morgan, P., Randle, E., Schofield, N., Varley, P. G., Fisher, J., Waltho, J. P., and Hunter, M. G. (1999). Identification of amino acid residues critical for aggregation of human CC chemokines macrophage inflammatory protein (MIP)-1alpha, MIP-1beta, and RANTES. Characterization of active disaggregated chemokine variants. J. Biol. Chem. 274, 16077–16084.
Das, S. T., Rajagopalan, L., Guerrero-Plata, A., Sai, J., Richmond, A., Garofalo, R. P., and Rajarathnam, K. (2010). Monomeric and dimeric CXCL8 are both essential for in vivo neutrophil recruitment. PLoS ONE 5, e11754. doi:10.1371/journal.pone.0011754
Delano, M. J., Kelly-Scumpia, K. M., Thayer, T. C., Winfield, R. D., Scumpia, P. O., Cuenca, A. G., Harrington, P. B., O’Malley, K. A., Warner, E., Gabrilovich, S., Mathews, C. E., Laface, D., Heyworth, P. G., Ramphal, R., Strieter, R. M., Moldawer, L. L., and Efron, P. A. (2011). Neutrophil mobilization from the bone marrow during polymicrobial sepsis is dependent on CXCL12 signaling. J. Immunol. 187, 911–918.
Deuel, T. F., Senior, R. M., Chang, D., Griffin, G. L., Heinrikson, R. L., and Kaiser, E. T. (1981). Platelet factor 4 is chemotactic for neutrophils and monocytes. Proc. Natl. Acad. Sci. U.S.A. 78, 4584–4587.
Di Stefano, A., Caramori, G., Gnemmi, I., Contoli, M., Bristot, L., Capelli, A., Ricciardolo, F. L., Magno, F., D’Anna, S. E., Zanini, A., Carbone, M., Sabatini, F., Usai, C., Brun, P., Chung, K. F., Barnes, P. J., Papi, A., Adcock, I. M., and Balbi, B. (2009). Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax 64, 968–975.
Diamond, M. S., and Springer, T. A. (1993). A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 120, 545–556.
Drury, L. J., Ziarek, J. J., Gravel, S., Veldkamp, C. T., Takekoshi, T., Hwang, S. T., Heveker, N., Volkman, B. F., and Dwinell, M. B. (2011). Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 108, 17655–17660.
Dubrac, A., Quemener, C., Lacazette, E., Lopez, F., Zanibellato, C., Wu, W. G., Bikfalvi, A., and Prats, H. (2010). Functional divergence between 2 chemokines is conferred by single amino acid change. Blood 116, 4703–4711.
Dudek, A. Z., Nesmelova, I., Mayo, K., Verfaillie, C. M., Pitchford, S., and Slungaard, A. (2003). Platelet factor 4 promotes adhesion of hematopoietic progenitor cells and binds IL-8: novel mechanisms for modulation of hematopoiesis. Blood 101, 4687–4694.
Elsner, J., Petering, H., Hochstetter, R., Kimmig, D., Wells, T. N., Kapp, A., and Proudfoot, A. E. (1997). The CC chemokine antagonist Met-RANTES inhibits eosinophil effector functions through the chemokine receptors CCR1 and CCR3. Eur. J. Immunol. 27, 2892–2898.
Fernando, H., Chin, C., Rosgen, J., and Rajarathnam, K. (2004). Dimer dissociation is essential for interleukin-8 (IL-8) binding to CXCR1 receptor. J. Biol. Chem. 279, 36175–36178.
Fong, A. M., Robinson, L. A., Steeber, D. A., Tedder, T. F., Yoshie, O., Imai, T., and Patel, D. D. (1998). Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J. Exp. Med. 188, 1413–1419.
Franceschini, N., Carty, C., Buzkova, P., Reiner, A. P., Garrett, T., Lin, Y., Vockler, J. S., Hindorff, L. A., Cole, S. A., Boerwinkle, E., Lin, D. Y., Bookman, E., Best, L. G., Bella, J. N., Eaton, C., Greenland, P., Jenny, N., North, K. E., Taverna, D., Young, A. M., Deelman, E., Kooperberg, C., Psaty, B., and Heiss, G. (2011). Association of genetic variants and incident coronary heart disease in multiethnic cohorts: the PAGE study. Circ. Cardiovasc. Genet. 4, 661–672.
Gangavarapu, P., Rajagopalan, L., Kolli, D., Guerrero-Plata, A., Garofalo, R. P., and Rajarathnam, K. (2012). The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leukoc. Biol. 91, 259–265.
Geissmann, F., Manz, M. G., Jung, S., Sieweke, M. H., Merad, M., and Ley, K. (2010). Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661.
Gilat, D., Hershkoviz, R., Mekori, Y. A., Vlodavsky, I., and Lider, O. (1994). Regulation of adhesion of CD4+ T lymphocytes to intact or heparinase-treated subendothelial extracellular matrix by diffusible or anchored RANTES and MIP-1 beta. J. Immunol. 153, 4899–4906.
Gilbert, J., Lekstrom-Himes, J., Donaldson, D., Lee, Y., Hu, M., Xu, J., Wyant, T., and Davidson, M. (2011). Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 107, 906–911.
Gleissner, C. A. (2012). Macrophage phenotype modulation by CXCL4 in atherosclerosis. Front. Physiol. 3:1. doi:10.3389/fphys.2012.00001
Goda, S., Imai, T., Yoshie, O., Yoneda, O., Inoue, H., Nagano, Y., Okazaki, T., Imai, H., Bloom, E. T., Domae, N., and Umehara, H. (2000). CX3C-chemokine, fractalkine-enhanced adhesion of THP-1 cells to endothelial cells through integrin-dependent and -independent mechanisms. J. Immunol. 164, 4313–4320.
Goodman, R. B., Strieter, R. M., Martin, D. P., Steinberg, K. P., Milberg, J. A., Maunder, R. J., Kunkel, S. L., Walz, A., Hudson, L. D., and Martin, T. R. (1996). Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 154, 602–611.
Gouwy, M., Struyf, S., Berghmans, N., Vanormelingen, C., Schols, D., and Van Damme, J. (2011). CXCR4 and CCR5 ligands cooperate in monocyte and lymphocyte migration and in inhibition of dual-tropic (R5/X4) HIV-1 infection. Eur. J. Immunol. 41, 963–973.
