

- Transplantation Research Immunology Group, Nuffield Department of Surgical Sciences, Level 6, John Radcliffe Hospital, University of Oxford, Oxford, UK
Manipulation of the immune system to prevent the development of a specific immune response is an ideal strategy to improve outcomes after transplantation. A number of experimental techniques exploiting central and peripheral tolerance mechanisms have demonstrated success, leading to the first early phase clinical trials for tolerance induction. The first major strategy centers on the facilitation of donor-cell mixed chimerism in the transplant recipient with the use of bone marrow or hematopoietic stem cell transplantation. The second strategy, utilizing peripheral regulatory mechanisms, focuses on cellular therapy with regulatory T cells. This review examines the key studies and novel research directions in the field of immunological tolerance.
Introduction
Strategies to prevent the development of a specific immune response are invaluable in the quest to achieve improved outcomes after solid organ transplantation (SOT), bone marrow and hematopoietic stem cell transplantation (BMT; HSCT), as well as for the treatment of autoimmune diseases. Specific immune unresponsiveness is the hallmark of clinical tolerance, which in turn may be defined as the long-term survival of an allograft with normal function and no evidence for rejection, in the absence of immunosuppressive drug therapy. The quest for tolerance began with the landmark paper by Billingham et al. (1953), in which tolerance was induced to a mouse skin allograft by injection of a recipient mouse with donor-derived F1 cells as a neonate. Current experimental and early clinical strategies to promote tolerance center on the induction of central tolerance by deletion of donor-reactive leukocytes, most commonly the induction of chimerism, or on peripheral tolerance, most commonly the induction or expansion of regulatory T cells (Treg; Wood et al., 2012).
Chimerism
During T cell development in the thymus, T cells with T cell receptors (TCRs) that are strongly reactive to host MHC molecules are deleted by a process termed negative selection (i.e., central deletion). This physiological process has been harnessed experimentally for the induction of tolerance to foreign antigens. The method used by Medawar to achieve tolerance to skin allografts over 60 years ago was in a fortunate strain combination with only a class I MHC mismatch (Billingham et al., 1953). More recently, similar methods have been used to achieve “central deletion” in fully MHC-mismatched models of transplantation (Cober et al., 1999; Butler et al., 2000; Petit et al., 2004; Mathes et al., 2005). Nevertheless, such strategies are neither consistently successful nor easily translatable to the clinic. Alternatively, hematopoietic complete chimerism through myeloablative therapy and donor-derived bone marrow transplantation results in the repopulation of the host thymus with donor-type dendritic cells (DCs) that delete donor-reactive T cells. Complete chimerism is the replacement of all host hematopoietic cells with donor-derived stem cells such as hematopoietic stem cells (HSCs). Because such donor-derived stem cells have the ability to replicate perpetually, they theoretically continue to provide donor-type DCs indefinitely.
A number of successful clinical cases in SOT have been reported whereby patients with hematological indications for bone marrow ablation who also require renal transplantation have received a BMT and a kidney transplant from the same donor, resulting in long-term donor-specific tolerance (Buhler et al., 2002; Fudaba et al., 2006; Spitzer et al., 2011). Nevertheless, the morbidity and mortality of myeloablative therapy and risk of graft-versus-host disease (GvHD) in most transplant recipients makes this mode of therapy unacceptable to those without a hematological indication for bone marrow ablation. On the other hand, mixed chimerism, where donor cells represent a varying proportion (but not 100%) of the total hematopoietic pool is a more promising area of research (Kawai et al., 2011). Mixed chimerism can be established using non-myeloablative conditioning regimens, therefore maintaining immunocompetence and reducing the risk of GvHD (Figure 1).
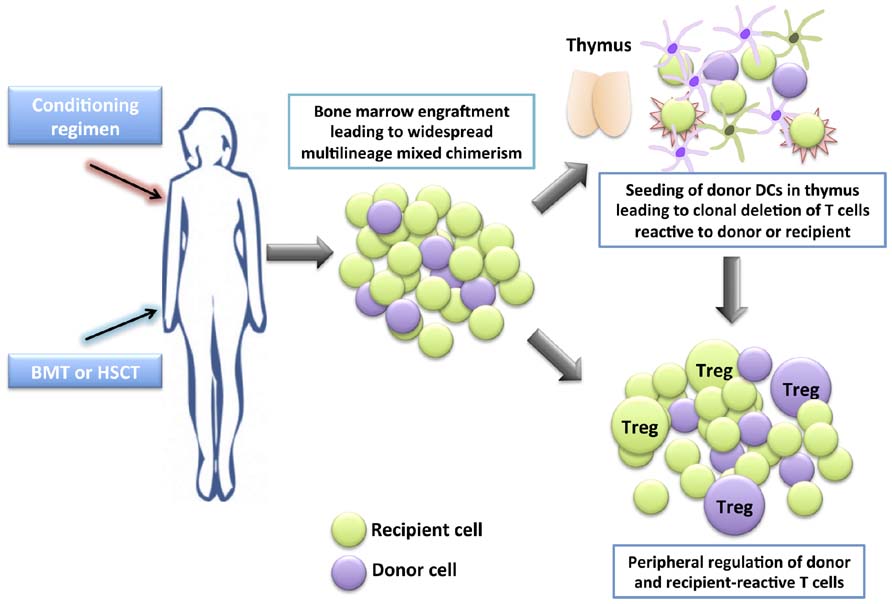
FIGURE 1. Development of tolerance through mixed chimerism. A conditioning regimen is administered which consists of a combination of drugs (occasionally together with irradiation) to allow the engraftment of allogeneic bone marrow (BM) or hematopoietic stem cells (HSCs). A bone marrow transplant (BMT) or hematopoietic stem cell transplant (HSCT) is given the to patient. The donor HSCs seed into the recipient’s BM niches together with the recipient’s HSCs, providing a self-renewing source of donor and recipient hematopoietic cells, leading to widespread multilineage mixed chimerism. Donor dendritic cells seed the thymus, and together with recipient dendritic cells, mediate central clonal deletion of newly developing donor-reactive and recipient-reactive thymocytes. Peripheral regulation also takes place whereby newly developing donor-reactive and recipient-reactive T cells that escape negative selection in the thymus are suppressed in the periphery by regulatory T cells (Treg).
There is evidence for the operation of both central deletional and peripheral regulatory mechanisms in mixed chimerism (Pilat and Wekerle, 2010; Sachs et al., 2011). In models where mixed chimerism is induced following total body irradiation (TBI), the specific depletion of donor cells is associated with the appearance of donor-reactive T cells in the periphery and the loss of tolerance (Khan et al., 1996). This loss of tolerance may be avoided by removal of the thymus before depletion of donor cells, highlighting the importance of the intrathymic chimerism in the maintenance of tolerance. In less intensive conditioning mechanisms where costimulatory blockade is used to facilitate mixed chimerism, intrathymic deletion remains an important mechanism contributing to tolerance and there is no evidence of a role for Treg (Wekerle et al., 2000; Fehr et al., 2008). Nevertheless, regimens that produce less complete deletion of pre-existing donor-reactive T cells may be dependent on peripheral tolerance mechanisms. For example, in a costimulation-based non-myeloablative BMT model, depletion of CD25+ cells at the time of BMT prevents the induction of tolerance (Bigenzahn et al., 2005). CD4+ T cells isolated from these chimeras display regulatory capabilities. In another model, the challenge of mixed chimeras with naïve T cells does not lead to the rejection of skin allografts, suggesting a role for peripheral regulatory mechanisms (Domenig et al., 2005). Moreover, mixed chimerism induction techniques that actively employ peripheral regulation, for example, by the infusion of Treg, may facilitate the development of mixed chimerism and lead to more robust tolerance (Seung et al., 2003; Pilat et al., 2010).
