

- 1 Kerkvliet Laboratory, Environmental and Molecular Toxicology, Oregon State University, Corvallis, OR, USA
- 2 Environmental Health Sciences Center, Oregon State University, Corvallis, OR, USA
Activation of the aryl hydrocarbon receptor (AhR) by its prototypic ligand, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), mediates potent suppression of T-cell dependent immune responses. The suppressive effects of TCDD occur early during CD4+ T-cell differentiation in the absence of effects on proliferation and have recently been associated with the induction of AhR-dependent regulatory T-cells (Treg). Since AhR functions as a ligand-activated transcription factor, changes in gene expression induced by TCDD during the early stages of CD4+ T-cell differentiation are likely to reflect fundamental mechanisms of AhR action. A custom panel of genes associated with T-cell differentiation was used to query changes in gene expression induced by exposure to 1 nM TCDD. CD4+ T-cells from AhR+/+ and AhR−/− mice were cultured with cytokines known to polarize the differentiation of T-cells to various effector lineages. Treatment with TCDD induced the expression of Cyp1a1, Cyp1b1, and Ahrr in CD4+ T-cells from AhR+/+ mice under all culture conditions, validating the presence and activation of AhR in these cells. The highest levels of AhR activation occurred under Th17 conditions at 24 h and Tr1 conditions at 48 h. Unexpectedly, expression levels of most genes associated with early T-cell differentiation were unaltered by AhR activation, including lineage-specific genes that drive CD4+ T-cell polarization. The major exception was AhR-dependent up-regulation of Il22 that was seen under all culture conditions. Independent of TCDD, AhR down-regulated the expression of Il17a and Rorc based on increased expression of these genes in AhR-deficient cells across culture conditions. These findings are consistent with a role for AhR in down-regulation of inflammatory immune responses and implicate IL-22 as a potential contributor to the immunosuppressive effects of TCDD.
Introduction
Activation of the transcription factor, aryl hydrocarbon receptor (AhR), by the prototypic ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) has been shown to exert potent immunosuppressive effects in a variety of T-cell dependent disease models. TCDD suppresses Th1-dependent autoimmune diseases such as Type 1 diabetes in the non-obese diabetic (NOD) mouse (Kerkvliet et al., 2009) and experimental autoimmune uveoretinitis (Zhang et al., 2010). TCDD also suppresses the Th17-mediated response in both experimental autoimmune encephalomyelitis (EAE) and colitis (Quintana et al., 2008; Benson and Shepherd, 2011). These potent immunosuppressive effects are also conserved in non-autoimmune disease models. The Th1-driven cytotoxic T-lymphocyte (CTL) and alloantibody responses to allogeneic P815 tumor cells are dose-dependently suppressed by TCDD treatment (Kerkvliet et al., 1996), as are CTL and antibody responses to influenza virus infection (Warren et al., 2000). Treatment with TCDD also suppresses Th2-dependent cytokine and OVA-specific antibody production (Shepherd et al., 2000; Nohara et al., 2002), as well as several Th2-dependent allergic diseases including atopic dermatitis (Fujimaki et al., 2002), dust mite-induced asthma (Luebke et al., 2001), and peanut allergy (Schulz et al., 2011).
One of the mechanisms by which TCDD suppresses adaptive immunity occurs via activation of AhR in differentiating CD4+ T-cells. This was demonstrated using an acute parent-into-F1 graft-versus-host (GVH) model wherein the suppression of donor CTL development was contingent upon the donor CD4+ T-cells expressing AhR (Kerkvliet et al., 2002). Furthermore, optimal suppression of the CTL response occurred when TCDD was administered within 3 days of alloantigen stimulation – the time during which donor CD4+ T-cells undergo differentiation (Kerkvliet et al., 1996). Phenotypic and functional analysis of the differentiating allospecific donor cells suggested that AhR activation was driving the development of an immunosuppressive regulatory T-cell (Treg), characterized by a CD4+CD25highFoxp3negCTLA-4+CD62LlowIL-10+ phenotype (Funatake et al., 2005; Marshall et al., 2008). Subsequently, TCDD has also been shown to induce Foxp3+ Tregs and Tr1 cells in vitro (Apetoh et al., 2010; Gandhi et al., 2010) and to increase the frequency of Foxp3+ CD4+ T-cells in vivo in several models of immune-mediated disease (Quintana et al., 2008; Kerkvliet et al., 2009; Takamura et al., 2010; Zhang et al., 2010; Benson and Shepherd, 2011; Schulz et al., 2011; Singh et al., 2011).
Based on its role as a transcription factor, activation of AhR in CD4+ T-cells may directly alter CD4+ T-cell differentiation by influencing gene expression during early differentiation events. The likelihood of such effects is high given the large number of immune-related genes that contain dioxin response elements (DRE; Sun et al., 2004; Frericks et al., 2008; Kerkvliet, 2009). In the present studies, we characterized the influence of TCDD-activated AhR on gene expression during CD4+ T-cell differentiation under Th0, Th1, Treg, Tr1, and Th17 polarizing conditions. We utilized a custom panel of 48 genes that have been associated with AhR activation, T-cell differentiation, and/or Treg induction (Table 1). CD4+ T-cells were obtained from AhR+/+ and AhR-deficient (AhR−/−) mice, allowing us to validate the AhR-dependence of TCDD’s effects. In addition, differences in gene expression between vehicle-treated cultures of AhR+/+ and AhR−/− CD4+ T-cells identified genes that are regulated by AhR during T-cell activation in the absence of an exogenous ligand.
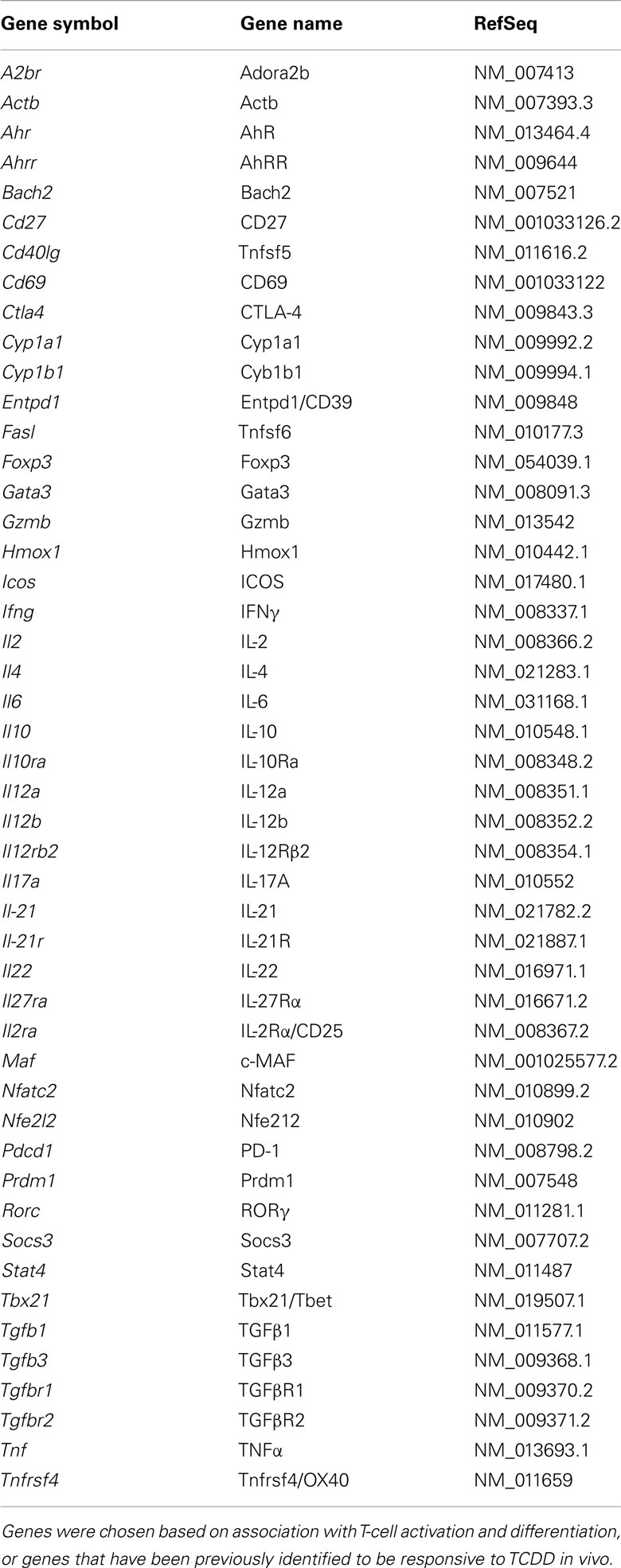
Table 1. Panel of genes used to evaluate AhR regulation of gene expression in CD4+ T-cells.