Gouwy, M., Struyf, S., Noppen, S., Schutyser, E., Springael, J. Y., Parmentier, M., Proost, P., and Van Damme, J. (2008). Synergy between coproduced CC and CXC chemokines in monocyte chemotaxis through receptor-mediated events. Mol. Pharmacol. 74, 485–495.
Gouwy, M., Struyf, S., Proost, P., and Van Damme, J. (2005). Synergy in cytokine and chemokine networks amplifies the inflammatory response. Cytokine Growth Factor Rev. 16, 561–580.
Gouwy, M., Struyf, S., Verbeke, H., Put, W., Proost, P., Opdenakker, G., and Van Damme, J. (2009). CC chemokine ligand-2 synergizes with the nonchemokine G protein-coupled receptor ligand fMLP in monocyte chemotaxis, and it cooperates with the TLR ligand LPS via induction of CXCL8. J. Leukoc. Biol. 86, 671–680.
Grommes, J., Alard, J. E., Drechsler, M., Wantha, S., Morgelin, M., Kuebler, W. M., Jacobs, M., Von Hundelshausen, P., Markart, P., Wygrecka, M., Preissner, K. T., Hackeng, T. M., Koenen, R. R., Weber, C., and Soehnlein, O. (2012). Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am. J. Respir. Crit. Care Med. 185, 628–636.
Haley, K. J., Lilly, C. M., Yang, J. H., Feng, Y., Kennedy, S. P., Turi, T. G., Thompson, J. F., Sukhova, G. H., Libby, P., and Lee, R. T. (2000). Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: using genomic technology to identify a potential novel pathway of vascular inflammation. Circulation 102, 2185–2189.
Harker, L. A., Ross, R., Slichter, S. J., and Scott, C. R. (1976). Homocystine-induced arteriosclerosis. The role of endothelial cell injury and platelet response in its genesis. J. Clin. Invest. 58, 731–741.
Hentzen, E. R., Neelamegham, S., Kansas, G. S., Benanti, J. A., Mcintire, L. V., Smith, C. W., and Simon, S. I. (2000). Sequential binding of CD11a/CD18 and CD11b/CD18 defines neutrophil capture and stable adhesion to intercellular adhesion molecule-1. Blood 95, 911–920.
Herder, C., Peeters, W., Illig, T., Baumert, J., De Kleijn, D. P., Moll, F. L., Poschen, U., Klopp, N., Muller-Nurasyid, M., Roden, M., Preuss, M., Karakas, M., Meisinger, C., Thorand, B., Pasterkamp, G., and Koenig, W. (2011). RANTES/CCL5 and risk for coronary events: results from the MONICA/KORA Augsburg case-cohort, Athero-Express and CARDIoGRAM studies. PLoS ONE 6, e25734. doi:10.1371/journal.pone.0025734
Huo, Y., Schober, A., Forlow, S. B., Smith, D. F., Hyman, M. C., Jung, S., Littman, D. R., Weber, C., and Ley, K. (2003). Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 9, 61–67.
Huo, Y., Weber, C., Forlow, S. B., Sperandio, M., Thatte, J., Mack, M., Jung, S., Littman, D. R., and Ley, K. (2001). The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J. Clin. Invest. 108, 1307–1314.
Johnson, Z., Kosco-Vilbois, M. H., Herren, S., Cirillo, R., Muzio, V., Zaratin, P., Carbonatto, M., Mack, M., Smailbegovic, A., Rose, M., Lever, R., Page, C., Wells, T. N., and Proudfoot, A. E. (2004). Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J. Immunol. 173, 5776–5785.
Kallel, A., Abdessalem, S., Sediri, Y., Mourali, M. S., Feki, M., Mechmeche, R., Jemaa, R., and Kaabachi, N. (2012). Polymorphisms in the CC-chemokine receptor-2 (CCR2) and -5 (CCR5) genes and risk of myocardial infarction among Tunisian male patients. Clin. Biochem. 45, 420–424.
Kanzler, I., Liehn, E. A., Koenen, R. R., and Weber, C. (2012). Anti-inflammatory therapeutic approaches to reduce acute atherosclerotic complications. Curr. Pharm. Biotechnol. 13, 37–45.
Karaali, Z. E., Sozen, S., Yurdum, M., Cacina, C., Toptas, B., Gok, O., and Agachan, B. (2010). Effect of genetic variants of chemokine receptors on the development of myocardial infarction in Turkish population. Mol. Biol. Rep. 37, 3615–3619.
Karin, N. (2010). The multiple faces of CXCL12 (SDF-1alpha) in the regulation of immunity during health and disease. J. Leukoc. Biol. 88, 463–473.