A series of promising clinical trials for SOT utilizing mixed chimerism for the induction of tolerance have been performed. An initial trial enrolled six patients with renal failure consequent to multiple myeloma (Fudaba et al., 2006). Patients received non-myeloablative BMTs and renal transplants from an HLA-identical sibling followed by a donor leukocyte infusion as treatment for both the multiple myeloma and renal failure. Four patients transiently developed mixed chimerism, which was later lost, while the other two patients eventually developed full donor chimerism. Interestingly, all patients successfully accepted their renal transplants long-term (up to >9 years) without any immunosuppression. Following this study, a similar approach was piloted in five patients without a hematological malignancy (Kawai et al., 2008; LoCascio et al., 2010). Patients received an HLA-mismatched haploidentical bone marrow transplant along with a renal transplant from the same donor. All patients developed transient mixed chimerism, but this was lost after day 21. Four patients in the trial currently maintain graft function after weaning from their initial immunosuppression (follow-up 2–5 years post-weaning). However, one kidney graft was lost due to acute antibody-mediated rejection, leading to a modification in the trial protocol to include B cell depletion with rituximab.
In another recent proof-of-concept study, 12 living donor HLA-matched kidney transplant recipients received a donor-cell infusion of 5–16 × 106/kg CD34+ cells mixed with 1–10 × 106/kg CD3+ T cells after conditioning with total lymphoid irradiation and five doses of rabbit antithymocyte globulin (Scandling et al., 2011), as per a protocol previously reported as a case report (Scandling et al., 2008). None of the patients developed GvHD. In 8 of the 12 patients, maintenance immunosuppression was eventually discontinued and patients have since been immunosuppression-free with good graft function and without evidence of acute or chronic rejection (follow-up 12–36 months). The remaining four patients experienced rejection episodes on weaning from immunosuppression and have therefore remained on immunosuppression. The authors now plan to apply the protocol to HLA-mismatched transplant recipients. Given the notable immunosuppression-free success rate and safety profile, it will be interesting to observe the efficacy of the protocol in this situation.
A recent Phase II clinical trial investigated the use of HSCT together with a facilitating cell (FC) infusion to promote the development of chimerism and subsequent tolerance in eight HLA-mismatched living donor renal transplant recipients (Leventhal et al., 2012). FCs were defined as CD8+ bone marrow-derived cells that did not express the TCR and primarily contained a plasmacytoid DC population (Kaufman et al., 1994; Grimes et al., 2004; Fugier-Vivier et al., 2005). In this clinical trial, combined FC and HSCT transplantation initially led to high levels of chimerism in all eight renal allograft recipients. Five of the recipients displayed stable chimerism and donor-specific tolerance and were subsequently weaned off maintenance immunosuppression one year post-transplant. Two patients developed only transient chimerism and were therefore maintained on low-dose tacrolimus monotherapy. One patient, although displaying robust chimerism, developed viral sepsis 2 months post-transplantation and subsequently lost the kidney graft due to renal artery thrombosis. Immunological monitoring of the patients enrolled in the trial showed a significant reduction in circulating CD4+ but not CD8+ cells post-transplantation. The significance of this observation is unclear, however it may be indicative of central deletion of alloreactive CD4+ T cells or peripheral regulation by Treg. Indeed, an increase in the Treg to effector T cell ratio was observed in chimeric recipients but not those that achieved only transient macrochimerism.
While the above approaches have demonstrated some success in living donor transplants, the induction of mixed chimerism in recipients of cadaveric organ transplants may prove more challenging. BMT has only been trialed on a small scale in cadaveric donor transplants, principally in the context of vascularized composite allograft (VCA) transplants. The first face transplant performed in France used a post-transplant donor-derived bone marrow infusion, although it does not appear that this approach accorded any clear benefit in terms of a reduction in episodes of rejection (Hequet et al., 2008). Furthermore, microchimerism was only detectable at a single point 2 months post-operatively and not thereafter (Hequet et al., 2008). Since then, five VCA transplants performed in Pittsburgh have employed the “Pittsburgh Protocol” in which a bone marrow infusion is given within 15 days of VCA transplantation (International Hand and Composite Tissue Allotransplantation Society Congress, Atlanta 2011). Early reports indicate that patients treated in this manner have been maintained successfully on single drug immunosuppression with tacrolimus.
Interestingly, the presence of vascularized bone marrow in many VCA transplants raises the possibility that chimerism may develop by nature of the simultaneous transplantation of HSCs within the VCA. To this end, various rat models of hindlimb transplantation using T cell depleting antibody along with immunosuppression have achieved long-term allograft survival (Siemionow et al., 2002a,b, 2003; Ozer et al., 2003, 2004; Siemionow and Klimczak, 2009), although mixed chimerism is not always readily detectable (Quatra et al., 2006). In these models, the bone marrow component of the VCA transplant is critical to the attainment of mixed chimerism and long-term allograft survival (Siemionow et al., 2005; Siemionow, 2011). Moreover, increased levels of chimerism are detectable with larger sized VCA allografts in rats, indicating the role of the transplant in providing donor cells (Nasir et al., 2008). In a rat model of facial allograft transplantation, mixed macrochimerism has been observed with the use of only cyclosporine monotherapy (Demir et al., 2004; Kulahci et al., 2010). Mixed chimerism has also been achieved using non-depleting CD4+ blockade and depleting CD8+ antibody in conjunction with rapamycin and α-CD154 costimulatory blockade, without a bone marrow transplant, relying on bone marrow in a mouse hindlimb VCA to provide donor cells for chimerism (Li et al., 2008). In general, clinical data have not been particularly encouraging, with no evidence for the development of chimerism in VCA transplantation. This may be due to only small amounts of bone marrow being transferred that have limited functionality in the adult (Granger et al., 2002; Petruzzo et al., 2003). Moreover, there is experimental evidence that the recipient thymus is necessary for peripheral chimerism to develop after transplantation of a bone marrow-containing VCA (Li et al., 2007). In humans the thymus involutes and becomes atrophic after puberty and is therefore less likely to support the development of chimerism. It is important to note that in theory, chimerism is a double-edged sword, whereby the greater the likelihood of chimerism, the greater the anti-host alloresponse and risk of GvHD (Wood, 2003).
