

- 1 Division of Allergic Diseases, Department of Medicine, Mayo Clinic, Rochester, MN, USA
- 2 Cellular and Molecular Immunology Laboratory, Division of Clinical Biochemistry and Immunology, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, USA
Common variable immunodeficiency (CVID) is a humoral immunodeficiency whose primary diagnostic features include hypogammaglobulinemia involving two or more immunoglobulin isotypes and impaired functional antibody responses in the majority of patients. While increased susceptibility to respiratory and other infections is a common thread that binds a large cross-section of CVID patients, the presence of autoimmune complications in this immunologically and clinically heterogeneous disorder is recognized in up to two-thirds of patients. Among the autoimmune manifestations reported in CVID (20–50%; Chapel et al., 2008; Cunningham-Rundles, 2008), autoimmune cytopenias are by far the most common occurring variably in 4–20% (Michel et al., 2004; Chapel et al., 2008) of these patients who have some form of autoimmunity. Association of autoimmune cytopenias with granulomatous disease and splenomegaly has been reported. The spectrum of autoimmune cytopenias includes thrombocytopenia, anemia, and neutropenia. While it may seem paradoxical “prima facie” that autoimmunity is present in patients with primary immune deficiencies, in reality, it could be considered two sides of the same coin, each reflecting a different but inter-connected facet of immune dysregulation. The expansion of CD21 low B cells in CVID patients with autoimmune cytopenias and other autoimmune features has also been previously reported. It has been demonstrated that this unique subset of B cells is enriched for autoreactive germline antibodies. Further, a correlation has been observed between various B cell subsets, such as class-switched memory B cells and plasmablasts, and autoimmunity in CVID. This review attempts to explore the most recent concepts and highlights, along with treatment of autoimmune hematological manifestations of CVID.
Introduction
Common variable immunodeficiency (CVID) is a highly heterogeneous immunodeficiency with varying complexity. The key diagnostic elements include low IgG (2 SD below mean of age) along with low IgA and/or IgM (Park et al., 2008; Resnick et al., 2011). CVID is considered the most commonly encountered and clinically relevant primary immunodeficiency in adults (Chapel et al., 2008; Park et al., 2008) and though the majority of patients are diagnosed between the age of 20 and 40 years, at least another 20% are diagnosed during childhood (>2 years) or adolescence (Cunningham-Rundles, 2010).
While recurrent sinopulmonary infections are one of the hallmarks of this disease, gastrointestinal, viral, and systemic bacterial infections have also been reported (Park et al., 2008; Resnick et al., 2011). Besides infections, CVID is associated with a variety of non-infectious complications including pulmonary disease, autoimmunity, granulomatous disease, gastrointestinal disease, and malignancy (Chapel et al., 2008; Resnick et al., 2011).
The clinical heterogeneity and complexity of CVID has led to renewed efforts over the past decade to identify causal genetic defects as well as correlate the “immuno-phenotype” with clinical phenotype (Warnatz et al., 2002; Piqueras et al., 2003; Wehr et al., 2008; Eibel et al., 2010). In the last 10 years, monogenic defects associated with antibody deficiency have been described in a small subset of CVID patients or patients with hypogammaglobulinemia, or single or few families with a history of consanguinity. These genetic defects include disease-causing mutations or polymorphisms in the TNFRSF13B (TACI), CD19, ICOS, TNFRSF13C (BAFF-R), CD81, CD20, MSH5, and CD21 genes (Grimbacher et al., 2003; Salzer et al., 2004, 2005, 2009; Castigli et al., 2005, 2007; Warnatz et al., 2005; van Zelm et al., 2006, 2010; Kanegane et al., 2007; Pan-Hammarstrom et al., 2007; Schaffer et al., 2007; Sekine et al., 2007; Zhang et al., 2007; Kuijpers et al., 2010; Frank, 2012; Thiel et al., 2012). However, single-gene defects were identified in only a relatively small subset of CVID patients raising the possibility that the majority (>75%) of CVID patients have oligogenic or polygenic defects. This was recently substantiated by a genome-wide association study of 363 CVID patients, which revealed that copy number variations (CNV), including gene duplications and/or deletions were present and this analysis led to the identification of several “novel” genes, which may play an important role in the immune response, and genetic variations therein could lead to a disease phenotype associated with CVID (Orange et al., 2011).
Paradoxical as it may seem, autoimmune manifestations are not uncommon in patients with primary immunodeficiencies (PIDDs) and at least 25% of all PIDDs described in the 2011 IUIS classification may have some form of autoimmune phenomenon (Bussone and Mouthon, 2009; Notarangelo, 2009; Al-Herz et al., 2011). The autoimmunity observed in PIDDs may be related either to a direct or indirect genetic effect, and includes defects in genes that regulate immunological self-tolerance as well as genetic variations that alter immune regulation. Not surprisingly, therefore, autoimmune features are identified relatively frequently in CVID patients (Brandt and Gershwin, 2006; Knight and Cunningham-Rundles, 2006; Cunningham-Rundles, 2008).
Autoimmunity in CVID
Autoimmune hematological abnormalities, specifically cytopenias, are the most common of all autoimmune manifestations in CVID and may present as thrombocytopenia, anemia or neutropenia. In the longitudinal study mentioned above, immune thrombocytopenia (ITP) was reported in 14% of patients, while autoimmune hemolytic anemia (AIHA) and neutropenia was less common with only 7 and <1%, respectively, of the cohort affected (Resnick et al., 2011). It should also be kept in mind that autoimmune cytopenias may in fact be the presenting symptom for a small subset of CVID patients, especially in children, where Evans syndrome (ES) has been reported to precede the clinical and immunological phenotype of CVID (Savasan et al., 2007). Other autoimmune presentations reported in CVID include rheumatoid arthritis, anti-IgA antibodies, vitiligo, and alopecia (Horn et al., 2007; Park et al., 2008; Resnick et al., 2011). A very recent longitudinal study assessing clinical complications that cause morbidity and mortality in CVID patients identified autoimmune complications in 29% of a cohort of 473 patients studied over 4 decades (Resnick et al., 2011). Interestingly, in the same study, the presence of autoimmunity was not associated with an increase in mortality.