Materials and Methods
Animals
B6.PL-Thy1a/CyJ mice (Thy1.1+/+, AhR+/+) and B6.129-AhRtm1Bra/J (Thy1.1+/+, AhR−/−) mice were bred and maintained under specific pathogen-free conditions at the Laboratory Animal Resource Center at Oregon State University (Corvallis, OR, USA). All animal procedures were approved by the Institutional Animal Care and Use Committee.
CD4+ T-cell Cultures
Spleens were aseptically removed and processed into single-cell suspensions via dissociation between the frosted ends of microscope slides. Red blood cells and dead cells were removed by hypotonic water lysis. CD4+ T-cells were isolated by negative selection using a CD4+ T-cell isolation kit and an autoMACS separator (Miltenyi Biotec; Auburn, CA, USA). T-cells were cultured in RPMI 1640 media (Invitrogen; Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (HyClone; Logan, UT, USA), 10 mM HEPES (Invitrogen), 50 μg/ml gentamicin (Invitrogen), and 50 μM 2-β-mercaptoethanol (Sigma; St. Louis, MO, USA). At the time of culture initiation, cells were treated with 1 nM TCDD (dissolved in DMSO) or 0.001% DMSO (vehicle). The 1 nM concentration of TCDD used in these studies was sufficient to induce maximum activation of AhR in T-cells as reflected in expression of known AhR-regulated genes Cyp1a1, Cyp1b1, and Ahrr (unpublished data).
CD4+ T-cells (1 × 106 cells/well) were activated with soluble anti-CD3 (0.5 μg/ml) and anti-CD28 (2.5 μg/ml) or plate-bound anti-CD3 (2 μg/ml) and anti-CD28 (2 μg/ml) in a 24-well plate. For Th1 conditions, anti-IL-4 (10 μg/ml) and IL-12 (3 ng/ml) was added to each well. For Treg polarizing conditions, TGFβ1 (3 ng/ml) was added. In addition to TGFβ1, IL-27 (25 ng/ml), or IL-6 (15 ng/ml) was added for Tr1 or Th17 polarizing conditions, respectively. All reagents for T-cell polarization were purchased from eBioscience. T-cells cultured under Th0 conditions received no exogenous cytokines.
For some genes (Il22, Il17a, Rorc), protein levels were also measured. Whole splenocytes or purified CFSE-labeled CD4+ T-cells were cultured under Th17 conditions. After 48 h, cells were treated for 6 h with a Cell Stimulation Cocktail (eBioscience) containing phorbol 12-myristate 13-acetate, ionomycin, Brefeldin A, and monensin. The cells were then stained for intracellular protein prior to flow cytometric analysis as described below. Supernatants were collected for ELISA analysis.
RNA Isolation and QPCR
CD4+ T-cells were harvested at 24 and 48 h and flash frozen in liquid nitrogen. Cell pellets were stored at −80°C. RNA was isolated using the RNeasy Mini Kit #74104 (Qiagen; Valencia, CA, USA), with on-column DNase digestion (Qiagen #79254) performed twice to ensure complete elimination of genomic DNA. RNA concentrations and purity were determined using the NanoDrop ND-1000 UV-Vis Spectrophotometer (ThermoScientific). The Reaction Ready First Strand cDNA synthesis kit #C-01/C-03 (SA Biosciences, formerly SuperArray; Frederick, MD, USA) was used to synthesize cDNA in a two-step PCR reaction. A minimum of 200 ng RNA was used for all reactions. Following reverse transcription, samples were stored at −80°C.
An ABI PRISM 7500 Real-Time PCR system (Applied Biosystems) was used for all qPCR reactions. SYBR Green/ROX qPCR Master Mix (SA Bioscience) was used to prepare a 25 μl reaction mix consisting of 12.5 μl master mix, cDNA equivalent to 2 ng RNA, 1 μl primer, and RNase-free H20 to obtain the final volume. All primers were obtained from SA Biosciences. Sequence accession numbers are shown in Table 1. The qPCR settings were as follows: 10 min at 95°C, followed by 40 cycles of 15 s at 95°C, followed by 1 min at 60°C. This was followed by a standard dissociation step of 15 s at 95°C to generate a melting curve. A no-reverse transcription (NRT) control was included to validate the absence of genomic DNA from all samples.
QPCR Data Analysis
Data were analyzed using ABI 7500 analysis software. Genes that were not detected within 40 cycles received a value of 40. Cycle numbers were then normalized to Actb to calculate ΔCt. The data were analyzed as either 1/ΔCt or 1/ΔCt × 100; all other data were presented as fold change. Fold changes were calculated by the following formulas:
When analyzing gene expression across polarizing conditions, fold changes were calculated relative to Th0 as follows:
Flow Cytometric Analysis
The following antibodies were purchased from eBioscience: CD4-PE (GK1.5), IL-22-PE (1H8PWSR), IL-17A-APC (eBio17B7), and RORγ-PE (AFKJS). CD4-APC (GK1.5) and carboxyfluorescein succinimidyl ester (CFSE) were purchased from Invitrogen. Splenocytes or purified CFSE-labeled CD4+ T-cells were resuspended in PAB (PBS, 1% BSA, 0.1% sodium azide), and incubated with PE- or APC-labeled anti-CD4 antibody for 10 min. After two washes, cells were resuspended in Fixable Viability Dye efluor 780 (eBioscience) for 30 min. The BD Cytofix/Cytoperm buffer set or the Foxp3 Staining Buffer set (eBioscience) was used for intracellular staining. Control samples that contained all of the antibodies except the one of interest (fluorescence minus one) were used as negative staining controls. A minimum of 150,000 viable CD4+ T-cells were collected per sample on a Beckman Coulter FC-500 flow cytometer. Compensation and analysis of data were performed in WinList (Verity Software, Version 6.0).
ELISA
Supernatants from purified CD4+ T-cells and splenocyte cultures were collected after 48 h. IL-22 production was quantified using the IL-22 Ready-Set-Go® ELISA kit from eBioscience, according to manufacturer’s instructions.
Statistical Analysis
All treatment groups consisted of a minimum of three biological replicates (two mice per replicate), except where indicated. Each biological sample was treated with vehicle or TCDD in separate wells. Differences due to TCDD treatment were determined using a paired Student’s t-test. For comparisons between AhR+/+ and AhR−/− CD4+ T-cells, or between culture conditions, a non-paired Student’s t-test was used. Values were considered statistically significant at p ≤ 0.05, in conjunction with a fold change of ≥2 between treatment groups. For Il22 analysis across treatments, a linear mixed model was utilized using the Mixed procedure of SAS (v. 9.2).
Results
Aryl Hydrocarbon Receptor is Present and Transcriptionally Active during Early Differentiation of CD4+ T-cells
Ahr message was detected in CD4+ T-cells cultured under all conditions. Cells activated for 24 h in the absence of exogenous cytokines (Th0 conditions) expressed Ahr at a level comparable to that of non-activated cells, indicating that expression of Ahr is not up-regulated due to T-cell receptor activation alone (Figure 1A). Likewise, neither Th1 nor Tr1 polarizing conditions produced a significant increase in Ahr expression. In contrast, Treg conditions induced a fourfold increase and Th17 conditions induced a 78-fold increase in Ahr expression at 24 h (Figure 1B).
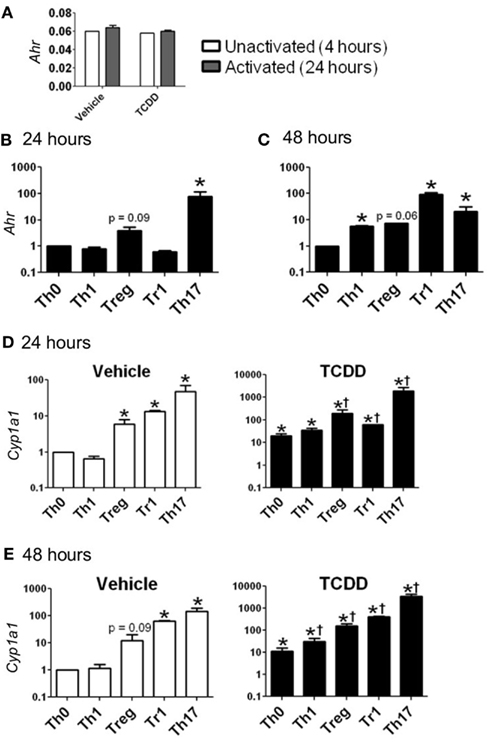
Figure 1. Expression of Ahr and Cyp1a1 in CD4+ T-cells cultured under various conditions. (A) Basal expression of Ahr (1/ΔCt) in CD4+ T-cells cultured for 4 h without stimulation or 24 h following stimulation with anti-CD3/anti-CD28. (B,C) Ahr expression in CD4+ T-cells cultured for 24 or 48 h with polarizing cytokines as described in Section “Materials and Methods.” Data are presented as fold change relative to Th0 vehicle conditions at 24 or 48 h. (D,E) Expression of Cyp1a1 in vehicle- and TCDD-treated CD4+ T-cells cultured for 24 or 48 h under different polarizing conditions. Data are presented as fold change relative to the vehicle-treated Th0 condition at 24 or 48 h. *p ≤ 0.05 relative to either vehicle or TCDD Th0 control within that time point. †p ≤ 0.05 relative to the TCDD-treated Th0 condition at 24 or 48 h. Error bars indicate the mean ± SEM; n = 3−4 biological replicates.
