 Alberto Anel1*
Alberto Anel1* Juan I. Aguiló1 Elena Catalán1 Johan Garaude2 Moeez G. Rathore2
Juan I. Aguiló1 Elena Catalán1 Johan Garaude2 Moeez G. Rathore2 Julián Pardo1,3 and Martín Villalba2
Julián Pardo1,3 and Martín Villalba2
- 1 Departamento de Bioquímica y Biología Molecular y Celular, Universidad de Zaragoza, Zaragoza, Spain
- 2 INSERM-U1040, Institut de Recherche en Biothérapie, Montpellier, France
- 3 ARAID/Gobierno de Aragón, Zaragoza, Spain
The protein kinase C-θ (PKCθ), which is essential for T cell function and survival, is also required for efficient anti-tumor immune surveillance. Natural killer (NK) cells, which express PKCθ, play a prominent role in this process, mainly by elimination of tumor cells with reduced or absent major histocompatibility complex class-I (MHC-I) expression. This justifies the increased interest of the use of activated NK cells in anti-tumor immunotherapy in the clinic. The in vivo development of MHC-I-deficient tumors is much favored in PKCθ−/− mice compared with wild-type mice. Recent data offer some clues on the mechanism that could explain the important role of PKCθ in NK cell-mediated anti-tumor immune surveillance: some studies show that PKCθ is implicated in signal transduction and anti-tumoral activity of NK cells elicited by interleukin (IL)-12 or IL-15, while others show that it is implicated in NK cell functional activation mediated by certain killer-activating receptors. Alternatively, the possibility that PKCθ is involved in NK cell degranulation is discussed, since recent data indicate that it is implicated in microtubule-organizing center polarization to the immune synapse in CD4+ T cells. The implication of PKC isoforms in degranulation has been more extensively studied in cytotoxic T lymphocyte, and these studies will be also summarized.
PKCθ in T Cells
The protein kinase C-θ (PKC-θ) was initially isolated as a PKC isoform expressed in T cells (Baier et al., 1993), although it was demonstrated afterward that its expression was not restricted to them (Isakov and Altman, 2002). Structurally, PKCθ is a member of the novel, Ca2+-independent, PKC subfamily (which also includes PKCδ, ε, and η). PKCθ rapidly translocated to the immunological synapse (IS), suggesting a central role in T lymphocyte signal transduction (Monks et al., 1997). Seminal discoveries by the group of Amnon Altman demonstrated that PKCθ was indeed essential in T cell activation through the triggering of key transcription factors such as AP-1 (activating protein-1; Baier-Bitterlich et al., 1996), nuclear factor-κB (NF-κB; Coudronniere et al., 2000), and nuclear factor of activated T cell (NF-AT; Altman et al., 2004). This was confirmed by two independent groups who developed two different strategies to knockout PKCθ mice (Sun et al., 2000; Pfeifhofer et al., 2003). These transcription factors control the expression of target genes implicated in proliferation, survival, and/or cytotoxicity. One of them, Fas Ligand (FasL), is also implicated in immune homeostasis maintenance through activation-induced cell death (AICD). PKCθ deficient mice show normal lymphocyte development but display a selective phenotype in their mature T cell compartment, characterized by impaired proliferation and interleukin (IL)-2 production in response to T cell receptor (TCR)/CD28 co-stimulation (Sun et al., 2000; Pfeifhofer et al., 2003). This phenotype is also linked to the regulation of genes essential for survival, such as B-cell lymphoma (Bcl)-2 family members (Bertolotto et al., 2000; Villalba et al., 2001b; Barouch-Bentov et al., 2005). The mechanism by which PKCθ contributes to the first steps of T cell activation has recently been uncovered. A motif in the V3 domain determines its localization to the IS through the direct interaction with the co-stimulation molecule CD28 (Kong et al., 2011). Other experts in this issue extensively describe the biochemical and functional aspects of PKCθ regulation in T cells.
Natural Killer Cells
The appearance of clinically detectable tumors may be the result of the proliferation of cells that have developed sophisticated strategies to escape the immune response. Arguably, the most relevant is the total or selective loss of expression of the major histocompatibility complex class-I (MHC-I). MHC-I, known as human leukocyte antigen (HLA) in humans, mediates self-recognition and thus present endogenously synthesized antigens to CD8+ cytotoxic T lymphocytes (CTLs). Changes in MHC-I allow tumor cells to avoid CTLs and thereby the adaptive immune response (Aptsiauri et al., 2007). However, a small amount of surface MHC-I must be maintained, because its absence would make tumor cells targets of natural killer (NK) cells.
Natural killer cells are members of the innate immune system with natural cytotoxicity against tumor cells. This lymphocyte lineage produces cytokines and shows cytotoxicity and effector functions (Lanier, 2008; Velardi, 2008). NK cells predominantly target cells lacking MHC-I, which include transformed or virus-infected cells, which down-regulate MHC-I expression to avoid recognition by CTLs. Therefore the “missing self” hypothesis proposes that NK cells discriminate target cells from other healthy “self” cells based on MHC-I expression. However, it is now clear that NK cell activation depends on a complex signaling process mediated by activating and inhibitory receptors, being the functional outcome the final result of the different activating and inhibitory signals received (see the schematic representation shown in Figure 1).
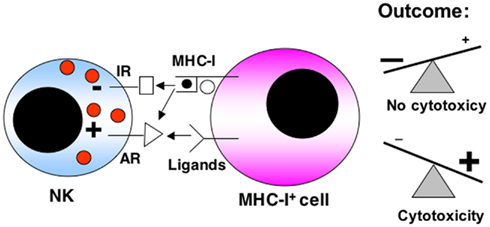
Figure 1. Schematic representation of the balance between activating (AR) and inhibitory receptors (IR) in NK cell anti-tumoral function. Receptors for MHC-I can be either inhibitory or activating. The expression levels of MHC-I or of the different ligands for the activating receptors on the target cell is of paramount importance in the final response. The final outcome between both types of signaling will determine if there is a cytotoxic response or not.
One of the most potent activating NK cell receptor (killer-activating receptor, KAR) is CD16, a receptor for the Fc domain of IgG, also termed FcγRIII, which is responsible for antibody-dependent cell-mediated cytotoxicity (ADCC), one of the main physiological functions of NK cells. CD16 signals through its association with the immunoreceptor tyrosine-based activation motif (ITAM)-containing adaptors FcεR1γ and CD3ζ, which finally activate ZAP-70 and Syk protein tyrosine kinases (López-Botet et al., 2000; Lanier, 2008; KEGGpathway, 2011). Other human KARs that share a similar signal transduction machinery are NKp46 and NKp30. Influenza virus hemagglutinin (HA) and B7-H6 expressed on the surface of tumors, respectively, have been identified as ligands for these KAR, but it is also possible that other unidentified endogenous ligands exist.
NKp44, which ligand is also unknown, completes, together with NKp30 and NKp46, the so-called natural cytotoxicity receptors (NCR) family. However, NKp44 signals through a different ITAM-containing adaptor, DAP12, that also results in the activation of ZAP-70 and Syk. This signaling adaptor is shared by several activating receptors belonging to the killer cell immunoglobulin-like receptors (KIR) family: KIR2DS1 and KIR2DS4, which ligands are specific haplotypes of HLA-C, and KIR2DS2 and KIR3DS1, which ligands are still unknown. Finally, the DAP12 adaptor is also used by NKG2C, a member of a different family of NK cell receptors, the C-lectin type family. This activating receptor is expressed as a heterodimer with CD94 and its ligand is HLA-E.
Another activating receptor of the C-lectin type is NKG2D. This protein is expressed as a homodimer, and uses a different adaptor for signaling, DAP10. This adaptor does not contain ITAM domains in its cytoplasmic tail, but it contains one YINM motif, that, upon tyrosine phosphorylation recruits both PI3K and the Grb2-Vav1-Sos1 complex. The downstream signaling of this receptor does not require ZAP-70 and/or Syk activation, but results also in ERK and PKC activation. NKG2D ligands in humans are members of the MIC and ULBP families of proteins, which are expressed by virus-infected or tumoral cells.
Another family of activating receptors is the SLAM family, which includes CD244 (also known as 2B4), CRACC (CD319), and NTB-A. CD244 ligand is CD48, while CRACC and NTB-A promote homophilic interactions. These receptors posses TIYXX motifs in their cytoplasmic tails (also called immunoreceptor tyrosine-based switch motif, ITSM), which, upon tyrosine phosphorylation recruit the adaptors SAP or EAT2. If SAP is recruited, then the signals generated, including the activation of the protein tyrosine kinase Fyn, result in activation. However, if EAT2 is recruited, then signals are rather inhibitory.
Finally, two other activating receptors are expressed in human NK cells, DNAM-1, also known as CD226, and NKp80, also known as CLEC5C. It seems that DNAM-1 signaling is dependent on specific domains present in its own cytoplasmic tail (Shibuya et al., 1998). On the other hand, it has been recently demonstrated that NKp80 transmits signals through a semi-ITAM domain present in its cytoplasmic tail, activating directly Syk (Dennehy et al., 2011). The ligands for DNAM-1 are CD112 and CD155, molecules implicated in leukocyte adhesion, and the ligand for NKp80 is AICL, a very close homolog to NKp80 expressed on the surface of solid tumors.
In mice, NK cell activating receptors belong rather to the C-lectin family, normally forming homodimers on the surface of NK cells, and transmitting signals through the DAP12 adaptor. These are Ly49D, Ly49H, and Ly49P, which ligands are, respectively, H-2Dd and murine cytomegalovirus antigens. Two other members of this family, NK1.1 and NKR-P1F signals instead through FcεR1γ and recognize members of a new C-type lectin-related (Clr) family.
PILRβ has been identified as an activating NK cell receptor in mice, being its signaling dependent also on DAP12 and its ligand CD99.
Activating NK cell receptors used by mice and humans include CD16, CD244, the heterodimer CD94/NKG2C, the homodimer NKG2D, and DNAM-1.