Regulatory T Cells
While the methods described above relate to exploiting the natural mechanisms used by the immune system to ensure self-tolerance through central mechanisms, several peripheral regulatory mechanisms also exist as a fail safe mechanism to maintain self-tolerance and to prevent an overshoot of the normal immune response (Issa and Wood, 2010; Wood and Goto, 2012). While most autoreactive cells are deleted centrally in the thymus, some autoreactive T cells escape this process and require peripheral regulation to prevent autoimmunity. CD4+ Treg are central to these mechanisms. Scurfy mice lacking the Treg-specific transcription factor forkhead box P3 (foxp3) develop a lymphoproliferative disorder (Brunkow et al., 2001) and humans with mutations in FOXP3 can develop IPEX (immunodysregulation, polyendocrinopathy, and enteropathy, X-linked; Bennett et al., 2001). FOXP3 is closely linked to suppressive activity and its sustained expression is required for the maintenance of regulatory activity (Josefowicz and Rudensky, 2009).
Treg can be divided into thymus-derived naturally occurring CD4+CD25hiFOXP3+ Treg (nTreg or tTreg; Hori et al., 2003) and induced or adaptive CD4+ Treg (iTreg), which upregulate FOXP3 in the periphery under defined conditions of antigen-exposure, for example, in the presence of transforming growth factor β (TGFβ; Kingsley et al., 2002; Karim et al., 2004, 2005). Type 1 Treg (Tr1) cells are a distinct population of peripherally induced Treg that develop in the presence of IL-10 and regulate responses through FOXP3-independent secretion of IL-10 and TGFβ, leading to bystander regulation of effector T cells (Battaglia et al., 2006a). nTreg represent 5–10% of the peripheral CD4+ pool and constitutively express high levels of surface CD25 although this is not a reliable marker due to its upregulation on recently activated T cells. Nevertheless, although CD25 appears on recently activated CD4+ T cells, some of these are true proliferating Treg. For example, during the secondary antigenic response that develops after human tuberculin purified protein derivative is injected into skin, CD4+CD25+ cells proliferate within the skin. Many of these proliferating cells are in fact FOXP3+ and display functional and phenotypic markers of Treg (Vukmanovic-Stejic et al., 2008). It is unclear how much of the peripheral CD4+ population iTreg represent, but given that these cells are induced in specific inflammatory environments it is likely that their number is location and time-dependent.
In transplantation, both direct and indirect allorecognition contribute to the immune response that results in graft destruction. However, with time after transplantation, passenger antigen-presenting cells are lost and organ parenchyma is less able to stimulate the host via the direct pathway. The indirect alloresponse therefore becomes of increasing importance and may be more relevant in chronic rejection (Baker et al., 2001). Interestingly, alloreactive T cells that respond by the indirect pathway are more resistant to inhibition by conventional immunosuppression and are detectable in the peripheral blood of transplant recipients years after transplantation (Sawyer et al., 1993; Vella et al., 1997). The alloreactivity of Treg may therefore be important in determining their ability to promote tolerance. Indeed, Treg that are both directly and indirectly alloreactive are able to prevent both acute and chronic rejection in mice, whereas those that are only directly alloreactive appear to only be able to prevent acute rejection (Joffre et al., 2008; Tsang et al., 2008).
Studies assessing the potential of nTreg, iTreg, and Tr1 cells to promote allograft survival in experimental transplantation have yielded promising results to date. In these studies Treg may be induced in vivo by employing costimulatory blockade or lymphocyte depletion around the time of transplantation, often together with an antigen challenge (Cobbold et al., 1986; Qin et al., 1993; Graca et al., 2000; Kingsley et al., 2007; Francis et al., 2011). Alternatively nTreg may be expanded ex vivo or converted from non-Treg cell types to iTreg in vitro.
Ex Vivo Expansion
Human Treg for cell therapy protocols are produced by isolation of cells from peripheral or umbilical cord blood (UCB) and subsequent ex vivo expansion or direct use in vivo. In order to isolate Treg efficiently and to a high purity, reliable markers of identification are required. Given the non-exclusivity of CD25 and FOXP3 expression, a number of other markers are in use. Of these, CD127 (the IL-7 receptor α-chain), CD49b (the α-chain of the integrin VLA-4 – a4b1), CD45RA, and latency-associated peptide (LAP) are particularly useful. Other Treg markers include CD152 (CTLA-4), GITR, CD69, and CD44 but these are less useful as they may also be expressed in almost identical patterns on non-regulatory activated T cells.
The use of the low expression of CD127 for the isolation of Treg was described approximately 5 years ago (Liu et al., 2006; Seddiki et al., 2006; Putnam et al., 2009) and is particularly helpful as it defines a highly suppressive population of Treg. In a humanized mouse model of vessel allograft rejection, human ex vivo-expanded CD25hiCD4+ or CD127loCD25+CD4+ nTreg were used to modulate immune responses in vivo to reduce neointimal expansion. Treg expressing low levels of CD127 were found to be five times more potent than those expressing only CD25. The same population of CD127loTreg have been shown to be active in the prevention of human skin graft rejection in a similar humanized mouse model (Issa et al., 2010). The absence of CD49b is another helpful marker for Treg identification, as together with CD127 it allows for Treg isolation by negative selection alone (Kleinewietfeld et al., 2009).
CD45RA allows cells to be divided into CD25+CD45RA+FOXP3lo (resting naïve Treg), CD25hiCD45RA-FOXP3hi (activated Treg), and CD25+CD45RA-FOXP3lo (non-suppressive T cells) populations (Miyara et al., 2009). Resting naïve and activated Treg are both suppressive in vitro, whilst only resting naïve Treg proliferate in vivo and evolve into suppressive CD45RA-Treg. UCB contains a high number of naïve CD45RA+ cells, and is therefore an attractive source of resting naïve Treg (Riley et al., 2009). However, UCB Treg are low in frequency and require either in vitro culture or pooling of multiple blood units. Furthermore, as UCB Treg are allogeneic to both the donor and recipient they are likely to be subject to an alloresponse therefore complicating their in vivo use.
LAP has been shown to define a population of Treg that express high levels of foxp3, secrete immunosuppressive TGFβ, and exhibit enhanced in vivo regulatory activity (Chen et al., 2008). Moreover, LAP itself is functionally suppressive independent of TGFβ (Ali et al., 2008). In Treg expansion cultures, the expression of LAP allows the distinction and selection of activated Treg from activated non-Treg cell types (Tran et al., 2009).
Ex vivo expansion of isolated Treg is largely performed by stimulation with αCD3/αCD28 microbeads in the presence of recombinant human (rh) IL-2 (Sagoo et al., 2008; Trzonkowski et al., 2009). The non-specific TCR stimulation in this system leads to the production of a polyclonally reactive population of Treg. Donor alloantigen-reactive Treg that have been expanded in the presence of donor-derived APC have been shown to be more potent suppressors in vitro and in vivo than polyclonally reactive Treg, and their specific reactivity implies that they are safer for in vivo use (Golshayan et al., 2007; Sagoo et al., 2011). Selection of alloantigen-stimulated Treg from a culture where allogeneic stimulators are used may be possible by enrichment of Treg that co-express the activation markers CD69 and CD71 (Sagoo et al., 2011; J. Hester and K.J. Wood, unpublished data). Other methods for the production of alloantigen-reactive Treg include retroviral vector transduction of Treg with genes that encode for TCRs with known antigen specificities (Jiang et al., 2006).