Kathiresan, S., Voight, B. F., Purcell, S., Musunuru, K., Ardissino, D., Mannucci, P. M., Anand, S., Engert, J. C., Samani, N. J., Schunkert, H., Erdmann, J., Reilly, M. P., Rader, D. J., Morgan, T., Spertus, J. A., Stoll, M., Girelli, D., Mckeown, P. P., Patterson, C. C., Siscovick, D. S., O’Donnell, C. J., Elosua, R., Peltonen, L., Salomaa, V., Schwartz, S. M., Melander, O., Altshuler, D., Merlini, P. A., Berzuini, C., Bernardinelli, L., Peyvandi, F., Tubaro, M., Celli, P., Ferrario, M., Fetiveau, R., Marziliano, N., Casari, G., Galli, M., Ribichini, F., Rossi, M., Bernardi, F., Zonzin, P., Piazza, A., Yee, J., Friedlander, Y., Marrugat, J., Lucas, G., Subirana, I., Sala, J., Ramos, R., Meigs, J. B., Williams, G., Nathan, D. M., Macrae, C. A., Havulinna, A. S., Berglund, G., Hirschhorn, J. N., Asselta, R., Duga, S., Spreafico, M., Daly, M. J., Nemesh, J., Korn, J. M., Mccarroll, S. A., Surti, A., Guiducci, C., Gianniny, L., Mirel, D., Parkin, M., Burtt, N., Gabriel, S. B., Thompson, J. R., Braund, P. S., Wright, B. J., Balmforth, A. J., Ball, S. G., Hall, A. S., Linsel-Nitschke, P., Lieb, W., Ziegler, A., Konig, I., Hengstenberg, C., Fischer, M., Stark, K., Grosshennig, A., Preuss, M., Wichmann, H. E., Schreiber, S., Ouwehand, W., Deloukas, P., Scholz, M., Cambien, F., Li, M., Chen, Z., Wilensky, R., Matthai, W., Qasim, A., Hakonarson, H. H., Devaney, J., Burnett, M. S., Pichard, A. D., Kent, K. M., Satler, L., Lindsay, J. M., Waksman, R., Knouff, C. W., Waterworth, D. M., Walker, M. C., Mooser, V., Epstein, S. E., Rader, D. J., Scheffold, T., Berger, K., Stoll, M., Huge, A., Girelli, D., Martinelli, N., Olivieri, O., Corrocher, R., Morgan, T., Spertus, J. A., McKeown, P., Patterson, C. C., Schunkert, H., Erdmann, E., Linsel-Nitschke, P., Lieb, W., Ziegler, A., König, I. R., Hengstenberg, C., Fischer, M., Stark, K., Grosshennig, A., Preuss, M., Wichmann, H. E., Schreiber, S., Hólm, H., Thorleifsson, G., Thorsteinsdottir, U., Stefansson, K., Engert, J. C., Do, R., Xie, C., Anand, S., Kathiresan, S., Ardissino, D., Mannucci, P. M., Siscovick, D., O’Donnell, C. J., Samani, N. J., Melander, O., Elosua, R., Peltonen, L., Salomaa, V., Schwartz, S. M., and Altshuler, D. (2009). Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 41, 334–341.
Kerfoot, S. M., Lord, S. E., Bell, R. B., Gill, V., Robbins, S. M., and Kubes, P. (2003). Human fractalkine mediates leukocyte adhesion but not capture under physiological shear conditions; a mechanism for selective monocyte recruitment. Eur. J. Immunol. 33, 729–739.
Kiechl, S., Laxton, R. C., Xiao, Q., Hernesniemi, J. A., Raitakari, O. T., Kahonen, M., Mayosi, B. M., Jula, A., Moilanen, L., Willeit, J., Watkins, H., Samani, N. J., Lehtimaki, T. J., Keavney, B., Xu, Q., and Ye, S. (2010). Coronary artery disease-related genetic variant on chromosome 10q11 is associated with carotid intima-media thickness and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 30, 2678–2683.
Kim, G. Y., Lee, J. W., Ryu, H. C., Wei, J. D., Seong, C. M., and Kim, J. H. (2010). Proinflammatory cytokine IL-1beta stimulates IL-8 synthesis in mast cells via a leukotriene B4 receptor 2-linked pathway, contributing to angiogenesis. J. Immunol. 184, 3946–3954.
Koenen, R. R., Von Hundelshausen, P., Nesmelova, I. V., Zernecke, A., Liehn, E. A., Sarabi, A., Kramp, B. K., Piccinini, A. M., Paludan, S. R., Kowalska, M. A., Kungl, A. J., Hackeng, T. M., Mayo, K. H., and Weber, C. (2009). Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 15, 97–103.
Koenen, R. R., and Weber, C. (2010a). Manipulating the chemokine system: therapeutic perspectives for atherosclerosis. Curr. Opin. Investig. Drugs 11, 265–272.
Koenen, R. R., and Weber, C. (2010b). Therapeutic targeting of chemokine interactions in atherosclerosis. Nat. Rev. Drug Discov. 9, 141–153.
Koenen, R. R., and Weber, C. (2011). Chemokines: established and novel targets in atherosclerosis. EMBO Mol. Med. 3, 713–725.
Kramp, B. K., Sarabi, A., Koenen, R. R., and Weber, C. (2011). Heterophilic chemokine receptor interactions in chemokine signaling and biology. Exp. Cell Res. 317, 655–663.
Kuscher, K., Danelon, G., Paoletti, S., Stefano, L., Schiraldi, M., Petkovic, V., Locati, M., Gerber, B. O., and Uguccioni, M. (2009). Synergy-inducing chemokines enhance CCR2 ligand activities on monocytes. Eur. J. Immunol. 39, 1118–1128.
Landsman, L., Bar-On, L., Zernecke, A., Kim, K. W., Krauthgamer, R., Shagdarsuren, E., Lira, S. A., Weissman, I. L., Weber, C., and Jung, S. (2009). CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 113, 963–972.
Lasagni, L., Grepin, R., Mazzinghi, B., Lazzeri, E., Meini, C., Sagrinati, C., Liotta, F., Frosali, F., Ronconi, E., Alain-Courtois, N., Ballerini, L., Netti, G. S., Maggi, E., Annunziato, F., Serio, M., Romagnani, S., Bikfalvi, A., and Romagnani, P. (2007). PF-4/CXCL4 and CXCL4L1 exhibit distinct subcellular localization and a differentially regulated mechanism of secretion. Blood 109, 4127–4134.
Lee, J. Y., Buzney, C. D., Poznansky, M. C., and Sackstein, R. (2009). Dynamic alterations in chemokine gradients induce transendothelial shuttling of human T cells under physiologic shear conditions. J. Leukoc. Biol. 86, 1285–1294.
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689.
Liehn, E. A., Piccinini, A. M., Koenen, R. R., Soehnlein, O., Adage, T., Fatu, R., Curaj, A., Popescu, A., Zernecke, A., Kungl, A. J., and Weber, C. (2010). A new monocyte chemotactic protein-1/chemokine CC motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J. Am. Coll. Cardiol. 56, 1847–1857.
Liehn, E. A., Tuchscheerer, N., Kanzler, I., Drechsler, M., Fraemohs, L., Schuh, A., Koenen, R. R., Zander, S., Soehnlein, O., Hristov, M., Grigorescu, G., Urs, A. O., Leabu, M., Bucur, I., Merx, M. W., Zernecke, A., Ehling, J., Gremse, F., Lammers, T., Kiessling, F., Bernhagen, J., Schober, A., and Weber, C. (2011). Double-edged role of the CXCL12/CXCR4 axis in experimental myocardial infarction. J. Am. Coll. Cardiol. 58, 2415–2423.