Immunological and Phenotypic Manifestations of Autoimmune Cytopenias in CVID
As alluded to previously, several clinical and immunological classifications have been posited in an attempt to stratify and may be even simplify the complex and heterogeneous phenotypes seen in CVID (Warnatz et al., 2002; Piqueras et al., 2003; Chapel et al., 2008; Wehr et al., 2008). The relatively more recent EUROclass study attempted to cohesively link the earlier Freiburg and Paris classifications by correlating B cell subset immunophenotypes with clinical presentation specifically providing correlation for autoimmunity, granulomatous disease, and splenomegaly (Warnatz et al., 2002; Piqueras et al., 2003; Wehr et al., 2008). Of particular relevance was the correlation of an expansion of CD21low/dim B cells with splenomegaly (Wehr et al., 2008). The CD21low/dim B cells have been previously reported to be a subset of anergic B cells with defective signaling that has the capacity to home to sites of inflammation (Rakhmanov et al., 2009, 2010; Foerster et al., 2010; Charles et al., 2011). Additionally, correlations were identified between an expansion of transitional B cells with lymphadenopathy and autoimmune cytopenias with reduced plasmablasts – pre-terminally differentiated plasma cells (Wehr et al., 2008).
Data from Sanchez-Ramon et al. (2008) and Vodjgani et al. (2007) provide independent substantiation of the association between low class-switched memory B cells and clinical features of autoimmunity and splenomegaly in CVID patients reported by the EUROclass and other classification studies (Warnatz et al., 2002; Piqueras et al., 2003; Wehr et al., 2008).
Martinez-Gamboa et al. (2009) showed that there was a numerical decrease in memory B cell numbers in ITP patients who underwent splenectomy and alluded to a potential role for the spleen in maintaining memory B cell homeostasis. However, a different study suggests that the age at which splenectomy is performed is more relevant to maintenance of marginal zone (memory) B cells numbers than consideration of splenectomy in isolation, regardless of age at which the procedure is done (Wasserstrom et al., 2008).
Besides the correlation of B cell subsets, specifically switched memory B cells, with autoimmunity, there is evidence from multiple human and mouse models on the significance and importance of regulatory T cells expressing FOXP3 in suppressing or controlling autoimmunity (Buckner, 2010; Long and Buckner, 2011). It has been shown in at least a subset of CVID patients, particularly those with autoimmune features, that there is a substantial decrease in relative frequency (%) but not absolute quantitation of FOXP3+ Tregs raising the possibility of abnormal immune regulation in these patients (Arumugakani et al., 2010), though the mechanism of immune dysregulation in this context may extend beyond numerical changes to possible functional alterations as well (Jang et al., 2011; Long and Buckner, 2011).
Another recent study demonstrated B cell receptor recombination bias in a subset of CVID patients and postulated that this may predispose to decreased secondary recombination with subsequent defective central tolerance leading ultimately to the escape of autoreactive clones (Romberg et al., 2011). Further, a biomarker (soluble BAFF/BLys) produced by monocytes and dendritic cells (DCs), which is a critical B cell survival and proliferation factor, and known to be abnormally increased in contexts of autoimmunity, especially in rheumatologic diseases (Becker-Merok et al., 2006) was also been shown to be elevated in CVID patients but there was no demonstrable correlation with the incidence of autoimmunity (Knight et al., 2007).
CVID: Overlap with Autoimmune Lymphoproliferative Syndrome and Evans Syndrome
Published data have demonstrated a clear immunologic and clinical overlap between CVID, ES, and autoimmune lymphoproliferative syndrome (ALPS). ES is characterized by the presence of autoimmune cytopenias in two or more hematopoietic lineages. A small study evaluating 12 pediatric patients with ES determined that half (6/12) also had elevated αβ TCR+ DNT T cells (CD3+CD4–8–) and defective Fas apoptosis characteristic of ALPS patients (Teachey et al., 2005). A subsequent larger study of 45 patients with ES substantiated the earlier finding by demonstrating diagnostic criteria for ALPS in 21/45 patients (Seif et al., 2010).
The correlation between ES, ALPS, and CVID was made in a different study, which though limited in sample size (n = 7), showed development of hypogammaglobulinemia, as seen in CVID in 5/7 patients with ES. These patients also had increased Fas expression (Savasan et al., 2007). A larger cohort study of 68 patients with ES showed that only a relatively small proportion, 4/68 had CVID (Michel et al., 2009).
In a separate study of ALPS patients (n = 66), an equally small number, 5/66 had hypogammaglobulinemia, suggesting a potential phenotypic overlap with CVID. The majority of the ALPS patients in this study had reduced class-switched memory B cells, similar to what has been reported in two-third or greater of CVID patients (Rensing-Ehl et al., 2010).
Mechanisms of Development of Autoreactivity
The development of self-reactive B cells is regulated both centrally (bone marrow) and peripherally through at least two independent check-points. It has been suggested that there may be a failure of both central and peripheral tolerance mechanisms in CVID due to immune dysregulation resulting in a flawed negative selection process. Logically, this would suggest that there would be an increased selection of autoreactive B cells prior to affinity maturation (somatic hypermutation) or memory B cell/plasma cell commitment in the secondary lymphoid organs (Haymore et al., 2008). This is a topic that is discussed in depth elsewhere in this journal series, and therefore, not addressed herein.