In Vivo Induction
In vivo approaches are based on increasing the frequency or potency of Treg by exposure to antigen, inducing an expansion of nTreg or converting non-Treg to iTreg (Long and Wood, 2009; Francis et al., 2011). Treg may be generated in vivo by pre-treating mice with a donor alloantigen (in the form of a donor-specific transfusion) along with a non-depleting α-CD4 mAb (Kingsley et al., 2002, 2007). Treg produced in this manner are capable of preventing allograft rejection in vivo (Wood et al., 1991; Bushell et al., 1994,1995; Saitovitch et al., 1995, 1996, 1997; Kingsley et al., 2002, 2007; Bushell et al., 2003; Karim et al., 2005; Warnecke et al., 2007). While this tolerance appears to be antigen-specific in nature, another “boosting” blood transfusion allows tolerance to develop to a third-party allograft (Karim et al., 2005). CD4+CD25+ Treg isolated from these animals may prevent allograft rejection in naïve mice by adoptive transfer (Hara et al., 2001; Kingsley et al., 2002) and are able to prevent skin graft rejection initiated by both CD4+ (Hara et al., 2001; Golshayan et al., 2007) and CD8+ (van Maurik et al., 2002; Jones et al., 2010) T cells. Alternatively, costimulatory blockade or lymphocyte depletion using monoclonal antibodies around the time of transplantation may also promote tolerance induction (Qin et al., 1990; Qin et al., 1993; Graca et al., 2000; Waldmann et al., 2006). Interestingly, even nTreg isolated from naïve animals may prevent rejection, although 10-fold more such Treg are required to attain long-term allograft survival compared to Treg isolated from tolerant mice treated with antigen exposure (Graca et al., 2002). The folate receptor 4 (FR4) allows the identification of these alloantigen-stimulated Treg (Yamaguchi et al., 2007). Alloantigen-stimulated FR4high Treg are significantly more effective at prolonging mouse skin allograft survival compared to FR4intermediate Treg.
Another method for in vivo generation is the injection of IL-2-IL-2 mAb complexes into mice, resulting in an over 10-fold expansion of Treg in vivo. Animals treated by this method are resistant to experimental autoimmune encephalomyelitis (EAE) induction and display tolerance to islet allografts (Webster et al., 2009). Injection of IL-2-IL-2 mAb complexes together with recombinant granulocyte-colony stimulating factor (G-CSF) induces expansion of Treg and myeloid-derived suppressor cells (MDSCs) in vivo, promoting mouse skin allograft survival in MHC Class II-mismatched models (Adeegbe et al., 2010).
There has been a great deal of discussion regarding the functional stability of Treg. Zhou et al. (2009) demonstrated that some Treg lose foxp3 (becoming exfoxp3 cells), developing an activated memory-type phenotype, and are pathogenic in vivo. This loss of foxp3 was linked to a proinflammatory microenvironment in which Treg acquire an effector T cell phenotype, secreting IL-17 and interferon γ (IFNγ; Yang et al., 2008; Ayyoub et al., 2009; Komatsu et al., 2009; Voo et al., 2009; Chadha et al., 2011). Importantly, Treg may not be particularly effective at suppressing IL-17 producing T cells (Heidt et al., 2010; Zhang et al., 2010; Chadha et al., 2011), although data from our own studies in kidney transplant patients treated with the leukocyte depleting monoclonal antibody alemtuzumab (Campath or anti-CD52 antibody) suggest that Treg present in these patients can regulate Th17 cells (Hester et al., 2011). Stability of Treg is important to consider in transplantation as there is evidence that Treg transfer into lymphopaenic mice may result in the loss of foxp3 expression in up to 50% of the adoptively transferred cells (Duarte et al., 2009). This is a particularly important point to consider if Treg cellular therapy is to be employed in patients who are lymphopaenic post-immunosuppressive induction therapy. However, a recent study by Miyao et al. (2012) has elegantly demonstrated that the plasticity of Treg is due to only a minor population of foxp3+ cells. In this study, suppressive foxp3+Treg do not develop effector cell function even in inflammatory or lymphopaenic environments. While such Treg may transiently lose foxp3 expression, on activation foxp3 is re-expressed and suppressive capabilities return. It is therefore only a small minority (2–3%) of the peripheral foxp3+ pool which are originally non-regulatory and which may then lose foxp3 to become pathogenic. Overgrowth of this small population may explain previous data demonstrating the plasticity of Treg. Importantly in this study, it is epigenetic control of Foxp3 that dictates whether foxp3+ cells are true Treg. Demethylation of the Treg cell-specific demethylated region (TSDR) indicates that cells are committed suppressive Treg, regardless of the ongoing expression of foxp3. Identification of the methylation status of the TSDR is therefore a valuable indicator of the purity of cell preparations produced for clinical use.
Regulatory B Cells
There are multiple reports of clinical operational tolerance, or long-term functioning allograft survival in the absence of any immunosuppression. This has most commonly been observed in liver transplantation (Lerut and Sanchez-Fueyo, 2006) but has also been reported in a small number of renal transplant recipients (Orlando et al., 2010). In a study where the immune profile of tolerant renal transplant recipients was analyzed, the most striking feature was a bias towards a differential expression of B cell-related genes and an expansion of peripheral blood B cells in tolerant patients (Newell et al., 2010; Sagoo et al., 2010). This latter observation raises the interesting possibility that regulatory B cells (Bregs) may be playing a role. Bregs express high levels of CD1d, CD21, CD24, and IgM, have an immature or transitional phenotype, and are active through the secretion of suppressive IL-10 (Mauri and Blair, 2010). IL-10-secreting B cells have been shown to regulate autoimmune responses in vivo (Fillatreau et al., 2002; Mauri et al., 2003). There are currently no clinical studies examining Bregs as a cellular therapy. Further work is required to determine the optimal methods for the production of a functionally suppressive population of Bregs that may be used clinically.
Dendritic Cells
While DCs are known to be pivotal in the development of the alloresponse, some populations of DCs may also be active in the promotion of tolerance (Morelli and Thomson, 2007; van Kooten et al., 2011). Immature myeloid-derived DCs have been shown to promote the survival of heart allografts in a donor-specific manner (Lutz et al., 2000) and regulatory DCs with low costimulatory ability may prevent the development of GvHD in mice (Sato et al., 2003). However, even mature DCs expressing normal or high levels of MHC and costimulatory molecules may promote the development of tolerance. These DCs prime CD4+ and CD8+ T cells that in turn develop regulatory activity in vitro (Albert et al., 2001; Verhasselt et al., 2004). In vivo, myeloid-derived DCs matured with tumor necrosis factor α (TNFα) and expressing high levels of MHC II are able to protect mice from CD4+ T cell-mediated EAE (Menges et al., 2002).
The tolerogenic effects of DCs can be potentiated by the administration of costimulatory blockade agents. For example, plasmacytoid DCs have been shown to promote the induction of IL-10-secreting Treg and may promote heart allograft survival in vivo (Gilliet and Liu, 2002; Abe et al., 2005). However, with the addition of anti-CD154 antibody, this effect is significantly enhanced (Bjorck et al., 2005). A similar effect may be observed with the administration of costimulatory blockade with immature myeloid-derived DCs (Lu et al., 1997). Plasmacytoid DCs may also be important in facilitating mixed chimerism as described earlier when in the form of FCs (Kaufman et al., 1994; Grimes et al., 2004; Fugier-Vivier et al., 2005; Leventhal et al., 2012). Importantly, FCs have been demonstrated to promote the generation of Treg and prevent GvHD development in mice (Colson et al., 2004; Taylor et al., 2007; Huang et al., 2011).