At 48 h, Ahr expression under Th0 conditions was equivalent to the level of expression seen at 24 h, while expression was up-regulated by 5.6-fold under Th1 conditions and 7.4-fold under Treg polarizing conditions (Figure 1C). Expression of Ahr declined under Th17 conditions but remained significantly up-regulated (21-fold) compared to Th0 conditions. Ahr expression was highest at 48 h under Tr1 conditions with a fold change of 92 relative to Th0 conditions.
To determine if the level of Ahr message present under different culture conditions was indicative of AhR protein, the ability of TCDD to increase expression of known AhR-regulated genes was examined. Addition of TCDD per se did not influence Ahr expression except for a small increase that was noted under Treg conditions at 24 h (Table 2). In contrast, addition of TCDD significantly up-regulated Cyp1a1 expression under all conditions tested (Figures 1D,E), with good correlation between the levels of expression of Cyp1a1 and Ahr at both 24 and 48 h (Figure 1). A similar correlation was observed between expression levels of Ahr and expression levels of the AhR-regulated genes Cyp1b1 and Ahrr (Figure A1 in Appendix). These results indicate that sufficient AhR is expressed in CD4+ T-cells cultured under any condition to be able to respond to 1 nM TCDD and alter expression of AhR-regulated genes.
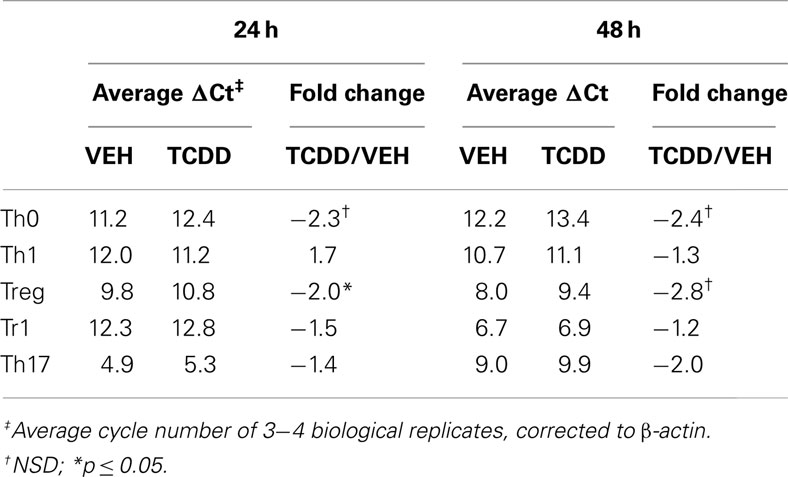
Table 2. Influence of TCDD on Ahr expression in CD4+ T-cells cultured under different polarizing conditions.
While TCDD-induced relatively high levels of Cyp1a1, low levels of Cyp1a1 were also seen in the absence of an exogenous ligand in CD4+ T-cells cultured under Treg, Tr1, and Th17 polarizing conditions at both 24 and 48 h (Figures 1D,E). These results suggest that the cytokine milieu either contains or is capable of inducing the formation of an AhR ligand in the responding T-cells. Interestingly very low but detectable Cyp1a1 expression was also seen in AhR-deficient cells especially at 48 h under all polarizing conditions (Figure 2). Addition of TCDD did not up-regulate this expression, validating the absence of functional AhR protein in AhR−/− cells (Figure 2), but implicating an AhR-independent pathway for Cyp1a1 expression.
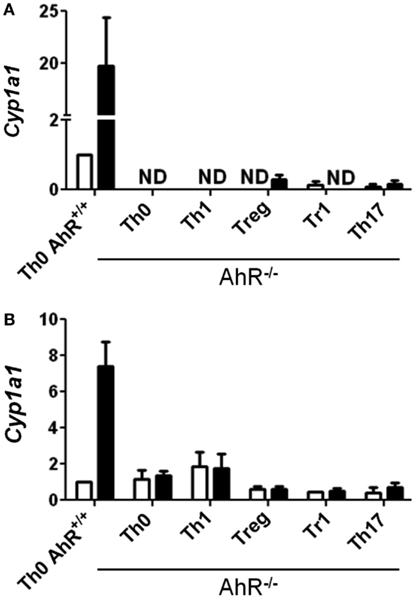
Figure 2. Expression of Cyp1a1 in AhR−/− CD4+ T-cells. Data are presented as fold change relative to AhR+/+ CD4+ T-cells cultured under vehicle-treated Th0 conditions for (A) 24 or (B) 48 h. Error bars indicate the mean ± SEM; n = 3−4 biological replicates, except for 24 h AhR−/− Treg and Th17 conditions, and 48 h AhR−/− Treg and Tr1 conditions, where n = 2. *p ≤ 0.05; ND = not detected within 40 cycles.
Effect of AhR Activation by TCDD on Expression of Genes Involved in CD4+ T-cell Differentiation
The pattern of gene expression observed under the various polarizing conditions was consistent with early differentiation events specific to different CD4+ T-cell subsets (Figure 3; Zhou et al., 2009; Hermann-Kleiter and Baier, 2010). Under Th1 polarizing conditions, the transcription factor Tbet was up-regulated at 48 h, and the associated cytokine Ifng was up-regulated at 24 and 48 h when compared to expression levels in non-polarized (Th0) cells. The up-regulation of these Th1-associated genes was concurrent with the down-regulation of Gata3, also consistent with Th1 differentiation. Similarly, the transcription factor for T-regulatory cells, Foxp3, and the associated Treg marker Cd39 were up-regulated under Treg polarizing conditions, while Tr1 polarizing conditions induced the expression of Maf and Il10. Foxp3 expression was also up-regulated under Tr1 polarizing conditions at 48 h, albeit to a lesser extent than that seen under Treg polarizing conditions. As expected Th17 polarizing conditions produced increased expression of Rorc and Il17 at 24 and 48 h.
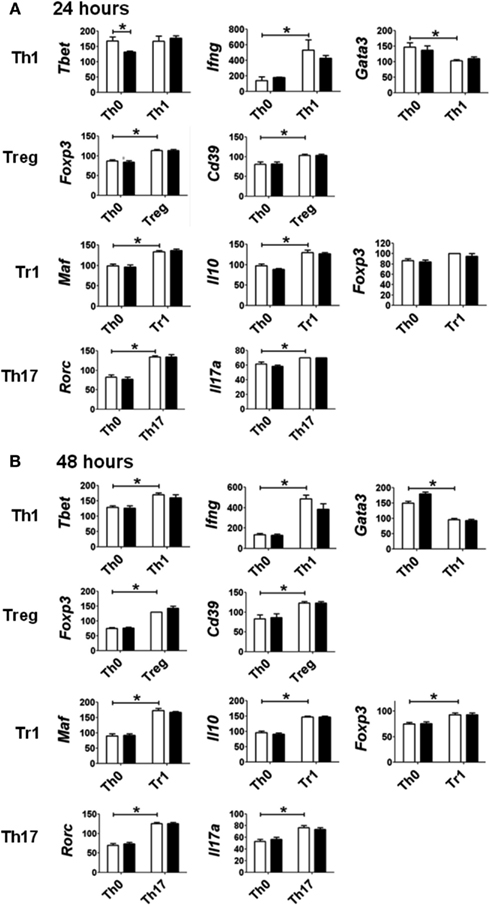
Figure 3. Expression of genes associated with specific polarization in CD4+ T-cells treated with vehicle (white bar) or TCDD (black bar) for (A) 24 or (B) 48 h. Data are presented as 1/ΔCt × 100. Gene expression under Th0 conditions (no polarizing cytokines) is presented for comparison with gene expression under specific polarizing conditions as described in Section “Materials and Methods.” Error bars represent the mean ± SEM; n = 3−4 biological replicates. *p ≤ 0.05.