Diagnosis and Treatment
Diagnosis
The evaluation of CVID patients for autoimmune cytopenias should include appropriate diagnostic work-up (Figure 1), however, in the case of ITP this may primarily be a diagnosis of exclusion. A presumptive diagnosis of ITP can be arrived at by ruling out alternative pathological mechanisms through clinical history, physical review, complete blood count (CBC) analysis, and peripheral blood smears (Provan et al., 2010). Confirmation of the diagnosis is usually determined by response to appropriate treatment. As per the previous discussion that autoimmune cytopenias may precede a diagnosis of CVID, it would be reasonable to evaluate both pediatric and adult patients for immunoglobulin levels on diagnosing ITP to rule out a possible CVID or selective IgA deficiency (Provan et al., 2010). Additionally, follow-up may be required with periodic evaluation and correlation with clinical history to document evolution of the disease process.
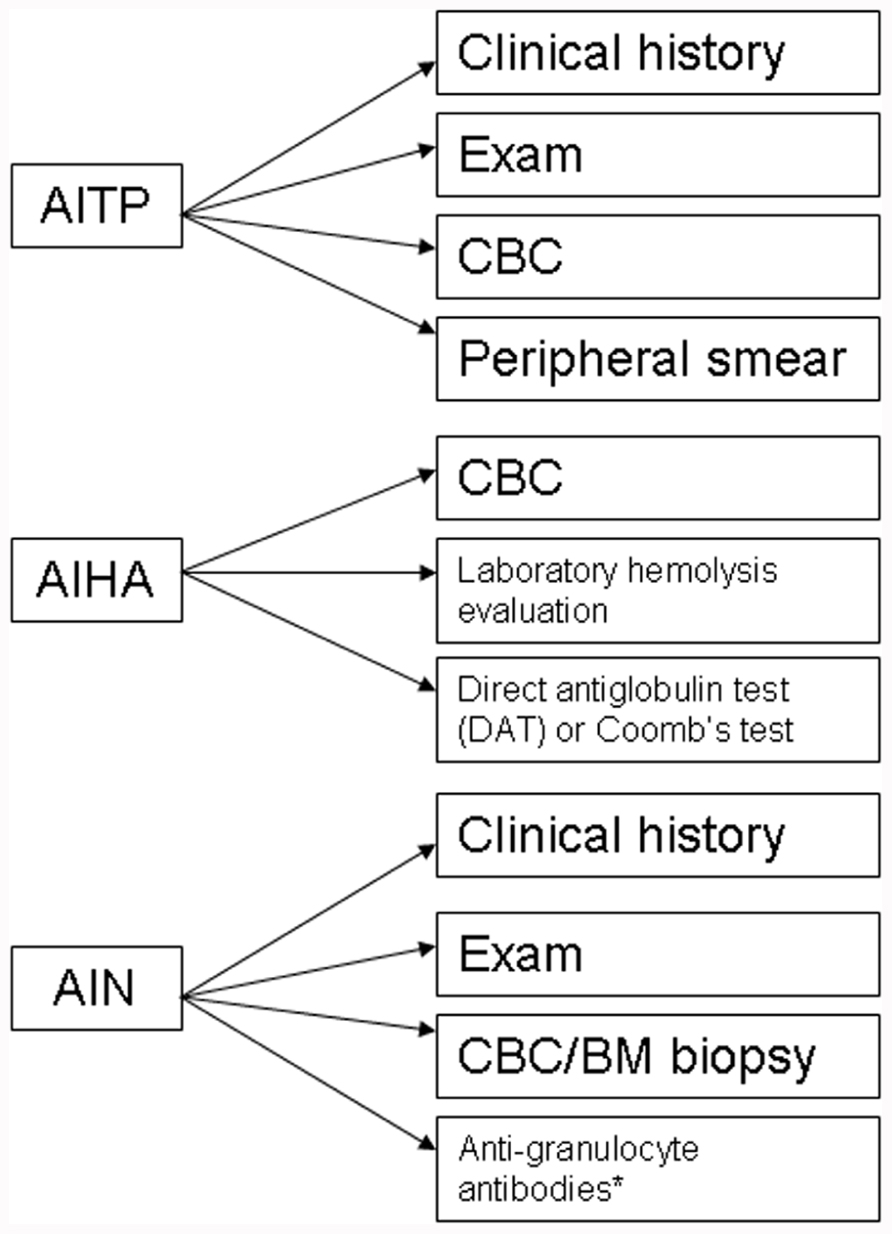
FIGURE 1. Diagnostic algorithm for autoimmune cytopenias in CVID. AITP, autoimmune thrombocytopenia purpura; AIHA, autoimmune hemolytic anemia; AIN, autoneutropenia; IVIG, intravenous immunoglobulin;. *non-exclusionary.
Likewise, the diagnosis of AIHA mandates evidence of hemolysis along with detection of an autoantibody. There are a number of laboratory markers for establishing hemolysis, including a CBC with peripheral smear, increased indirect bilirubin, increased lactate dehydrogenase (LDH), and decreased haptoglobin. Autoantibodies can be detected by a direct antiglobulin test (DAT) or Coomb’s test (Gehrs and Friedberg, 2002).
The diagnosis of autoimmune neutropenia (AIN) is similar to ITP in that it is a diagnosis of exclusion. In some cases, detection of anti-granulocyte antibodies may be useful but the lack of detectable autoantibodies does not exclude a diagnosis of AIN (Bope and Kellerman, 2012). Most cases of AIN are associated with normal marrow reserve and pathogenesis is related to antibody-mediated destruction and in some cases, sequestration. The diagnosis can include a bone marrow biopsy, which would reveal a hypercellular marrow and usually a late maturational arrest, though in some cases, an early arrest can also be seen. AIN may be associated with ITP and/or AIHA in CVID patients. Besides, the possible presence of anti-neutrophil antibodies, circulating immune complexes may also be present in a subset of patients with AIN (Dinauer and Coates, 2009).