Myeloid-Derived Suppressor Cells and Regulatory Macrophages
Myeloid-derived suppressor cells are a heterogeneous population of cells with both innate and adaptive immune targets, which include T, B and NK cells (Boros et al., 2010). Common phenotypic markers among MDSCs include GR1 and CD11b in mice and CD33, CD11b, CD34, and low MHC Class II expression in humans. Experimentally, mouse MDSCs induced by costimulatory blockade in vivo migrate to heart transplants where they prevent the development of alloresponses and promote the development of Treg (Garcia et al., 2010). Similarly, anti-CD28 antibody-induced rat kidney allograft tolerance leads to the accumulation of MDSCs in the blood. These MDSCs inhibit effector T cell proliferation in vitro through the activity of inducible nitric oxide (NO) synthase. Interestingly, however, the adoptive transfer of MDSC in this model does not induce kidney allograft tolerance.
Regulatory macrophages (Mregs) are a population of macrophages which produce large amounts of IL-10 and are able to suppress T cell proliferation in vitro (Fleming and Mosser, 2011). A population of cells termed “transplant acceptance inducing cells” (TAICs) has also been shown to promote the survival of heart and lung transplants in animal models (Fandrich et al., 2002a,b,c). TAICs are impure populations of macrophages contaminated with other leukocytes, whereas Mreg preparations are a uniform population of macrophages (Hutchinson et al., 2012).
Clinical Trials of Cellular Therapy for Peripheral Regulation
Several studies have investigated the use of Treg for the treatment of GvHD post-HSC transplantation. These studies are paving the way for Treg therapy in SOT. Trzonkowski et al. (2009) reported the “first-in-man” trial of ex vivo-expanded recipient-derived Treg in two patients: in one case of chronic GvHD a significant alleviation of symptoms and a reduction of required immunosuppression was achieved, whereas in one case of severe grade IV acute GvHD only a transient improvement in symptoms and signs was reported.
Significantly, two major Phase I/II trials have been carried out at the University of Minnesota and in Italy. Blazar’s group in Minnesota evaluated the safety profile of human UCB-derived partially HLA-matched ex vivo-expanded Treg (Brunstein et al., 2011). The study was designed as a Phase I dose-escalation trial and reported a reduced incidence of grades II–IV acute GvHD in the test group of 23 patients compared to 108 identically treated historical controls not receiving Treg therapy. Doses of Treg ranged from 1 × 105/kg to 30 × 105/kg and there was no reported increase in infectious complications. The Italian study was performed to assess the safety and efficacy of expanded CD4+CD25+ human nTreg in prevention of GvHD in 28 patients with high-risk acute leukaemias undergoing HLA-haploidentical HSC transplants. Patients were also given donor conventional T cells to enhance immune reconstitution. Treg were derived from the same HLA-haploidentical donor by apheresis followed by large scale CD4+CD25+ magnetic bead selection. Despite no GvHD prophylaxis being given, chronic GvHD did not develop in 26 out of 28 patients in whom full donor-type engraftment was achieved. However, in two of the 26 patients acute GvHD of grade II or above developed, which may be due to these two patients being given the highest dose of conventional T cells. Patients in the Italian trial displayed an overall faster post-transplant immune reconstitution as well as a reduction in the risk of CMV reactivation compared to those not receiving Treg (Di Ianni et al., 2011). While Brunstein et al. (2011) used UCB-derived Treg, Di Ianni et al. (2011) used adult expanded Treg. The difference in efficacy between these two populations on a cell-by-cell basis is unclear from these studies. However, as discussed earlier, UCB-derived Treg may contain a higher proportion of “naïve” CD45RA+ Treg and therefore a greater number of Treg which may readily proliferate in vivo. This would theoretically represent an advantage in terms of the cell dose required to prevent disease.
Other ongoing trials not yet published include one being conducted by Matthias Edinger at the University Hospital in Regensburg using CD25hi magnetically isolated non-expanded Treg infused into post-HSCT recipients (Edinger and Hoffmann, 2011). Trials using Tr1 cells for GvHD are also ongoing at the San Raffaele Hospital in Milan. Early results have been promising with no adverse side effects (Roncarolo and Battaglia, 2007; Allan et al., 2008; Battaglia and Roncarolo, 2011).
Two Phase I/II trials of TAICs assessed the safety of administration of these cells in 5–12 kidney transplant patients (Hutchinson et al., 2008a,b, 2009). The studies aimed to determine the possibility of immunosuppression withdrawal. The infusion of TAICs appeared safe but did not promote tolerance, with acute rejection developing in several patients on withdrawal of immunosuppression. Nevertheless, renal function was maintained in four out of five patients that were tapered to low-dose tacrolimus monotherapy. Moreover, one patient achieved complete immunosuppression withdrawal for 8 months before experiencing a rejection episode. Moving on from this approach, more uniform Mreg populations have been trialed in two renal transplant recipients, leading to a reduction in the required dose of immunosuppression with good graft function at 3 years post-transplantation (Hutchinson et al., 2011).
The European Union is currently funding the first study for the evaluation of immunomodulatory cellular therapy in SOT (www.onestudy.org). The ONE Study, a multicenter Phase I/II clinical trial, will evaluate the safety and feasibility of various types of cell therapy including expanded nTreg, Tr1 cells, Mregs, and tolerogenic DCs in living-donor kidney transplantation. All centers will utilize a common adjunctive immunosuppressive protocol in order to provide a true comparison of the various cellular therapies. Control patients will be transplanted in 2013 and cell therapy groups in 2014, providing a follow-up period of 12 months. Table 1 summarizes the concluded and ongoing clinical trials of cellular therapy.