Lievens, D., and von Hundelshausen, P. (2011). Platelets in atherosclerosis. Thromb. Haemost. 106, 827–838.
Lodi, P. J., Garrett, D. S., Kuszewski, J., Tsang, M. L., Weatherbee, J. A., Leonard, W. J., Gronenborn, A. M., and Clore, G. M. (1994). High-resolution solution structure of the beta chemokine hMIP-1 beta by multidimensional NMR. Science 263, 1762–1767.
Loike, J. D., Sodeik, B., Cao, L., Leucona, S., Weitz, J. I., Detmers, P. A., Wright, S. D., and Silverstein, S. C. (1991). CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the A alpha chain of fibrinogen. Proc. Natl. Acad. Sci. U.S.A. 88, 1044–1048.
Luan, B., Han, Y., Zhang, X., Kang, J., and Yan, C. (2010). Association of the SDF1-3′A polymorphism with susceptibility to myocardial infarction in Chinese Han population. Mol. Biol. Rep. 37, 399–403.
Lum, A. F., Green, C. E., Lee, G. R., Staunton, D. E., and Simon, S. I. (2002). Dynamic regulation of LFA-1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM-1) in shear flow. J. Biol. Chem. 277, 20660–20670.
Luo, B. H., Carman, C. V., and Springer, T. A. (2007). Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647.
Luster, A. D., Alon, R., and Von Andrian, U. H. (2005). Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 6, 1182–1190.
Mantovani, A., Bonecchi, R., and Locati, M. (2006). Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat. Rev. Immunol. 6, 907–918.
Massberg, S., Konrad, I., Schurzinger, K., Lorenz, M., Schneider, S., Zohlnhoefer, D., Hoppe, K., Schiemann, M., Kennerknecht, E., Sauer, S., Schulz, C., Kerstan, S., Rudelius, M., Seidl, S., Sorge, F., Langer, H., Peluso, M., Goyal, P., Vestweber, D., Emambokus, N. R., Busch, D. H., Frampton, J., and Gawaz, M. (2006). Platelets secrete stromal cell-derived factor 1alpha and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo. J. Exp. Med. 203, 1221–1233.
Mause, S. F., Von Hundelshausen, P., Zernecke, A., Koenen, R. R., and Weber, C. (2005). Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler. Thromb. Vasc. Biol. 25, 1512–1518.
McDermott, D. H., Halcox, J. P., Schenke, W. H., Waclawiw, M. A., Merrell, M. N., Epstein, N., Quyyumi, A. A., and Murphy, P. M. (2001). Association between polymorphism in the chemokine receptor CX3CR1 and coronary vascular endothelial dysfunction and atherosclerosis. Circ. Res. 89, 401–407.
McDermott, D. H., Yang, Q., Kathiresan, S., Cupples, L. A., Massaro, J. M., Keaney, J. F. Jr., Larson, M. G., Vasan, R. S., Hirschhorn, J. N., O’Donnell, C. J., Murphy, P. M., and Benjamin, E. J. (2005). CCL2 polymorphisms are associated with serum monocyte chemoattractant protein-1 levels and myocardial infarction in the Framingham Heart Study. Circulation 112, 1113–1120.
Mehta, N. N., Li, M., William, D., Khera, A. V., Derohannessian, S., Qu, L., Ferguson, J. F., Mclaughlin, C., Shaikh, L. H., Shah, R., Patel, P. N., Bradfield, J. P., He, J., Stylianou, I. M., Hakonarson, H., Rader, D. J., and Reilly, M. P. (2011). The novel atherosclerosis locus at 10q11 regulates plasma CXCL12 levels. Eur. Heart J. 32, 963–971.
Meiron, M., Zohar, Y., Anunu, R., Wildbaum, G., and Karin, N. (2008). CXCL12 (SDF-1alpha) suppresses ongoing experimental autoimmune encephalomyelitis by selecting antigen-specific regulatory T cells. J. Exp. Med. 205, 2643–2655.
Middleton, J., Neil, S., Wintle, J., Clark-Lewis, I., Moore, H., Lam, C., Auer, M., Hub, E., and Rot, A. (1997). Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell 91, 385–395.
Moatti, D., Faure, S., Fumeron, F., Amara Mel, W., Seknadji, P., Mcdermott, D. H., Debre, P., Aumont, M. C., Murphy, P. M., De Prost, D., and Combadiere, C. (2001). Polymorphism in the fractalkine receptor CX3CR1 as a genetic risk factor for coronary artery disease. Blood 97, 1925–1928.
Möller, A., Lippert, U., Lessmann, D., Kolde, G., Hamann, K., Welker, P., Schadendorf, D., Rosenbach, T., Luger, T., and Czarnetzki, B. M. (1993). Human mast cells produce IL-8. J. Immunol. 151, 3261–3266.
Mortier, A., Gouwy, M., Van Damme, J., and Proost, P. (2011). Effect of posttranslational processing on the in vitro and in vivo activity of chemokines. Exp. Cell Res. 317, 642–654.
Mueller, A., Meiser, A., Mcdonagh, E. M., Fox, J. M., Petit, S. J., Xanthou, G., Williams, T. J., and Pease, J. E. (2008). CXCL4-induced migration of activated T lymphocytes is mediated by the chemokine receptor CXCR3. J. Leukoc. Biol. 83, 875–882.
Neelamegham, S., Taylor, A. D., Burns, A. R., Smith, C. W., and Simon, S. I. (1998). Hydrodynamic shear shows distinct roles for LFA-1 and Mac-1 in neutrophil adhesion to intercellular adhesion molecule-1. Blood 92, 1626–1638.
Nesmelova, I. V., Sham, Y., Dudek, A. Z., Van Eijk, L. I., Wu, G., Slungaard, A., Mortari, F., Griffioen, A. W., and Mayo, K. H. (2005). Platelet factor 4 and interleukin-8 CXC chemokine heterodimer formation modulates function at the quaternary structural level. J. Biol. Chem. 280, 4948–4958.
Niessner, A., Marculescu, R., Haschemi, A., Endler, G., Zorn, G., Weyand, C. M., Maurer, G., Mannhalter, C., Wojta, J., Wagner, O., and Huber, K. (2005). Opposite effects of CX3CR1 receptor polymorphisms V249I and T280M on the development of acute coronary syndrome. A possible implication of fractalkine in inflammatory activation. Thromb. Haemost. 93, 949–954.
