 Nicolas Kluger1
Nicolas Kluger1 Annamari Ranki1 and Kai Krohn2*
Annamari Ranki1 and Kai Krohn2*- 1 Department of Dermatology, Allergology and Venereology, Institute of Clinical Medicine, Skin and Allergy Hospital, Helsinki University Central Hospital, University of Helsinki, Helsinki, Finland
- 2 Clinical Research Institute HUCH Ltd, Helsinki, Finland
In APECED, the key abnormality is in the T cell defect that may lead to tissue destruction chiefly in endocrine organs. Besides, APECED is characterized by high-titer antibodies against a wide variety of cytokines that could partly be responsible for the clinical symptoms during APECED, mainly chronic mucocutaneous candidiasis, and linked to antibodies against Th17 cells effector molecules, IL-17 and IL-22. On the other hand, the same antibodies, together with antibodies against type I interferons may prevent the patients from other immunological diseases, such as psoriasis and systemic lupus erythematous. The same effector Th17 cells, present in the lymphocytic infiltrate of target organs of APECED, could be responsible for the tissue destruction. Here again, the antibodies against the corresponding effector molecules, anti-IL-17 and anti-IL-22 could be protective. The occurrence of several effector mechanisms (CD4+ Th17 cell and CD8+ CTL and the effector cytokines IL-17 and IL-22), and simultaneous existence of regulatory mechanisms (CD4+ Treg and antibodies neutralizing the effect of the effector cytokines) may explain the polymorphism of APECED. Almost all the patients develop the characteristic manifestations of the complex, but temporal course and severity of the symptoms vary considerably, even among siblings. The autoantibody profile does not correlate with the clinical picture. One could speculate that a secondary homeostatic balance between the harmful effector mechanisms, and the favorable regulatory mechanisms, finally define both the extent and severity of the clinical condition in the AIRE defective individuals. The proposed hypothesis that in APECED, in addition to strong tissue destructive mechanisms, a controlling regulatory mechanism does exist, allow us to conclude that APECED could be treated, and even cured, with immunological manipulation.
Introduction
Autoimmune polyendocrinopathy syndrome type 1 (APS-1) or autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy syndrome (APECED; OMIM 240300) is a rare recessively inherited disorder (Perheentupa, 2002; Betterle and Zanchetta, 2003; Perheentupa, 2006; Husebye et al., 2009). It is caused by mutations in the autoimmune regulator (AIRE) gene located on locus 21q22.3 (Bjorses et al., 1996; Nagamine et al., 1997; The Finnish–German APECED Consortium, 1997). APECED displays a worldwide distribution, but specific clusters of high prevalence of the disease are observed among Finns (1:25,000; Ahonen et al., 1990) and Sardinians 1 (1:14,500; Rosatelli et al., 1998; Meloni et al., 2012). It is characterized by the variable association of autoimmune endocrine [hypoparathyroidism (HP), Addison’s disease (AD), hypothyroidism, gonadal insufficiency, insulin-dependent diabetes mellitus, atrophic gastritis, and Biermer’s disease] and non-endocrine disorders (keratitis, malabsorption, vitiligo, and alopecia areata) and a specific predisposition to chronic mucocutaneous candidiasis (CMC). A definite diagnosis of APECED is made upon one of the following criteria: (i) the presence of at least two of three major clinical features: CMC, HP, and AD, or (ii) one disease component if a sibling has already a definite diagnosis, or (iii) disease-causing mutations in both alleles of the AIRE gene. However, APECED being highly variable in its presentation, the classical triad may be complete only after years of evolution and diagnose may be therefore missed. Besides, APECED may appear during adolescence or in the young adult (Husebye et al., 2009). Therefore, criteria for a probable APECED have been defined as follows: (i) presence of one of CMC, HP, AD (before 30 years of age) and at least one of the minor components chronic diarrhea, keratitis, periodic rash with fever, severe constipation, autoimmune hepatitis, vitiligo, alopecia, enamel hypoplasia, (ii) any component and anti-interferon antibodies, or (iii) any component and antibodies against NACHT leucine-rich repeat protein 5 (NALP5), AADC, tryptophan hydroxylase (TPH), or TH (Husebye et al., 2009).
From Circulating Autoimmune Antibodies to AIRE, FOXP3, APECED, and IPEX
Our knowledge of the nature of the condition now called APS-1 or APECED has increased simultaneously with the general development of immunology and autoimmunity. Since the condition was clearly defined in the end of 1950s and early 1960s, the characteristic clinical picture, the immunological abnormalities and the relationship to other autoimmune endocrine diseases were defined in late 1960s and early 1970s. Furthermore, the genetics of APECED, and the fact that the syndrome was caused by a recessive gene defect – as opposed to the HLA-linked genetics seen in the other solitarily occurring endocrine diseases – were characterized in the 1980s and the target antigens in the organs affected by APECED were molecularly defined in 1990s. A landmark stage in the study of APECED was reached in 1997, when the long sought APECED gene was cloned by two independent groups (Nagamine et al., 1997; The Finnish–German APECED Consortium, 1997). Finally, a new phase in APECED research occurred during the first decennium of 2000, when the autoantibodies toward soluble mediators if immune response were characterized (Meager et al., 2006; Kisand et al., 2011).
The notion that several diseases affecting endocrine organs and earlier defined as idiopathic, were in fact caused by an autoimmune response toward self antigens, became apparent when novel immunological methods became available in 1950s and early 1960s (Blizzard et al., 1963). The association of the three conditions, candidiasis, HP, and AD that were later judged to be the hallmarks of APECED was clearly stated by the groups of Blizzard and Maclaren (Blizzard et al., 1963; Brun, 1978; Neufeld et al., 1981). These groups also defined two clearly distinct syndromes with several associated autoimmune diseases: autoimmune polyglandular syndrome type 1 (PGS-1) and polyglandular syndrome type 2 (PGS-2). The nomenclature was later changed to APS-1 and APS-2, and the former further to APECED (Ahonen et al., 1990; Perheentupa, 2002, 2006; Betterle and Zanchetta, 2003).
Pioneering studies in this field were made especially with the use of immunohistochemistry, demonstrating antibodies reacting with gastric parietal cells in chronic gastritis (Walder et al., 1963; Irvine et al., 1965) and intrinsic factor (IF) in pernicious anemia (Schwartz, 1961; Jeffries et al., 1962), with thyroid epithelial cells in various forms of thyroid diseases (Witebsky et al., 1957; Irvine et al., 1962; Doniach and Roitt, 1964), with the beta cells of Langerhans islands in diabetes mellitus (Kaldany, 1979; Bottazzo et al., 1980), and adrenal cortical cells in AD (Blizzard et al., 1962).
Blizzard’s group noticed that the two polyglandular syndromes, APS-1 and APS-2, differed in their HLA haplotypes (Neufeld et al., 1981). Further studies on the HLA haplotypes revealed that the genetic basis of APS-2, but also of the other isolated forms of endocrine autoimmune diseases found in APS-1, were in the HLA haplotype of the patients. In contrast, APS-1 was shown not to be linked to HLA, and studies with large patient material, collected by Perheentupa’s group in Finland, clearly stated that APS-1 was linked to a recessively inherited gene defect (Ahonen et al., 1990; Perheentupa, 2002, 2006). The autoimmune endocrinopathies could thus be grouped on the basis of their genetic background in two distinct categories: those linked to HLA variation and the one, APS-1 caused by a single mutated gene (Table 1). At that stage, however, the responsible gene, the APECED gene, was not yet identified. Once identified, the APECED gene was renamed as AIRE in 1997 (Nagamine et al., 1997; The Finnish–German APECED Consortium, 1997).