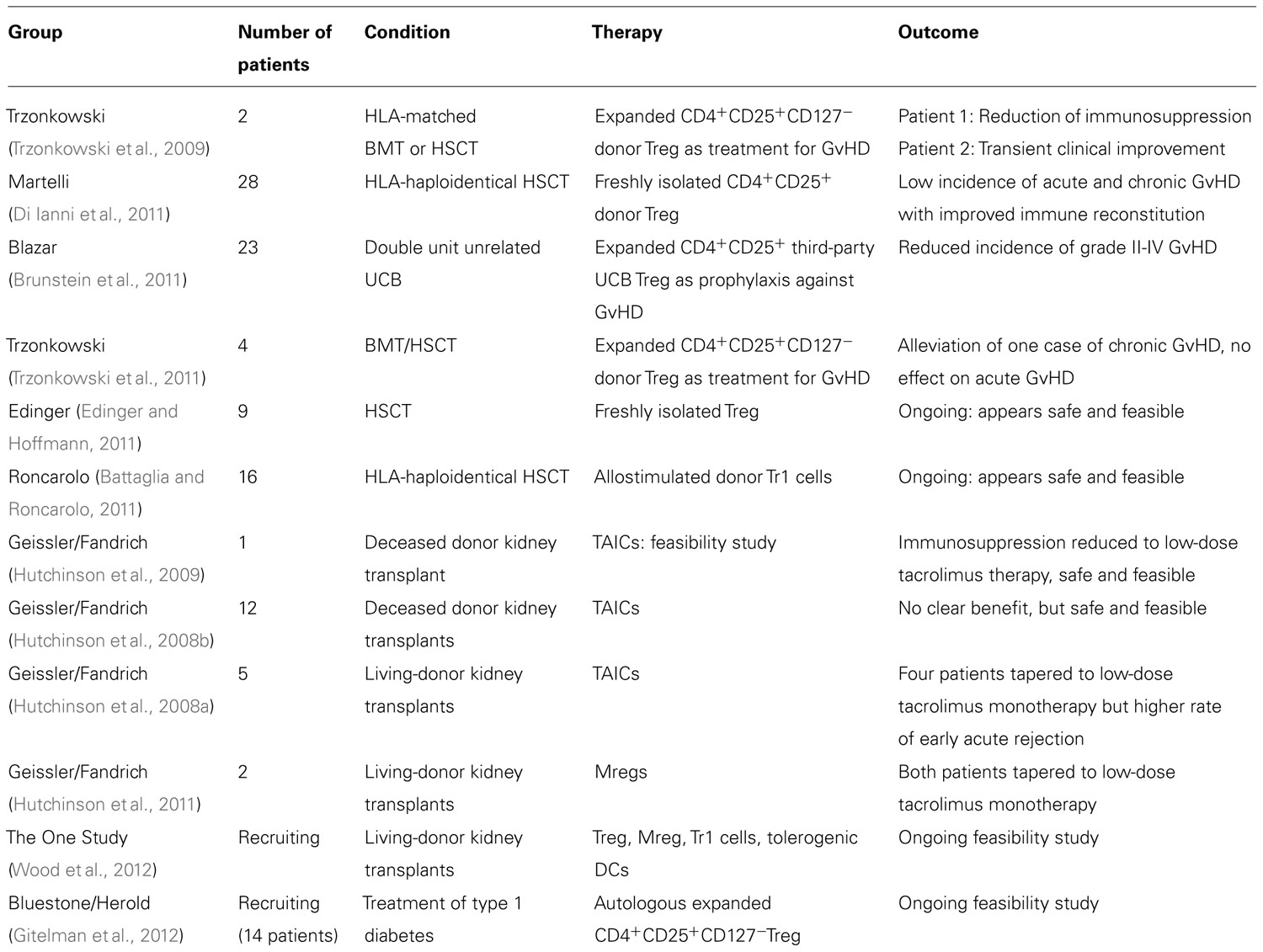
TABLE 1. Concluded and ongoing clinical studies using cellular therapy for peripheral regulation.
Immunotherapy for the Promotion of Tolerance
Treg and effector T cells preferentially employ different intracellular activation pathways. Treg utilize IL-2-dependent STAT-5 (Burchill et al., 2007; Vogtenhuber et al., 2010), whereas effector T cells utilize the phosphoinositide 3-kinase/Akt/mTOR pathway (Delgoffe et al., 2009). Rapamycin, an mTOR inhibitor, takes advantage of this distinction. The beneficial effects of rapamycin on Treg survival and proliferation have been demonstrated in vitro and in vivo (Battaglia et al., 2005, 2006b,c; Gao et al., 2007; Zeiser et al., 2008; Hendrikx et al., 2009). Experimental work in mouse allograft models has demonstrated that rapamycin inhibits chronic cardiac allograft rejection and that this effect is potentiated when used in combination with an α-CCR5 antibody (Li et al., 2009). In this study, an increase in intragraft numbers of CD4+CD25+foxp3+ Treg was observed. A similar effect has also been observed clinically, with rapamycin increasing the frequency of CD62Lhigh Treg in the peripheral blood of lung transplant recipients (Lange et al., 2010). Experimentally, the adoptive transfer of a small number of alloantigen-specific Treg along with low dose rapamycin treatment has been shown to induce long-term survival of cardiac allografts in mice (Raimondi et al., 2010). Moreover, alloantigen-pulsed rapamycin-conditioned DCs have been shown to promote long-term engraftment of vascularized skin allografts in rats with an associated expansion of CD4+foxp3+ Treg (Horibe et al., 2008).
Rabbit anti-murine thymocyte globulin (mATG), a T cell depleting polyclonal antibody has also shown promise. ATG promotes the generation of Treg (Lopez et al., 2006), and when combined with CTLA4-Ig and rapamycin, mATG shifts the effector memory T cell-Treg balance in favor of Treg, prolonging the survival of skin allografts in a fully MHC-mismatched mouse model (D’Addio et al., 2010).
Interestingly, glucocorticoids may act on human Langerhans cells to promote a phenotype that favors the induction of Treg in vitro (Stary et al., 2011). Some patients treated with glucocorticoids have increased numbers of dermal FOXP3+CD25+ Treg as well as increased numbers of epidermal Langerhans cells that display upregulated expression of TGFβ mRNA. However, there is no clear clinical evidence that ATG or glucocorticoids are beneficial in terms of increasing Treg numbers in transplant recipients.
Alemtuzumab may favor Treg survival, with evidence from one study demonstrating a higher proportional depletion of T effector cells than Treg (Bloom et al., 2008). However, data in this study are confounded by the introduction of rapamycin in patients early after transplantation. Indeed, in a separate study Treg numbers in alemtuzumab-treated patients remained low until the late introduction of rapamycin (Trzonkowski et al., 2008). Interestingly, Bregs have been identified in renal transplant recipients treated with alemtuzumab (Heidt et al., 2012). Alemtuzumab induction has been trialed at the University of Wisconsin for the minimization of immunosuppression (Knechtle et al., 2009). In this study, induction with alemtuzumab together with rapamycin maintenance monotherapy successfully led to long-term graft survival in nine of 10 patients although five patients developed anti-donor antibodies and graft C4d deposition.
Blockade of the IL-2-CD25 or CD28-CD80/CD86 pathways is an effective method of producing T cell anergy experimentally (Vincenti, 2008), however these pathways are also essential for the survival of Treg. Indeed, in mouse models where these pathways are targeted, there is a reduction in the survival and function of Treg with an associated exacerbation of autoimmunity in vivo (Tang et al., 2003; Wing et al., 2008). Clinically, however, there is no difference in circulating Treg numbers between renal transplant recipients treated with both belatacept (a second-generation CTLA-4-related. Ig fusion protein) and basiliximab (an α-CD25 monoclonal antibody, mAb) compared to those treated with calcineurin inhibitors (CNIs; Bluestone et al., 2008). Nevertheless, CNIs such as cyclosporine have a detrimental effect on Treg (Ma et al., 2009; Presser et al., 2009), thus confounding this observation. In this same study patients receiving belatacept displayed higher levels of intragraft FOXP3+ T cells during acute rejection (Bluestone et al., 2008). Belatacept has proven to be an effective immunosuppressant, but has not yet demonstrated any efficacy in the promotion of transplant tolerance in clinical transplantation. This may be related to the blockade of CD80/86-CTLA-4 interaction by CTLA-4.Ig. In a study investigating a novel CD28 antagonist for use in transplantation, there was an increase in the number and activity of Treg in a non-human primate (NHP) renal transplantation model (Poirier et al., 2010). The benefit of this costimulatory blockade, unlike CTLA-4.Ig, is that it allows physiological immune regulation through CD80/86 to continue (Wing et al., 2008). The effects α-CD25 mAb on Treg are not entirely clear. In the study by Bluestone et al. (2008), basiliximab was shown to deplete all CD25-bearing cells, including Treg. However, in another study examining daclizumab (a humanized α-CD25 mAb) in cardiac transplant patients, Treg generation in the periphery was not affected (Vlad et al., 2007). The timing of treatment with α-CD25 antibodies or CTLA-4.Ig may be of critical importance and may explain some of the differences between data from animal and human studies. Early use of these molecules may target Treg, resulting in deleterious effects in models dependent on Treg function, whereas later use post-transplantation may preferentially target activated effector T cells. Other possibilities include a lower sensitivity of human Treg to CD28 blockade or the presence of other costimulatory molecules on human Treg that may substitute for the absence of CD28 costimulation.