Treatment
A treatment algorithm for autoimmune cytopenias in CVID is provided in Figure 2. The American Society of Hematology has provided guidelines for the treatment of patients with ITP and these include initiation of treatment in adult patients if the platelets are below 30 × 109/L. However, in pediatric patients, the current guidelines state that treatment is based on clinical symptoms associated with thrombocytopenia regardless of the platelet counts (Neunert et al., 2011). Pediatric patients are far more likely to experience spontaneous remissions. The treatment of choice as first-line therapy for ITP is the use of steroids at 1 mg/kg for a duration of at least three weeks with subsequent dose reduction and eventual withdrawal. Alternative therapeutic options could include a single dose of intravenous immunoglobulin (IVIG) at 1 g/kg. Further use of IVIG is dependent on clinical response to the initial dose. A combination of the above two therapies may be utilized if a rapid response is required. Rho(D) immune globulin is an option for Rh-positive individuals who have not undergone a splenectomy and are unable to tolerate steroid treatment (Neunert et al., 2011). Splenectomy is recommended as a therapeutic option only for those patients that fail corticosteroid therapy. CVID patients undergoing splenectomy or receiving immunosuppressive medication may be at increased risk for infection given their intrinsic immunological defects.
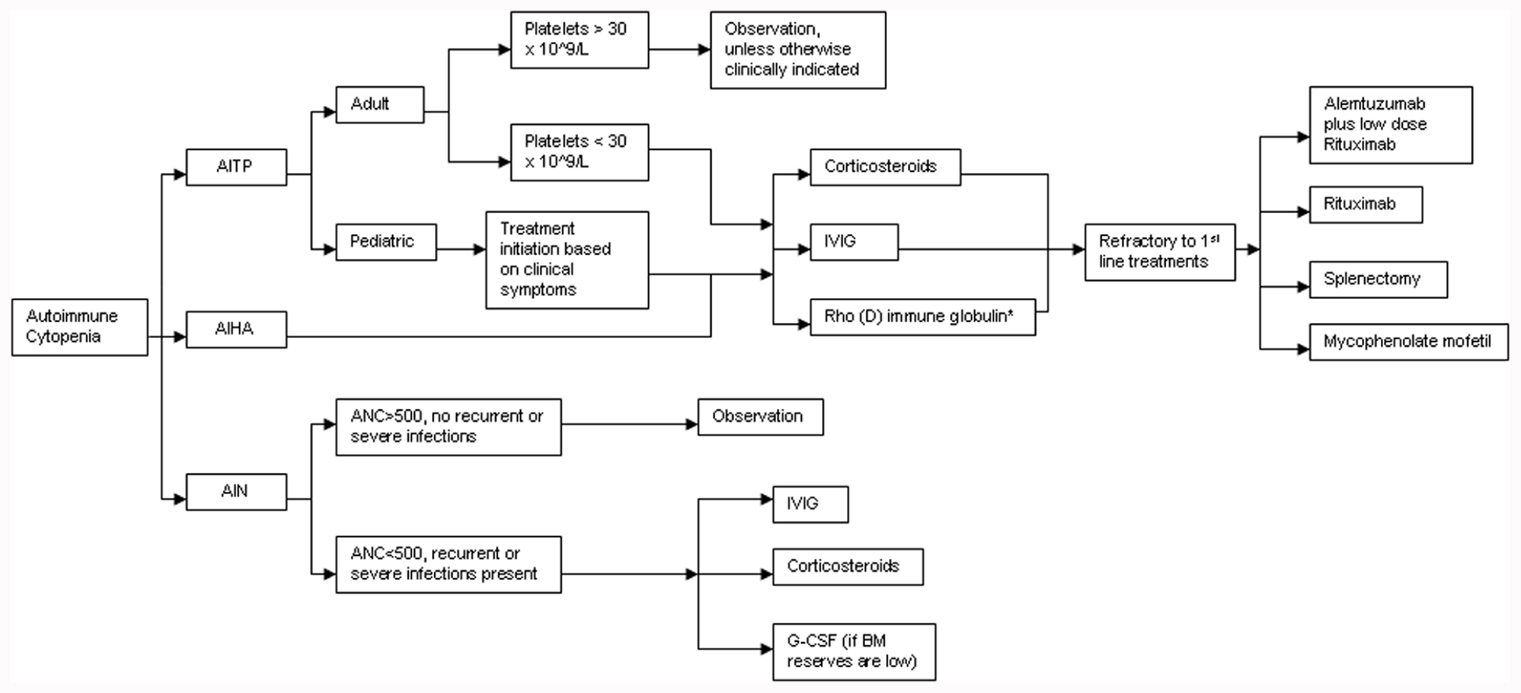
FIGURE 2. Treatment algorithm for autoimmune cytopenias in CVID. AITP, autoimmune thrombocytopenia; AIHA, autoimmune hemolytic anemia; AIN, autoneutropenia; IVIG, intravenous immunoglobulin; ANC, absolute neutrophil count; G-CSF, granulocyte colony stimulating factor. *Rh+, non-splenectomized individuals only.