Inhibitory NK cell receptors posses ITIM domains in their cytoplasmic tails, and its ligation results in activation of protein tyrosine phosphatases, mainly SHP-1 and SHP-2, that counteract the signals given by activating receptors, initiated by tyrosine phosphorylation.
Most of the killer cell immunoglobulin-like receptors (KIR) have inhibitory activity, being their ligands specific or broad HLA haplotypes. Among them, KIR2DL1 recognizes HLA-Cw4 and related, “group2” alleles. KIR2DL2 and KIR2DL3 recognize HLA-Cw3 and related, “group1” alleles. KIR3DL1 is the receptor for HLA-B allotypes with Bw4 motifs. Finally, KIR3DL2 is the receptor for HLA-A3/11 and KIR2DL5 ligands are unknown.
Some members of the C-lectin type family of NK cell receptors are inhibitory. These are the heterodimer CD94/NKG2A, which ligand is HLA-E, and the homodimers Mafa and NKR-P1 (CD161), which ligands, are, respectively, cadherins and lectin-like transcript 1 (LLT1), respectively.
Other human NK cell inhibitory receptors are immunoglobulin-like transcript 2 (ILT2), which binds to most HLA-I haplotypes; LAIR-1, which binds to collagen; CEACAM-1, that binds to the different CD66 variants; and SIGLEC7, that binds to sialic acids.
Finally, KIR2DL4 (CD158d) is a member of the KIR family, which ligand is the non-classical HLA protein HLA-G, that has both activating and inhibitory capacities, similar to the described situation with CD244 (2B4).
Again, most of the inhibitory receptors in mouse NK cells belong to the C-type lectin family, Ly49a, c, e, f, g, and i which ligands are different MHC-I haplotypes (see Table 1 for the specific ligands of each receptor). Other inhibitory receptors in mouse NK cells are gp49b1, that binds to integrins, and PILRα, which ligand is CD99. Inhibitory receptors shared by mouse and human NK cells are Mafa, CD94/NKG2A, and LAIR-1. See Table 1 for a summary of all receptors, ligands, and adaptors.
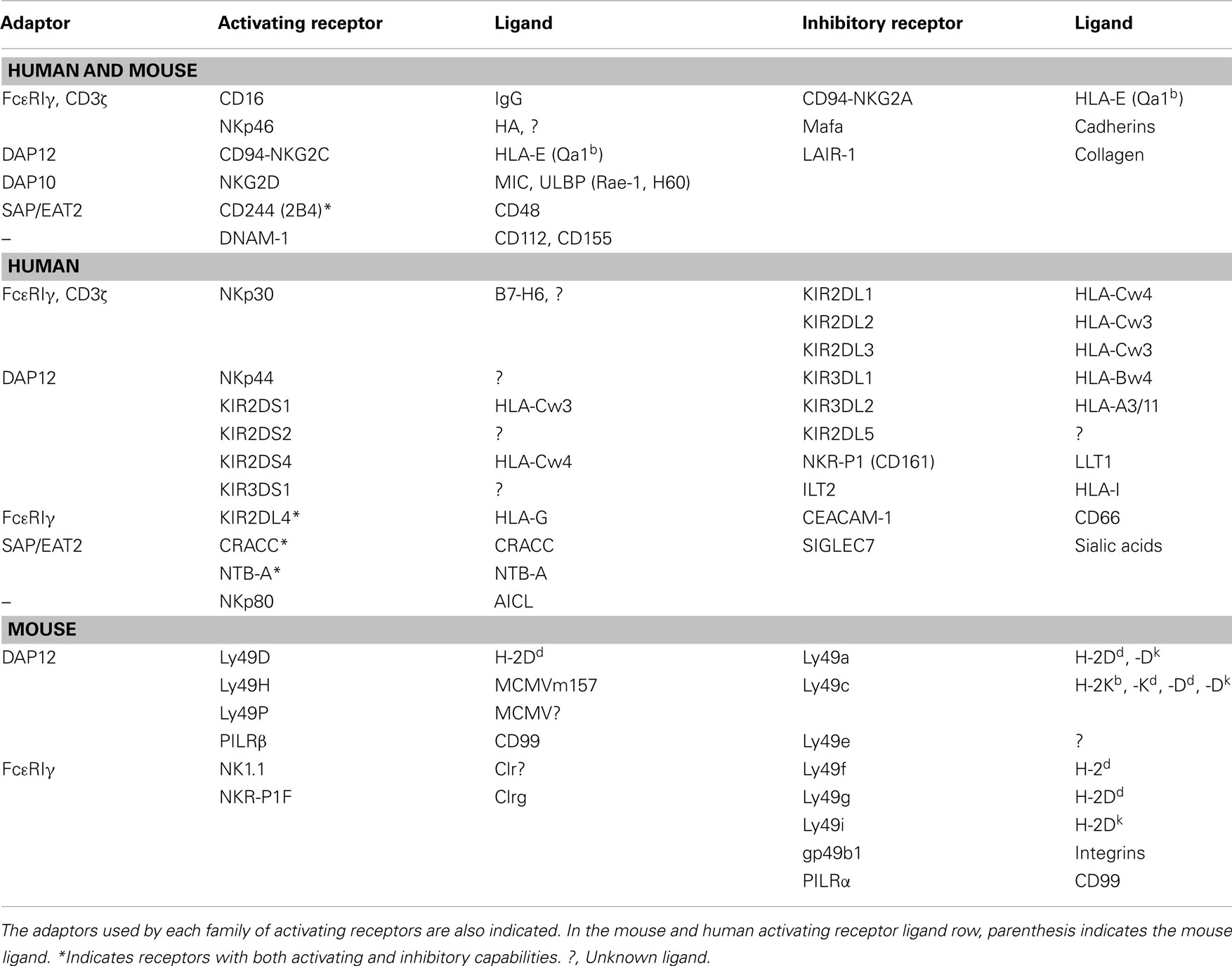
Table 1. Natural killer cell activating and inhibitory receptors in mice and humans and their ligands.
NK Cells in Tumor Immunotherapy
A large interest in NK cells is currently coming from the field of cancer immunotherapy, which tries to increase the anti-tumoral immune response of cancer patients. Data from several laboratories suggest that exploiting NK cell alloreactivity could be largely beneficial independently of the NK cell source for the treatment of blood-borne cancers (Terme et al., 2008; Velardi, 2008; Cho and Campana, 2009). Donor-versus-recipient NK cell reactivity is mediated by KIRs, which sense missing expression of donor KIR-ligand(s) in the recipient and mediate alloreactions. KIR-ligand mismatching is a prerequisite for NK cell alloreactivity because in 20 donor–recipient pairs that were not KIR-ligand mismatched in the graft-versus-host direction, no donor alloreactive NK clones were found (Ruggeri et al., 2007b).
The most advanced clinical use of NK cells is related to hematological cancers in which current clinical protocols fail inducing long-term survival in a significant number of patients. Those refractory to standard treatment, i.e., radio- and/or chemotherapy are often subjected to a myeloablative regimen followed by allogenic hematopoietic stem cell transplantation (HSCT). However, the mortality linked to this treatment approaches to 20%. In addition, Graft-versus-Host (GvH) and HvG diseases and opportunistic infections hamper this procedure. Moreover, relapse has become the leading cause of death following allogenic HSCT: the relapse rate has not decreased over the past 20 years (Kroger, 2011). In general, prognosis is poor for patients who relapsed to an allograft since effective treatment options are limited. These include donor lymphocyte infusions, withdrawal of immunosuppressive medication and second allogeneic HSCT. However, new specific cellular approaches are under investigation. In particular, the use of alloreactive NK cells after umbilical cord blood transplantation (UCBT) seems promising. KIR-ligand incompatibility in the GvH direction improves outcomes after UCBT in the clinic (Stern et al., 2008; Willemze et al., 2009). Moreover, NK cells: (1) are not responsible of GvHD; (2) can be injected as “differentiated” cells without the need of long time survival on patients body; (3) protect from opportunist infections (Willemze et al., 2009), probably through their immunoregulatory effects on B cells, T cells and macrophages, and more importantly on polymorphonuclear cells (PMNs; Bhatnagar et al., 2010). Finally, NK cell-mediated therapy after hematopoietic cell transplantation seems safe (Miller et al., 2005; Ruggeri et al., 2007a; Rubnitz et al., 2010). Immunotherapy, in particular NK cell-mediated, is probably the only approach to eliminate these highly resistant tumor cells.
In vitro studies on primary lympho-hematopoietic lineage tumor cells showed that alloreactive NK cells kill acute and chronic myeloid leukemia, as well as T cell acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia, non-Hodgkin’s lymphoma, and multiple myeloma. The only non-susceptible target was common ALL, however, KIR-ligand incompatibility in the GvH direction improves outcomes after UCBT in ALL patients. Despite their source, alloreactive NK cells home to lympho-hematopoietic sites and ablate recipient lympho-hematopoietic cells, including leukemic cells, while sparing other healthy organs. This explains why NK cells immunotherapy would be mainly useful in blood-borne cancers (Stern et al., 2008; Willemze et al., 2009).
New strategies are being developed to use NK cells in the treatment of solid tumors in the clinic. Several tumor-targeted monoclonal antibodies (mAbs) are included in the clinical care for certain tumors. Besides inducing antibody-dependent cell-mediated cytotoxicity (ADCC), these mAbs can kill their targets through activation of the complement, which in certain cases could be associated to clinical toxicity, i.e., anti-GD2 therapy (Sorkin et al., 2010). Nowadays the clinical use of mAbs tries to mainly exploit ADCC, which induces a more satisfactory clinical response and is mainly mediated by NK cells in vivo (Alderson and Sondel, 2011). Between the several activating or inhibitory Fc receptors for IgG (FcγR), NK cells express almost exclusively the activating FcγRIIIa (CD16). The importance of this receptor in the clinic is highlighted by the fact that patients with a valine at position 158 of FcγRIIIa (FcγRIIIa158v) respond better to mAbs-mediated therapy. This is linked to the higher affinity for IgG of FcγRIIIa158v NK cells, leading to a more sensitivity activating receptor and increase ADCC (Alderson and Sondel, 2011).