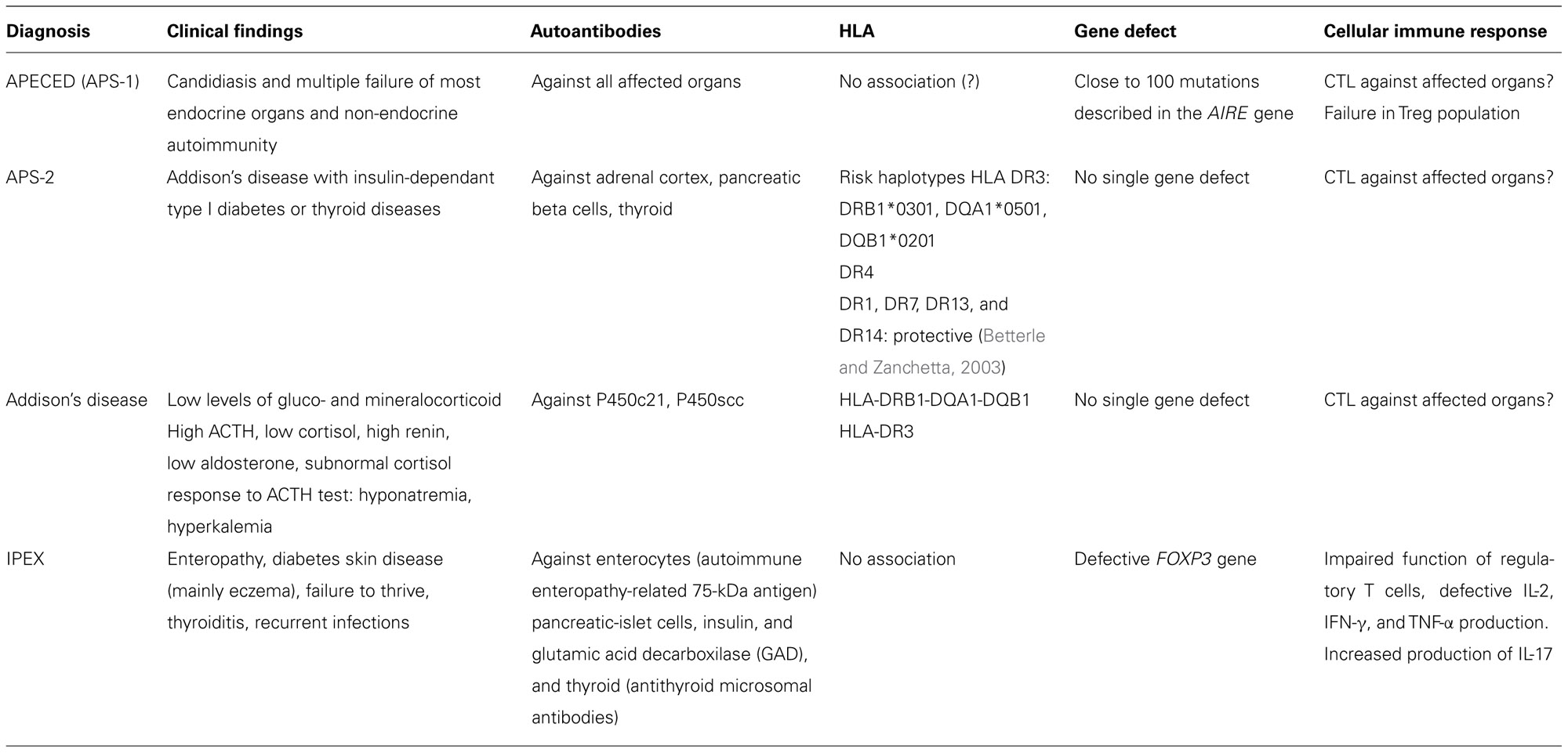
TABLE 1. Key laboratory findings in the different autoimmune endocrine diseases.
Another immunopathy, termed originally as autoimmune enteropathy (AIE) and later identified as immune dysregulation, polyendocrinopathy, enteropathy and X-linked (IPEX), was described in the 1980s and 1990s (Powell et al., 1982). This disorder was later shown to be caused by a defect in a single gene, FOXP3 (Bennett et al., 2000). IPEX and APECED are two examples of immune deficiency diseases disclosing both disturbed tolerance and autoimmune phenomena (Moraes-Vasconcelos et al., 2008). Traditionally, reviews tend to associate both IPEX and APECED because of common features. However, both clinical manifestations and predisposition to infections are rather different when comparing both diseases (Moraes-Vasconcelos et al., 2008).
AIRE Gene, Mutations, and Mechanism of Action
AIRE is expressed in thymus, lymph nodes, and fetal liver, and encodes a protein with two putative zinc fingers and other motifs suggestive of a transcriptional regulator (Nagamine et al., 1997; The Finnish–German APECED Consortium, 1997). The AIRE gene, approximately 13 kb in length, contains 14 exons that encode a polypeptide of 545 amino acids. The AIRE protein functions as a transcription factor (Fierabracci, 2011; Gardner et al., 2009). AIRE is expressed in the thymic medullary epithelial cells (mTECs, Figure 1) and in cells of the monocyte/dendritic cell lineage (Kogawa et al., 2002). mTECs through the expression of MHC class II express a wide array of tissue-restricted antigens (TRAs) derived from different organs in the body. TRAs include self-proteins with patterns of expression restricted to a single or small handful of organs. Thymic expression of TRA serves as an important source of self-antigens to allow the negative selection of autoreactive T cells. Collectively, mTEC and thymic monocyte/dendritic cells play a crucial role in establishing self-tolerance by eliminating autoreactive T cells (negative selection) and/or by producing immunoregulatory FOXP3+ T cells, which prevent CD4+ T cell-mediated organ-specific autoimmune diseases. Collectively, several studies in mouse and man have shown that AIRE regulates thymic expression of several genes of ectopic peripheral proteins including many TRAs. Thus, AIRE dysfunction leads to a decrease in the expression of TRAs in the thymus, and consequently, autoreactive T cell clones escape into the periphery (Derbinski et al., 2005; Moraes-Vasconcelos et al., 2008; Gardner et al., 2009; Fierabracci, 2011)
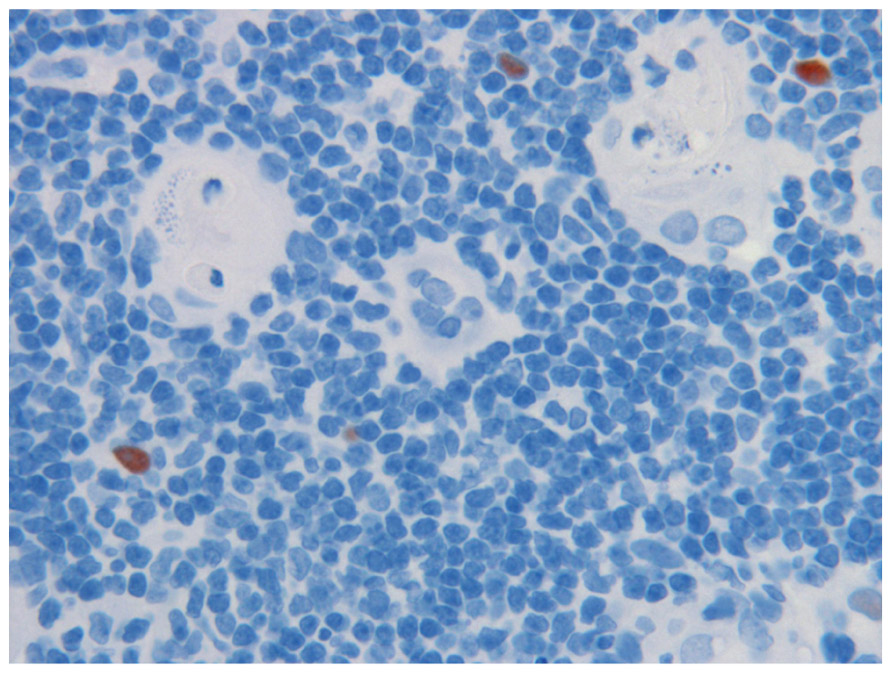
FIGURE 1. Medullary epithelia cells in thymus, expressing the AIRE proteins (reddish brown), in close vicinity of the Hassall’s corpuscles (HC) where auto-reactive T cells are thought to be destroyed. Note cell debris in HC. Magnification 1:40. AIRE was demonstrated with specific monoclonal antibody at 1:2,000 dilution.
The most common AIRE mutation, the “Finnish mutation,” R257X, affects 82% of Finnish APECED alleles (Nagamine et al., 1997; The Finnish–German APECED Consortium, 1997). Interestingly, this mutation occurs also in 70% of the Russian APECED patients studied (Orlova et al., 2010). The same mutation, R257X was also detected in Swiss patients on a different haplotype with closely linked polymorphic markers (Nagamine et al., 1997) and in northern Italian APECED patients. Nonsense mutation R139X was found as the predominant haplotype among Sardinian patients (18/20 independent alleles; Rosatelli et al., 1998). Other hotspots have been identified such as the Y85C missense mutation in an isolated Iranian Jewish community (Zlotogora and Shapiro, 1992; Björses et al., 2000). A 13-bp deletion in exon 8 [1085–1097(del)] is ubiquitous and can be found in Norwegians, but also Anglo–Saxons descendant (Zlotogora and Shapiro, 1992) and south Americans (Moraes-Vasconcelos et al., 2008). Today, over 60 different mutations have been described throughout the coding region of AIRE (Akirav et al., 2011).
Clinical Picture of APECED
The clinical picture of APECED is characterized by sequentially occurring diseases, with great variation among the patients as to the severity and time course of the various conditions. In most cases, the affected individual starts suffering from CMC in early infancy or childhood. In most cases, the next organs to be affected are the parathyroid glands, followed by AD and at puberty, hypogonadism mainly in female teens or young adults. Additional clinical features are less common, and include diabetes type I, hypothyroidism, atrophic gastritis with or without pernicious anemia (Biermer’s disease), cutaneous manifestations (alopecia areata, vitiligo, transient skin rash during fever episodes, non-infectious nail dysplasia), ocular symptoms (keratoconjunctivitis, dry eye, iridocyclitis, cataract, retinal detachment, and optic atrophy; Merenmies and Tarkkanen, 2000), enamel dysplasia, hyposplenism/asplenia (implying vaccination against Streptococcus pneumonia, Haemophilus influenzae, and Hepatitis B as well as antibiotic prophylaxis), autoimmune hepatitis, tubulo-interstitial nephritis, or organized pneumonitis. Involvement of the gastro-intestinal (GI) tract may be responsible for chronic diarrhea, constipation, and malabsorption leading sometimes to malnutrition. GI involvement is difficult to assess as it can be due to numerous various causes that may be associated or follow each other during the life of the patients.