While AIHA is treated much like ITP, it may be more challenging to manage, particularly in patients with ES (Cunningham-Rundles, 2002; Wang and Cunningham-Rundles, 2005). For refractory cases of ITP, AIHA, or both, Rituximab, a chimeric monoclonal anti-CD20 B cell-depleting agent, has been effectively used. In a modest-size cohort of CVID patients (n = 33) with refractory autoimmune cytopenias (failure of at least 2–6 treatments prior to initiation of Rituximab), the initial response rate was remarkably high at 84% (Gobert et al., 2011). Severe infection was an unfortunate consequence in almost a quarter of these patients (8/33) over a mean follow-up period of 39 months. Of note, half the patients (4/8) were not on replacement immunoglobulin therapy at the time of infectious diagnosis. An earlier study reports similar rates of infection in patients with ITP who received standard treatment (Michel et al., 2004).
The treatment of AIN is primarily dictated by the severity of neutropenia-associated clinical symptoms and the underlying disease context. Treatment with high-dose IVIG or steroids may be used if there is very profound neutropenia (ANC < 500/mm3) in conjunction with recurrent or fulminant infections. G-CSF therapy is only of value if bone marrow reserves are depleted. Splenectomy has little value in reversing neutropenia, especially if it is isolated, since the effect is transient, and can ultimately increase overall infection risk (Dinauer and Coates, 2009).
A separate study of 19 adult patients with steroid-refractory autoimmune cytopenias, reported a 100% initial response rate to a combination of low-dose Rituximab and Alemtuzumab (anti-CD52 humanized monoclonal antibody). Infection occurred in 6/19 patients after a median period of 70 weeks (Gomez-Almaguer et al., 2010). Other reports have documented an initial response rate of 78–92% for refractory autoimmune cytopenias treated with mycophenolate mofetil with no significant adverse events reported (Kotb et al., 2005; Rao et al., 2005). Thus, the approach to treating autoimmune cytopenias in CVID is not dissimilar to the treatment of immune competent patients (Wang and Cunningham-Rundles, 2005).
Summary
This minireview, which is limited in scope, provides an encapsulated discussion on the incidence and presentation of autoimmunity in CVID, specifically autoimmune cytopenias, their overlap with other clinical entities, some notable immunological hallmarks, laboratory diagnosis and an overview of standard and new therapies. As mentioned in the text, a more exhaustive treatment of autoimmunity in CVID, focusing on mechanistic aspects, is provided elsewhere.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Al-Herz, W., Bousfiha, A., Casanova, J.-L., Chapel, H., Conley, M. E., Cunningham-Rundles, C., Etzioni, A., Fischer, A., Franco, J. L., Geha, R., Hammarstrom, L., Nonoyama, S., Notarangelo, L. D., Ochs, H. D., Puck, J., Roifman, C., Seger, R., and Tang, M. L. K. (2011). Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front. Immunol. 2:54. doi: 10.3389/fimmu.2011.00054
Arumugakani, G., Wood, P. M., and Carter, C. R. (2010). Frequency of Treg cells is reduced in CVID patients with autoimmunity and splenomegaly and is associated with expanded CD21lo B lymphocytes. J. Clin. Immunol. 30, 292–300.
Becker-Merok, A., Nikolaisen, C., and Nossent, H. C. (2006). B-lymphocyte activating factor in systemic lupus erythematosus and rheumatoid arthritis in relation to autoantibody levels, disease measures and time. Lupus 15, 570–576.
Brandt, D., and Gershwin, M. E. (2006). Common variable immune deficiency and autoimmunity. Autoimmun. Rev. 5, 465–470.
Buckner, J. H. (2010). Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat. Rev. Immunol. 10, 849–859.
Bussone, G., and Mouthon, L. (2009). Autoimmune manifestations in primary immune deficiencies. Autoimmun. Rev. 8, 332–336.
Castigli, E., Wilson, S., Garibyan, L., Rachid, R., Bonilla, F., Schneider, L., Morra, M., Curran, J., and Geha, R. (2007). Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat. Genet. 39, 430–431.
Castigli, E., Wilson, S. A., Garibyan, L., Rachid, R., Bonilla, F., Schneider, L., and Geha, R. S. (2005). TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat. Genet. 37, 829–834.
Chapel, H., Lucas, M., Lee, M., Bjorkander, J., Webster, D., Grimbacher, B., Fieschi, C., Thon, V., Abedi, M. R., and Hammarstrom, L. (2008). Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 112, 277–286.
Charles, E. D., Brunetti, C., Marukian, S., Ritola, K. D., Talal, A. H., Marks, K., Jacobson, I. M., Rice, C. M., and Dustin, L. B. (2011). Clonal B cells in patients with hepatitis C virus-associated mixed cryoglobulinemia contain an expanded anergic CD21low B-cell subset. Blood 117, 5425–5437.
Cunningham-Rundles, C. (2002). Hematologic complications of primary immune deficiencies. Blood Rev. 16, 61–64.
Cunningham-Rundles, C. (2008). Autoimmune manifestations in common variable immunodeficiency. J. Clin. Immunol. 28(Suppl. 1), S42–S45.
Dinauer, M. C., and Coates, T. D. (2009). Disorders of Phagocyte Function and Number, Chap. 50. Philadelphia, PA: Churchill Livingstone Elsevier.
Eibel, H., Salzer, U., and Warnatz, K. (2010). Common variable immunodeficiency at the end of a prospering decade: towards novel gene defects and beyond. Curr. Opin. Allergy Clin. Immunol. 10, 526–533.
Foerster, C., Voelxen, N., Rakhmanov, M., Keller, B., Gutenberger, S., Goldacker, S., Thiel, J., Feske, S., Peter, H. H., and Warnatz, K. (2010). B cell receptor-mediated calcium signaling is impaired in B lymphocytes of type Ia patients with common variable immunodeficiency. J. Immunol. 184, 7305–7313.
Frank, M. M. (2012). CD21 deficiency, complement, and the development of common variable immunodeficiency. J. Allergy Clin. Immunol. 129, 811–813.