PKCθ in NK Cells
Compared to T cells, much less is known about the role of PKCθ in NK cells. PKCθ is expressed in NK cells (Balogh et al., 1999; Vyas et al., 2002b) and it has been demonstrated that it mediates the phosphorylation of WASP-interacting protein (WIP) during NK cell activation (Krzewski et al., 2006). In addition, it has been shown that PKCθ translocates to the IS during NK recognition of target cells (Davis et al., 1999).
It was shown that NK cells from PKCθ deficient mice had impaired IL-12-stimulated interferon (IFN)-γ production, without affecting their cytotoxic potential on YAC-1 cells (Page et al., 2008), which are extremely sensitive to NK cells, including naïve, ex vivo obtained, NK cells. However, in a different study (Tassi et al., 2008), no effect of PKCθ deficiency was observed on IFN-γ secretion induced by IL-12, IL-18, or a combination of both cytokines. In this study no effect of PKCθ deficiency was either observed on cytotoxicity against YAC-1 cells or against tumoral cells over-expressing KAR ligands (Tassi et al., 2008). It is also possible that the protocol used to generate NK cells, ex vivo culture in the presence of IL-2, may have masked the effect of PKCθ during the cellular cytotoxicity assays, since the tumor cells used in that study are especially sensitive to NK cells (Van den Broek et al., 1995; Screpanti et al., 2001; Pardo et al., 2002). Nevertheless, PKCθ was required to induce IFN-γ and TNF-α secretion by KAR that contain ITAMs such as NK1.1 and Ly49D. Finally, the early control of murine cytomegalovirus infection, that is dependent on NK cell activity, was not affected by the absence of PKCθ (Tassi et al., 2008).
In human γδ T cells the co-stimulation mediated by NKG2D was dependent on PKCθ (Nedellec et al., 2010). However, this study mostly relies on the use of the PKCθ inhibitor rottlerin, which has been shown to also inhibit other cellular kinases (Davies et al., 2000).
In addition, our group has demonstrated that PKCθ plays a prominent role in tumor immune surveillance mediated by NK cells (see below, Aguiló et al., 2009).
PKCθ in Anti-Tumor Immune Surveillance
The cancer immuno-surveillance hypothesis proposes that the immune system detects and eliminates cells undergoing tumor transformation. Immuno-deficient mice develop more tumors than immuno-competent mice and clinical data support the notion that cancer immuno-surveillance also occurs in humans (Dunn et al., 2002, 2006; Aptsiauri et al., 2007). In addition, the adaptive immune system is thought to maintain small cancer lesions in an equilibrium state (Koebel et al., 2007; Melief, 2007). Therefore, the relevant cellular effectors of immuno-surveillance must perform two critical tasks to eradicate developing tumors: directly kill tumor cells and produce cytokines such as IFN-γ to stimulate the host immune response (Dunn et al., 2006).
We tested the role of PKCθ in T cell leukemia progression by inducing the disease in wild-type (wt) and PKCθ-deficient mice with moloney murine leukemia virus (M-MuLV). In line with the above-mentioned studies, we found that disease incidence and onset were enhanced in PKCθ−/− mice. Transfer of leukemic T cells from wt donors into PKCθ-deficient and wt recipients induced leukemia in 100 and 40% of the mice, respectively. Interestingly, leukemic cells from PKCθ−/− donors were less efficient at forming tumor since only 50% of the PKCθ-deficient and 10% of the wt recipients developed the disease. Consistent with these observations, intravenous injection of low numbers of the murine lymphoma T cell line EL4 induced tumors more rapidly in PKCθ−/− mice compared to their wt counterpart. These results showed that PKCθ was essential for the immune response to leukemia in mice. This response probably involved CTL function, since both M-MuLV-induced tumors and EL4 cells expressed MHC-I (Garaude et al., 2008). These results also suggest a role of PKCθ in cancer immune surveillance, since tumors generated in the absence of this protein were less aggressive than those generated in the presence of PKCθ. In this connection, it has been also described that PKCθ, by mediating the activation of NF-κB by pre-TCR in immature thymocytes, contributes to the development of Notch3-dependent T cell lymphoma (Felli et al., 2005). This indicates that PKCθ could be somehow required for lymphoma cell “fitness,” and the results obtained in PKCθ−/− mice could reflect both effects.
Cells of the innate immune system (γδ T, NK, and NK T cells) can also mediate anti-tumor responses. Specifically, NK cells play an important role in tumor immune surveillance (Ljunggren and Karre, 1985; Terme et al., 2008; Vivier et al., 2008), as they control progression of MHC-I-deficient tumors using perforin/granzyme- and FasL-mediated cytotoxicity (Van den Broek et al., 1995; Screpanti et al., 2001; Pardo et al., 2002). Moreover, NK cells also function as mediators between innate and adaptive immunity in anti-tumor responses (Moretta et al., 2008).
Since most tumors cells down-regulate MHC-I expression to escape the CTL-mediated response (Garrido et al., 2010), it is important to study the role of NK cells in this context.
The in vivo development of a MHC-I-deficient tumor (RMA-S) is favored in PKCθ−/− mice compared with wild-type mice (Aguiló et al., 2009). Previous studies clearly demonstrated that the in vivo development of this tumor is not dependent on T cells and that it is controlled by NK cell activity (Kärre et al., 1986; Smyth et al., 1998, 2000; Kelly et al., 2002; Vosshenrich et al., 2005). The enhanced tumor growth in PKCθ−/− mice was associated with a deficient recruitment of NK cells to the site of tumor development and with a decreased activation status of recruited NK cells. This correlated with a reduced ex vivo cytotoxic potential of NK cells isolated from PKCθ−/− mice on RMA-S cells after poly I:C treatment (Aguiló et al., 2009). Interestingly, and confirming previous data (Page et al., 2008; Tassi et al., 2008), no difference was observed on the killing of YAC-1 cells suggesting that in addition to the absence of MHC-I, other regulatory events might be required for the tight control of NK cytotoxicity. YAC-1 cells are sensitive to naïve NK cells and are not able to induce tumors in syngeneic mice because of their extreme sensitivity to NK cell-mediated lysis, mediated by both perforin/granzymes and FasL (Aguiló et al., 2009). However, NK cells do not target RMA-S cells unless they are previously activated, i.e., by in vivo injection of poly I:C. Interestingly, only perforin/granzymes, but not FasL, are responsible for the elimination of RMA-S cells mediated by activated NK cells (Pardo et al., 2002; Aguiló et al., 2009). Adoptive transfer of naïve NK cells from wt or PKCθ−/− mice to PKCθ−/− mice was also performed to demonstrate that the defect in activation was intrinsic to NK cells, and not due to any other cellular component. Hence, PKCθ seems to be implicated in NK cell-mediated anti-tumor immunity at least by acting on the cytolytic potential of activated NK cells.
Poly I:C has been extensively used as an indirect in vivo NK cell activator, through the secretion of cytokines by macrophages and/or dendritic cells (Djeu et al., 1979). Poly I:C, mimics viral double-stranded RNA, and is recognized by the member of the Toll-like receptor family TLR3, which is expressed by antigen-presenting cells (Alexopoulou et al., 2001), and also by cytosolic receptors such as MDA5 and RIG-I (Kato et al., 2006). Poly I:C treatment increased the level of expression and the activation status of PKCθ in NK cells, both in vivo and in vitro. In the latter case, the presence of the whole splenocyte population was needed, being the effect presumably mediated by macrophages and/or dendritic cells (Aguiló et al., 2009). This was in agreement with previous studies demonstrating the increase in anti-tumor activity of NK cells activated by poly I:C (Akazawa et al., 2007). The increase in PKCθ expression depended on cell-to-cell contact, while its activation was mediated by a soluble factor. This soluble factor should be one of the cytokines secreted by macrophages and/or DCs that are known to activate NK cells. Since IL-12 signal transduction was reported to be affected in NK cells from PKCθ−/− mice (Page et al., 2008), this cytokine was tested first. IL-15 was also included in those studies, since it is important in regulating NK cell function and survival (Carson et al., 1994; Cooper et al., 2002), and for efficient anti-tumoral NK cell activity (Liu et al., 2012). Indeed, both, IL-12 and IL-15, activated PKCθ in NK cells, with IL-15 being more potent at inducing PKCθ phosphorylation. More importantly, neutralizing antibodies to IL-15, but not those blocking IL-12, reduced substantially NK cell PKCθ phosphorylation induced in vitro by poly I:C treatment of a mixed splenocyte population (Aguiló et al., 2009). How could IL-15 be coupled to PKCθ activation? Interestingly, PKCθ is the only T cell expressed PKC isoform that is activated through a PI3K-dependent pathway, which is also activated by TCR ligation (Villalba et al., 2002). Although the main signaling pathway elicited by cytokine receptors is mediated by JAK and STATs, cytokines such as IL-2 are also able to activate the PI3K pathway (Brennan et al., 1997). It is interesting to note that IL-2 and IL-15 share the same signaling receptors suggesting that IL-15 could also trigger the PI3K pathway, as has been suggested in some studies in T cells (Ben Ahmed et al., 2009).
Therefore, IL-15 looked as a very feasible candidate to be a mediator in PKCθ-dependent anti-tumoral NK cell activation. We have performed additional experiments, analyzing the effect of IL-15 on different NK cell functions. We have found that, although IL-15 is able to signal through PKCθ, this signaling is not needed for the main functional effects of IL-15 on NK cells (Aguiló et al., in preparation). Hence, IL-15 probably is not the main cytokine implicated in PKCθ-dependent NK cell anti-tumoral activity. Additional experiments will be required to uncover cytokines that mediate dendritic cell-mediated NK cell activation.