While CNIs are normally detrimental to Treg, there is some evidence that low-dose cyclosporine may enhance the number of Treg in the skin of patients with atopic dermatitis (Brandt et al., 2009). This appears to be related to the retained ability of patients on low-dose cyclosporine to produce IL-2, which is necessary for Treg survival and expansion (Baumgrass et al., 2010; Brandt et al., 2010).
Memory T cells present a formidable barrier to the induction of tolerance in higher mammals (Brook et al., 2006; Ford and Larsen, 2011). A solution to overcoming this barrier is the use of immunosuppressants that target memory T cell responses while promoting the generation of immunoregulatory elements. In this respect, targeting adhesion molecules such as CD2 or LFA-1 is a promising strategy. Alefacept, an LFA-3.Ig fusion protein binds to and polymerizes CD2, leading to selective elimination of memory T cells. Treatment together with CTLA-4.Ig prevents acute rejection and allows prolonged engraftment of kidney transplants in a NHP model (Weaver et al., 2009). Efalizumab, an anti-LFA-1 antibody, initially displayed promise in early clinical trials of islet transplantation (Badell et al., 2010; Posselt et al., 2010; Setoguchi et al., 2011). Its use however is no longer possible due to withdrawal from the market after the development of progressive multifocal leukoencephalopathy in four patients treated for psoriasis with efalizumab (Tavazzi et al., 2011).
Conclusion
With the trailblazing work of Medawar, clinical tolerance appeared to be eminently within reach. Yet 70 years on, tolerance has been achieved in only a small number of patients in whom full or mixed chimerism was generated. We propose that achieving tolerance in each and every transplant recipient will require a more complete understanding of the biovariability between patients that allows tolerance to be easily induced in some but not others. The attainment of tolerance in a heterogeneous population of transplant recipients may therefore require a tailored approach, with the balanced use of both central and peripheral tolerance induction techniques.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work from the authors’ own group referred to in this review was supported by grants from The Wellcome Trust, Medical Research Council UK, European Union Integrated Projects, RISET and the ONE Study, the Royal College of Surgeons of England, the Dunhill Medical Trust and the Oxford Health Services Research Committee Charitable Trust.
References
Abe, M., Wang, Z., De Creus, A., and Thomson, A. W. (2005). Plasmacytoid dendritic cell precursors induce allogeneic T-cell hyporesponsiveness and prolong heart graft survival. Am. J. Transplant. 5, 1808–1819.
Adeegbe, D., Serafini, P., Bronte, V., Zoso, A., Ricordi, C., and Inverardi, L. (2010). In vivo induction of myeloid suppressor cells and CD4+Foxp3+ T regulatory cells prolongs skin allograft survival in mice. Cell Transplant. 20, 941–954.
Albert, M. L., Jegathesan, M., and Darnell, R. B. (2001). Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat. Immunol. 2, 1010–1017.
Ali, N. A., Gaughan, A. A., Orosz, C. G., Baran, C. P., Mcmaken, S., Wang, Y., Eubank, T. D., Hunter, M., Lichtenberger, F. J., Flavahan, N. A., Lawler, J., and Marsh, C. B. (2008). Latency associated peptide has in vitro and in vivo immune effects independent of TGF-beta1. PLoS ONE 3, e1914. doi: 10.1371/journal.pone.0001914
Allan, S. E., Broady, R., Gregori, S., Himmel, M. E., Locke, N., Roncarolo, M. G., Bacchetta, R., and Levings, M. K. (2008). CD4+ T-regulatory cells: toward therapy for human diseases. Immunol. Rev. 223, 391–421.
Ayyoub, M., Deknuydt, F., Raimbaud, I., Dousset, C., Leveque, L., Bioley, G., and Valmori, D. (2009). Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc. Natl. Acad. Sci. U.S.A. 106, 8635–8640.
Badell, I. R., Russell, M. C., Thompson, P. W., Turner, A. P., Weaver, T. A., Robertson, J. M., Avila, J. G., Cano, J. A., Johnson, B. E., Song, M., Leopardi, F. V., Swygert, S., Strobert, E. A., Ford, M. L., Kirk, A. D., and Larsen, C. P. (2010). LFA-1-specific therapy prolongs allograft survival in rhesus macaques. J. Clin. Invest. 120, 4520–4531.
Baker, R. J., Hernandez-Fuentes, M. P., Brookes, P. A., Chaudhry, A. N., Cook, H. T., and Lechler, R. I. (2001). Loss of direct and mainenance of indirect alloresponses in renal allograft recipients: implications for the pathogenesis of chronic allograft nephropathy. J. Immunol. 7199–7206.
Battaglia, M., Gregori, S., Bacchetta, R., and Roncarolo, M. G. (2006a). Tr1 cells: from discovery to their clinical application. Semin. Immunol. 18, 120–127.
Battaglia, M., Stabilini, A., Draghici, E., Gregori, S., Mocchetti, C., Bonifacio, E., and Roncarolo, M. G. (2006b). Rapamycin and interleukin-10 treatment induces T regulatory type 1 cells that mediate antigen-specific transplantation tolerance. Diabetes 55, 40–49.
Battaglia, M., Stabilini, A., Migliavacca, B., Horejs-Hoeck, J., Kaupper, T., and Roncarolo, M. G. (2006c). Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J. Immunol. 177, 8338–8347.
Battaglia, M., and Roncarolo, M. G. (2011). Immune intervention with T regulatory cells: past lessons and future perspectives for type 1 diabetes. Semin. Immunol. 23, 182–194.
Battaglia, M., Stabilini, A., and Roncarolo, M. G. (2005). Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 105, 4743–4748.
Baumgrass, R., Brandt, C., Wegner, F., Abdollahnia, M., and Worm, M. (2010). Low-dose, but not high-dose, cyclosporin A promotes regulatory T-cell induction, expansion, or both. J. Allergy Clin. Immunol. 126, 183–184; author reply 184.
Bennett, C. L., Christie, J., Ramsdell, F., Brunkow, M. E., Ferguson, P. J., Whitesell, L., Kelly, T. E., Saulsbury, F. T., Chance, P. F., and Ochs, H. D. (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21.
Bigenzahn, S., Blaha, P., Koporc, Z., Pree, I., Selzer, E., Bergmeister, H., Wrba, F., Heusser, C., Wagner, K., Muehlbacher, F., and Wekerle, T. (2005). The role of non-deletional tolerance mechanisms in a murine model of mixed chimerism with costimulation blockade. Am. J. Transplant. 5, 1237–1247.