Nomiyama, H., Hieshima, K., Osada, N., Kato-Unoki, Y., Otsuka-Ono, K., Takegawa, S., Izawa, T., Yoshizawa, A., Kikuchi, Y., Tanase, S., Miura, R., Kusuda, J., Nakao, M., and Yoshie, O. (2008). Extensive expansion and diversification of the chemokine gene family in zebrafish: identification of a novel chemokine subfamily CX. BMC Genomics 9, 222. doi:10.1186/1471-2164-9-222
Nomiyama, H., Osada, N., and Yoshie, O. (2010). The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. 21, 253–262.
Nomiyama, H., Osada, N., and Yoshie, O. (2011). A family tree of vertebrate chemokine receptors for a unified nomenclature. Dev. Comp. Immunol. 35, 705–715.
O’Boyle, G., Mellor, P., Kirby, J. A., and Ali, S. (2009). Anti-inflammatory therapy by intravenous delivery of non-heparan sulfate-binding CXCL12. FASEB J. 23, 3906–3916.
Ortlepp, J. R., Schmitz, F., Mevissen, V., Weiss, S., Huster, J., Dronskowski, R., Langebartels, G., Autschbach, R., Zerres, K., Weber, C., Hanrath, P., and Hoffmann, R. (2004). The amount of calcium-deficient hexagonal hydroxyapatite in aortic valves is influenced by gender and associated with genetic polymorphisms in patients with severe calcific aortic stenosis. Eur. Heart J. 25, 514–522.
Ortlepp, J. R., Vesper, K., Mevissen, V., Schmitz, F., Janssens, U., Franke, A., Hanrath, P., Weber, C., Zerres, K., and Hoffmann, R. (2003). Chemokine receptor (CCR2) genotype is associated with myocardial infarction and heart failure in patients under 65 years of age. J. Mol. Med. (Berl.) 81, 363–367.
Paoletti, S., Petkovic, V., Sebastiani, S., Danelon, M. G., Uguccioni, M., and Gerber, B. O. (2005). A rich chemokine environment strongly enhances leukocyte migration and activities. Blood 105, 3405–3412.
Petersen, F., Bock, L., Flad, H. D., and Brandt, E. (1999). Platelet factor 4-induced neutrophil-endothelial cell interaction: involvement of mechanisms and functional consequences different from those elicited by interleukin-8. Blood 94, 4020–4028.
Petrkova, J., Cermakova, Z., Drabek, J., Lukl, J., and Petrek, M. (2003). CC chemokine receptor (CCR)2 polymorphism in Czech patients with myocardial infarction. Immunol. Lett. 88, 53–55.
Piccinini, A. M., Knebl, K., Rek, A., Wildner, G., Diedrichs-Mohring, M., and Kungl, A. J. (2010). Rationally evolving MCP-1/CCL2 into a decoy protein with potent anti-inflammatory activity in vivo. J. Biol. Chem. 285, 8782–8792.
Pitsilos, S., Hunt, J., Mohler, E. R., Prabhakar, A. M., Poncz, M., Dawicki, J., Khalapyan, T. Z., Wolfe, M. L., Fairman, R., Mitchell, M., Carpenter, J., Golden, M. A., Cines, D. B., and Sachais, B. S. (2003). Platelet factor 4 localization in carotid atherosclerotic plaques: correlation with clinical parameters. Thromb. Haemost. 90, 1112–1120.
Postea, O., Koenen, R. R., Hristov, M., Weber, C., and Ludwig, A. (2008). Homocysteine up-regulates vascular transmembrane chemokine CXCL16 and induces CXCR6+ lymphocyte recruitment in vitro and in vivo. J. Cell. Mol. Med. 12, 1700–1709.
Postea, O., Vasina, E. M., Cauwenberghs, S., Projahn, D., Liehn, E. A., Lievens, D., Theelen, W., Kramp, B. K., Butoi, E. D., Soehnlein, O., Heemskerk, J. W., Ludwig, A., Weber, C., and Koenen, R. R. (2012). Contribution of platelet CX(3)CR1 to platelet-monocyte complex formation and vascular recruitment during hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 32, 1186–1193.
Poznansky, M. C., Olszak, I. T., Foxall, R., Evans, R. H., Luster, A. D., and Scadden, D. T. (2000). Active movement of T cells away from a chemokine. Nat. Med. 6, 543–548.
Proost, P., Vynckier, A. K., Mahieu, F., Put, W., Grillet, B., Struyf, S., Wuyts, A., Opdenakker, G., and Van Damme, J. (2003). Microbial Toll-like receptor ligands differentially regulate CXCL10/IP-10 expression in fibroblasts and mononuclear leukocytes in synergy with IFN-gamma and provide a mechanism for enhanced synovial chemokine levels in septic arthritis. Eur. J. Immunol. 33, 3146–3153.
Proudfoot, A. E., Buser, R., Borlat, F., Alouani, S., Soler, D., Offord, R. E., Schroder, J. M., Power, C. A., and Wells, T. N. (1999). Amino-terminally modified RANTES analogues demonstrate differential effects on RANTES receptors. J. Biol. Chem. 274, 32478–32485.
Proudfoot, A. E., De Souza, A. L., and Muzio, V. (2008). The use of chemokine antagonists in EAE models. J. Neuroimmunol. 198, 27–30.
Proudfoot, A. E., Power, C. A., Hoogewerf, A. J., Montjovent, M. O., Borlat, F., Offord, R. E., and Wells, T. N. (1996). Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J. Biol. Chem. 271, 2599–2603.
Pruenster, M., Mudde, L., Bombosi, P., Dimitrova, S., Zsak, M., Middleton, J., Richmond, A., Graham, G. J., Segerer, S., Nibbs, R. J., and Rot, A. (2009). The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat. Immunol. 10, 101–108.
Qi, L., Ma, J., Qi, Q., Hartiala, J., Allayee, H., and Campos, H. (2011). Genetic risk score and risk of myocardial infarction in Hispanics. Circulation 123, 374–380.
Ransohoff, R. M. (2009). Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity 31, 711–721.