As a summary (Figure 2), we propose that poly I:C, through TLR3 activation on macrophages or dendritic cells, elicits the secretion of cytokines that activate NK cells, including IL-15. During tumor development, danger signals are generated that activate antigen-presenting cells, which in turn ensure tumor antigen presentation and secrete a set of cytokines that regulate NK cells. Some of these cytokines may signal through PKCθ to control NK cell activation. This activated state is needed to counteract the growing of MHC-I negative tumors such as RMA-S, while other tumors, extremely sensitive to NK cell-mediated cytotoxicity, such as YAC-1, do not grow in syngenic mice. Once KARs are activated through the ligands expressed on tumoral cells, activated NK cells would be able to eliminate the tumor in a defined cytokine environment. On the other hand, KAR ligation induces the NK cell secretion of TNF-α and IFN-γ from activated NK cells, a process in which PKCθ is implicated (Tassi et al., 2008). TNF-α is important for the recruitment of more NK cells to the tumor environment (Smyth et al., 1998), which could explain the reduced recruitment of NK cells observed in PKCθ−/− mice (Aguiló et al., 2009).
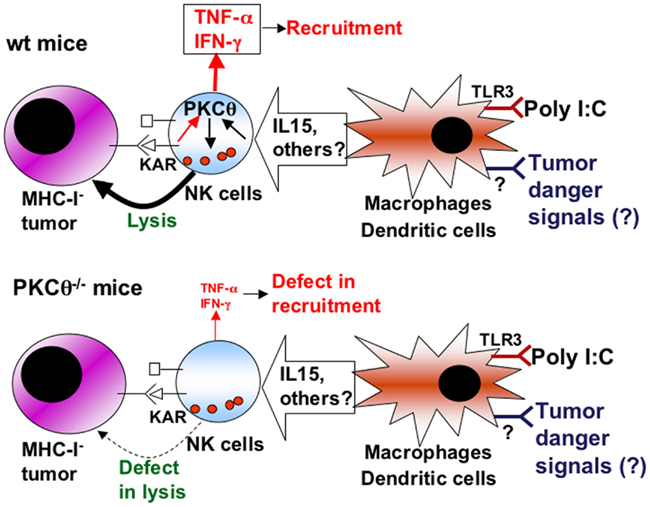
Figure 2. Schematic showing the role of PKCθ in anti-tumoral NK cell activation. Upper panel, situation in NK cells from wild-type (wt) mice: cytokines produced by macrophages and/or dendritic cells upon poly I:C activation, or upon the sensing of tumor “danger” signals, including IL-15, induce the activation of NK cells through signaling dependent on PKC-θ (black arrows). This activation increases the cytotoxic potential of NK cells that can lyse tumoral target cells, especially those that are negative for MHC-I expression, and prevent tumor development. In addition, the production of TNF-α and IFN-γ by NK cells induced by certain killer-activating receptors (KAR) is also dependent on PKC-θ (red arrows), allowing the recruitment of more immune cells and sustaining immune activation. Lower panel, situation in NK cells from PKC-θ−/− mice: as a consequence of the absence of PKC-θ, cytokines produced by macrophages and/or dendritic cells do not activate properly NK cells, which then show a defect in lysis of tumoral cells, allowing tumoral development. In addition, the production of TNF-α and IFN-γ by NK cells is decreased, causing defects in recruitment.
A Role for PKCθ in NK Cell Degranulation?
Natural killer cells from PKCθ−/− mice had a reduced capacity to degranulate against RMA-S cells, which correlated with the impairment in their lytic ability (Aguiló et al., 2009). It has been recently shown that PKC-θ, together with other members of the novel PKC family, is implicated in the formation of the IS in CD4+ T cells, probably through a function in microtubule-organizing center (MTOC) polarization (Quann et al., 2011). However, this study did not address the possible involvement of this mechanism in lytic granule secretion and on cytotoxicity.
Immunological Synapse Formation
Most of the studies performed to determine the relevant molecular components in lytic granule secretion have been realized on CTL. The IS of CTL is quite distinct from that of CD4+ T cells since it should allow both signaling and degranulation. The description of the CTL immune synapse showed the presence of a “cleft” that allowed the secretion of lytic granules components (Stinchcombe et al., 2001b). Hence, the signaling regulating synapse formation could be different in CD4+ and CD8+ T cells, and even if this signaling is shared, the regulation of lytic granule secretion should be particular to cytotoxic cells (CTL and NK cells). At the functional level, not all cytokines are secreted into the synapse of CD4+ T cells: IL-2 and IFN-γ follow a synapse-mediated secretion, while TNF-α and some chemokines do not follow a directional type of secretion (Huse et al., 2006). On the other hand, cytokine release and degranulation proceed by different pathways also in NK cells (Reefman et al., 2010).
The IS of NK cells is similar to the one described for CTLs although it has some particularities (Davis et al., 1999; Krzewski and Strominger, 2008). Two different IS have been named, the activating IS and the inhibitory IS. Activating IS is formed by a central supramolecular activating cluster (SMAC) where all receptors involved in signal transduction accumulate and a peripheral SMAC where the adhesion molecule LFA-1 is placed. Similarly, inhibitory receptors accumulate in the cSMIC and LFA-1 in the pSMIC (Carlin et al., 2001). However, given the different number of activating and inhibitory receptors (see Natural Killer Cells), the molecular mechanisms regulating signaling from the synapse is more complex than in the case of other lymphocytes subsets like CTLs (Vyas et al., 2002a; Krzewski and Strominger, 2008). Specifically, formation and signaling through IS in NK cells is dynamically regulated by activation thresholds dictated by the balance between activating and inhibitory receptors. Most of the studies have characterized the IS formed after engagement of target cells by the activating NKG2D receptor (Krzewski and Strominger, 2008). However, as explained above, signaling from NKG2D receptors is regulated differentially from other receptors. Formation of the NK cell IS is regulated at different stages (Orange et al., 2003). The first stage implies an activation process after target cell engagement, followed by signal amplification and the rearrangement of the actin cytoskeleton in the pSMAC. This process is regulated by the WASP protein. Subsequently, MTOC is polarized and granule content is released through the cSMAC, as in the case of CTLs. All these activation steps are negatively regulated by signaling transduced by SHP phosphatases from inhibitory receptors (Krzewski and Strominger, 2008).
Molecules Regulating CTL and NK Cell Degranulation
The molecular defects responsible of several diseases have allowed the identification of important molecules implicated in CTL degranulation (Clark and Griffiths, 2003; Stinchcombe and Griffiths, 2007; de Saint Basille et al., 2010). Granule movement through tubulin filaments is regulated by the adaptor protein-4 (AP-3; Clark et al., 2003), docking onto the plasma membrane is regulated by Rab27a (Haddad et al., 2001; Stinchcombe et al., 2001a), and priming or fusion with the membrane by Munc-13-4 (Feldmann et al., 2003). Finally, ASMAse is involved in the proper shrinkage of secretory granules after docking and priming steps to efficiently release granule content into target cells (Herz et al., 2009).
Regarding NK cells, it has been demonstrated that myosin IIA and WIP are required for granule polarization and NK cell-mediated cytotoxicity (Krzewski et al., 2006). Moreover, it could be suggested that Munc-13-4 is also involved in NK cell granule exocytosis since patients deficient in this protein present deficient NK cell-mediated cytotoxicity (Marcenaro et al., 2006; Bryceson et al., 2007). Syntaxin-11 also participates in the fusion of granules with the membrane of human NK cells (Arneson et al., 2007; Bryceson et al., 2007).
TCR Early Signaling during CTL Degranulation. Implication of PKC Isoforms
Although the most important regulators of lytic granule exocytosis are well described, at least in CTL, the connection between activating receptor-triggered signal transduction and lytic granule secretion is not completely understood. In the case of CTL, TCR signal transduction is initiated through protein tyrosine kinases of the src family that, together with the action of recruited ZAP-70 results in the phosphorylation and activation of phospholipase C-γ1 (Mustelin et al., 1990; Chan et al., 1992). CTL degranulation depends on these tyrosine kinase-mediated signaling pathways (Secrist et al., 1991; O’Rourke and Mescher, 1992; Anel and Kleinfeld, 1993). PLC-γ activation produces diacyl glycerol (DAG), which activates PKCs, and inositol-trisphosphate (IP3), which increases the intracellular Ca++ concentration. A combination of the phorbol ester phorbol myristate-acetate (PMA) and the calcium ionophore ionomycin induces CTL degranulation. Granule movement was dependent on Ca++ and CTL shape change was dependent on PKC activity (Takayama and Sitkovsky, 1987; Haverstick et al., 1991). Depletion of PMA-sensitive PKC isoforms by prolonged PMA exposure prevented TCR-induced degranulation in CTL clones (Nishimura et al., 1987; Anel et al., 1994). In addition, PKC inhibition prevented MTOC polarization in CTL (Nesic et al., 1998).
A closer look at the role of individual PKC isoforms in CTL function revealed that PKCθ was implicated in the induction of FasL expression at a transcriptional level (Villalba et al., 1999; Villunger et al., 1999; Pardo et al., 2003). This is probably due to the role of PKCθ in NF-AT and NF-κB activation (Sun et al., 2000; Pfeifhofer et al., 2003), transcription factors which are involved in the control of FasL gene transcription (Latinis et al., 1997; Kasibhlatla et al., 1999). As already commented above, PKCθ is the only PKC isoform that is activated through a PI3K-dependent pathway that is triggered by TCR ligation (Villalba et al., 2002). This is in agreement with the fact that functional FasL expression is also prevented by PI3K inhibitors in long-term CTL clones (Anel et al., 1995; Pardo et al., 2003). Perforin/granzyme mediated lysis of Fas-negative target cells and CTL degranulation is sensitive to broad-spectrum PKC inhibitors, and also to Gö-6976, a specific inhibitor of the classical PKC isoforms (α, β, γ), but not to low doses of rottlerin, an inhibitor that prevents PKCθ activation (Villalba et al., 1999; Pardo et al., 2003). In addition, transfection with constitutively active PKCα, but not with PKCθ, cooperated with ionomycin to promote degranulation in murine CTL clones (Pardo et al., 2003). Later works demonstrated that both constitutively active PKCα and PKCθ cooperated with thapsigargin to induce degranulation in a human CTL tumoral cell line, although PKCα seemed more efficient (Grybko et al., 2007).