Surprisingly, activation of AhR by TCDD did not significantly alter expression of any of the master regulator genes associated with CD4+ T-cell polarization or other genes associated with polarization (Figure 3). In fact, expression of only one gene in the panel of genes tested, Il22, was clearly altered by treatment with TCDD, and this effect was seen under all conditions except Tr1 (Table 3). When analyzed over all conditions except Tr1, expression of Il22 was significantly increased in the presence of TCDD at both 24 (p = 0.03) and 48 h (p < 0.001). The level of Il22 expression was highest under Th1 conditions in vehicle-treated cells, and addition of TCDD further increased expression (Figure 4), while the greatest induction of Il22 expression by TCDD (17.6-fold) occurred under Th17 conditions at 48 h (Table 3). The minimal expression of Il22 under Tr1 conditions was surprising given the high level of Ahr expressed in these cells at 48 h and suggested that the presence of IL-27 as a polarizing cytokine inhibits Il22 transcription. In fact, Rutz et al. (2011) recently showed that the induction of c-Maf by IL-27 results in downstream repression of Il22. Analysis of AhR−/− CD4+ T-cells showed no significant increase in Il22 expression upon treatment with TCDD at either 24 or 48 h under any polarizing condition, verifying that the up-regulation of Il22 by TCDD was dependent on AhR activation (Table 3).
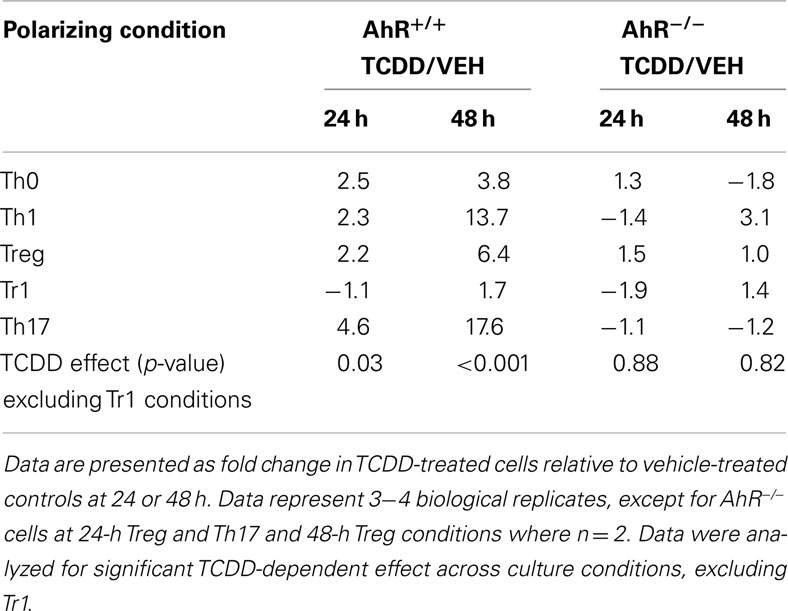
Table 3. Influence of TCDD on Il22 expression in AhR+/ + and AhR−/ − CD4+ T-cells activated under different polarizing conditions.
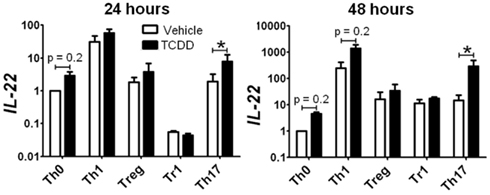
Figure 4. Influence of TCDD on Il22 expression under various polarizing conditions. Data are presented as fold change relative to expression in Th0 vehicle controls at 24 or 48 h. Error bars represent the mean ± SEM; n = 3−4 biological replicates. *p ≤ 0.05.
To determine if AhR-mediated regulation of Il22 was conserved at the level of protein, purified CD4+ T-cells or whole splenocytes were cultured with or without TCDD for 48 h under Th17 polarizing conditions. IL-22 was measured in CD4+ T-cells and in culture supernatants. As shown in Figure 5A, a small population of CD4+ T-cells was found to express IL-22 in vehicle-treated cultures. Addition of TCDD increased the percentage of IL-22+ cells (p = 0.06) and the amount of IL-22 produced on a per cell basis (p = 0.08) as measured by the median fluorescence intensity (MFI). IL-22 was also detected in supernatants from cultures of purified CD4+ T-cells. TCDD-treated samples had an average of 28 pg/ml compared to <8 pg/ml (the reporting limit) in vehicle-treated cultures (Figure 5B). Likewise, when whole splenocytes were cultured, treatment with TCDD increased the frequency of IL-22+ CD4+ T-cells (p = 0.05) and the MFI (p < 0.001; Figure 5C). Significantly more IL-22 was found in supernatants from whole splenocytes consistent with a previous report (Alam et al., 2010). Treatment with TCDD significantly increased the amount of IL-22 present (p = < 0.001) from 65 pg/ml in vehicle-treated cultures to 374 pg/ml in TCDD-treated cultures (Figure 5D).
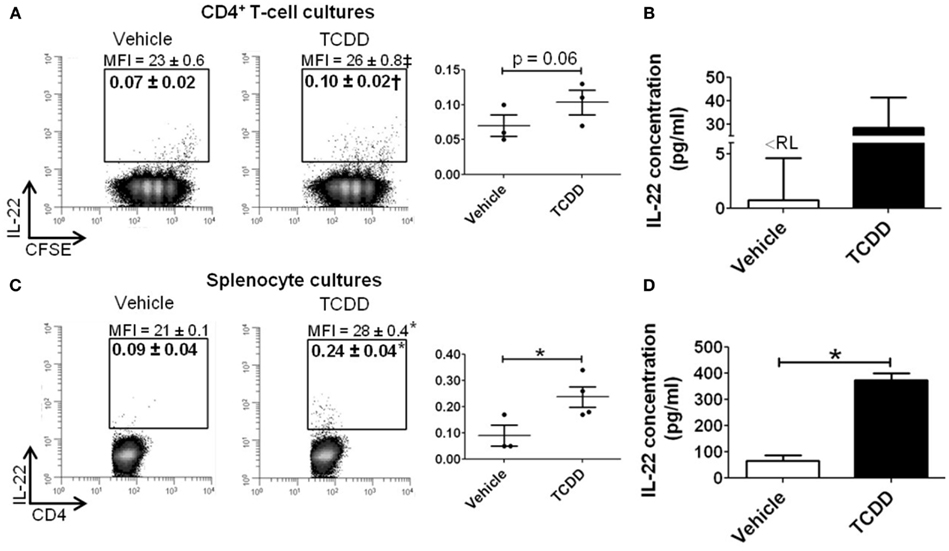
Figure 5. Influence of TCDD on IL-22 protein in CD4+ T-cells cultured under Th17 polarizing conditions for 48 h. Data shown are gated on viable CD4+ T-cells. (A) CD4+ T-cells were purified and labeled with CFSE prior to activation. Percent CD4+IL-22+ T-cells are shown. MFI, median fluorescence intensity. Individual biological replicates are represented graphically. Histograms shown are representative of 3−4 biological replicates. (B) Concentration of IL-22 in supernatant from purified CD4+ T-cell cultures.<RL = below reporting limit. (C) Whole spleen cell suspensions were activated and cultured under Th17 polarizing conditions, as in (A). (D) Concentration of IL-22 in supernatant from whole splenocyte cultures. Error bars represent the mean ± SEM; n = 3−4 biological replicates. †p = 0.06; ‡p = 0.08; *p ≤ 0.05.
Other statistically significant changes in gene expression induced by TCDD in differentiating CD4+ T-cells are summarized in Table 4. Increased expression of Ctla4 (2.3-fold), Cd40lg (2.2-fold), and Il12rb2 (2.9-fold) was associated with TCDD treatment at 24 h under Th1 conditions. Il12rb2 was also increased (7.1-fold) by TCDD under Th17 conditions at 48 h. Under Treg conditions, only Stat4 expression was altered by TCDD and it was down-regulated (−2.7-fold), while no changes were seen under Tr1 conditions. Under Th0 conditions, TCDD significantly suppressed expression of Tbet (2.7-fold) and Il17a (2.1-fold).