Ray, P., Lewin, S. A., Mihalko, L. A., Lesher-Perez, S. C., Takayama, S., Luker, K. E., and Luker, G. D. (2012). Secreted CXCL12 (SDF-1) forms dimers under physiological conditions. Biochem. J. 442, 433–442.
Rek, A., Krenn, E., and Kungl, A. J. (2009). Therapeutically targeting protein-glycan interactions. Br. J. Pharmacol. 157, 686–694.
Ren, M., Guo, Q., Guo, L., Lenz, M., Qian, F., Koenen, R. R., Xu, H., Schilling, A. B., Weber, C., Ye, R. D., Dinner, A. R., and Tang, W. J. (2010). Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme. EMBO J. 29, 3952–3966.
Sachais, B. S., Turrentine, T., Dawicki Mckenna, J. M., Rux, A. H., Rader, D., and Kowalska, M. A. (2007). Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE−/− mice. Thromb. Haemost. 98, 1108–1113.
Saeki, H., and Tamaki, K. (2006). Thymus and activation regulated chemokine (TARC)/CCL17 and skin diseases. J. Dermatol. Sci. 43, 75–84.
Samani, N. J., Erdmann, J., Hall, A. S., Hengstenberg, C., Mangino, M., Mayer, B., Dixon, R. J., Meitinger, T., Braund, P., Wichmann, H. E., Barrett, J. H., Konig, I. R., Stevens, S. E., Szymczak, S., Tregouet, D. A., Iles, M. M., Pahlke, F., Pollard, H., Lieb, W., Cambien, F., Fischer, M., Ouwehand, W., Blankenberg, S., Balmforth, A. J., Baessler, A., Ball, S. G., Strom, T. M., Braenne, I., Gieger, C., Deloukas, P., Tobin, M. D., Ziegler, A., Thompson, J. R., and Schunkert, H. (2007). Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 357, 443–453.
Sarabi, A., Kramp, B. K., Drechsler, M., Hackeng, T. M., Soehnlein, O., Weber, C., Koenen, R. R., and Von Hundelshausen, P. (2011). CXCL4L1 inhibits angiogenesis and induces undirected endothelial cell migration without affecting endothelial cell proliferation and monocyte recruitment. J. Thromb. Haemost. 9, 209–219.
Sarmiento, J., Shumate, C., Suetomi, K., Ravindran, A., Villegas, L., Rajarathnam, K., and Navarro, J. (2011). Diverging mechanisms of activation of chemokine receptors revealed by novel chemokine agonists. PLoS ONE 6, e27967. doi:10.1186/1471-2164-9-222
Schall, T. J., and Proudfoot, A. E. (2011). Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat. Rev. Immunol. 11, 355–363.
Schenk, B. I., Petersen, F., Flad, H. D., and Brandt, E. (2002). Platelet-derived chemokines CXC chemokine ligand (CXCL)7, connective tissue-activating peptide III, and CXCL4 differentially affect and cross-regulate neutrophil adhesion and transendothelial migration. J. Immunol. 169, 2602–2610.
Schober, A., Manka, D., Von Hundelshausen, P., Huo, Y., Hanrath, P., Sarembock, I. J., Ley, K., and Weber, C. (2002). Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation 106, 1523–1529.
Schulz, C., Schafer, A., Stolla, M., Kerstan, S., Lorenz, M., Von Bruhl, M. L., Schiemann, M., Bauersachs, J., Gloe, T., Busch, D. H., Gawaz, M., and Massberg, S. (2007). Chemokine fractalkine mediates leukocyte recruitment to inflammatory endothelial cells in flowing whole blood: a critical role for P-selectin expressed on activated platelets. Circulation 116, 764–773.
Schunkert, H., Konig, I. R., Kathiresan, S., Reilly, M. P., Assimes, T. L., Holm, H., Preuss, M., Stewart, A. F., Barbalic, M., Gieger, C., Absher, D., Aherrahrou, Z., Allayee, H., Altshuler, D., Anand, S. S., Andersen, K., Anderson, J. L., Ardissino, D., Ball, S. G., Balmforth, A. J., Barnes, T. A., Becker, D. M., Becker, L. C., Berger, K., Bis, J. C., Boekholdt, S. M., Boerwinkle, E., Braund, P. S., Brown, M. J., Burnett, M. S., Buysschaert, I., Carlquist, J. F., Chen, L., Cichon, S., Codd, V., Davies, R. W., Dedoussis, G., Dehghan, A., Demissie, S., Devaney, J. M., Diemert, P., Do, R., Doering, A., Eifert, S., Mokhtari, N. E., Ellis, S. G., Elosua, R., Engert, J. C., Epstein, S. E., De Faire, U., Fischer, M., Folsom, A. R., Freyer, J., Gigante, B., Girelli, D., Gretarsdottir, S., Gudnason, V., Gulcher, J. R., Halperin, E., Hammond, N., Hazen, S. L., Hofman, A., Horne, B. D., Illig, T., Iribarren, C., Jones, G. T., Jukema, J. W., Kaiser, M. A., Kaplan, L. M., Kastelein, J. J., Khaw, K. T., Knowles, J. W., Kolovou, G., Kong, A., Laaksonen, R., Lambrechts, D., Leander, K., Lettre, G., Li, M., Lieb, W., Loley, C., Lotery, A. J., Mannucci, P. M., Maouche, S., Martinelli, N., Mckeown, P. P., Meisinger, C., Meitinger, T., Melander, O., Merlini, P. A., Mooser, V., Morgan, T., Muhleisen, T. W., Muhlestein, J. B., Munzel, T., Musunuru, K., Nahrstaedt, J., Nelson, C. P., Nothen, M. M., Olivieri, O., Patel, R. S., Patterson, C. C., Peters, A., Peyvandi, F., Qu, L., Quyyumi, A. A., Rader, D. J., Rallidis, L. S., Rice, C., Rosendaal, F. R., Rubin, D., Salomaa, V., Sampietro, M. L., Sandhu, M. S., Schadt, E., Schäfer, A., Schillert, A., Schreiber, S., Schrezenmeir, J., Schwartz, S. M., Siscovick, D. S., Sivananthan, M., Sivapalaratnam, S., Smith, A., Smith, T. B., Snoep, J. D., Soranzo, N., Spertus, J. A., Stark, K., Stirrups, K., Stoll, M., Tang, W. H., Tennstedt, S., Thorgeirsson, G., Thorleifsson, G., Tomaszewski, M., Uitterlinden, A. G., van Rij, A. M., Voight, B. F., Wareham, N. J., Wells, G. A., Wichmann, H. E., Wild, P. S., Willenborg, C., Witteman, J. C., Wright, B. J., Ye, S., Zeller, T., Ziegler, A., Cambien, F., Goodall, A. H., Cupples, L. A., Quertermous, T., März, W., Hengstenberg, C., Blankenberg, S., Ouwehand, W. H., Hall, A. S., Deloukas, P., Thompson, J. R., Stefansson, K., Roberts, R., Thorsteinsdottir, U., O’Donnell, C. J., McPherson, R., Erdmann, J., and CARDIoGRAM Consortiumand Samani, N. J. (2011). Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 43, 333–338.