Gehrs, B. C., and Friedberg, R. C. (2002). Autoimmune hemolytic anemia. Am. J. Hematol. 69, 258–271.
Gobert, D., Bussel, J. B., Cunningham-Rundles, C., Galicier, L., Dechartres, A., Berezne, A., Bonnotte, B., Derevel, T., Auzary, C., Jaussaud, R., Larroche, C., Lequellec, A., Ruivard, M., Seve, P., Smail, A., Viallard, J. F., Godeau, B., Hermine, O., and Michel, M. (2011). Efficacy and safety of rituximab in common variable immunodeficiency-associated immune cytopenias: a retrospective multicentre study on 33 patients. Br. J. Haematol. 155, 498–508.
Gomez-Almaguer, D., Solano-Genesta, M., Tarin-Arzaga, L., Herrera-Garza, J. L., Cantu-Rodriguez, O. G., Gutierrez-Aguirre, C. H., and Jaime-Perez, J. C. (2010). Low-dose rituximab and alemtuzumab combination therapy for patients with steroid-refractory autoimmune cytopenias. Blood 116, 4783–4785.
Grimbacher, B., Hutloff, A., Schlesier, M., Glocker, E., Warnatz, K., Drager, R., Eibel, H., Fischer, B., Schaffer, A. A., Mages, H. W., Kroczek, R. A., and Peter, H. H. (2003). Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat. Immunol. 4, 261–268.
Haymore, B. R., Mikita, C. P., and Tsokos, G. C. (2008). Common variable immune deficiency (CVID) presenting as an autoimmune disease: role of memory B cells. Autoimmun. Rev. 7, 309–312.
Horn, J., Thon, V., Bartonkova, D., Salzer, U., Warnatz, K., Schlesier, M., Peter, H. H., and Grimbacher, B. (2007). Anti-IgA antibodies in common variable immunodeficiency (CVID): diagnostic workup and therapeutic strategy. Clin. Immunol. 122, 156–162.
Jang, E., Cho, W. S., Cho, M. L., Park, H. J., Oh, H. J., Kang, S. M., Paik, D. J., and Youn, J. (2011). Foxp3+ regulatory T cells control humoral autoimmunity by suppressing the development of long-lived plasma cells. J. Immunol. 186, 1546–1553.
Kanegane, H., Agematsu, K., Futatani, T., Sira, M. M., Suga, K., Sekiguchi, T., van Zelm, M. C., and Miyawaki, T. (2007). Novel mutations in a Japanese patient with CD19 deficiency. Genes Immun. 8, 663–670.
Knight, A. K., and Cunningham-Rundles, C. (2006). Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmun. Rev. 5, 156–159.
Knight, A. K., Radigan, L., Marron, T., Langs, A., Zhang, L., and Cunningham-Rundles, C. (2007). High serum levels of BAFF, APRIL, and TACI in common variable immunodeficiency. Clin. Immunol. 124, 182–189.
Kotb, R., Pinganaud, C., Trichet, C., Lambotte, O., Dreyfus, M., Delfraissy, J. F., Tchernia, G., and Goujard, C. (2005). Efficacy of mycophenolate mofetil in adult refractory auto-immune cytopenias: a single center preliminary study. Eur. J. Haematol. 75, 60–64.
Kuijpers, T. W., Bende, R. J., Baars, P. A., Grummels, A., Derks, I. A., Dolman, K. M., Beaumont, T., Tedder, T. F., Van Noesel, C. J., Eldering, E., and Van Lier, R. A. (2010). CD20 deficiency in humans results in impaired T cell-independent antibody responses. J. Clin. Invest. 120, 214–222.
Long, S. A., and Buckner, J. H. (2011). CD4+FOXP3+ T regulatory cells in human autoimmunity: more than a numbers game. J. Immunol. 187, 2061–2066.
Martinez-Gamboa, L., Mei, H., Loddenkemper, C., Ballmer, B., Hansen, A., Lipsky, P. E., Emmerich, F., Radbruch, A., Salama, A., and Dorner, T. (2009). Role of the spleen in peripheral memory B-cell homeostasis in patients with autoimmune thrombocytopenia purpura. Clin. Immunol. 130, 199–212.
Michel, M., Chanet, V., Dechartres, A., Morin, A. S., Piette, J. C., Cirasino, L., Emilia, G., Zaja, F., Ruggeri, M., Andres, E., Bierling, P., Godeau, B., and Rodeghiero, F. (2009). The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood 114, 3167–3172.
Michel, M., Chanet, V., Galicier, L., Ruivard, M., Levy, Y., Hermine, O., Oksenhendler, E., Schaeffer, A., Bierling, P., and Godeau, B. (2004). Autoimmune thrombocytopenic purpura and common variable immunodeficiency: analysis of 21 cases and review of the literature. Medicine (Baltimore) 83, 254–263.
Neunert, C., Lim, W., Crowther, M., Cohen, A., Solberg, L. Jr., and Crowther, M. A. (2011). The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 117, 4190–4207.
Notarangelo, L. D. (2009). Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematology Am. Soc. Hematol. Educ. Program 139–143.
Orange, J. S., Glessner, J. T., Resnick, E., Sullivan, K. E., Lucas, M., Ferry, B., Kim, C. E., Hou, C., Wang, F., Chiavacci, R., Kugathasan, S., Sleasman, J. W., Baldassano, R., Perez, E. E., Chapel, H., Cunningham-Rundles, C., and Hakonarson, H. (2011). Genome-wide association identifies diverse causes of common variable immunodeficiency. J. Allergy Clin. Immunol. 127, 1360–1367, e1366.