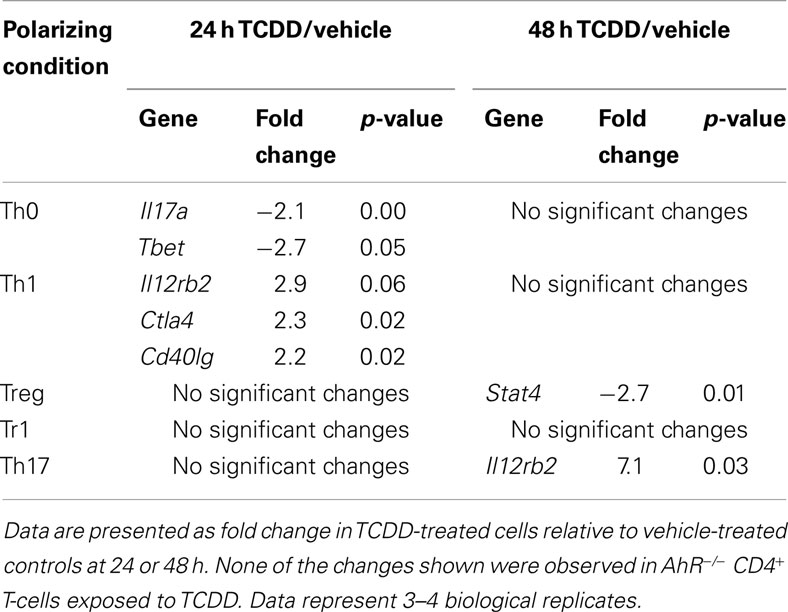
Table 4. Summary of changes in expression of immune-related genes induced by TCDD under different polarizing conditions.
Influence of AhR Deficiency on Gene Expression in Activated CD4+ T-cells
Significant changes in gene expression were observed between vehicle-treated AhR+/+ and AhR−/− CD4+ T-cells cultured under different polarizing conditions (Figure 6). The most significant effect of AhR deficiency was increased expression of Il17a at both 24 and 48 h under all culture conditions (Figure 6A). The greatest up-regulation of Il17a was noted under Treg conditions, with a 53-fold increase, whereas 16- and 4-fold changes were observed under Th17 and Tr1 conditions, respectively, at 24 h, with a similar trend for Th1 and Th0 conditions. At 48 h, Il17a expression was significantly up-regulated in AhR−/− CD4+ T-cells under Th1, Treg, and Tr1 conditions.
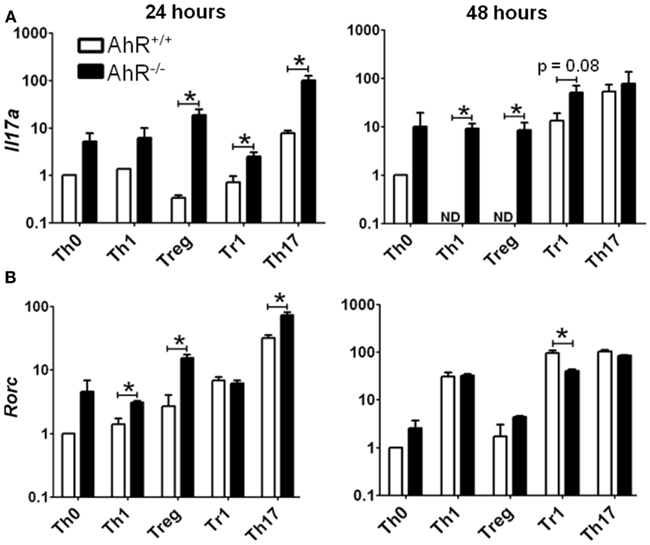
Figure 6. Influence of AhR deficiency on expression of (A) Il17a or (B) Rorc in AhR+/+ or AhR−/− CD4+ T-cells cultured under different polarizing conditions for 24 or 48 h. Data are expressed as fold change relative to AhR+/+ T-cells cultured under vehicle-treated Th0 conditions. Error bars represent the mean ± SEM; n = 3 biological replicates, except for 24 or 48 h AhR−/− Treg conditions, and 24 h Th17 conditions, where n = 2. *p ≤ 0.05. ND = not detected within 40 cycles.
Aryl hydrocarbon receptor deficiency also resulted in increased expression of Rorc, the master transcription factor for Th17 differentiation (Figure 6B). At 24 h, AhR−/− T-cells expressed a sevenfold increase in Rorc under Treg conditions, and a twofold increase under Th1 and Th17 conditions. Rorc expression was also increased in AhR−/− cells under Th0 conditions (p = 0.06), while expression was unaltered under Tr1 polarizing conditions. AhR regulation of Rorc was no longer seen at 48 h, with the exception of Tr1 conditions, where Rorc was down-regulated twofold in AhR−/− CD4+ T-cells.
To determine if the increase in expression of Il17a and Rorc was observed at the protein level, IL-17A, and RORγ were measured by flow cytometry in cells cultured under Th17 conditions for 48 h. As shown in Figure 7A, 1.9% of the AhR+/+ CD4+ T-cells expressed IL-17A compared to 1.0% of the AhR−/− CD4+ T-cells (p = 0.05). MFI values were also lower in AhR−/− cells (p = 0.1). Similarly, a smaller percentage of AhR−/− CD4+ T-cells stained positive for RORγ compared to AhR+/+ CD4+ T-cells, but the changes were not statistically significant (p = 0.2; Figure 7B). The basis for the lack of correlation between gene and protein expression is not known but may be related to the transient nature of the up-regulation of Il17a and Rorc expression seen in AhR-deficient cells. Under Th17 conditions, this effect was most pronounced at 24 h but gone by 48 h. As expected, there was no effect of AhR status on CD4+ T-cell proliferation, assessed by CFSE dilution.
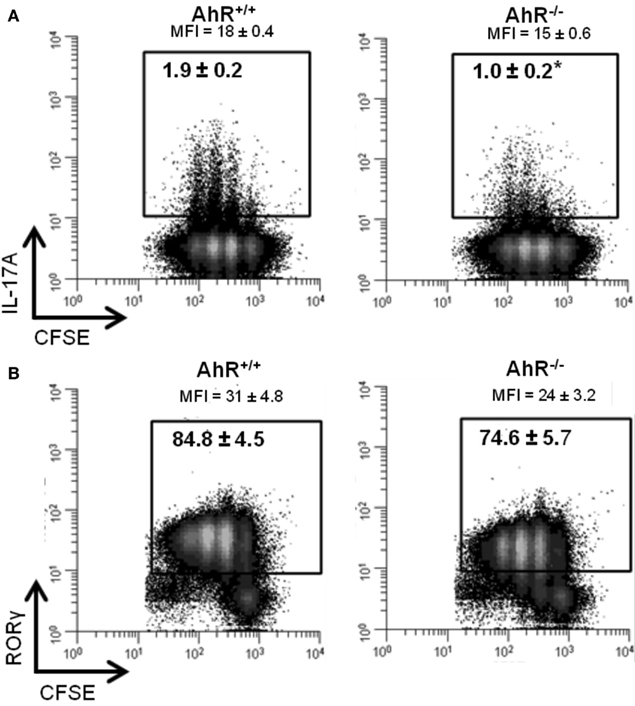
Figure 7. Influence of AhR deficiency on (A) IL-17A or (B) RORγ protein in AhR+/+ or AhR−/− CD4+ T-cells cultured under Th17 polarizing conditions for 48 h. Data shown are gated on viable CD4+ T-cells. Percent IL-17A+ or RORγ+ T-cells are shown. MFI, median fluorescence intensity. Histograms shown are representative of 3−4 biological replicates. *p ≤ 0.05. Changes in RORγ expression were not statistically significant.
Aryl hydrocarbon receptor deficiency had no significant effect on expression of Il22 or Foxp3 under any of the polarizing conditions tested (Figure A2 in Appendix). Tgfb3 and Gzmb were up-regulated under Treg and Th0 conditions and Il6 was up-regulated under Treg and Tr1 conditions in AhR-deficient cells, while other changes were unique to specific culture conditions (Table 5). In general, the results suggest that AhR deficiency in CD4+ T-cells favors increased expression of genes associated with inflammation, consistent with the pro-inflammatory phenotype of AhR-deficient mice.