Schwartz, D., Andalibi, A., Chaverri-Almada, L., Berliner, J. A., Kirchgessner, T., Fang, Z. T., Tekamp-Olson, P., Lusis, A. J., Gallegos, C., Fogelman, A. M., and Territo, M. C.. (1994). Role of the GRO family of chemokines in monocyte adhesion to MM-LDL-stimulated endothelium. J. Clin. Invest. 94, 1968–1973.
Schwartzkopff, F., Petersen, F., Grimm, T. A., and Brandt, E. (2012). CXC chemokine ligand 4 (CXCL4) down-regulates CC chemokine receptor expression on human monocytes. Innate Immun. 18, 124–139.
Sebastiani, S., Danelon, G., Gerber, B., and Uguccioni, M. (2005). CCL22-induced responses are powerfully enhanced by synergy inducing chemokines via CCR4: evidence for the involvement of first beta-strand of chemokine. Eur. J. Immunol. 35, 746–756.
Seizer, P., Stellos, K., Selhorst, G., Kramer, B. F., Lang, M. R., Gawaz, M., and May, A. E. (2011). CXCL16 is a novel scavenger receptor on platelets and is associated with acute coronary syndrome. Thromb. Haemost. 105, 1112–1114.
Shahrara, S., Proudfoot, A. E., Park, C. C., Volin, M. V., Haines, G. K., Woods, J. M., Aikens, C. H., Handel, T. M., and Pope, R. M. (2008). Inhibition of monocyte chemoattractant protein-1 ameliorates rat adjuvant-induced arthritis. J. Immunol. 180, 3447–3456.
Sharp, C. D., Huang, M., Glawe, J., Patrick, D. R., Pardue, S., Barlow, S. C., and Kevil, C. G. (2008). Stromal cell-derived factor-1/CXCL12 stimulates chemorepulsion of NOD/LtJ T-cell adhesion to islet microvascular endothelium. Diabetes 57, 102–112.
Sheikine, Y. A., and Hansson, G. K. (2006). Chemokines as potential therapeutic targets in atherosclerosis. Curr. Drug Targets 7, 13–27.
Shulman, Z., Cohen, S. J., Roediger, B., Kalchenko, V., Jain, R., Grabovsky, V., Klein, E., Shinder, V., Stoler-Barak, L., Feigelson, S. W., Meshel, T., Nurmi, S. M., Goldstein, I., Hartley, O., Gahmberg, C. G., Etzioni, A., Weninger, W., Ben-Baruch, A., and Alon, R. (2011). Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat. Immunol. 13, 67–76.
Simeoni, E., Winkelmann, B. R., Hoffmann, M. M., Fleury, S., Ruiz, J., Kappenberger, L., Marz, W., and Vassalli, G. (2004). Association of RANTES G-403A gene polymorphism with increased risk of coronary arteriosclerosis. Eur. Heart J. 25, 1438–1446.
Simi, M., Leardi, S., Tebano, M. T., Castelli, M., Costantini, F. M., and Speranza, V. (1987). Raised plasma concentrations of platelet factor 4 (PF4) in Crohn’s disease. Gut 28, 336–338.
Singh, N., Sinha, N., Kumar, S., Pandey, C. M., and Agrawal, S. (2012). Polymorphism in chemokine receptor genes and risk of acute myocardial infarction in North Indian population. Mol. Biol. Rep. 39, 2753–2759.
Sohy, D., Yano, H., De Nadai, P., Urizar, E., Guillabert, A., Javitch, J. A., Parmentier, M., and Springael, J. Y. (2009). Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J. Biol. Chem. 284, 31270–31279.
Stellos, K., Bigalke, B., Langer, H., Geisler, T., Schad, A., Kogel, A., Pfaff, F., Stakos, D., Seizer, P., Muller, I., Htun, P., Lindemann, S., and Gawaz, M. (2009). Expression of stromal-cell-derived factor-1 on circulating platelets is increased in patients with acute coronary syndrome and correlates with the number of CD34+ progenitor cells. Eur. Heart J. 30, 584–593.
Szalai, C., Duba, J., Prohaszka, Z., Kalina, A., Szabo, T., Nagy, B., Horvath, L., and Csaszar, A. (2001). Involvement of polymorphisms in the chemokine system in the susceptibility for coronary artery disease (CAD). Coincidence of elevated Lp(a) and MCP-1 -2518 G/G genotype in CAD patients. Atherosclerosis 158, 233–239.
Tanaka, Y., Adams, D. H., Hubscher, S., Hirano, H., Siebenlist, U., and Shaw, S. (1993). T-cell adhesion induced by proteoglycan-immobilized cytokine MIP-1 beta. Nature 361, 79–82.
Thelen, M., Munoz, L. M., Rodriguez-Frade, J. M., and Mellado, M. (2010). Chemokine receptor oligomerization: functional considerations. Curr. Opin. Pharmacol. 10, 38–43.
Traves, S. L., Smith, S. J., Barnes, P. J., and Donnelly, L. E. (2004). Specific CXC but not CC chemokines cause elevated monocyte migration in COPD: a role for CXCR2. J. Leukoc. Biol. 76, 441–450.
Tsai, J. J., Liao, E. C., Hsu, J. Y., Lee, W. J., and Lai, Y. K. (2010). The differences of eosinophil- and neutrophil-related inflammation in elderly allergic and non-allergic chronic obstructive pulmonary disease. J. Asthma 47, 1040–1044.
van Gils, J. M., Zwaginga, J. J., and Hordijk, P. L. (2009). Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J. Leukoc. Biol. 85, 195–204.