Candidiasis
Chronic mucocutaneous candidiasis infection by Candida albicans is one of the major characteristic of APECED, usually one of first symptoms and most likely the most disabling features of APECED. CMC is naturally not specific for APECED but any child with CMC should be suspected of APECED. According to the Finnish experience, almost all adults with APECED display symptoms of CMC, up to 70% of the patients at the age of 10, up to 94% at age of 20, and 97% at the age of 30 (Perheentupa, 2002, 2006; Betterle and Zanchetta, 2003; Husebye et al., 2009). However, the course and severity vary widely. Oral candidiasis affects the tongue, the buccal mucosa, the gingival, and the pharynx. It ranges in severity from mild form with redness, soreness, angular cheilitis, pseudomembranous lesions, erosions, ulceration and pain to severe chronic inflammation with dysphagia, and development of hyperkeratotic plaques. In the absence of active antimycotic treatment and careful follow-up, chronic oral candidiasis may lead to the development of squamous cell carcinoma with potential lethality by metastatic dissemination. Candida esophagitis has been reported to affect 15–22% of the patients (Perheentupa, 2006; Kisand et al., 2011) with pain while swallowing, retrosternal pain, and dysphagia (Ahonen et al., 1990; Husebye et al., 2009). Chronic esophagitis can lead to local stricture and exceptional esophageal cancer (Rautemaa et al., 2007).
Intestinal candidiasis may cause chronic diarrhea. It should be stressed that esophageal and intestinal candidiasis may occur without any active oral candidiasis. Genital candidiasis affects mainly women with pruritus and vaginal whitish discharge while genital candidiasis seems less frequent in males, possibly underreported due to discrete signs of balanitis. Lastly, Candida may affect the nails with chronic paronychia and onychomycosis (Collins et al., 2006). Fingernails are more commonly affected than toenails and the thumbs are the commonest digit affected. This can be explained as infection occurs during the “thumbsucking” period. Management of candidiasis in APECED patients implies an excellent oral hygiene with a careful and regular dental follow-up. Candidiasis should be treated aggressively with antimicrobial therapy and regular prophylaxis should be given.
Any clinically suspicious, chronic thickening or erosion of the mucosa that does not heal should be biopsied to rule out a potential underlying lesion of squamous cell carcinoma. Any difficulties in swallowing or eating or retrosternal pain should prompt to perform esophagoscopy (Rautemaa et al., 2007).
Hypoparathyoidism
Hypoparathyroidism is one of the first endocrine features of APECED. Symptoms are related to hypocalcemia, muscle cramps, paresthesia, clumsiness, seizures, and diarrhea. The diagnosis is simply based on blood calcium, phosphorus, and parathormone (PTH) levels: hypocalcemia, hyperphosphatemia, inadapted normal/low PTH without any kidney failure. It is considered that APECED should be systematically considered in cases of primary HP (Husebye et al., 2009). Antibodies against NALP5 as well as against the calcium-sensing receptor of parathyroid epithelial cell have been identified in APECED patients (Gavalas et al., 2007; Kemp et al., 2009, 2010). Patients who are free of HP need an annual monitoring of blood calcium and phosphorus levels. Management of HP relies on daily oral supplementation of vitamin D derivatives and calcium.
Gastritis and Pernicious Anemia
Chronic gastritis, with or without concomitant pernicious anemia belongs to the APECED complex but is found only in a fraction of cases. In non-APECED population, two types of chronic gastritis occur, divided by Strickland into type A and B gastritis. Type A gastritis was known to be caused by autoimmunity while the B gastritis was suspected to be the results of environmental factors. In early 1980s, it was shown by Warren and Marshall (1984) that the major environmental factor was in fact a chronic infection with Helicobacterium pylori.
The type A chronic gastritis, with and without pernicious anemia that occur in non-APECED individuals, is clearly linked to certain HLA risk haplotypes, in analogy to isolated AD. In APECED patients, the chronic gastritis differs from the above also in time of occurrence and the speed of the progression. In non-APECED patients, the time needed for progression from the early stage of gastritis, the superficial form to diffuse gastritis, to atrophic gastric and to full gastric atrophy is a slow process, taking up several decennia. Also, the process usually starts in the adult life. In contrast, an APECED-associated gastritic process is much faster and can start in the first decennium of life. Thus, one of the authors of this review was able to follow such a gastric process in two 8-year-old girls with sequential gastric biopsies and could see how, within the time period of only 2 months, the superficial process lead to complete gastric atrophy of the fundus and corpus (K. Krohn, personal experience).
The target molecule for the parietal cell antibodies were shown to be the sodium-potassium channel molecule of the parietal cells on corpus and fundal part of the stomach (Karlsson et al., 1988). In antral gastritis, the antigen are the gastrin-producing cells (Uibo and Krohn, 1984).
Pernicious anemia is the end stage of the gastric immunological destruction, caused partly by the lack of IF, that in addition to the hydrochloric acid is the main product of parietal cells, but also by the autoantibodies recognizing this vitamin B12-binding protein. There are two types of antibodies to IF: one blocking vitamin B12 binding to IF and another type, binding to the IF molecule without interfering with vitamin B12 binding (Toh et al., 1997). Both antibody types prevent the binding of IF to its receptor on the ileal mucosa and subsequent translocation of the vitamin B12 from ileum to circulation.
Addison’s Disease
Adrenocortical failure or AD, described by Thomas Addison in the ninetieth century, is considered one of the three hallmarks of APECED, but it occurs also as a solitary disease, or as part of the APS-2 complex. Today, in western word, most cases of AD are caused by autoimmunity, but adrenal cortical destruction and subsequent cortical failure can be caused by several other factors, notably by secondary tuberculosis or other chronic infections. In retrospect, the cases described by Thomas Addison were most likely caused by tuberculosis.
The clinical signs and symptoms of AD are mostly similar in APECED and in solitary AD as well as in APS-2 complex. These include decreased levels of gluco- and mineralocorticoids and elevated ACTH concentrations. The most severe consequence of AD is the life-threatening Addisonian crisis, characterized by general fatigue, dizziness, diarrhea, and death, if the patient is not quickly substituted with corticosteroids, mainly hydrocortisol.
Autoantibodies to adrenal cortex are the characteristic immunological feature in AD, be it part of APECED or APS-2 or the solitary form. These antibodies can be easily demonstrated by immunofluorescence. However, in APECED, but not in the other forms of AD, the autoantibodies are precipitating, and this phenomenon can be demonstrated by Ouchterlony’s immunodiffusion (Andrada et al., 1968; Krohn et al., 1974; Heinonen et al., 1976). In immunodiffusion with APECED serum against adrenal homogenate three precipitating lines were observed, and one of these were shown to represent a mitochondrial antigen while the two others were microsomal.
The nature of the adrenal cortical autoantigens were revealed in early 1990s and shown to be the three main steroidogenic enzymes, P450c17, P450c21, and P450scc (Krohn et al., 1974; Winqvist et al., 1993; Uibo et al., 1994a,b). These three enzymes were also shown to be the ones that could be precipitated by immunodiffusion. The autoantibodies against these three steroidogenic enzymes clearly distinguish the three clinical conditions with adrenal failure: antibodies to all three can be found only in APECED, while in solitary AD and in APS-1, only antibodies to P450c21 are seen. Furthermore, in non-autoimmune AD, caused by tumors or chronic infections, such antibodies do not occur.
Gonadal Functions
Autoimmune oophoritis is responsible for an ovarian insufficiency that may be dramatic for female patients as insufficiency starting in teenagers and young adults. Patients may have either a primary amenorrhea with no or arrested puberty. Other patients develop premature menopause. The diagnosis is confirmed by sexual hormones status; elevated plasma levels of follicle stimulating hormone (FSH) and luteinizing hormone (LH) and low estrogen levels. Autoantibodies against side-chain cleavage enzyme have been related to ovarian insufficiency (Soderbergh et al., 2004) and also steroidogenic enzymes antibodies against cytochrome p450 21-hydroxylase (CYP21A2), cytochrome p450 17α-hydroxylase (CYP17), and cytochrome p450 side-chain cleavage enzyme (CYP11A1).
In female patients, hormonal substitution by estrogen needs to be initiated during puberty. It is strongly advised not to delay pregnancy. In case of hypogonadism, embryo donation has been tried with success.
In males, testicular failure is less common and occurs later. The prevalence of hypogonadism in males is three times lower (8–28%) than in females (35–70%; Perheentupa, 2006). It leads to clinical hypogonadism or isolated azoospermia (Husebye et al., 2009). It has been hypothesized that the blood–testis barrier protects the Leydig cells from an autoimmune attack. However, the physiopathogenic link between circulating autoantibodies and hypogonadism is far from being clear. The two steroidogenic enzymes, p450scc and p450c17, are the main antigens in gonadal failure linked to APECED, but other potential antigenic targets have been identified such as testis-expressed protein TSGA10 (Reimand et al., 2008). However, despite autoantibodies directed against TSGA10 in 7% of the APECED patients, no correlation could be found with gonadal failure (Reimand et al., 2008). One should not forget that the origin of gonadal dysfunction may be related to an authentic-specific autoimmune attack but also be related to other hormonal dysfunction such as AD, pituitary insufficiency, dysthyroidism, or diabetes for instance. Besides, Schaller et al. (2008) suggested that lack of AIRE might affect fertility by disrupting scheduled apoptosis of testicular germ cells. In this respect, the recent hypothesis presented by Matsumoto (2011) that the function of AIRE in thymus would not be in the regulation of transcription but rather in the development and differentiation of the medullary epithelial cells is of primary interest
Other Endocrine Disorders
Various other endocrine disorders have been described such diabetes type I mellitus, hypothyroidism, and pituitary failure, the latter diagnosed by a growth hormone deficiency. The diagnosis and management of these conditions does not differ from the standard guidelines for each disorder separately (Perheentupa, 2002; Betterle and Zanchetta, 2003; Husebye et al., 2009).