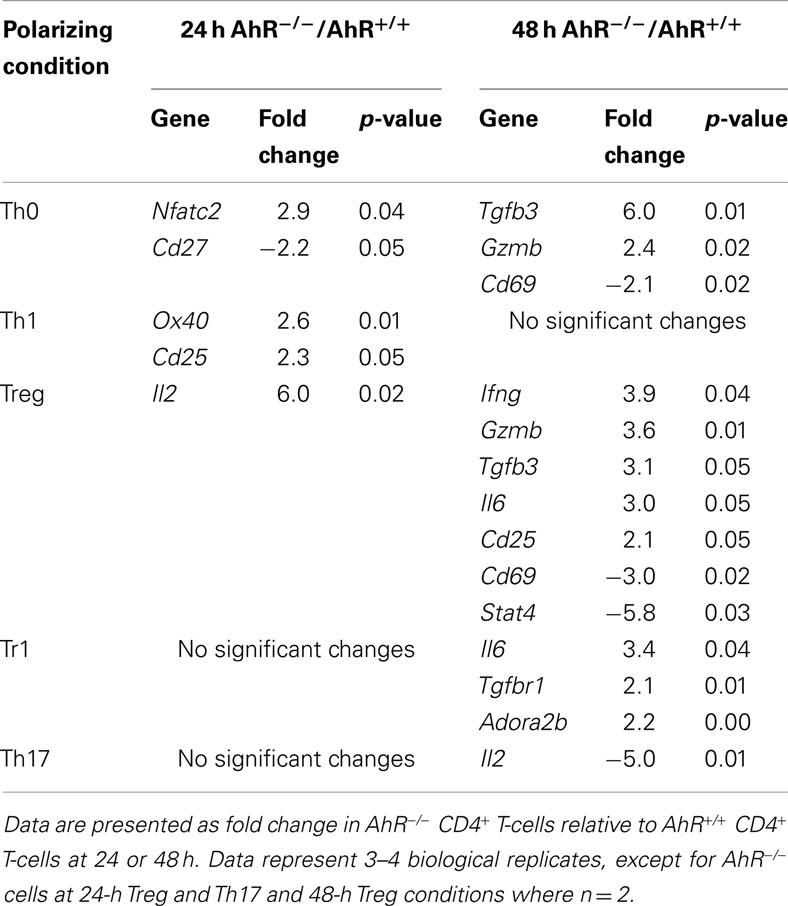
Table 5. Summary of changes in expression in immune-related genes related to AhR deficiency in CD4+ T-cells cultured under different polarizing conditions.
Discussion
The AhR is a ligand-activated transcription factor that has been shown to play a role in CD4+ T-cell differentiation, yet little is known about the changes in gene expression that occur in these cells following AhR activation. Previous studies have examined gene expression in donor CD4+ T-cells isolated from the spleen of TCDD-treated mice during a GVH response, however the identification of direct gene targets was confounded by the presence of contaminating host cells that also express AhR (Marshall et al., 2008). Identification of gene targets has become more important as recent reports indicate that there may be ligand-specific effects of AhR activation on CD4+ T-cell differentiation (Quintana et al., 2008; Veldhoen et al., 2008). Furthermore, the initial report of highly increased AhR expression in CD4+ T-cells cultured under Th17 conditions (Kimura et al., 2008; Veldhoen et al., 2008) led to an early misconception that AhR was functional only in Th17 cells in the mouse (Ho and Steinman, 2008; Veldhoen et al., 2008; Stockinger, 2009; Ramirez et al., 2010). However, more recently, AhR was shown to be significantly up-regulated in mouse and human CD4+ T-cells under Tr1 conditions as well (Apetoh et al., 2010; Gandhi et al., 2010). Given that TCDD suppresses Th1-, Th2-, and Th17-mediated responses in vivo, it implicates a role for AhR in the differentiation of many T-cell subsets. The present studies were designed to determine if genes involved in CD4+ T-cell differentiation are selectively regulated by AhR when the cells are activated in the presence of TCDD under a variety of Th-polarizing culture conditions.
The results of our studies show that Ahr is expressed in CD4+ T-cells under all conditions tested, including un-activated cells, while elevated levels of Ahr are seen under Treg, Tr1, and Th17 conditions. The highest level of Ahr expression was seen under Th17 conditions at 24 h and Tr1 conditions at 48 h. Functional AhR protein was also present under all culture conditions based on the ability of TCDD to induce the expression of known AhR-regulated genes (Cyp1a1, Cyp1b1, and Ahrr). The level of induced gene expression was generally proportional to the amount of Ahr present with the highest induction seen under Th17 conditions. Since many putative endogenous AhR ligands have anti-inflammatory functions, it suggests that up-regulation of AhR may be a protective response during highly inflammatory Th17 conditions, allowing low-affinity endogenous AhR ligands to down-regulate the response, consistent with the effects seen with TCDD. Examples of low-affinity ligands with immunosuppressive activity include lipoxin A4, bilirubin, and kynurenine (Sinal and Bend, 1997; Schaldach et al., 1999; Liu et al., 2008; Mezrich et al., 2010). Indeed, bilirubin and kynurenine have been shown to suppress EAE and induce CD4+Foxp3+ T-cells, respectively (Liu et al., 2008; Mezrich et al., 2010). Elevated levels of AhR in CD4+ T-cells undergoing Tr1 and Treg differentiation would also facilitate AhR signaling to promote the development of these anti-inflammatory cells.
TCDD did not appear to alter early polarization of activated CD4+ T-cells despite strong activation of AhR. Both vehicle- and TCDD-treated cells showed the expected increase in expression of Tbet, Foxp3, Maf, and Rorc under Th1, Treg, Tr1, and Th17 polarizing conditions, respectively. Likewise, the expression of other genes that are associated with specific polarizing conditions was unaffected by TCDD, including Ifng, Cd39, Il10, and Il17a. The lack of effect of TCDD on Foxp3 expression was somewhat surprising since TCDD has been previously reported to directly regulate Foxp3 expression in vitro and increase the generation of Foxp3+ cells (Kimura et al., 2008; Quintana et al., 2008). However, the lack of effect of TCDD on Foxp3 expression is consistent with the Foxp3neg Treg phenotype induced by TCDD in vivo (Funatake et al., 2005; Marshall et al., 2008).
Only a few of the other genes in our 48-gene panel showed altered expression as a result of the presence of TCDD during CD4+ T-cell activation. Ctla4 expression was increased by TCDD under Th1 conditions, a change that is consistent with the AhR-dependent increase in expression of CTLA-4 on donor CD4+ T-cells in vivo during the GVH response (Funatake et al., 2005). Likewise, increased expression of Il12rb2 under both Th1 and Th17 conditions is consistent with the increased responsiveness of donor CD4+ T-cells from TCDD-treated mice to IL-12, resulting in enhanced STAT4 phosphorylation (Marshall et al., 2008). Under Th0 conditions, TCDD also suppressed the expression of Tbet and Il17a, providing a mechanism to promote Treg differentiation at the expense of effector Th1 or Th17 cells.
The most noteworthy effect of TCDD in CD4+ T-cells was the AhR-dependent increase in expression of Il22 that was seen across all culture conditions except Tr1. AhR-mediated up-regulation of Il22 was also observed at the protein level in CD4+ T-cells cultured under Th17 conditions. AhR signaling has been implicated in IL-22 production in previous studies (Veldhoen et al., 2008; Ramirez et al., 2010; Rutz et al., 2011). Additionally, Notch signaling was shown to create AhR ligands that enhance IL-22 production (Alam et al., 2010). Based on the fact that IL-22 can be produced in the absence of IL-17A or IFNγ, a unique Th22 subset of CD4+ T-cells has been proposed (Trifari et al., 2009). The putative transcription factor for this Th22 subset is the AhR (Trifari et al., 2009). IL-22 has complex functions and has been shown to produce both inflammatory (psoriasis, rheumatoid arthritis) and protective (irritable bowel disease) responses (reviewed in Zenewicz and Flavell, 2011). Enhanced production of IL-22 by CD4+ T-cells following AhR activation by TCDD may play a role in the immunoregulatory effects of TCDD.
Apart from its role in mediating effects of exogenous ligands such as TCDD, the consequences of AhR activation via endogenous ligands is also of interest. Although in vivo studies indicate that AhR−/− mice generate normal adaptive immune responses to model antigens (Vorderstrasse et al., 2001) and that AhR−/− donor CD4+ T-cells differentiate normally during an acute GVH response (Kerkvliet et al., 2002; Funatake et al., 2005), AhR-deficient mice have been reported to express a hypersensitive phenotype in experimental colitis, and following challenge with either LPS or cigarette smoke (Thatcher et al., 2007; Sekine et al., 2009; Furumatsu et al., 2011). Since several putative endogenous AhR ligands have been associated with the induction of immunosuppressive Tregs, the absence of AhR could impair Treg differentiation resulting in hyper-inflammation. Several pro-inflammatory genes were up-regulated in AhR-deficient cells at 48 h under Treg polarizing conditions including Ifng, Gzmb, Tgfb3, and Il6 suggesting that the absence of AhR signaling in CD4+ T-cells alters Treg differentiation. Also consistent with a hypersensitive phenotype, CD4+ T-cells from AhR−/− mice showed a transient increase in expression of genes associated with Th17 differentiation, Rorc, and Il17a, which was seen under Th1, Treg, Tr1, and Th17 conditions at 24 h. However, neither transcript nor protein levels were affected at 48 h when measured under Th17 conditions. These results suggest that the impairment of Th17 differentiation in the absence of AhR (Kimura et al., 2008; Veldhoen et al., 2009) results from post-transcriptional events. Similarly, whereas Kimura et al. (2008) reported that fewer Foxp3+ cells were generated when AhR−/− cells were cultured under Treg conditions, AhR deficiency did not influence Foxp3 expression under any of the conditions tested in our studies.