Downstream Signaling during CTL Degranulation
Cytotoxic T lymphocyte and NK cell degranulation is dependent on both actin cytoskeleton and on tubulin microtubules (Gomez and Billadeau, 2008). The proximal signaling described above connects initially with remodeling of the actin cytoskeleton mainly through the adaptors Vav1 and SLP76 (Villalba et al., 2001a; Zeng et al., 2003). These actin cytoskeleton remodeling events are needed for immune synapse formation in CD4+ T cells, in CTL and also in NK cells. Once the immune synapse is formed, the next step for degranulation to occur is MTOC polarization (Kuhn and Poenie, 2002). Once MTOC is polarized, granules should move on tubulin rails, dock to the plasma membrane, and fuse to release their content in the IS. For these steps to occur, the granule and membrane proteins described above are needed, but signaling events are not completely elucidated. A role for ERK activation in CTL degranulation was clearly established (Berg et al., 1998). This activation was dependent on PI3K, and paxillin has been identified as the ERK substrate that mediates MTOC polarization to the IS (Robertson et al., 2005; Robertson and Ostergaard, 2011). However, a role for PKCθ in ERK activation was not clearly demonstrated, and primary CTL from PKCθ−/− mice degranulated normally (Puente et al., 2006).
Early Signaling during NK Cell Degranulation
Regarding NK cell degranulation elicited by KARs, signal transduction pathways are similar to those described in CTL in the case of those receptors that use FcεR1γ, CD3ζ, or DAP12 adaptors, but different in receptors that use DAP10 or SAP adaptors, such as NKG2D and CD244, respectively. However, by activating the PI3K pathway and the PLCγ-mediated increase in intracellular calcium concentration in the case of NKG2D and by the SAP-mediated activation of the protein tyrosine kinase Fyn in the case of CD244, these KAR arrive to generate similar downstream signaling that leads finally to degranulation (Cerwenka and Lanier, 2001; Lanier, 2008). In fact, it has been demonstrated that mouse NK cells do not require Syk and ZAP-70 kinases to kill different types of target cells, even those that do not express ligands for NKG2D, and that only abrogating at the same time the activity of src kinases and PI3K, degranulation was prevented (Colucci et al., 2002). In addition, the consequences of signaling transduced by the different receptors do not seem to be redundant. It has been previously shown that not a single activating receptor is sufficient to induce NK cell-mediated cytotoxicity against target cells (Bryceson et al., 2006). Only certain synergistic combinations of receptors are able to trigger this process. For example, it has been shown that engagement of CD16 induces degranulation in a non-polarized manner, meanwhile engagement of LFA-1 was able to polarize granules toward the IS. However, only after simultaneous engagement of both molecules NK cells were able to kill target cells (Bryceson et al., 2005), indicating also the importance of adhesión receptors such as LFA-1.
Downstream Signaling during NK Cell Degranulation. Implication of PKC
Regarding downstream signaling in NK cell degranulation, it has been clearly demonstrated that the PI3K-mediated activation of ERK is involved in NK cell degranulation elicited by CD16 (Bonnema et al., 1994), by KARs that use DAP12 (Jiang et al., 2000, 2002) and also by those using DAP10 as adaptor (Billadeau et al., 2003; Upshaw et al., 2006). ERK activation results finally in MTOC polarization, similarly to what was observed in CTL degranulation (Chen et al., 2006). In the case of NKG2D, the PI3K pathway also results in the recruitment of the adaptor CrkL, needed for efficient MTOC polarization (Segovis et al., 2009). A role for JNK activation has been also demonstrated in NKG2D-mediated cytotoxicity (Li et al., 2008).
The role of PKC in NK cell degranulation was initially demonstrated since a combination of PMA and ionomycin is able to induce degranulation in these cells (Bonnema et al., 1994). It was also demonstrated that DNAM-1 signal transduction is dependent on PKC expression (Shibuya et al., 1998). Less is known about the role of different PKC isoforms in NK cell degranulation. As mentioned above, PKCθ is expressed by NK cells. During activation of the human NK cell line YTS with target cells with no HLA-I expression, a PKCθ pseudo-substrate inhibitor prevented WIP phosphorylation (Krzewski et al., 2006). WIP forms a complex with WASP and myosin IIA that regulates actin cytoskeleton dynamics during NK cell activation, and WIP knockdown prevents NK cell degranulation (Krzewski et al., 2008). However, it was not demonstrated that the formation of this complex was dependent on WIP phosphorylation by PKCθ (Krzewski et al., 2006). Interestingly, it has been recently published the kinome of NK cells activated by CD16 or simultaneous CD244 and DNAM-1 ligation, being PKC-θ identified as the only PKC isoform that increase phosphorylation upon receptor ligation (König et al., 2012).
The results described above, most of them obtained in CTL, suggest that PKCθ is not mandatory for granule exocytosis. However, it has not been studied yet in detail its possible involvement in NK cell immune synapse signaling and in NK cell granule polarization and/or secretion.
Conclusion
PKCθ plays an important role in tumor immune surveillance in vivo (Garaude et al., 2008; Aguiló et al., 2009). In the case of CTL-mediated tumor control, this can be explained by its role as a CTL survival factor (Barouch-Bentov et al., 2005), the impaired cytokine response observed in PKCθ deficient animals (Sun et al., 2000) and its implication in FasL expression (Villalba et al., 1999; Villunger et al., 1999; Pardo et al., 2003).
In the case of NK cell-mediated tumor control, several functional consequences of PKCθ deficiency can also contribute to the final outcome. As depicted in the schematic Figure 2, KAR-induced TNF-α and IFN-γ secretion is defective in PKCθ−/− mice (Tassi et al., 2008), and this can contribute to a defective recruitment of effector cells to the site of tumor development (Aguiló et al., 2009). Also, PKCθ could be implicated in the induction of FasL expression (Pardo et al., 2003), although this has not been demonstrated in NK cells. In addition, the possibilities shown in Figure 3 may be considered, although they should be taken as working hypothesis that need to be better characterized:
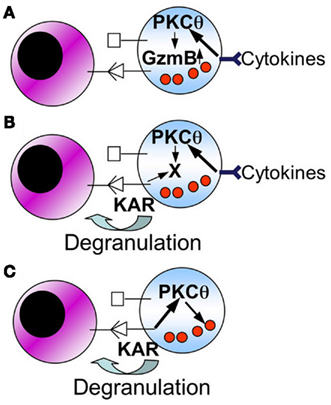
Figure 3. Schematic showing possible mechanisms for the role of PKCθ in NK cell-mediated anti-tumor immunity. The following working hypothesis are formulated (see the text for details): (A) cytokines produced by macrophages and/or dendritic cells, by signaling through PKC-θ, increase granzyme B expression. An increase in granzyme B expression is directly associated with the augmentation of the cytolytic potential of NK cells; (B) cytokines produced by macrophages and/or dendritic cells, by signaling through PKC-θ, induce or increase the expression of a protein (X) implicated in degranulation of NK cells; (C) PKC-θ could be directly implicated in degranulation of NK cells through specific KARs.
a) Macrophage or dendritic cell derived cytokines, through a pathway that implicates PKCθ, induce an increase in granzyme B expression.
b) Macrophage or dendritic cell derived cytokines, through a pathway that implicates PKCθ, induces or increases the expression of a cellular component (X) that is needed to increase the degranulation potential of NK cells.
c) PKCθ is directly implicated in NK cell degranulation elicited by specific KARs.
These three possibilities are not mutually exclusive, and all of them can contribute to the regulation of NK cell anti-tumoral potential. In addition, different signaling and/or functional responses can be activated by different KARs, and PKC-θ could be relevant for signaling elicited by some KARs but not by others. Undoubtedly, uncovering the precise molecular mechanisms that direct these processes will allow a rational improvement of NK cell-mediated anti-tumor immunotherapy protocols.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work reflected in this review was supported by the CliNK project from Sudoe (UE), by SAF2010-15341 grant to Alberto Anel, SAF2008-02139 and SAF2011-2539 grants to Julián Pardo, from Ministerio de Economía y Competitividad (Spain). Elena Catalán was supported by an FPI fellowship from MEC. This work was also supported by the program “Chercheur d’avenir” from the Region Languedoc-Rousillon (Martín Villalba), a scientific program from the “Communauté de Travail des Pyrénées” (CTPP10/09 to Martín Villalba and Alberto Anel), the Association pour la Recherche contre le Cancer (Martín Villalba), the Fondation pour la Recherche Medicale (Martín Villalba), a grant FEDER Objectif competitivite (Martín Villalba), and a fellowship from the ARC and Higher Education Commission, Pakistan (M.G. Rathore).
References
Aguiló, J. I., Garaude, J., Pardo, J., Villalba, M., and Anel, A. (2009). Protein kinase C-theta is required for NK cell activation and in vivo control of tumor progression. J. Immunol. 182, 1972–1981.
Akazawa, T., Ebihara, T., Okuno, M., Okuda, Y., Shingai, M., Tsujimura, K., Takahashi, T., Ikawa, M., Okabe, M., Inoue, N., Okamoto-Tanaka, M., Ishizaki, H., Miyoshi, J., Matsumoto, M., and Seya, T. (2007). Antitumor NK activation induced by the Toll-like receptor 3-TICAM-1 (TRIF) pathway in myeloid dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 104, 252–257.
Alderson, K. L., and Sondel, P. M. (2011). Clinical cancer therapy by NK cells via antibody-dependent cell-mediated cytotoxicity. J. Biomed. Biotechnol. 2011, 379123.
Alexopoulou, L., Holt, A. C., Medzhitov, R., and Flavell, R. A. (2001). Recognition of double-stranded RNA and activation of NF-κappaB by Toll-like receptor 3. Nature 413, 732–738.
Altman, A., Kaminski, S., Busuttil, V., Droin, N., Hu, J., Tadevosyan, Y., Hipskind, R. A., and Villalba, M. (2004). Positive feedback regulation of PLCγ1/Ca2+ signaling by PKCθ in restimulated T cells via a Tec kinase-dependent pathway. Eur. J. Immunol. 34, 2001–2011.