Other Non-Endocrine Disorders
Enamel hypoplasia affect mainly permanent teeth (Perheentupa, 2006), but also deciduous teeth (Pavlic and Waltimo-Sirén, 2009). Pavlic and Waltimo-Sirén (2009) recently suggested that an inadequate process of enamel formation might affect all ameloblasts in phase. Ameloblasts have an epithelial origin with parenchymal cells of endocrine origin. It is speculated that ameloblasts or secreted protein in the extracellular matrix may be the target of autoantibodies leading to hypoplasia. Thereby, APECED would be the first model of dental hard tissue autoimmune disease (Pavlic and Waltimo-Sirén, 2009).
Ocular manifestations affect 25% of the patients and include mainly keratitis that can lead to blindness. It is assumed that the origin of keratitis is the result of autoimmunity against corneal epithelium (Merenmies and Tarkkanen, 2000; Perheentupa, 2006). However, to our knowledge no specific antibodies have been identified in APECED patients. Only antibodies against OBP1 have been found in the AIRE mouse model against lacrimal glands (DeVoss et al., 2010).
Hyposplenism or asplenia is often diagnosed upon the development of thrombocytosis, circulating Howell–Jolly bodies, abdominal ultrasound imaging or in case of severe S. pneumoniae infection (Pollak et al., 2009). Destruction of the spleen in APECED has been related to an autoimmune attack against the spleen (Perheentupa, 2002, 2006; Betterle and Zanchetta, 2003; Husebye et al., 2009) although the exact mechanism remains obscure. Again, the mechanisms proposed by Schaller et al. (2008) and by Matsumoto (2011) are of interest, as AIRE expression has also been described in lymphoid tissue and skin. A speculative hypothesis to the evolution of splenic atrophy could thus be disturbance of differentiation, due to lack of AIRE expression.
Various types of GI manifestations are common in APECED patients. These include chronic diarrhea that can be related to HP, severe constipation. Intestinal infection by candida and giardia especially, pancreatic insufficiency and autoimmune enteropathy. Several autoreactive circulating antibodies directed toward intestinal components have been described. Ekwall et al. (1998) identified TPH as an intestinal autoantigen in APECED patients. TPH is expressed in serotonin-producing cells in the central nervous system and in the intestine. In their series of 80 patients, they were able to relate “GI symptoms” to the presence of circulating TPH antibodies and also to the total absence of enterochromaffin cells in the mucosa of small bowel. These enteroendocrine cells (EECs) are scattered through the intestinal mucosa, from the gastric body and antrum to the rectum. They play a key role in growth of the gut, blood flow, motility, secretion of pancreatic enzymes, bile, and bicarbonate-rich fluid (Posovszky et al., 2012). TPH antibodies were found in 89% of the APECED patients with GI symptoms and in 34% of those without (Ekwall et al., 1998). Antibodies can precede clinical symptoms (Ekwall et al., 1998). Conversely, TPH autoantibodies are absent in other inflammatory or autoimmune intestinal diseases. Additionally, Sköldberg et al, (2003) identified also autoantibodies against histidine decarboxylase expressed by EEC – like cells in the gastric mucosa. It is noteworthy, that it is not a routine procedure to perform EECs staining on intestinal biopsies in case of diarrhea or malabsorption, as stressed by Ohsie et al. (2009). Besides, several studies showed repeatedly that EECs were lacking in the intestinal mucosa and were related to chronic diarrhea (Padeh et al., 1997; Ward et al., 1999; Oliva-Hemker et al., 2006; Posovszky et al., 2012).
Tubulo-interstitial nephritis, life-threatening autoimmune bronchiolitis and other rare manifestations have also been reported in APECED (Perheentupa, 2002, 2006; Betterle and Zanchetta, 2003; Husebye et al., 2009). The main identified autoantibodies are summarized in Table 2.
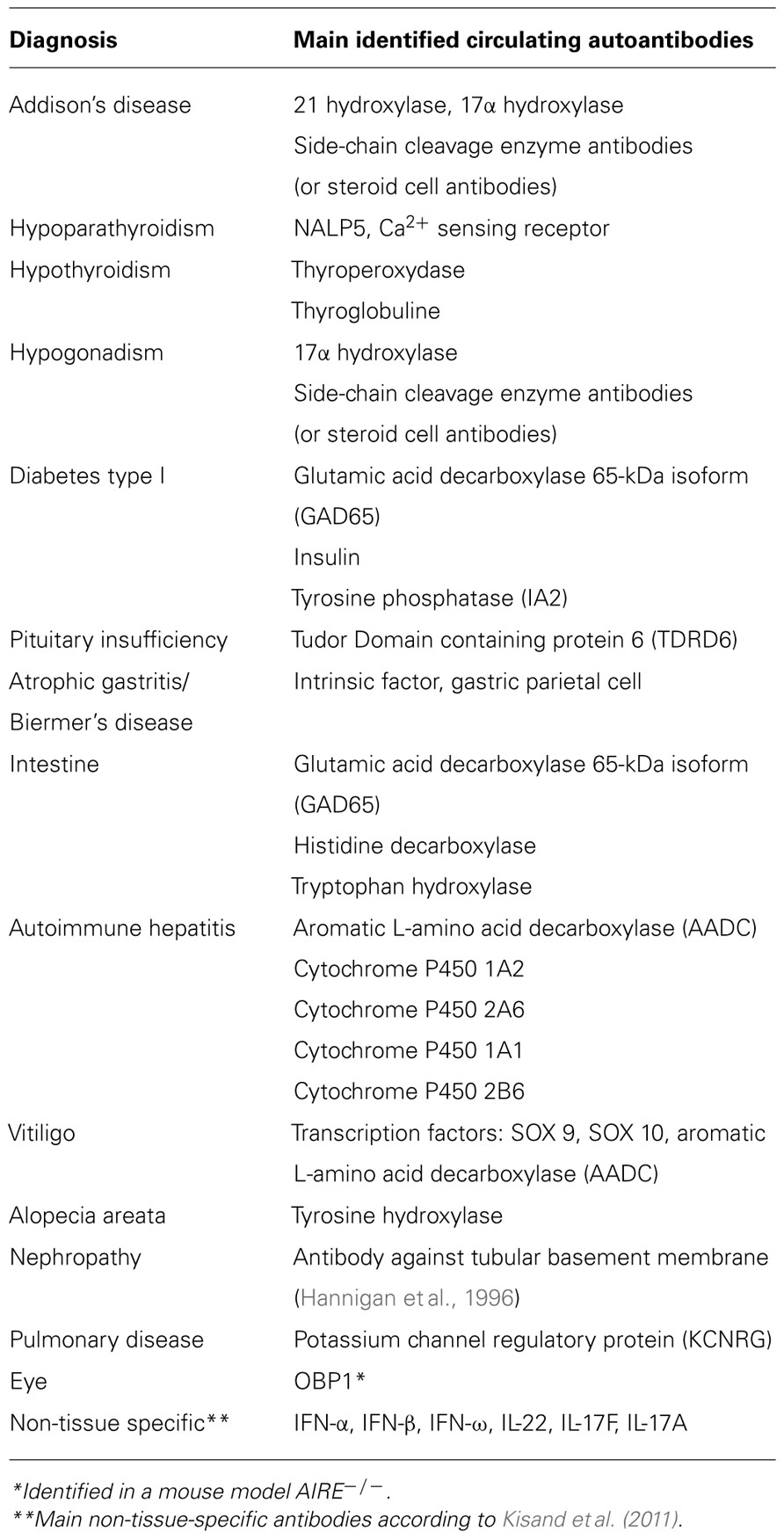
TABLE 2. Main identified target of autoimmune antibodies in APECED patients.
Treatment
Management of APECED relies in education of the patients to know his disease, education of the local physician, and the knowledge that new components of the disease may develop during life. Psychological support is strongly recommended as this disease impairs greatly the quality of life of the patients (Perheentupa, 2006). Except candidiasis treatment that has been explained previously, treatment of APECED relies mostly on hormone replacement therapy according to affected organs (thyroid, parathyroid, pancreas, etc.). In some rare and potentially lethal situations, however, patients may require corticosteroid treatment in association with immunosuppressive therapies. These rare situations include autoimmune hepatitis, especially its fulminant form, which may be lethal and therefore prompt immunosuppressive therapy is needed (Obermayer-Straub et al., 2001). The same is true for interstitial nephritis and bronchiolitis in association to APECED. Immunosuppressive therapies have been also proposed in case of severe intestinal malabsorption with efficacy (Padeh et al., 1997; Ward et al., 1999). Very recently, Rituximab, a chimeric monoclonal antibody targeting B cell lymphocytes expressive CD20 has been successfully used in a young patient with bronchiolitis (Popler et al., 2012). The rationale for Rituximab use in APECED is supported by the presence of B cell infiltrates in the affected organs (Gavanescu et al., 2008).
Autoantibodies Toward Interferons and Cytokines
At the beginning of this millennium, the antibody responses to the main target organs affected in APECED, and the responsible target antigens were fairly well characterized. A new period in APECED studies started along the publication by Meager et al. (2006), describing high-titer antibodies to several type I interferons in practically all APECED patients studied. This anti-interferon response was exceptionally strong, since serum titers up to 1:1,000,000, and clearly exceeding the titers seen against organ-specific antigens, were found.