Given the emerging role of AhR in IL-22 production and the significant up-regulation of Il22 by TCDD, we were surprised to find that AhR deficiency did not impair expression of the Il22 gene. This finding was in direct contrast to the results of Veldhoen et al. (2008) who first reported that IL-22 expression was absent in AhR-deficient cells (Veldhoen et al., 2008). One possible explanation for these divergent findings may derive from the type of media used for T-cell culture. All of our studies utilized RPMI 1640 whereas Veldhoen et al. (2008) may have used a specific medium (IMDM) that contains high levels of aromatic amino acids including tryptophan, a source of several AhR ligands (Veldhoen et al., 2009). If IMDM was used, the expression of Il22 may have been up-regulated in WT cells due to the presence of endogenous AhR ligands, while AhR-deficient cells would be unable to respond. We saw this effect when we compared IL-22 expression in Th17-polarized cultures of AhR+/+ and AhR−/− CD4+ T-cells treated with TCDD. Expression of Il22 was more than 50-fold higher in AhR+/+ cells at 48 h (unpublished observations). Our data suggest that AhR is not needed for constitutive IL-22 expression.
In summary, strong activation of AhR by TCDD in differentiating CD4+ T-cells failed to influence the expression of numerous genes associated with T-cell activation and differentiation. Since many of these genes have been shown to be altered by TCDD in differentiating CD4+ T-cells in vivo, the results cast doubt on the ability of in vitro conditions to recapitulate in vivo events. This could be due to lack of accessory cell signals not mimicked by antibodies to CD3 and CD28, or to the presence of high levels of polarizing cytokines that might dominate the signaling pathways. Other than known AhR-regulated genes (Cyp1a1, Cyp1b1, Ahrr), the only immune-related gene that was clearly regulated by AhR in CD4+ T-cells was Il22, a gene that has been recently associated with the AhR pathway. The possible role that IL-22 plays in the immunoregulatory effects of TCDD awaits future studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Jamie Pennington and Sumit Punj for critical reading of the manuscript, as well as Sam Bradford for assistance with experiments involving flow cytometry. We alo thank Cliff Pereira for help with statistical analysis. We also acknowledge the services provided by the Cell and Tissue Analysis Facilities and Services Core of the Environmental Health Sciences Center, Oregon State University, NIEHS Grant number P30ES000210. This work was supported by the National Institute of Environmental Health Sciences, National Institutes of Health [Grant numbers 5R01ES016651 and T32ES007060].
References
Alam, M. S., Maekawa, Y., Kitamura, A., Tanigaki, K., Yoshimoto, T., Kishihara, K., and Yasutomo, K. (2010). Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U.S.A. 107, 5943–5948.
Apetoh, L., Quintana, F., Pot, C., Joller, N., Xiao, S., Kumar, D., Burns, E., Sherr, D., Weiner, H., and Kuchroo, V. (2010). The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 11, 854–861.
Benson, J. M., and Shepherd, D. M. (2011). Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease. Toxicol. Sci. 120, 68–78.
Frericks, M., Burgoon, L. D., Zacharewski, T. R., and Esser, C. (2008). Promoter analysis of TCDD-inducible genes in a thymic epithelial cell line indicates the potential for cell-specific transcription factor crosstalk in the AhR response. Toxicol. Appl. Pharmacol. 232, 268–279.
Fujimaki, H., Nohara, K., Kobayashi, T., Suzuki, K., Eguchi-Kasai, K., Tsukumo, S., Kijima, M., and Tohyama, C. (2002). Effect of a single oral dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin on immune function in male NC/Nga mice. Toxicol. Sci. 66, 117–124.
Funatake, C. J., Marshall, N. B., Steppan, L. B., Mourich, D. V., and Kerkvliet, N. I. (2005). Cutting Edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+CD25+ cells with characteristics of regulatory T Cells. J. Immunol. 175, 4184–4188.
Furumatsu, K., Nishiumi, S., Kawano, Y., Ooi, M., Yoshie, T., Shiomi, Y., Kutsumi, H., Ashida, H., Fujii-Kuriyama, Y., Azuma, T., and Yoshida, M. (2011). “A role of the aryl hydrocarbon receptor in attenuation of colitis,” in Digestive Diseases and Sciences, Vol. 56 (Dordrecht: Springer), 2532–2544.
Gandhi, R., Kumar, D., Burns, E. J., Nadeau, M., Dake, B., Laroni, A., Kozoriz, D., Weiner, H. L., and Quintana, F. J. (2010). Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3+ regulatory T cells. Nat. Immunol. 11, 846–853.
Hermann-Kleiter, N., and Baier, G. (2010). NFAT pulls the strings during CD4+ T helper cell effector functions. Blood 115, 2989–2997.
Ho, P. P., and Steinman, L. (2008). The aryl hydrocarbon receptor: a regulator of Th17 and Treg cell development in disease. Cell Res. 18, 605–608.
Kerkvliet, N. I. (2009). AHR-mediated immunomodulation: the role of altered gene transcription. Biochem. Pharmacol. 77, 746–760.
Kerkvliet, N. I., Baecher-Steppan, L., Shepherd, D. M., Oughton, J. A., Vorderstrasse, B. A., and DeKrey, G. K. (1996). Inhibition of TC-1 cytokine production, effector cytotoxic T lymphocyte development and alloantibody production by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Immunol. 157, 2310–2319.
Kerkvliet, N. I., Shepherd, D. M., and Baecher-Steppan, L. (2002). T lymphocytes are direct, aryl hydrocarbon receptor (AhR)-dependent targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): AhR expression in both CD4+ and CD8+ T cells is necessary for full suppression of a cytotoxic T lymphocyte response by TCDD. Toxicol. Appl. Pharmacol. 185, 146–152.
Kerkvliet, N. I., Steppan, L. B., Vorachek, W., Oda, S., Farrer, D., Wong, C. P., Pham, D., and Mourich, D. V. (2009). Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy 1, 539–547.
Kimura, A., Naka, T., Nohara, K., Fujii-Kuriyama, Y., and Kishimoto, T. (2008). Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. U.S.A. 105, 9721–9726.
Liu, Y., Li, P., Lu, J., Xiong, W., Oger, J., Tetzlaff, W., and Cynader, M. (2008). Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J. Immunol. 181, 1887–1897.
Luebke, R. W., Copeland, C. B., Daniels, M., Lambert, A. L., and Gilmour, M. I. (2001). Suppression of allergic immune responses to house dust mite (HDM) in rats exposed to 2,3,7,8-TCDD. Toxicol. Sci. 62, 71–79.
Marshall, N. B., Vorachek, W. R., Steppan, L. B., Mourich, D. V., and Kerkvliet, N. I. (2008). Functional characterization and gene expression analysis of CD4+ CD25+ regulatory T cells generated in mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Immunol. 181, 2382–2391.
Mezrich, J. D., Fechner, J. H., Zhang, X., Johnson, B. P., Burlingham, W. J., and Bradfield, C. A. (2010). An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T Cells. J. Immunol. 185, 3190–3198.
Nohara, K., Fujimaki, H., Tsukumo, S. I., Inouye, K., Sone, H., and Tohyama, C. (2002). Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on T cell-derived cytokine production in ovalbumin (OVA)-immunized C57Bl/6 mice. Toxicology 172, 49–58.
Quintana, F. J., Basso, A. S., Iglesias, A. H., Korn, T., Farez, M. F., Bettelli, E., Caccamo, M., Oukka, M., and Weiner, H. L. (2008). Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71.
Ramirez, J.-M., Brembilla, N. C., Sorg, O., Chicheportiche, R., Matthes, T., Dayer, J.-M., Saurat, J.-H., Roosnek, E., and Chizzolini, C. (2010). Activation of the aryl hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells. Eur. J. Immunol. 40, 2450–2459.
Rutz, S., Noubade, R., Eidenschenk, C., Ota, N., Zeng, W., Zheng, Y., Hackney, J., Ding, J., Singh, H., and Ouyang, W. (2011). Transcription factor c-Maf mediates the TGF-[beta]-dependent suppression of IL-22 production in TH17 cells. Nat. Immunol. 12, 1238–1245.
Schaldach, C. M., Riby, J., and Bjeldanes, L. F. (1999). Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry 38, 7594–7600.
Schulz, V. J., Smit, J. J., Willemsen, K. J., Fiechter, D., Hassing, I., Bleumink, R., Boon, L., van den Berg, M., van Duursen, M. B. M., and Pieters, R. H. H. (2011). Activation of the aryl hydrocarbon receptor suppresses sensitization in a mouse peanut allergy model. Toxicol. Sci. 123, 491–500.