Veillard, N. R., Kwak, B., Pelli, G., Mulhaupt, F., James, R. W., Proudfoot, A. E., and Mach, F. (2004). Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 94, 253–261.
Venetz, D., Ponzoni, M., Schiraldi, M., Ferreri, A. J., Bertoni, F., Doglioni, C., and Uguccioni, M. (2010). Perivascular expression of CXCL9 and CXCL12 in primary central nervous system lymphoma: T-cell infiltration and positioning of malignant B cells. Int. J. Cancer 127, 2300–2312.
von Hundelshausen, P., Koenen, R. R., Sack, M., Mause, S. F., Adriaens, W., Proudfoot, A. E., Hackeng, T. M., and Weber, C. (2005). Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 105, 924–930.
von Hundelshausen, P., Koenen, R. R., and Weber, C. (2009). Platelet-mediated enhancement of leukocyte adhesion. Microcirculation 16, 84–96.
von Hundelshausen, P., Petersen, F., and Brandt, E. (2007). Platelet-derived chemokines in vascular biology. Thromb. Haemost. 97, 704–713.
von Hundelshausen, P., Weber, K. S., Huo, Y., Proudfoot, A. E., Nelson, P. J., Ley, K., and Weber, C. (2001). RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 103, 1772–1777.
Walz, A., and Baggiolini, M. (1990). Generation of the neutrophil-activating peptide NAP-2 from platelet basic protein or connective tissue-activating peptide III through monocyte proteases. J. Exp. Med. 171, 449–454.
Weber, C., and Koenen, R. R. (2006). Fine-tuning leukocyte responses: towards a chemokine “interactome.” Trends Immunol. 27, 268–273.
Weber, C., Meiler, S., Doring, Y., Koch, M., Drechsler, M., Megens, R. T., Rowinska, Z., Bidzhekov, K., Fecher, C., Ribechini, E., Van Zandvoort, M. A., Binder, C. J., Jelinek, I., Hristov, M., Boon, L., Jung, S., Korn, T., Lutz, M. B., Forster, I., Zenke, M., Hieronymus, T., Junt, T., and Zernecke, A. (2011). CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J. Clin. Invest. 121, 2898–2910.
White, G. E., Tan, T. C., John, A. E., Whatling, C., Mcpheat, W. L., and Greaves, D. R. (2010). Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signalling. Cardiovasc. Res. 85, 825–835.
Wuttge, D. M., Zhou, X., Sheikine, Y., Wagsater, D., Stemme, V., Hedin, U., Stemme, S., Hansson, G. K., and Sirsjo, A. (2004). CXCL16/SR-PSOX is an interferon-gamma-regulated chemokine and scavenger receptor expressed in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 24, 750–755.
Wuyts, A., Proost, P., Lenaerts, J. P., Ben-Baruch, A., Van Damme, J., and Wang, J. M. (1998). Differential usage of the CXC chemokine receptors 1 and 2 by interleukin-8, granulocyte chemotactic protein-2 and epithelial-cell-derived neutrophil attractant-78. Eur. J. Biochem. 255, 67–73.
Zarbock, A., Kempf, T., Wollert, K. C., and Vestweber, D. (2012). Leukocyte integrin activation and deactivation: novel mechanisms of balancing inflammation. J. Mol. Med. (Berl.) 90, 353–359.
Zee, R. Y., Cook, N. R., Cheng, S., Erlich, H. A., Lindpaintner, K., Lee, R. T., and Ridker, P. M. (2004). Threonine for alanine substitution in the eotaxin (CCL11) gene and the risk of incident myocardial infarction. Atherosclerosis 175, 91–94.
Zeiffer, U., Schober, A., Lietz, M., Liehn, E. A., Erl, W., Emans, N., Yan, Z. Q., and Weber, C. (2004). Neointimal smooth muscle cells display a proinflammatory phenotype resulting in increased leukocyte recruitment mediated by P-selectin and chemokines. Circ. Res. 94, 776–784.
Zhang, X., Zhang, B., Zhang, M., Han, Y., Zhao, Y., Meng, Z., Li, X., Kang, J., and Yan, C. (2011). Interleukin-8 gene polymorphism is associated with acute coronary syndrome in the Chinese Han population. Cytokine 56, 188–191.
Zhou, Z., Subramanian, P., Sevilmis, G., Globke, B., Soehnlein, O., Karshovska, E., Megens, R., Heyll, K., Chun, J., Saulnier-Blache, J. S., Reinholz, M., Van Zandvoort, M., Weber, C., and Schober, A. (2011). Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 13, 592–600.
Zineh, I., Beitelshees, A. L., Welder, G. J., Hou, W., Chegini, N., Wu, J., Cresci, S., Province, M. A., and Spertus, J. A. (2008). Epithelial neutrophil-activating peptide (ENA-78), acute coronary syndrome prognosis, and modulatory effect of statins. PLoS ONE 3, e3117. doi:10.1371/journal.pone.0003117
Keywords: chemokine, chemokine receptor, arrest, oligomerization, vascular inflammatory diseases, leukocyte trafficking, therapeutics
Citation: Blanchet X, Langer M, Weber C, Koenen RR and von Hundelshausen P (2012) Touch of chemokines. Front. Immun. 3:175. doi: 10.3389/fimmu.2012.00175
Received: 20 March 2012; Paper pending published: 11 April 2012;
Accepted: 09 June 2012; Published online: 12 July 2012.
Edited by:
Klaus Ley, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Klaus Ley, La Jolla Institute for Allergy and Immunology, USATatsuo Kinashi, Kansai Medical University, Japan
Copyright: © 2012 Blanchet, Langer, Weber, Koenen and von Hundelshausen. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Xavier Blanchet and Marcella Langer, Institute for Cardiovascular Prevention, Ludwig-Maximilians-University of Munich, Pettenkoferstrasse 9, 80336 Munich, Germany. e-mail:eGF2aWVyLmJsYW5jaGV0QG1lZC51bmktbXVlbmNoZW4uZGU=;bWFyY2VsbGEubGFuZ2VyQG1lZC51bmktbXVlbmNoZW4uZGU=