Furthermore, high-titer antibodies were seen against the two main mediators secreted by Th17 cells, interleukin-17 and interleukin-22 (IL-17 and IL-22). Responses with lower titers were occasionally seen against other interleukins, too. In our own as yet unpublished observations we have detected occasional high-titer responses against several other interleukins and chemokines, as well, but in contrast to the aforementioned responses, these responses are not characteristic to all APECED patients but rather occur occasionally in only a few patients.
The significance of these novel findings are still unclear, but some information concerning the role of IL-17/IL-22 antibodies in the chronic candida infections, characteristic for APECED, has been obtained. Th17 cells secrete IL-17 and IL-22, which are cytokines with potent antifungal properties (Engelhardt and Grimbacher, 2012) and the occurrence of autoantibodies against IL-17/IL-22 were reported to closely correlate to the presence of candida infection (Kisand et al., 2011; Engelhardt and Grimbacher, 2012). However, recent evidence points to a new interaction between AIRE and dectin-1, a pattern-recognition receptor that is important in antifungal innate immunity. Pedroza et al. (2012) recently showed that AIRE participates in the dectin-1 signaling pathway, and thus, missing AIRE activity could contribute to fungal susceptibility through this pathway. Dectin-1 is expressed on phagocytes and was recently shown to induce a non-canonical caspase-8 inflammasome in response to fungal and mycobacterial infection (Gringhuis et al., 2012). The activation of the dectin signaling pathway also leads to expression of IL-17 and 22 and defensins, however. Besides, other mechanisms such Dominant-negative mutations in STAT3, gain of mutation of STAT1, mutations in IL-17F and IL-17R may be alternate causes of CMC (Engelhardt and Grimbacher, 2012).
The significance of the antibody response toward interferons and other cytokines is presently also unclear. One could speculate that some of these antibodies against type I interferons as well as reacting with IL-17 and IL-22 might have a protective function. As pointed out by Waterfield and Anderson (2011), antibodies to type I interferons do not seem to lead to increased susceptibility to viral infections. This resistance might be due to redundancy and it has to be seen whether this anti-interferon response is directed only toward certain members of the interferon family. While Th17 cell response and the release of soluble IL-17 and IL-22 are evidently necessary for the defense against mucocutaneous candida infection, the same cytokines have a role in the development of psoriasis. Similarly, interferons are known to be involved in the pathogenesis of several conditions, and one such chronic ailment is the autoimmune diseases belonging to the systemic lupus erythematosus (SLE) complex. Anti-interferon alpha antibodies are currently being tested as a therapeutic mean against SLE (Merrill et al., 2011). In order to be able to find out if the antibodies against interferons and other cytokines could have a protective role in APECED, large APECED patient cohorts have to be studied in order to find out whether, e.g., psoriasis and SLE are significantly less common in APECED patients than in the general population.
The reason for the antibody response toward soluble immune mediators is still unclear, and we do not yet know what exactly elicits them and thus, only speculative scenarios can be presented. It is conceivable to hypothesize, however, that the tissue destruction preceding the failure of the endocrine organs may have a role. Tissue destruction, be it caused by trauma, viral infection or autoimmune attack, would probably lead not only to the release of potential tissue-specific autoantigens and thus, to autoantibody formation against these proteins, but could also lead to an inflammatory response and production of several mediators of inflammation. One key group of molecules in this respect is the acute phase proteins, notably those belonging to the IL-1 group.
It is generally believed that the destruction of the endocrine organs in APECED is caused by the autoreactive CD8+ cytotoxic T cells, although definitive evidence for this mechanism is still lacking (Betterle and Zanchetta, 2003; Moraes-Vasconcelos et al., 2008). This hypothesis is reinforced by the examination of microscopic examinations of samples, sometimes obtained post-mortem. Indeed, parathyroid, adrenal glands, or ovaries pathology disclosed also atrophy and lymphocytic infiltration that suggest lymphocytic aggression of the organs leading to atrophy and dysfunction (Betterle and Zanchetta, 2003). This is also stressed, indirectly, by the analysis of the AIRE-deficient mouse model, who develop also a lymphocytic infiltration in some inner organs along with atrophy (Ramsey et al., 2002).
However, cell destruction caused by an immune response against the endocrine organ would in fact lead to a similar situation that is thought to happen in viral infections. In fact, several autoimmune diseases, such as diabetes type I or chronic autoimmune liver diseases are thought to be a consequence of preceding viral infection: enterovirus infection in the case of diabetes type I and hepatitis B in the case of chronic active hepatitis. In viral infections, a specific group of intracellular regulatory molecules, TRIMs (tripartite motif-containing proteins), have been shown to have a key role in eliciting an autoimmune or auto inflammatory consequence (Jefferies et al., 2011).
The TRIM protein family is a form of RING domain containing E3 ligases and they exert a variety of biological functions, related to immunity and inflammation (Jefferies et al., 2011). Specifically, of the more than 20 different TRIM proteins, some seem to up-regulate the expression of type I interferons and proinflammatory cytokines, notable interleukin-1beta (IL-1beta). Furthermore, the same mediators of immune response and inflammation are in some cases known to up-regulate the expression of TRIMs. Thus, a vicious circle can theoretically occur and this in turn could lead to autoimmunity. So far, overexpression of TRIMs, or an autoimmune response toward them, has been shown to be linked to autoimmune and autoinflammatory processes in Sjögren’s syndrome or rheumatoid arthritis (Jefferies et al., 2011).
Presently, we have no information how the occurrence of autoantibodies toward the interferons and other mediators of immune response might affect the aforementioned vicious circle, but it is conceivable to speculate that such an antibody response could have an balancing effect. One could thus form a hypothesis, that in APECED, the primary defect outside thymus, where the autoreactive T cells are not destroyed, would be the cell destruction of the endocrine organs by cell-mediated immune response, followed by release of cellular components taken up by professional antigen presenting cells and further stimulating the activation of CD4+ Th-cells and finally resulting in an autoantibody response to these organ-specific antigens. However, simultaneous overexpression of TRIMs and subsequent up-regulation of a variety of soluble mediators of immune response and inflammation, such as interferons and members of the IL-1 family would lead to autoantibody formation also against these cytokines. Lastly, one reason for the break of tolerance to immune mediators, and subsequent production of autoantibodies could be related the fact that AIRE expression seems to occur, in addition to thymic epithelial cells also outside thymus, notably in dendritic cells, that normally express also such mediators (Heath and Carbone, 2009)
The consequences of such cytokine-directed antibody response are still an open question. In case of the Th17 type interleukins (IL-17 and IL-22) there is convincing evidence that such antibodies are linked to the CMC. However, at least in some cases, the antibodies may have a balancing, down-regulating effect on the expression of the corresponding biologically active molecules but also, by regulating the immune response to target organs. Thus, it is possible to presume, that especially the antibodies to type I interferons might have a protective effect, as some chronic immune diseases, such as psoriasis and SLE, are rare or non-existing among APECED patients.
Cell-Mediated Immune Responses
Although it is now a generally accepted view that the consequence of the AIRE defect in APECED will lead to the escape of the potentially autoreactive T cells, there is in fact rather little direct evidence to show that the tissue destruction in the endocrine organs affected in APECED is caused by cytotoxic CD8+ T cells. Furthermore, most studies describing the phenotype of the lymphocytes infiltrating affected organs is not from APECED patients directly, but from patients suffering of solitary lesions that are similar to the ones seen in APECED, such as solitary AD or diabetes. However, the solitary endocrine diseases, such as isolated thyroid disease or AD are remarkably similar in their clinical picture as well as immunological findings as those of APECED. Thus, in solitary AD and in APECED with adrenocortical failure, autoantibodies recognize the p450c21 steroidogenic enzyme. Interestingly, in this disease complex CD8+ T cells that reach against specific T cell epitopes in p450c21 has been demonstrated (Bratland et al., 2009; Rottembourg et al., 2010) Likewise, in thyroid diseases, thyroglobulin and thyroid peroxidase are recognized by the autoantibodies, irrespective if the condition is occurring alone, in association of APS-2 or as part of the APECED complex. The similar synergism in terms of the nature of autoantigens occurs in chronic immunological liver diseases, too.
In chronic aggressive hepatitis the lymphocytic infiltrating cell population has been shown to be of the CD8+ lineage (Si et al., 1984). In a murine model of Graves’ disease, the CD8+ cell population contains also the recently identified CD8+CD122+ T cells that are functionally similar to the CD4+CD25+ regulatory T cells (Ryan et al., 2005). Furthermore, studies in thyroid and other affected organs show that one of the main cell population in the lymphocytic infiltrate are in fact the CD4+ Th17 cells that secrete as effector molecules, the cytokines IL-17 and IL-22. In experimental autoimmune diseases, the balance between the Th17 effector cells and the two regulatory T cells, CD8+CD122+ and CD4+CD25+, seems to regulate both the occurrence and severity of tissue destruction and functional failure.
There could thus be two distinct mechanisms operating in the pathogenesis of autoimmunity in the endocrinopathies: one mediated by soluble effector molecules, such as IL-17 and IL-22 as well as type I interferons, and an other one mediated by effector T cells, which are either of the CD8+ CTL cell or of the Th17 effector cell lineage. To counteract these, again two distinct biological processes would occur: the production of autoantibodies and secondly, the emergency of the regulatory T cells. As to the regulatory T cell response, it is to note that one key immunological failure in APECED, is the dysregulation of the Treg cell maturation (Ryan et al., 2005; Kekäläinen et al., 2007; Saitoh et al., 2007; Laakso et al., 2010, 2011; Wolff et al., 2010).