Sekine, H., Mimura, J., Oshima, M., Okawa, H., Kanno, J., Igarashi, K., Gonzalez, F. J., Ikuta, T., Kawajiri, K., and Fujii-Kuriyama, Y. (2009). Hypersensitivity of aryl hydrocarbon receptor-deficient mice to Lipopolysaccharide-induced septic shock. Mol. Cell. Biol. 29, 6391–6400.
Shepherd, D. M., Dearstyne, E. A., and Kerkvliet, N. I. (2000). The effects of TCDD on the activation of ovalbumin (OVA)-specific DO11.10 transgenic CD4(+) T cells in adoptively transferred mice. Toxicol. Sci. 56, 340–350.
Sinal, C. J., and Bend, J. R. (1997). Aryl hydrocarbon receptor-dependent induction of cyp1a1 by bilirubin in mouse hepatoma hepa 1c1c7 cells. Mol. Pharmacol. 52, 590–599.
Singh, N. P., Singh, U. P., Singh, B., Price, R. L., Nagarkatti, M., and Nagarkatti, P. S. (2011). Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of Foxp3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE 6, e23522. doi:10.1371/journal.pone.0023522
Stockinger, B. M. V. (2009). Beyond toxicity: aryl hydrocarbon receptor-mediated functions in the immune system. J. Biol. 8, 61.
Sun, Y. V., Boverhof, D. R., Burgoon, L. D., Fielden, M. R., and Zacharewski, T. R. (2004). Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res. 32, 4512–4523.
Takamura, T., Harama, D., Matsuoka, S., Shimokawa, N., Nakamura, Y., Okumura, K., Ogawa, H., Kitamura, M., and Nakao, A. (2010). Activation of the aryl hydrocarbon pathway may ameliorate dextran sodium sulfate-induced colitis in mice. Immunol. Cell Biol. 88, 685–689.
Thatcher, T. H., Maggirwar, S. B., Baglole, C. J., Lakatos, H. F., Gasiewicz, T. A., Phipps, R. P., and Sime, P. J. (2007). Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-[kappa]B component RelB. Am. J. Pathol. 170, 855–864.
Trifari, S., Kaplan, C. D., Tran, E. H., Crellin, N. K., and Spits, H. (2009). Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from TH-17, TH1 and TH2 cells. Nat. Immunol. 10, 864–871.
Veldhoen, M., Hirota, K., Christensen, J., O’Garra, A., and Stockinger, B. (2009). Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 206, 43–49.
Veldhoen, M., Hirota, K., Westendorf, A. M., Buer, J., Dumoutier, L., Renauld, J.-C., and Stockinger, B. (2008). The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109.
Vorderstrasse, B. A., Steppan, L. B., Silverstone, A. E., and Kerkvliet, N. I. (2001). Aryl hydrocarbon receptor-deficient mice generate normal immune responses to model antigens and are resistant to TCDD-induced immune suppression. Toxicol. Appl. Pharmacol. 171, 157–164.
Warren, T. K., Mitchell, K. A., and Lawrence, B. P. (2000). Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) suppresses the humoral and cell-mediated immune responses to Influenza A virus without affecting cytolytic activity in the lung. Toxicol. Sci. 56, 114–123.
Zenewicz, L. A., and Flavell, R. A. (2011). Recent advances in IL-22 biology. Int. Immunol. 23, 159–163.
Zhang, L., Ma, J., Takeuchi, M., Usui, Y., Hattori, T., Okunuki, Y., Yamakawa, N., Kezuka, T., Kuroda, M., and Goto, H. (2010). Suppression of experimental autoimmune uveoretinitis by inducing differentiation of regulatory T Cells via activation of aryl hydrocarbon receptor. Invest. Ophthalmol. Vis. Sci. 51, 2109–2117.
Zhou, L., Chong, M. M. W., and Littman, D. R. (2009). Plasticity of CD4+ T cell lineage differentiation. Immunity 30, 646–655.
Appendix
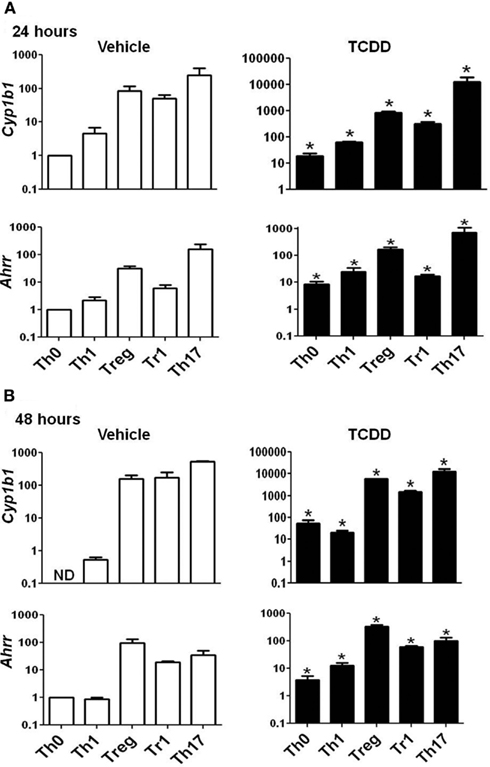
Figure A1. Expression of Cyp1b1 and Ahrr in AhR+/+ CD4+ T-cells treated with vehicle or TCDD. Data are presented as fold change calculated relative to Th0 vehicle at (A) 24 or (B) 48 h. Expression of Cyp1b1 at 48 h in Th0 vehicle was greater than 40 cycles; a value of 40 was used to calculate fold changes. Error bars represent the mean ± SEM; n = 3−4 biological replicates. *p ≤ 0.05 relative to the vehicle control. ND = not detected.
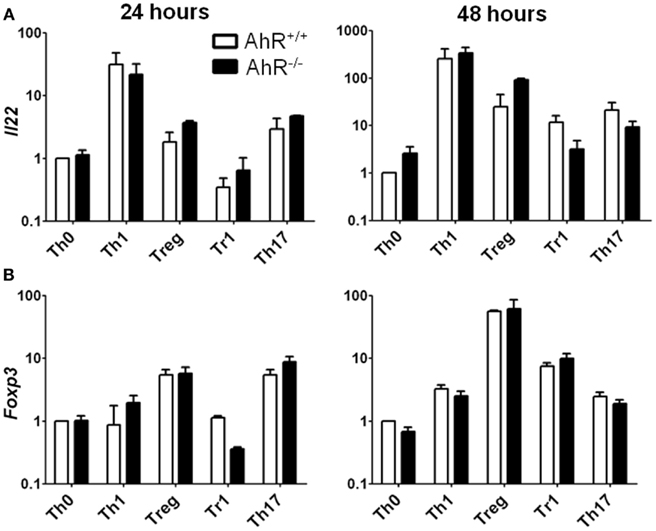
Figure A2. Expression of Il22 and Foxp3 in AhR+/+ and AhR−/− CD4+ T-cells cultured under different polarizing conditions. Data are presented as fold change calculated relative to AhR+/+ T-cells cultured under vehicle-treated Th0 conditions. Expression of (A) Il22 and (B) Foxp3 in AhR+/+ and AhR−/− cells at 24 and 48 h. Error bars represent the mean ± SEM; n = 3 biological replicates except for 24 and 48 h AhR−/− Treg conditions, and 24 h Th17 polarizing conditions, where n = 2. *p ≤ 0.05.
Keywords: aryl hydrocarbon receptor, 2,3,7,8-tetrachlorodibenzo-p-dioxin, CD4+ T-cell differentiation, gene expression, IL-22
Citation: Rohlman D, Pham D, Yu Z, Steppan LB and Kerkvliet NI (2012) Aryl hydrocarbon receptor-mediated perturbations in gene expression during early stages of CD4+ T-cell differentiation. Front. Immun. 3:223. doi: 10.3389/fimmu.2012.00223
Received: 03 May 2012; Accepted: 10 July 2012;
Published online: 06 August 2012.
Edited by:
Barbara Fazekas De St. Groth, Centenary Institute of Cancer Medicine and Cell Biology, AustraliaReviewed by:
António Castro, University of Minho, PortugalKoji Yasutomo, University of Tokushima, Japan
Copyright: © 2012 Rohlman, Pham, Yu, Steppan and Kerkvliet. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Nancy I. Kerkvliet, Environmental and Molecular Toxicology, Oregon State University, 1007 Agricultural and Life Sciences Building, Corvallis, OR 97331, USA. e-mail:bmFuY3kua2Vya3ZsaWV0QG9yZWdvbnN0YXRlLmVkdQ==
†Present address: Duy Pham, Department of Microbiology and Immunology, Indiana University School of Medicine, Indianapolis, IN, USA