Anel, A., and Kleinfeld, A. M. (1993). Tyrosine phosphorylation of a 100 kDa protein is correlated with cytotoxic T-lymphocyte function. Evidence from cis unsaturated fatty acid and phenylarsineoxide inhibition. J. Biol. Chem. 268, 17578–17587.
Anel, A., Richieri, G. V., and Kleinfeld, A. M. (1994). A tyrosine phosphorylation requirement for cytotoxic T-lymphocyte degranulation. J. Biol. Chem. 269, 9506–9513.
Anel, A., Simon, A. K., Auphan, N., Buferne, M., Boyer, C., Golstein, P., and Schmitt-Verhulst, A. M. (1995). Two signalling pathways can lead to Fas ligand expression in CD8+ cytotoxic T lymphocyte clones. Eur. J. Immunol. 25, 3381–3387.
Aptsiauri, N., Cabrera, A., García-Lora, A., López-Nevoti, M. A., Ruiz-Cabello, F., and Garrido, F. (2007). MHC class I antigens and immune surveillance in transformed cells. Int. Rev. Cytol. 256, 139–189.
Arneson, L., Brickshawana, A., Segovis, C., Schoon, R., Dick, C., and Leibson, P. J. (2007). Syntaxin 11 regulates lymphocyte-mediated secretion and cytotoxicity. J. Immunol. 179, 3397–3401.
Baier, G., Telford, D., Giampa, K. M., Coggeshall, K. M., Baier-Bitterlich, G., Isakov, N., and Altman, A. (1993). Molecular cloning and characterization of PKCθ, a human novel member of the protein kinase C (PKC) gene family expressed predominantly in hematopoietic cells. J. Biol. Chem. 268, 4997–5004.
Baier-Bitterlich, G., Uberall, F., Bauer, B., Fresser, F., Wachter, H., Grunicke, H., Utermann, G., Altman, A., and Baier, G. (1996). Protein kinase C-θ isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol. Cell. Biol. 16, 1842–1850.
Balogh, G., de Boland, A. R., Boland, R., and Barja, P. (1999). Effect of 1,25[OH](2)-vitamin D(3) on the activation of natural killer cells: role of protein kinase C and extracellular calcium. Exp. Mol. Pathol. 67, 63–74.
Barouch-Bentov, R., Lemmens, E. E., Hu, J., Janssen, E. M., Droin, N. M., Song, J., Schoenberger, S. P., and Altman, A. (2005). Protein kinase C-θ is an early survival factor required for differentiation of effector CD8+ T cells. J. Immunol. 175, 5126–5134.
Ben Ahmed, M., Belhadj Hmida, N., Moes, N., Buyse, S., Abledadhim, M., Louzir, H., and Cerf-Bensussan, N. (2009). IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway. J. Immunol. 182, 6763–6770.
Berg, N. N., Puente, L. G., Dawicki, W., and Ostergaard, H. L. (1998). Sustained TCR signaling is required for MAPK activation and degranulation by CTL. J. Immunol. 161, 2919–2924.
Bertolotto, C., Maulon, L., Filippa, N., Baier, G., and Auberger, P. (2000). Protein kinase C theta and epsilon promote T-cell survival by a rsk-dependent phosphorylation and inactivation of BAD. J. Biol. Chem. 275, 37246–37250.
Bhatnagar, N., Hong, H. S., Krishnaswamy, J. K., Haghikia, A., Behrens, G. M., Schmidt, R. E., and Jacobs, R. (2010). Cytokine-activated NK cells inhibit PMN apoptosis and preserve their functional capacity. Blood 116, 1308–1316.
Billadeau, D. D., Upshaw, J. L., Schoon, R. A., Dick, C. J., and Leibson, P. J. (2003). NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 4, 557–564.
Bonnema, J. D., Karnitz, L. M., Schoon, R. A., Abraham, R. T., and Leibson, P. J. (1994). Fc receptor stimulation of phosphatidylinositol 3-kinase in natural killer cells is associated with protein kinase C-independent granule release and cell-mediated cytotoxicity. J. Exp. Med. 180, 1427–1435.
Brennan, P., Babage, J. W., Burgering, B. M. T., Gromer, B., Reif, K., and Cantrell, D. A. (1997). Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity 7, 679–689.
Bryceson, Y., March, M., Ljunggren, H., and Long, E. (2006). Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107, 159–166.
Bryceson, Y., Rudd, E., Zheng, C., Edner, J., Ma, D., Wood, S., Bechensteen, A., Boelens, J. J., Celkan, T., Farah, R. A., Hultenby, K., Winiarski, J., Roche, P. A., Nordenskjöld, M., Henter, J. I., Long, E. O., and Ljunggren, H. G. (2007). Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood 110, 1906–1915.
Bryceson, Y. T., March, M., Barber, D. F., Ljunggren, H. G., and Long, E. O. (2005). Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J. Exp. Med. 202, 1001–1012.
Carlin, L., Eleme, K., McCann, F., and Davis, D. (2001). Intercellular transfer and supramolecular organization of human leukocyte antigen C at inhibitory natural killer cell immune synapses. J. Exp. Med. 194, 1507–1517.
Carson, W. E., Giri, J. G., Lindemann, M. J., Linett, M. L., Ahdieh, M., Paxton, R., Anderson, D., Eisenmann, J., Grabstein, K., and Caligiuri, M. A. (1994). Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J. Exp. Med. 180, 1395–1403.
Cerwenka, A., and Lanier, L. L. (2001). Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 1, 41–49.
Chan, A. C., Iwashima, M., Turck, C. W., and Weiss, A. (1992). ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR ζ chain. Cell 71, 649.
Chen, X., Allan, D. S., Krzewski, K., Ge, B., Koopcow, H., and Strominger, J. L. (2006). CD28-stimulated ERK2 phosphorylation is required for polarization of the microtubule organizing center and granules in YTS NK cells. Proc. Natl. Acad. Sci. U.S.A. 103, 10346–10351.
Cho, D., and Campana, D. (2009). Expansion and activation of natural killer cells for cancer immunotherapy. Korean J. Lab. Med. 29, 89–96.
Clark, R., and Griffiths, G. M. (2003). Lytic granules, secretory lysosomes and disease. Curr. Opin. Immunol. 15, 516–521.
Clark, R. H., Stinchcombe, J. C., Day, A., Blott, E., Booth, S., Bossi, G., Hamblin, T., Davies, E. G., and Griffiths, G. M. (2003). AP3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat. Immunol. 4, 1111–1120.
Colucci, F., Schweighoffer, E., Tomasello, E., Turner, M., Ortaldo, J., Vivier, E., Tybulewicz, V., and Di Santo, J. (2002). Natural cytotoxicity uncoupled from the Syk and ZAP-70 intracellular kinases. Nat. Immunol. 3, 288–294.
Cooper, M. A., Fehniger, T. A., VanDeusen, J. B., Waite, R. E., Liu, Y., Aguila, H. L., and Caligiuri, M. A. (2002). In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood 100, 3633–3638.
Coudronniere, N., Villalba, M., Englund, N., and Altman, A. (2000). NF-kB activation induced by TCR/CD28 costimulation is mediated by PKC-θ. Proc. Natl. Acad. Sci. U.S.A. 97, 3394–3399.
Davies, S. P., Reddy, H., Caivano, M., and Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105.
Davis, D., Chiu, I., Fassett, M., Cohen, G., Mandelboim, O., and Strominger, J. (1999). The human natural killer cell immune synapse. Proc. Natl. Acad. Sci. U.S.A. 96, 15062–15067.
de Saint Basille, G., Ménasché, G., and Fischer, A. (2010). Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat. Rev. Immunol. 11, 568–579.
Dennehy, K. M., Klimosch, S. N., and Steinle, A. (2011). NKp80 uses an atypical hemi-ITAM to trigger NK cytotoxicity. J. Immunol. 186, 657–661.
Djeu, J. Y., Heinbaugh, J. A., Holden, H. T., and Herberman, R. B. (1979). Role of macrophages in the augmentation of mouse NK cell activity by Poly I:C and interferon. J. Immunol. 122, 182–188.
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., and Schreiber, R. D. (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3, 991–998.
Dunn, G. P., Koebel, C. M., and Schreiber, R. D. (2006). Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 6, 836–848.
Feldmann, J., Callebaut, I., Raposo, G., Certain, S., Bacq, D., Dumont, C., Lambert, N., Ouachee-Chardin, M., Chedeville, G., Tamary, H., Minard-Colin, V., Vilmer, E., Blanche, S., Le Deist, F., Fischer, A., and de Saint Basile, G. (2003). Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 115, 461–473.
Felli, M., Vacca, A., Calce, A., Bellavia, D., Campese, A., Grillo, R., Di Giovine, M., Checquolo, S., Talora, C., Palermo, R., Di Mario, G., Frati, L., Gulino, A., and Screpanti, I. (2005). PKC theta mediates pre-TCR signaling and contributes to Notch3-induced T-cell leukemia. Oncogene 24, 992–1000.
Garaude, J., Kaminski, S., Cherni, S., Aguiló, J. I., Jacquet, C., Plays, M., Rodriguez, F., Hernández, J., Hipskind, R. A., Anel, A., and Villalba, M. (2008). Impaired anti-leukemic immune response in PKCθ-deficient mice. Mol. Immunol. 45, 3463–3469.
Garrido, F., Algarra, I., and García-Lora, A. (2010). The escape of cancer from T lymphocytes: immunoselection of MHC class I loss variants harboring structural-irreversible “hard” lesions. Cancer Immunol. Immunother. 59, 1601–1606.
Gomez, T. S., and Billadeau, D. D. (2008). T cell activation and the cytoskeleton: you can’t have one without the other. Adv. Immunol. 97, 1–64.
Grybko, M. J., Pores-Fernando, A. T., Wurth, G. A., and Zweifach, A. (2007). Protein kinase C activity is required for cytotoxic T cell lytic granule exocytosis, but the θ isoform does not play a preferential role. J. Leukoc. Biol. 81, 509–519.