In normal thymus, Treg maturation follows a preprogrammed scheme, and the immature CD8+CD4+FOXP3+ seems to be prone to apoptosis, whereas the more mature form CD4+CD8-FOXP3+ cells form the active Treg population (Lehtoviita et al., 2009). According to Endharti et al. (2011) the CD8+CXCR3+ Tregs in humans are functionally similar to murine CD8+CD122+ Tregs. Furthermore, in APECED patients the recent thymic emigrant (RTE) pool of Treg cells shift to the activated pool and the RTE reservoir is depleted. Most importantly, in APECED patients these cells express less FOXP3 than in the healthy controls (Laakso et al., 2010). Thus, in APECED the newly formed Treg cells have a developmental defect and their function is therefore impaired. Data concerning the CD8+ regulatory T cells in APECED patients is missing, however.
The finding that the regulatory T cell population in APECED is functionally defective and that the expression of the key molecule for Treg function, the FOXP3 is impaired, is consistent with clinical findings in IPEX syndrome, caused by a defect in the function of the FOXP3 gene. However, it should be noted that the effect of FOXP3 mutations in Treg population also in the IPEX patients is highly variable. Also, in contrast to APECED there seems to be a genotype–phenotype correlation in IPEX, as different mutations are associated in variable clinical picture, that show differences in severity as well in the types of clinical components that are present (Torgerson et al., 2007; d’Hennezel et al., 2009) A consistent finding in IPEX is however the inability of the CD4+CD25 high Tregs to suppress the function of autologous effector T cells (Bacchetta et al., 2006). There are, thus, several differences in the clinical picture of APECED and IPEX, but both conditions show clear immune destruction of at least some endocrine organs. Both conditions also share some similarities in the GI symptoms.
The proposed hypothesis that in APECED both tissue destructive mechanisms and controlling regulatory mechanisms exist raises a question whether APECED could be treated or even cured by immunological manipulations. To find an answer for this question is one of the further challenges for APECED research
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was financially supported by the European Science Foundation (ESF) and the Sigrid Juselius foundation (to Nicolas Kluger). We thank Professors Jaakko Perheentupa, Adrian Hayday, and Pärt Peterson, as well as Drs Kai Kisand, Annalisa Macagno, and Edward Stuart for stimulating discussions.
References
Ahonen, P., Myllarniemi, S., Sipila, I., and Perheentupa, J. (1990). Clinical variation of autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med. 322, 1829–1836.
Akirav, E. M., Ruddle, N. H., and Herold, K. C. (2011). The role of AIRE in human autoimmune disease. Nat. Rev. Endocrinol. 7, 25–33.
Andrada, J. A., Bigazzi, P. L., Andrada, E., Milgrom, F., and Witebsky, E. (1968). Serological investigations on Addison’s disease. JAMA 206, 1535–1541.
Bacchetta, R., Passerini, L., Gambineri, E., Dai, M., Allan, S. E., Perroni, L., Dagna-Bricarelli, F., Sartirana, C., Matthes-Martin, S., Lawitschka, A., Azzari, C., Ziegler, S. F., Levings, M. K., and Roncarolo, M. G. (2006). Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J. Clin. Invest. 116, 1713–1722.
Bennett, C. L., Yoshioka, R., Kiyosawa, H., Barker, D. F., Fain, P. R., Shigeoka, A. O., and Chance, P. F. (2000). X-Linked syndrome of polyendocrinopathy, immune dysfunction, and diarrhea maps to Xp11.23–Xq13.3. Am. J. Hum. Genet. 66, 461–468.
Betterle, C., and Zanchetta, R. (2003). Update on autoimmune polyendocrine syndromes (APS). Acta Biomed. 74, 9–33.
Bjorses, P., Alltonen, J., Vikman, A., Perheentupa, J., Ben-Zion, G., Chiumello, G., Dahl, N., Heidemen, P., Hoorweg-Nijman, J. J., Mathivon, L., Mullis, P. E., Pohl, M., Ritzen, M., Romeo, G., Shapiro, M. S., Smith, C. S., Solyom, J., Zlotogora, J., and Peltonen, L. (1996). Genetic homogeneity of autoimmune polyglandular disease type I. Am. J. Hum. Genet. 59, 879–886.
Björses, P., Halonen, M., Palvimo, J. J., Kolmer, M., Aaltonen, J., Ellonen, P., Perheentupa, J., Ulmanen, I., and Peltonen, L. (2000). Mutations in the AIRE gene: effects on subcellular location and transactivation function of the autoimmune polyendo-crinopathy-candidiasis-ectodermal dystrophy protein. Am. J. Hum. Genet. 66, 378–392.
Blizzard, R. M., Tomasi, T. B., and Christy, N. P. (1963). Autoantibodies against thyroid and adrenal tissue in a patient with mutiple, primary endocrine deficiencies. J. Clin. Endocrinol. Metab. 23, 1179–1180.
Blizzard, R. M., Chandler, R. W., Kyle, M. A., and Hung, W. (1962). Adrenal antibodies in Addison’s disease. Lancet 2, 901–903.
Bottazzo, G. F., Dean, B. M., Gorsuch, A. N., Cudworth, A. G., and Doniach, D. (1980). Complement-fixing islet-cell antibodies in type-I diabetes: possible monitors of active beta-cell damage. Lancet 1, 668–672.
Bratland, E., Skinningsrud, B., Undlien, D. E., Mozes, E., and Husebye, E. S. (2009). T cell responses to steroid cytochrome P450 21-hydroxylase in patients with autoimmune primary adrenal insufficiency. J. Clin. Endocrinol. Metab. 12, 5117–5124.
Brun, J. M. (1978). Auto-immune juvenile polyendocrinopathy (author’s transl). Ann. Endocrinol. (Paris) 39, 463–481.
Collins, S. M., Dominguez, M., Ilmarinen, T., Costigan, C., and Irvine, A. D. (2006). Dermatological manifestations of autoimmune polyendo-crinopathy-candidiasis-ectodermal dystrophy syndrome. Br. J. Dermatol. 154, 1088–1093.
Derbinski, J., Gäbler, J., Brors, B., Tierling, S., Jonnakuty, S., Hergenhahn, M., Peltonen, L., Walter, J., and Kyewski, B. (2005). Promiscuous gene expression in thymic epithelial cells is regulated at multiple levels. J. Exp. Med. 202, 33–45.
DeVoss, J. J., LeClair, N. P., Hou, Y., Grewal, N. K., Johannes, K. P., Lu, W., Yang, T., Meagher, C., Fong, L., Strauss, E. C., and Anderson, M. S. (2010). An autoimmune response to odorant binding protein 1a is associated with dry eye in the Aire-deficient mouse. J. Immunol. 184, 4236–4246.
d’Hennezel, E., Ben-Shoshan, M., Ochs, H. D., Torgerson, T. R., Russell, L. J., Lejtenyi, C., Noya, F. J., Jabado, N., Mazer, B., and Piccirillo, C. A. (2009). FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. N. Engl. J. Med. 361, 1710–1713.
Doniach, D., and Roitt, I. M. (1964). An evaluation of gastric and thyroid auto-immunity in relatation to hematologic auto-immunity. Semin. Hematol. 1, 313–343.
Ekwall, O., Hedstrand, H., Grimelius, L., Haavik, J., Perheentupa, J., Gustafsson, J., Husebye, E., Kämpe, O., and Rorsman, F. (1998). Identification of tryptophan hydroxylase as an intestinal autoantigen. Lancet 352, 279–283.
Endharti, A. T., Okuno, Y., Shi, Z., Misawa, N., Toyokuni, S., Ito, M., Isobe, K., and Suzuki, H. (2011). CD8+CD122+ regulatory T cells (Tregs) and CD4+ Tregs cooperatively prevent and cure CD4+ cell-induced colitis. J. Immunol. 186, 41–52.
Engelhardt, K. R., and Grimbacher, B. (2012). Mendelian traits causing susceptibility to mucocutaneous fungal infections in human subjects. J. Allergy Clin. Immunol. 129, 294–305.
Fierabracci, A. (2011). Recent insights into the role and molecular mechanisms of the autoimmune regulator (AIRE) gene in autoimmunity. Autoimmu. Rev. 10, 137–143.
Gardner, J. M., Fletcher, A. L., Anderson, M. S., and Turley, S. J. (2009). AIRE in the thymus and beyond. Curr. Opin. Immunol. 21, 582–589.
Gavalas, N. G., Kemp, E. H., Krohn, K. J., Brown, E. M., Watson, P. F., and Weetman, A. P. (2007). The calcium-sensing receptor is a target of autoantibodies in patients with autoimmune polyendocrine syndrome type 1. J. Clin. Endocrinol. Metab. 92, 2107–2114.
Gavanescu, I., Benoist, C., and Mathis, D. (2008). B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: a therapeutic approach for APECED patients. Proc. Natl. Acad. Sci. U.S.A. 105, 13009–13014.
Gringhuis, S. I., Kaptein, T. M., Wevers, B. A., Theelen, B., van der Vlist, M., Boekhout, T., and Geijtenbeek, T. B. (2012). Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 13, 246–254.
Hannigan, N. R., Jabs, K., Perez-Atayde, A. R., and Rosen, S. (1996). Autoimmune interstitial nephritis and hepatitis in polyglandular autoimmune syndrome. Pediatr. Nephrol. 10, 511–514.
Heath, W. R., and Carbone, F. R. (2009). Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 10, 1237–1244.