Pan-Hammarstrom, Q., Salzer, U., Du, L., Bjorkander, J., Cunningham-Rundles, C., Nelson, D. L., Bacchelli, C., Gaspar, H. B., Offer, S., Behrens, T. W., Grimbacher, B., and Hammarstrom, L. (2007). Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat. Genet. 39, 429–430.
Park, M. A., Li, J. T., Hagan, J. B., Maddox, D. E., and Abraham, R. S. (2008). Common variable immunodeficiency: a new look at an old disease. Lancet 372, 489–502.
Piqueras, B., Lavenu-Bombled, C., Galicier, L., Bergeron-Van Der Cruyssen, F., Mouthon, L., Chevret, S., Debre, P., Schmitt, C., and Oksenhendler, E. (2003). Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J. Clin. Immunol. 23, 385–400.
Provan, D., Stasi, R., Newland, A. C., Blanchette, V. S., Bolton-Maggs, P., Bussel, J. B., Chong, B. H., Cines, D. B., Gernsheimer, T. B., Godeau, B., Grainger, J., Greer, I., Hunt, B. J., Imbach, P. A., Lyons, G., McMillan, R., Rodeghiero, F., Sanz, M. A., Tarantino, M., Watson, S., Young, J., and Kuter, D. J. (2010). International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 115, 168–186.
Rakhmanov, M., Gutenberger, S., Keller, B., Schlesier, M., Peter, H. H., and Warnatz, K. (2010). CD21low B cells in common variable immunodeficiency do not show defects in receptor editing, but resemble tissue-like memory B cells. Blood 116, 3682–3683.
Rakhmanov, M., Keller, B., Gutenberger, S., Foerster, C., Hoenig, M., Driessen, G., Van Der Burg, M., Van Dongen, J. J., Wiech, E., Visentini, M., Quinti, I., Prasse, A., Voelxen, N., Salzer, U., Goldacker, S., Fisch, P., Eibel, H., Schwarz, K., Peter, H. H., and Warnatz, K. (2009). Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc. Natl. Acad. Sci. U.S.A. 106, 13451–13456.
Rao, V. K., Dugan, F., Dale, J. K., Davis, J., Tretler, J., Hurley, J. K., Fleisher, T., Puck, J., and Straus, S. E. (2005). Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br. J. Haematol. 129, 534–538.
Rensing-Ehl, A., Warnatz, K., Fuchs, S., Schlesier, M., Salzer, U., Draeger, R., Bondzio, I., Joos, Y., Janda, A., Gomes, M., Abinun, M., Hambleton, S., Cant, A., Shackley, F., Flood, T., Waruiru, C., Beutel, K., Siepermann, K., Dueckers, G., Niehues, T., Wiesel, T., Schuster, V., Seidel, M. G., Minkov, M., Sirkia, K., Kopp, M. V., Korhonen, M., Schwarz, K., Ehl, S., and Speckmann, C. (2010). Clinical and immunological overlap between autoimmune lymphoproliferative syndrome and common variable immunodeficiency. Clin. Immunol. 137, 357–365.
Resnick, E. S., Moshier, E. L., Godbold, J. H., and Cunningham-Rundles, C. (2011). Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 119, 1650–1657.
Romberg, N., Ng, Y.-S., Cunningham-Rundles, C., and Meffre, E. (2011). Common variable immunodeficiency patients with increased CD21-/lo B cells suffer from altered receptor editing and defective B cell tolerance. Blood 118, 5977–5978.
Salzer, U., Bacchelli, C., Buckridge, S., Pan-Hammarstrom, Q., Jennings, S., Lougaris, V., Bergbreiter, A., Hagena, T., Birmelin, J., Plebani, A., Webster, A. D., Peter, H. H., Suez, D., Chapel, H., Mclean-Tooke, A., Spickett, G. P., Anover-Sombke, S., Ochs, H. D., Urschel, S., Belohradsky, B. H., Ugrinovic, S., Kumararatne, D. S., Lawrence, T. C., Holm, A. M., Franco, J. L., Schulze, I., Schneider, P., Gertz, E. M., Schaffer, A. A., Hammarstrom, L., Thrasher, A. J., Gaspar, H. B., and Grimbacher, B. (2009). Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood 113, 1967–1976.
Salzer, U., Chapel, H. M., Webster, A. D., Pan-Hammarstrom, Q., Schmitt-Graeff, A., Schlesier, M., Peter, H. H., Rockstroh, J. K., Schneider, P., Schaffer, A. A., Hammarstrom, L., and Grimbacher, B. (2005). Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat. Genet. 37, 820–828.
Salzer, U., Maul-Pavicic, A., Cunningham-Rundles, C., Urschel, S., Belohradsky, B. H., Litzman, J., Holm, A., Franco, J. L., Plebani, A., Hammarstrom, L., Skrabl, A., Schwinger, W., and Grimbacher, B. (2004). ICOS deficiency in patients with common variable immunodeficiency. Clin. Immunol. 113, 234–240.
Sanchez-Ramon, S., Radigan, L., Yu, J. E., Bard, S., and Cunningham-Rundles, C. (2008). Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin. Immunol. 128, 314–321.
Savasan, S., Warrier, I., Buck, S., Kaplan, J., and Ravindranath, Y. (2007). Increased lymphocyte Fas expression and high incidence of common variable immunodeficiency disorder in childhood Evans’ syndrome. Clin. Immunol. 125, 224–229.
Schaffer, A. A., Salzer, U., Hammarstrom, L., and Grimbacher, B. (2007). Deconstructing common variable immunodeficiency by genetic analysis. Curr. Opin. Genet. Dev. 17, 201–212.
Seif, A. E., Manno, C. S., Sheen, C., Grupp, S. A., and Teachey, D. T. (2010). Identifying autoimmune lymphoproliferative syndrome in children with Evans syndrome: a multi-institutional study. Blood 115, 2142–2145.