Haddad, E. K., Wu, X., Hammer, J. A., and Henkart, P. A. (2001). Defective granule exocytosis in Rab27a-deficient lymphocytes from Ashen mice. J. Cell Biol. 152, 835–842.
Haverstick, D. M., Engelhard, V. H., and Gray, L. S. (1991). Three intracellular signals for cytotoxic T lymphocyte mediated killing. Independent roles for protein kinase C, Ca2+ influx, and Ca2+ release from internal stores. J. Immunol. 146, 3306–3313.
Herz, J., Pardo, J., Kashkar, H., Schramm, M., Kuzmenkina, E., Bos, E., Wiegmann, K., Wallich, R., Peters, P. J., Herzig, S., Schmelzer, E., Krönke, M., Simon, M. M., and Utermöhlen, O. (2009). Acid sphingomyelinase is a key regulator of cytotoxic granule secretion by primary T lymphocytes. Nat. Immunol. 10, 761–768.
Huse, M., Lillemeier, B. F., Kuhns, M. S., Chen, D. S., and Davis, M. M. (2006). T cells use two directionally distinct pathways for cytokine secretion. Nat. Immunol. 7, 247–255.
Isakov, N., and Altman, A. (2002). Protein kinase C θ in T cell activation. Annu. Rev. Immunol. 20, 761–794.
Jiang, K., Zhong, B., Gilvary, D. L., Corliss, B. C., Hong-Geller, E., Wei, S., and Djeu, J. Y. (2000). Pivotal role of PI-3 kinase in regulation of cytotoxicity in natural killer cells. Nat. Immunol. 1, 419–425.
Jiang, K., Zhong, B., Gilvary, D. L., Corliss, B. C., Vivier, E., Hong-Geller, E., Wei, S., and Djeu, J. Y. (2002). Syk regulation of phosphoinositide 3-kinase-dependent NK cell function. J. Immunol. 168, 3155–3164.
Kärre, K., Ljunggren, H. G., Piontek, G., and Kiessling, R. (1986). Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319, 675–678.
Kasibhlatla, S., Genestier, L., and Green, D. R. (1999). Regulation of Fas ligand expression during activation-induced cell death in T lymphocytes via nuclear factor κB. J. Biol. Chem. 274, 987–992.
Kato, H., Takeuchi, O., Sato, S., Yoneyama, M., Yamamoto, M., Matsui, K., Uematsu, S., Jung, A., Kawai, T., Ishii, K., Yamaguchi, O., Otsu, K., Tsujimura, T., Koh, C. S., Reis e Sousa, C., Matsuura, Y., Fujita, T., and Akira, S. (2006). Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105.
KEGGpathway. (2011). Natural Killer Cell Mediated Cytotoxicity – Homo sapiens. Available at: http://www.genome.jp/kegg/pathway/hsa/hsa04650.html
Kelly, J. M., Darcy, P. K., Markby, J. L., Godfrey, D. I., Takeda, K., Yagita, H., and Smyth, M. J. (2002). Induction of tumor-specific T cell memory by NK cell–mediated tumor rejection. Nat. Immunol. 3, 83–90.
Koebel, C. M., Vermi, W., Swann, J. B., Zerafa, N., Rodig, S. J., Old, L. J., Smyth, M. J., and Schreiber, R. D. (2007). Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907.
Kong, K. F., Yokosuka, T., Canonigo-Balancio, A. J., Isakov, N., Saito, T., and Altman, A. (2011). A motif in the V3 domain of the kinase PKC-θ determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat. Immunol. 12, 1105–1112.
König, S., Nimtz, M., Scheiter, M., Ljunggren, H. G., Bryceson, Y. T., and Jänsch, L. (2012). Kinome analysis of receptor-induced phosphorylation in human natural killer cells. PLoS ONE 7, e29672. doi:10.1371/journal.pone.0029672
Kroger, N. (2011). Approaches to relapse after allogeneic stem cell transplantation. Curr. Opin. Oncol. 23, 203–208.
Krzewski, K., Chen, X., Orange, J. S., and Stromminger, J. L. (2006). Formation of a WIP-, WASp-, actin-, and myosin IIA–containing multiprotein complex in activated NK cells and its alteration by KIR inhibitory signaling. J. Cell Biol. 173, 121–132.
Krzewski, K., Chen, X., and Strominger, J. L. (2008). WIP is essential for lytic granule polarization and NK cell cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 2568–2573.
Krzewski, K., and Strominger, J. L. (2008). The killer’s kiss: the many functions of NK cell immunological synapses. Curr. Opin. Cell Biol. 2008, 597–605.
Kuhn, J. R., and Poenie, M. (2002). Dynamic polarization of the microtubule cytoskeleton during CTL-mediated killing. Immunity 16, 111–121.
Lanier, L. L. (2008). Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 9, 495–502.
Latinis, K. M., Norian, L. A., Eliason, S. L., and Koretzky, G. A. (1997). Two NFAT transcription factor binding sites participate in the regulation of CD95 (Fas) ligand expression in activated human T cells. J. Biol. Chem. 272, 31427–31434.
Li, C., Ge, B., Nicotra, M., Stern, J. N., Kopcow, H. D., Chen, X., and Strominger, J. L. (2008). JNK MAP kinase activation is required for MTOC and granule polarization in NKG2D-mediated NK cell cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 3017–3022.
Liu, R. B., Engels, B., Arina, A., Schreiber, K., Hyjek, E., Schietinger, A., Binder, D. C., Butz, E., Krausz, T., Rowley, D. A., Jabri, B., and Schreiber, H. (2012). Densely granulated murine NK cells eradicate large solid tumors. Cancer Res. 72, 1964–1974.
Ljunggren, H. G., and Karre, K. (1985). Host resistance directed selectively gainst H-2-deficient lymphoma variants. Analysis of the mechanism. J. Exp. Med. 162, 1745–1795.
López-Botet, M., Bellón, T., Llano, M., Navarro, F., García, P., and de Miguel, M. (2000). Paired inhibitory and triggering NK cell receptors for HLA class I molecules. Hum. Immunol. 61, 7–17.
Marcenaro, S., Gallo, F., Martini, S., Santoro, A., Griffiths, G., Aricó, M., Moretta, L., and Pende, D. (2006). Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13-4 defect and discriminates between genetic subtypes of the disease. Blood 108, 2316–2323.
Miller, J. S., Soignier, Y., Panoskaltsis-Mortari, A., McNearney, S. A., Yun, G. H., Fautsch, S. K., McKenna, D., Le, C., Defor, T. E., Burns, L. J., Orchard, P. J., Blazar, B. R., Wagner, J. E., Slungaard, A., Weisdorf, D. J., Okazaki, I. J., and McGlave, P. B. (2005). Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 105, 3051–3057.
Monks, C. R. F., Kupfer, H., Tamir, I., Barlow, A., and Kupfer, A. (1997). Selective modulation of protein kinase C-θ during T-cell activation. Nature 385, 83–86.
Moretta, A., Marcenaro, E., Parolini, S., Ferlazzo, G., and Moretta, L. (2008). NK cells at the interface between innate and adaptive immunity. Cell Death Differ. 15, 226–233.
Mustelin, T., Coggeshall, K. M., Isakov, N., and Altman, A. (1990). T cell antigen receptor-Mediated activation of phospholipase C requires tyrosine phosphorylation. Science 247, 1584–1587.
Nedellec, S., Sabourin, C., Bonneville, M., and Scotet, E. (2010). NKG2D costimulates human Vγ9Vδ2 T cell antitumor cytotoxicity through protein kinase Cθ-dependent modulation of early TCR-induced calcium and transduction signals. J. Immunol. 185, 55–63.
Nesic, D., Henderson, S., and Vukmanovic, S. (1998). Prevention of antigen-induced MTOC reorientation in cytotoxic T cells by modulation of PKC activity. Int. Immunol. 10, 1741–1746.
Nishimura, T., Burakoff, S. J., and Herrmann, S. H. (1987). Protein kinase C required for cytotoxic T lymphocyte triggering. J. Immunol. 139, 2888–2891.
Orange, J., Harris, K., Andzelm, M., Valter, M., Geha, R., and Strominger, J. (2003). The mature activating natural killer cell immunologic synapse is formed in distinct stages. Proc. Natl. Acad. Sci. U.S.A. 100, 14151–14156.
O’Rourke, A. M., and Mescher, M. F. (1992). Cytotoxic T-lymphocyte activation involves a cascade of signalling and adhesion events. Nature 358, 253–255.
Page, K. M., Chaudhary, D., Goldman, S. J., and Kasaian, M. T. (2008). Natural killer cells from protein kinase C þeta–/– mice stimulated with interleukin-12 are deficient in production of interferon-γ. J. Leukoc. Biol. 83, 1267–1276.
Pardo, J., Balkow, S., Anel, A., and Simon, M. M. (2002). Granzymes are critically involved in NK-mediated control of RMA-S tumor growth in vivo. Eur. J. Immunol. 32, 2881–2886.
Pardo, J., Buferne, M., Martínez-Lorenzo, M. J., Naval, J., Schmitt-Verhulst, A. M., Boyer, C., and Anel, A. (2003). Differential implication of PKC isoforms in TCR/CD3-induced Fas ligand expression and in CTL degranulation. Int. Immunol. 15, 1441–1450.
Pfeifhofer, C., Kofler, K., Gruber, T., Tabrizi, N. G., Lutz, C., Maly, K., Leitges, M., and Baier, G. (2003). Protien kinase C θ affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J. Exp. Med. 197, 1525–1535.
Puente, L. G., He, J. S., and Ostergaard, H. L. (2006). A novel PKC regulates ERK activation and degranulation of cytotoxic T lymphocytes: plasticity in PKC regulation of ERK. Eur. J. Immunol. 36, 1009–1018.
Quann, E. J., Liu, X., Altan-Bonnet, G., and Huse, M. (2011). A cascade of protein kinase C isozymes promotes cytoskeletal polarization in T cells. Nat. Immunol. 12, 647–654.