Heinonen, E., Krohn, K., Perheentupa, J., Aro, A., and Pelkonen, R. (1976). Association of precipitating anti-adrenal anti-adrenal antibodies with moniliasis-polyendocrinopathy syndrome. Ann. Clin. Res. 8, 262–265.
Husebye, E. S., Perheentupa, J., Rautemaa, R., and Kämpe, O. (2009). Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J. Intern. Med. 265, 514–529.
Irvine, W. J., Davies, S. H., Teitelbaum, S., Delamore, I. W., and Williams, A. W. (1965). The clinical and pathological significance of gastric parietal cell antibody. Ann. N. Y. Acad. Sci. 124, 657–691.
Irvine, W. J., MacGregor, A. G., and Stuart, A. E. (1962). The prognostic significance of thyroid antibodies in the management of thyrotoxicosis. Lancet 2, 843–847.
Jefferies, C., Wynne, C., and Higgs, R. (2011). Antiviral TRIMs: friend or foe in autoimmune and autoinflammatory disease? Nat. Rev. Immunol. 11, 617–625.
Jeffries, G. H., Hoskins, D. W., and Sleisenger, M. H. (1962). Antibody to intrinsic factor in serum from patients with pernicious anemia. J. Clin. Invest. 41, 1106–1115.
Karlsson, F. A., Burman, P., Lööf, L., and Mårdh, S. (1988). Major parietal cell antigen in autoimmune gastritis with pernicious anemia is the acid-producing H+,K+-adenosine triphosphatase of the stomach. J. Clin. Invest. 81, 475–479.
Kekäläinen, E., Tuovinen, H., Joensuu, J., Gylling, M., Franssila, R., Pöntynen, N., Talvensaari, K., Perheentupa, J., Miettinen, A., and Arstila, T. P. (2007). A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Immunol. 178, 1208–1215.
Kemp, E. H., Gavalas, N. G., Akhtar, S., Krohn, K. J., Pallais, J. C., Brown, E. M., Watson, P. F., and Weetman, A. P. (2010). Mapping of human autoantibody binding sites on the calcium-sensing receptor. J. Bone Miner. Res. 25, 132–140.
Kemp, E. H., Gavalas, N. G., Krohn, K. J., Brown, E. M., Watson, P. F., and Weetman, A. P. (2009). Activating autoantibodies against the calcium-sensing receptor detected in two patients with autoimmune polyendocrine syndrome type 1. J. Clin. Endocrinol. Metab. 94, 4749–4756.
Kisand, K., Lilic, D., Casanova, J. L., Peterson, P., Meager, A., and Willcox, N. (2011). Mucocutaneous candidiasis and autoimmunity against cytokines in APECED and thymoma patients: clinical and pathogenetic implications. Eur. J. Immunol. 41, 1517–1527.
Kogawa, K., Nagafuchi, S., Katsuta, H., Kudoh, J., Tamiya, S., Sakai, Y., Shimizu, N., and Harada, M. (2002). Expression of AIRE gene in peripheral monocyte/dendritic cell lineage. Immunol. Lett. 80, 195–198.
Krohn, K., Perheentupa, J., and Heinonen, E. (1974). Precipitating anti-adrenal antibodies in Addison’s disease. Clin. Immunol. Immunopathol. 3, 59–68.
Laakso, S. M., Laurinolli, T. T., Rossi, L. H., Lehtoviita, A., Sairanen, H., Perheentupa, J., Kekäläinen, E., and Arstila, T. P. (2010). Regulatory T cell defect in APECED patients is associated with loss of naive FOXP3(+) precursors and impaired activated population. J. Autoimmun. 35, 351–357.
Laakso, S. M., Kekäläinen, E., Rossi, L. H., Laurinolli, T. T., Mannerström, H., Heikkilä, N., Lehtoviita, A., Perheentupa, J., Jarva, H., and Arstila, T. P. (2011). IL-7 dysregulation and loss of CD8+ T cell homeostasis in the monogenic human disease autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Immunol. 187, 2023–2030.
Lehtoviita, A., Rossi, L. H., Kekäläinen, E., Sairanen, H., and Arstila, T. P. (2009). The CD4(+)CD8(+) and CD4(+) subsets of FOXP3(+) thymocytes differ in their response to growth factor deprivation or stimulation. Scand. J. Immunol. 70, 377–383.
Matsumoto, M. (2011). Contrasting models for the roles of Aire in the differentiation program of epithelial cells in the thymic medulla. Eur. J. Immunol. 41, 12–17.
Meager, A., Visvalingam, K., Peterson, P., Möll, K., Murumägi, A., Krohn, K., Eskelin, P., Perheentupa, J., Husebye, E., Kadota, Y., and Willcox, N. (2006). Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. 3, e289. doi: 10.1371/journal.pmed.0030289
Meloni, A., Willcox, N., Meager, A., Atzeni, M., Wolff, A. S., Husebye, E. S., Furcas, M., Rosatelli, M. C., Cao, A., and Congia, M. (2012). Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J. Clin. Endocrinol. Metab. 97, 1114–1124.
Merenmies, L., and Tarkkanen, A. (2000). Chronic bilateral keratitis in autoimmune polyendocrinopathy-candidiadis-ectodermal dystrophy (APECED). A long-term follow-up and visual prognosis. Acta Ophthalmol. Scand. 78, 532–535.
Merrill, J. T., Wallace, D. J., Petri, M., Kirou, K. A., Yao, Y., White, W. I., Robbie, G., Levin, R., Berney, S. M., Chindalore, V., Olsen, N., Richman, L,. Le, C., Jallal, B., and White, B.; Lupus Interferon Skin Activity (LISA) Study Investigators. (2011). Safety profile and clinical activity of sifalimumab, a fully human anti-interferon α monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann. Rheum. Dis. 70, 1905–1913.
Moraes-Vasconcelos, D., Costa-Carvalho, B. T., Torgerson, T. R., and Ochs, H. D. (2008). Primary immune deficiency disorders presenting as autoimmune diseases: IPEX and APECED. J. Clin. Immunol. 28, S11–S19.
Nagamine, K., Peterson, P., Scott, H. S., Kudoh, J., Minoshima, S., Heino, M., Krohn, K. J., Lalioti, M. D., Mullis, P. E., Antonarakis, S. E., Kawasaki, K., Asakawa, S., Ito, F., and Shimizu, N. (1997). Positional cloning of the APECED gene. Nat. Genet. 17, 393–398.
Neufeld, M., Maclaren, N. K., and Blizzard, R. M. (1981). Two types of autoimmune Addison’s disease associated with different polyglandular autoimmune (PGA) syndromes. Medicine (Baltimore) 60, 355–362.
Obermayer-Straub, P., Perheentupa, J., Braun, S., Kayser, A., Barut, A., Loges, S., Harms, A., Dalekos, G., Strassburg, C. P., and Manns, M. P. (2001). Hepatic autoantigens in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Gastroenterology 121, 668–677.
Ohsie, S., Gerney, G., Gui, D., Kahana, D., Martín, M. G., and Cortina, G. (2009). A paucity of colonic enteroendocrine and/or enterochromaffin cells characterizes a subset of patients with chronic unexplained diarrhea/malabsorption. Hum. Pathol. 40, 1006–1014.
Oliva-Hemker, M., Berkenblit, G. V., Anhalt, G. J., and Yardley, J. H. (2006). Pernicious anemia and widespread absence of gastrointestinal endocrine cells in a patient with autoimmune polyglandular syndrome type I and malabsorption. J. Clin. Endocrinol. Metab. 91, 2833–2838.
Orlova, E. M., Bukina, A. M., Kuznetsova, E. S., Kareva, M. A., Zakharova, E. U., Peterkova, V. A., and Dedov, I. I. (2010). Autoimmune polyglandular syndrome type 1 in Russian patients: clinical variants and autoimmune regulator mutations. Horm. Res. Paediatr. 73, 449–457.
Padeh, S., Theodor, R., Jonas, A., and Passwell, J. H. (1997). Severe malabsorption in autoimmune polyendo-crinopathy-candidosis-ectodermal dystrophy syndrome successfully treated with immunosuppression. Arch. Dis. Child. 76, 532–534.
Pavlic, A., and Waltimo-Sirén, J. (2009). Clinical and microstructural aberrations of enamel of deciduous and permanent teeth in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Arch. Oral Biol. 54, 658–665.
Pedroza, L. A., Kumar, V., Sanborn, K. B., Mace, E. M., Niinikoski, H., Nadeau, K., Vasconcelos Dde, M., Perez, E., Jyonouchi, S., Jyonouchi, H., Banerjee, P. P., Ruuskanen, O., Condino-Neto, A., and Orange, J. S. (2012). Autoimmune regulator (AIRE) contributes to Dectin-1-induced TNF-α production and complexes with caspase recruitment domain-containing protein 9 (CARD9), spleen tyrosine kinase (Syk), and Dectin-1. J. Allergy Clin. Immunol. 129, 464–472, 472.e1–3.
Perheentupa, J. (2002). APS-I/APECED: the clinical disease and therapy. Endocrinol. Metab. Clin. North Am. 31, 295–320, vi.
Perheentupa, J. (2006). Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Clin. Endocrinol. Metab. 91, 2843–2850.
Pollak, U., Bar-Sever, Z., Hoffer, V., Marcus, N., Scheuerman, O., and Garty, B. Z. (2009). Asplenia and functional hyposplenism in autoimmune polyglandular syndrome type 1. Eur. J. Pediatr. 168, 233–235.