Sekine, H., Ferreira, R. C., Pan-Hammarstrom, Q., Graham, R. R., Ziemba, B., De Vries, S. S., Liu, J., Hippen, K., Koeuth, T., Ortmann, W., Iwahori, A., Elliott, M. K., Offer, S., Skon, C., Du, L., Novitzke, J., Lee, A. T., Zhao, N., Tompkins, J. D., Altshuler, D., Gregersen, P. K., Cunningham-Rundles, C., Harris, R. S., Her, C., Nelson, D. L., Hammarstrom, L., Gilkeson, G. S., and Behrens, T. W. (2007). Role for Msh5 in the regulation of Ig class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 104, 7193–7198.
Teachey, D. T., Manno, C. S., Axsom, K. M., Andrews, T., Choi, J. K., Greenbaum, B. H., Mcmann, J. M., Sullivan, K. E., Travis, S. F., and Grupp, S. A. (2005). Unmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS). Blood 105, 2443–2448.
Thiel, J., Kimmig, L., Salzer, U., Grudzien, M., Lebrecht, D., Hagena, T., Draeger, R., Volxen, N., Bergbreiter, A., Jennings, S., Gutenberger, S., Aichem, A., Illges, H., Hannan, J. P., Kienzler, A. K., Rizzi, M., Eibel, H., Peter, H. H., Warnatz, K., Grimbacher, B., Rump, J. A., and Schlesier, M. (2012). Genetic CD21 deficiency is associated with hypogammaglobulinemia. J. Allergy Clin. Immunol. 129, 801–810 e806.
van Zelm, M. C., Reisli, I., Van Der Burg, M., Castano, D., Van Noesel, C. J., Van Tol, M. J., Woellner, C., Grimbacher, B., Patino, P. J., Van Dongen, J. J., and Franco, J. L. (2006). An antibody-deficiency syndrome due to mutations in the CD19 gene. N. Engl. J. Med. 354, 1901–1912.
van Zelm, M. C., Smet, J., Adams, B., Mascart, F., Schandene, L., Janssen, F., Ferster, A., Kuo, C. C., Levy, S., Van Dongen, J. J., and Van Der Burg, M. (2010). CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J. Clin. Invest. 120, 1265–1274.
Vodjgani, M., Aghamohammadi, A., Samadi, M., Moin, M., Hadjati, J., Mirahmadian, M., Parvaneh, N., Salavati, A., Abdollahzade, S., Rezaei, N., and Srrafnejad, A. (2007). Analysis of class-switched memory B cells in patients with common variable immunodeficiency and its clinical implications. J. Investig. Allergol. Clin. Immunol. 17, 321–328.
Wang, J., and Cunningham-Rundles, C. (2005). Treatment and outcome of autoimmune hematologic disease in common variable immunodeficiency (CVID). J. Autoimmun. 25, 57–62.
Warnatz, K., Denz, A., Drager, R., Braun, M., Groth, C., Wolff-Vorbeck, G., Eibel, H., Schlesier, M., and Peter, H. H. (2002). Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood 99, 1544–1551.
Warnatz, K., Salzer, U., and Gutenberger, S. (2005). Finally found: human BAFF-R deficiency causes hypogammaglobulinemia. Clin. Immunol. 115, 820.
Wehr, C., Kivioja, T., Schmitt, C., Ferry, B., Witte, T., Eren, E., Vlkova, M., Hernandez, M., Detkova, D., Bos, P. R., Poerksen, G., Von Bernuth, H., Baumann, U., Goldacker, S., Gutenberger, S., Schlesier, M., Bergeron-Van Der Cruyssen, F., Le Garff, M., Debre, P., Jacobs, R., Jones, J., Bateman, E., Litzman, J., Van Hagen, P. M., Plebani, A., Schmidt, R. E., Thon, V., Quinti, I., Espanol, T., Webster, A. D., Chapel, H., Vihinen, M., Oksenhendler, E., Peter, H. H., and Warnatz, K. (2008). The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood 111, 77–85.
Wasserstrom, H., Bussel, J., Lim, L. C., and Cunningham-Rundles, C. (2008). Memory B cells and pneumococcal antibody after splenectomy. J. Immunol. 181, 3684–3689.
Zhang, L., Radigan, L., Salzer, U., Behrens, T. W., Grimbacher, B., Diaz, G., Bussel, J., and Cunningham-Rundles, C. (2007). Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: clinical and immunologic outcomes in heterozygotes. J. Allergy Clin. Immunol. 120, 1178–1185.
Keywords: common variable immunodeficiency (CVID), autoimmune cytopenias, immune thrombocytopenia, autoimmune hemolytic anemia, autoimmune lymphoproliferative syndrome, Evans syndrome
Citation: Podjasek JC and Abraham RS (2012) Autoimmune cytopenias in common variable immunodeficiency. Front. Immun. 3:189. doi: 10.3389/fimmu.2012.00189
Received: 22 February 2012; Accepted: 18 June 2012;
Published online: 24 July 2012.
Edited by:
Rosa Bacchetta, Fondazione Centro San Raffaele Del Monte Tabor, ItalyReviewed by:
Rosa Bacchetta, Fondazione Centro San Raffaele Del Monte Tabor, ItalyAntonio Condino-Neto, University of São Paulo, Brazil
Copyright: © 2012 Podjasek and Abraham. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Roshini S. Abraham, Cellular and Molecular Immunology Laboratory, Division of Clinical Biochemistry and Immunology, Department of Laboratory Medicine and Pathology, Mayo Clinic, Hilton 210e, 200 First Street Southwest, Rochester, MN 55905, USA. e-mail:YWJyYWhhbS5yb3NoaW5pQG1heW8uZWR1