Reefman, E., Kay, J. G., Wood, S. M., Offenhäuser, C., Brown, D. L., Roy, S., Stanley, A. C., Low, P. C., Manderson, A. P., and Stow, J. L. (2010). Cytokine secretion is distinct from secretion of cytotoxic granules in NK cells. J. Immunol. 184, 4852–4862.
Robertson, L. K., Mireau, L. R., and Ostergaard, H. L. (2005). A role for phosphatidylinositol 3-kinase in TCR-stimulated ERK activation leading to paxillin phosphorylation and CTL degranulation. J. Immunol. 175, 8138–8145.
Robertson, L. K., and Ostergaard, H. L. (2011). Paxillin associates with the microtubule cytoskeleton and the immunological synapse of CTL through its leucine-aspartic acid domains and contributes to microtubule organizing center reorientation. J. Immunol. 187, 5824–5833.
Rubnitz, J. E., Inaba, H., Ribeiro, R. C., Pounds, S., Rooney, B., Bell, T., Pui, C. H., and Leung, W. (2010). NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. 28, 955–959.
Ruggeri, L., Mancusi, A., Burchielli, E., Aversa, F., Martelli, M. F., and Velardi, A. (2007a). Natural killer cell alloreactivity in allogeneic hematopoietic transplantation. Curr. Opin. Oncol. 19, 142–147.
Ruggeri, L., Mancusi, A., Capanni, M., Urbani, E., Carotti, A., Aloisi, T., Stern, M., Pende, D., Perruccio, K., Burchielli, E., Topini, F., Bianchi, E., Aversa, F., Martelli, M. F., and Velardi, A. (2007b). Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood 110, 433–440.
Screpanti, V., Wallin, R. P. A., Ljunggren, H. G., and Grandien, A. (2001). A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J. Immunol. 167, 2068–2073.
Secrist, J. P., Karnitz, L., and Abraham, R. T. (1991). T-cell antigen receptor ligation induces tyrosine phosphorylation of phospholipase C-γ1. J. Biol. Chem. 266, 12135–12139.
Segovis, C. M., Schoon, R. A., Dick, C. J., Nacusi, L. P., Leibson, P. J., and Billadeau, D. D. (2009). PI3K links NKG2D signaling to a CrkL pathway involved in natural killer cell adhesion, polarity, and granule secretion. J. Immunol. 182, 6933–6942.
Shibuya, A., Lanier, L. L., and Phillips, J. H. (1998). Protein kinase C is involved in the regulation of both signaling and adhesion mediated by DNAX accessory molecule-1 receptor. J. Immunol. 161, 1671–1676.
Smyth, M. J., Kelly, J. M., Baxter, A. G., Körner, H., and Sedgwick, J. D. (1998). An essential role for tumor necrosis factor in natural killer cell–mediated tumor rejection in the peritoneum. J. Exp. Med. 188, 1611–1619.
Smyth, M. J., Thia, K. Y. T., Street, S. E. A., Cretney, E., Trapani, J. A., Taniguchi, M., Kawano, T., Pelikan, S. B., Crowe, N. Y., and Godfrey, D. I. (2000). Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 191, 661–668.
Sorkin, L. S., Otto, M., Baldwin, W. M., Vail, E., Gillies, S. D., Handgretinger, R., Barfield, R. C., Ming Yu, H., and Yu, A. L. (2010). Anti-GD(2) with an Fc point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain 149, 135–142.
Stern, M., Ruggeri, L., Mancusi, A., Bernardo, M. E., de Angelis, C., Bucher, C., Locatelli, F., Aversa, F., and Velardi, A. (2008). Survival after T cell-depleted haploidentical stem cell transplantation is improved using the mother as donor. Blood 112, 2990–2995.
Stinchcombe, J. C., Barral, D. C., Mules, E. H., Booth, S., Hume, A. N., Machesky, L. M., Seabra, M. C., and Griffiths, G. M. (2001a). Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J. Cell Biol. 152, 825–834.
Stinchcombe, J. C., Bossi, G., Booth, S., and Griffiths, G. M. (2001b). The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 15, 751–761.
Stinchcombe, J. C., and Griffiths, G. M. (2007). Secretory mechanisms in cell-mediated cytotoxicity. Annu. Rev. Cell Dev. Biol. 23, 495–517.
Sun, Z., Arendt, C. W., Ellmeier, W., Schaeffer, E. M., Sunshine, M. J., Gandhi, L., Annes, J., Petrzilka, D., Kupfer, A., Schwartzberg, P. L., and Littman, D. R. (2000). PKC-θ is required for TCR-induced NF-κB activation in mature but not in immature T lymphocytes. Nature 404, 402–407.
Takayama, H., and Sitkovsky, M. V. (1987). Antigen receptor-regulated exocytosis in cytotoxic T lymphocytes. J. Exp. Med. 166, 725–743.
Tassi, I., Cella, M., Presti, R., Colucci, A., Gilfillan, S., Littman, D. R., and Colonna, M. (2008). NK cell activating receptors require PKCθ for sustained signaling, transcriptional activation and IFN-γ secretion. Blood 112, 4109–4116.
Terme, M., Ullrich, E., Delahaye, N. F., Chaput, N., and Zitvogel, L. (2008). Natural killer cell–directed therapies: moving from unexpected results to successful strategies. Nat. Immunol. 9, 486–494.
Upshaw, J. L., Arneson, L. N., Schoon, R. A., Dick, C. J., Billadeau, D. D., and Leibson, P. J. (2006). NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 7, 524–532.
Van den Broek, M. F., Kägi, D., Zinkernagel, R. M., and Hengartner, H. (1995). Perforin dependence of natural killer cell-mediated tumor control in vivo. Eur. J. Immunol. 25, 3514–3516.
Velardi, A. (2008). Role of KIRs and KIR ligands in hematopoietic transplantation. Curr. Opin. Immunol. 20, 581–587.
Villalba, M., Bi, K., Hu, J., Altman, Y., Bushway, P., Reits, E., Neefjes, J., Baier, G., Abraham, R. T., and Altman, A. (2002). Translocation of PKCθ in T cells is mediated by a nonconventional, PI3-K- and Vav-dependent pathway, but does not absolutely require phospholipase C. J. Cell Biol. 157, 253–263.
Villalba, M., Bi, K., Rodriguez, F., Tanaka, Y., Schoenberger, S., and Altman, A. (2001a). Vav1/Rac-dependent actin cytoskeleton reorganization is required for lipid raft clustering in T cells. J. Cell Biol. 155, 331–338.
Villalba, M., Bushway, P., and Altman, A. (2001b). PKCθ mediates a selective T cell survival signal via phosphorylation of BAD. J. Immunol. 166, 5955–5963.
Villalba, M., Kasibhatla, S., Genestier, L., Mahboubi, A., Green, D. R., and Altman, A. (1999). Protein kinase C-θ cooperates with calcineurin to induce Fas ligand expression during activation-induced T cell death. J. Immunol. 163, 5813–5819.
Villunger, A., Ghaffari-Tabrizi, N., Tinhofer, I., Krumböck, N., Bauer, B., Schneider, T., Kasibhatla, S., Greil, R., Baier-Bitterlich, G., Überall, F., Green, D. R., and Baier, G. (1999). Synergistic action of protein kinase C θ and calcineurin is sufficient for Fas ligand expression and induction of a CrmA-sensitive apoptosis pathway in Jurkat T cells. Eur. J. Immunol. 29, 3549–3561.
Vivier, E., Tomasello, E., Baratin, M., Walzer, T., and Ugolini, S. (2008). Functions of natural killer cells. Nat. Immunol. 9, 503–510.
Vosshenrich, C. A. J., Ranson, T., Samson, S. I., Corcuff, E., Colucci, F., Rosmaraki, E. E., and DiSanto, J. P. (2005). Roles for common cytokine receptor γ-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J. Immunol. 174, 1213–1221.
Vyas, Y., Maniar, H., and Dupont, B. (2002a). Visualization of signaling pathways and cortical cytoskeleton in cytolytic and noncytolytic natural killer cell immune synapses. Immunol. Rev. 189, 161–178.
Vyas, Y. M., Maniar, H., and Dupont, B. (2002b). Differential segregation of the SRC homology 2-containing protein tyrosine phosphatase-1 within the early NK cell immune synapse distinguishes noncytolytic from cytolytic interactions. J. Immunol. 168, 3150–3154.
Willemze, R., Rodrigues, C. A., Labopin, M., Sanz, G., Michel, G., Socie, G., Rio, B., Sirvent, A., Renaud, M., Madero, L., Mohty, M., Ferra, C., Garnier, F., Loiseau, P., Garcia, J., Lecchi, L., Kögler, G., Beguin, Y., Navarrete, C., Devos, T., Ionescu, I., Boudjedir, K., Herr, A. L., Gluckman, E., Rocha, V., Eurocord-Netcord and Acute Leukaemia Working Party of the EBMT. (2009). KIR-ligand incompatibility in the graft-versus-host direction improves outcomes after umbilical cord blood transplantation for acute leukemia. Leukemia 23, 492–500.
Keywords: PKC-θ, NK cells, anti-tumor immunity, CTL
Citation: Anel A, Aguiló JI, Catalán E, Garaude J, Rathore MG, Pardo J and Villalba M (2012) Protein kinase C-θ (PKC-θ) in natural killer cell function and anti-tumor immunity. Front. Immun. 3:187. doi: 10.3389/fimmu.2012.00187
Received: 28 March 2012; Accepted: 15 June 2012;
Published online: 05 July 2012.
Edited by:
Amnon Altman, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Karsten Sauer, The Scripps Research Institute, USAJose Alberola-Ila, Oklahoma Medical Research Foundation, USA
Copyright: © 2012 Anel, Aguiló, Catalán, Garaude, Rathore, Pardo and Villalba. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Alberto Anel, Departamento de Bioquímica y Biología Molecular y Celular, Facultad de Ciencias, Universidad de Zaragoza, Campus Pza. San Francisco, Zaragoza E-50009, Spain. e-mail:YW5lbEB1bml6YXIuZXM=