Popler, J., Alimohammadi, M., Kämpe, O., Dalin, F., Dishop, M. K., Barker, J. M., Moriarty-Kelsey, M., Soep, J. B., and Deterding, R. R. (2012). Autoimmune polyendocrine syndrome type Utility of KCNRG autoantibodies as a marker of active pulmonary disease and successful treatment with rituximab. Pediatr. Pulmonol. 47, 84–87.
Posovszky, C., Lahr, G., von Schnurbein, J., Buderus, S., Findeisen, A., Schröder, C., Schütz, C., Schulz, A, Debatin, K. M., Wabitsch, M., and Barth, T. F. (2012). Loss of enteroendocrine cells in autoimmune-poly-endocrine-candidiasis-ectodermal-dystrophy (APECED) syndrome with gastrointestinal dysfunction. J. Clin. Endocrinol. Metab. 97, E292–E300.
Powell, B. R., Buist, N. R., and Stenzel, P. (1982). A X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J. Pediatr. 100, 731–737.
Ramsey, C., Winqvist, O., Puhakka, L., Halonen, M., Moro, A., Kämpe, O., Eskelin, P., Pelto-Huikko, M., and Peltonen, L. (2002). Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum. Mol. Genet. 11, 397–409.
Rautemaa, R., Hietanen, J., Niissalo, S., Pirinen, S., and Perheentupa, J. (2007). Oral and oesophageal squamous cell carcinoma – a complication or component of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED, APS-I). Oral Oncol. 43, 607–613.
Reimand, K., Perheentupa, J., Link, M., Krohn, K., Peterson, P., and Uibo, R. (2008). Testis-expressed protein TSGA10 an auto-antigen in autoimmune polyendocrine syndrome type I. Int. Immunol. 20, 39–44.
Rosatelli, M. C., Meloni, A., Meloni, A., Devoto, M., Cao, A., Scott, H. S., Peterson, P., Heino, M., Krohn, K. J., Nagamine, K., Kudoh, J., Shimizu, N., and Antonarakis, S. E. (1998). A common mutation in Sardinian autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. Hum. Genet. 103, 428–434.
Rottembourg, D., Deal, C., Lambert, M., Mallone, R., Carel, J. C., Lacroix, A., Caillat-Zucman, S., and le Deist, F. (2010). 21-Hydroxylase epitopes are targeted by CD8 T cells in autoimmune Addison’s disease. J. Autoimmun. 35, 309–315.
Ryan, K. R., Lawson, C. A., Lorenzi, A. R., Arkwright, P. D., Isaacs, J. D., and Lilic, D. (2005) CD4+CD25+ T-regulatory cells are decreased in patients with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy. J. Allergy Clin. Immunol. 116, 1158–1159.
Saitoh, O., Abiru, N., Nakahara, M., and Nagayama, Y. (2007). CD8+CD122+ T cells, a newly identified regulatory T subset, negatively regulate Graves’ hyperthyroidism in a murine model. Endocrinology 148, 6040–6046.
Schaller, C. E., Wang, C. L., Beck-Engeser, G., Goss, L., Scott, H. S., Anderson, M. S., and Wabl, M. (2008). Expression of Aire and the early wave of apoptosis in spermatogenesis. J. Immunol. 180, 1338–1343.
Si, L., Whiteside, T. L., Van Thiel, D. H., and Rabin, B. S. (1984). Lymphocyte subpopulations at the site of “piecemeal” necrosis in end stage chronic liver diseases and rejecting liver allografts in cyclosporine-treated patients. Lab. Invest. 50, 341–347.
Sköldberg, F., Portela-Gomes, G. M., Grimelius, L., Nilsson, G., Perheentupa, J., Betterle, C., Husebye, E. S., Gustafsson, J., Rönnblom, A., Rorsman, F., and Kämpe, O. (2003). Histidine decarboxylase, a pyridoxal phosphate-dependent enzyme, is an autoantigen of gastric enterochromaffin-like cells. J. Clin. Endocrinol. Metab. 88, 1445–1452.
Soderbergh, A., Myhre, A. G., Ekwall, O., Gebre-Medhin, G., Hedstrand, H., Landgren, E., Miettinen, A., Eskelin, P., Halonen, M., Tuomi, T., Gustafsson, J., Husebye, E. S., Perheentupa, J., Gylling, M., Manns, M. P., Rorsman, F., Kämpe, O., and Nilsson, T. (2004). Prevalence and clinical associations of 10 defined autoantibodies in autoimmune polyendocrine syndrome type I. J. Clin. Endocrinol. Metab. 89, 557–562.
The Finnish–German APECED Consortium. (1997). An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet. 17, 399–403.
Toh, B. H., van Driel, I. R., and Gleeson, P. A. (1997). Pernicious anemia. N. Engl. J. Med. 337, 1441–1448.
Torgerson, T. R., Linane, A., Moes, N., Anover, S., Mateo, V., Rieux-Laucat, F., Hermine, O., Vijay, S., Gambineri, E., Cerf-Bensussan, N., Fischer, A., Ochs, H. D., Goulet, O., and Ruemmele, F. M. (2007). Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology 132, 1705–1717.
Uibo, R., Aavik, E., Peterson, P., Perheentupa, J., Aranko, S., Pelkonen, R., and Krohn, K. J. (1994a). Autoantibodies to cytochrome P450 enzymes P450scc, P450c17, and P450c21 in autoimmune polyglandular disease types I and II and in isolated Addison’s disease. J. Clin. Endocrinol. Metab. 78, 323–328.
Uibo, R., Perheentupa, J., Ovod, V., and Krohn, K. J. (1994b). Characterization of adrenal autoantigens recognized by sera from patients with autoimmune polyglandular syndrome (APS) type I. J. Autoimmun. 7, 399–411.
Uibo, R. M., and Krohn, K. J. (1984). Demonstration of gastrin cell autoantibodies in antral gastritis with avidin-biotin complex antibody technique. Clin. Exp. Immunol. 58, 341–347.
Walder, A. I., Lunseth, J. B., and Peter, E. T. (1963). Antibodies to the gastric parietal cell. Surg. Forum 14, 327–328.
Ward, L., Paquette, J., Seidman, E., Huot, C., Alvarez, F., Crock, P., Delvin, E., Kämpe, O., and Deal, C. (1999). Severe autoimmune poly-endocrinopathy-candidiasis-ecto-dermal dystrophy in an adolescent girl with a novel AIRE mutation: response to immunosuppressive therapy. J. Clin. Endocrinol. Metab. 84, 844–852.
Warren, J. R., and Marshall, B. J. (1984). Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311–1315.
Waterfield, M., and Anderson, M. S. (2011). Autoimmunity’s collateral damage: Immunodeficiency hints at autoreactivity to cytokines. Nat. Med. 17, 1054–1055.
Winqvist, O., Gustafsson, J., Rorsman, F., Karlsson, F. A., and Kämpe, O. (1993). Two different cytochrome P450 enzymes are the adrenal antigens in autoimmune polyendocrine syndrome type I and Addison’s disease. J. Clin. Invest. 92, 2377–2385.
Witebsky, E., Rose, N. R., Paine, J. R., and Egan, R. W. (1957). Thyroid-specific autoantibodies. Ann. N. Y. Acad. Sci. 69, 669–677.
Wolff, A. S., Oftedal, B. E., Kisand, K., Ersvaer, E., Lima, K., and Husebye, E. S. (2010). Flow cytometry study of blood cell subtypes reflects autoimmune and inflammatory processes in autoimmune polyendocrine syndrome type I. Scand. J. Immunol. 71, 459–467.
Keywords: AIRE, APECED, endocrine disorders, interleukin 17, interleukin 22, IPEX, T regulatory cells
Citation: Kluger N, Ranki A and Krohn K (2012) APECED: is this a model for failure of T cell and B cell tolerance? Front. Immun. 3:232. doi: 10.3389/fimmu.2012.00232
Received: 24 April 2012; Accepted: 15 July 2012;
Published online: 02 August 2012.
Edited by:
Rosa Bacchetta, Fondazione Centro San Raffaele Del Monte Tabor, ItalyReviewed by:
Rosa Bacchetta, Fondazione Centro San Raffaele Del Monte Tabor, ItalyMario Abinun, Newcastle upon Tyne Hospitals NHS Foundation Trust, UK
Copyright: © 2012 Kluger, Ranki and Krohn. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Kai Krohn, Clinical Research Institute HUCH Ltd, Salmentaantie 751, 36450 Salmentaka, Finland. e-mail:a2FpLmtyb2huQG5pYy5maQ==
Abbreviations: AADC, aromatic L-amino acid decarboxylase; AD, Addison’s disease; AE, autoimmune enteropathy; APS-1, autoimmune polyendocrinopathy syndrome type 1; APECED, autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy; CMC, chronic mucocutaneous candidiasis; EECs, enteroendocrine cells; GI, gastro-intestinal; HP, hypoparathyroidism; IPEX, immune dysregulation, polyendocrinopathy, enteropathy and X-linked; IF, intrinsic factor; IL-1, interleukin-1; IL-17, interleukin-17; IL-22, interleukin-22; mTECs, thymic medullary epithelial cells; NALP-5, NACHT leucine-rich-repeat protein 5; PE, promiscuous expression; PTH, parathormone; SLE, systemic lupus erythematosus; TH, tyrosine hydroxylase; TPH, tryptophan hydroxylase; TRIMs, tripartite motif-containing proteins; TSA, tissue-specific antigens.