Billingham, R. E., Brent, L., and Medawar, P. B. (1953). Actively acquired tolerance of foreign cells. Nature 172, 603–606.
Bjorck, P., Coates, P. T., Wang, Z., Duncan, F. J., and Thomson, A. W. (2005). Promotion of long-term heart allograft survival by combination of mobilized donor plasmacytoid dendritic cells and anti-CD154 monoclonal antibody. J. Heart Lung Transplant. 24, 1118–1120.
Bloom, D. D., Chang, Z., Fechner, J. H., Dar, W., Polster, S. P., Pascual, J., Turka, L. A., and Knechtle, S. J. (2008). CD4+ CD25+ FOXP3+ regulatory T cells increase de novo in kidney transplant patients after immunodepletion with Campath-1H. Am. J. Transplant. 8, 793–802.
Bluestone, J. A., Liu, W., Yabu, J. M., Laszik, Z. G., Putnam, A., Belingheri, M., Gross, D. M., Townsend, R. M., and Vincenti, F. (2008). The effect of costimulatory and interleukin 2 receptor blockade on regulatory T cells in renal transplantation. Am. J. Transplant. 8, 2086–2096.
Boros, P., Ochando, J. C., Chen, S. H., and Bromberg, J. S. (2010). Myeloid-derived suppressor cells: natural regulators for transplant tolerance. Hum. Immunol. 71, 1061–1066.
Brandt, C., Liman, P., Bendfeldt, H., Mueller, K., Reinke, P., Radbruch, A., Worm, M., and Baumgrass, R. (2010). Whole blood flow cytometric measurement of NFATc1 and IL-2 expression to analyze cyclosporine A-mediated effects in T cells. Cytometry A 77, 607–613.
Brandt, C., Pavlovic, V., Radbruch, A., Worm, M., and Baumgrass, R. (2009). Low-dose cyclosporine A therapy increases the regulatory T cell population in patients with atopic dermatitis. Allergy 64, 1588–1596.
Brook, M. O., Wood, K. J., and Jones, N. D. (2006). The impact of memory T cells on rejection adn the induction of tolerance. Transplantation 82, 1–9.
Brunkow, M. E., Jeffery, E. W., Hjerrild, K. A., Paeper, B., Clark, L. B., Yasayko, S. A., Wilkinson, J. E., Galas, D., Ziegler, S. F., and Ramsdell, F. (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27, 68–73.
Brunstein, C. G., Miller, J. S., Cao, Q., Mckenna, D. H., Hippen, K. L., Curtsinger, J., Defor, T., Levine, B. L., June, C. H., Rubinstein, P., Mcglave, P. B., Blazar, B. R., and Wagner, J. E. (2011). Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 117, 1061–1070.
Buhler, L. H., Spitzer, T. R., Sykes, M., Sachs, D. H., Delmonico, F. L., Tolkoff-Rubin, N., Saidman, S. L., Sackstein, R., Mcafee, S., Dey, B., Colby, C., and Cosimi, A. B. (2002). Induction of kidney allograft tolerance after transient lymphohematopoietic chimerism in patients with multiple myeloma and end-stage renal disease. Transplantation 74, 1405–1409.
Burchill, M. A., Yang, J., Vogtenhuber, C., Blazar, B. R., and Farrar, M. A. (2007). IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 178, 280–290.
Bushell, A., Karim, M., Kingsley, C. I., and Wood, K. (2003). Pretransplant blood transfusion without additional immunotherapy generates CD25+CD4+ regulatory T cells: a potential explanation for the blood-transfusion effect. Transplantation 76, 449–455.
Bushell, A., Morris, P. J., and Wood, K. J. (1994). Induction of operational tolerance by random blood transfusion combined with anti-CD4 antibody therapy. A protocol with significant clinical potential. Transplantation 58, 133–139.
Bushell, A., Morris, P. J., and Wood, K. J. (1995). Transplantation tolerance induced by antigen pretreatment and depleting anti-CD4 antibody depends on CD4+ T cell regulation during the induction phase of the response. Eur. J. Immunol. 25, 2643–2649.
Butler, P. E., Lee, W. P., Van De Water, A. P., and Randolph, M. A. (2000). Neonatal induction of tolerance to skeletal tissue allografts without immunosuppression. Plast. Reconstr. Surg. 105, 2424–2430; discussion 2431–2422.
Chadha, R., Heidt, S., Jones, N. D., and Wood, K. J. (2011). Th17: contributors to allograft rejection and a barrier to the induction of transplantation tolerance? Transplantation 91, 939–945.
Chen, M. L., Yan, B. S., Bando, Y., Kuchroo, V. K., and Weiner, H. L. (2008). Latency-associated peptide identifies a novel CD4+CD25+ regulatory T cell subset with TGFbeta-mediated function and enhanced suppression of experimental autoimmune encephalomyelitis. J. Immunol. 180, 7327–7337.
Cobbold, S. P., Martin, G., Qin, S., and Waldmann, H. (1986). Monoclonal antibodies to promote marrow engraftment and tissue graft tolerance. Nature 323, 164–166.
Cober, S. R., Randolph, M. A., and Lee, W. P. (1999). Skin allograft survival following intrathymic injection of donor bone marrow. J. Surg. Res. 85, 204–208.
Colson, Y. L., Christopher, K., Glickman, J., Taylor, K. N., Wright, R., and Perkins, D. L. (2004). Absence of clinical GVHD and the in vivo induction of regulatory T cells after transplantation of facilitating cells. Blood 104, 3829–3835.
D’Addio, F., Yuan, X., Habicht, A., Williams, J., Ruzek, M., Iacomini, J., Turka, L. A., Sayegh, M. H., Najafian, N., and Ansari, M. J. (2010). A novel clinically relevant approach to tip the balance toward regulation in stringent transplant model. Transplantation 90, 260–269.
Delgoffe, G. M., Kole, T. P., Zheng, Y., Zarek, P. E., Matthews, K. L., Xiao, B., Worley, P. F., Kozma, S. C., and Powell, J. D. (2009). The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844.
Demir, Y., Ozmen, S., Klimczak, A., Mukherjee, A. L., and Siemionow, M. (2004). Tolerance induction in composite facial allograft transplantation in the rat model. Plast. Reconstr. Surg. 114, 1790–1801.
Di Ianni, M., Falzetti, F., Carotti, A., Terenzi, A., Castellino, F., Bonifacio, E., Del Papa, B., Zei, T., Ostini, R. I., Cecchini, D., Aloisi, T., Perruccio, K., Ruggeri, L., Balucani, C., Pierini, A., Sportoletti, P., Aristei, C., Falini, B., Reisner, Y., Velardi, A., Aversa, F., and Martelli, M. F. (2011). Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 117, 3921–3928.
Domenig, C., Sanchez-Fueyo, A., Kurtz, J., Alexopoulos, S. P., Mariat, C., Sykes, M., Strom, T. B., and Zheng, X. X. (2005). Roles of deletion and regulation in creating mixed chimerism and allograft tolerance using a nonlymphoablative irradiation-free protocol. J. Immunol. 175, 51–60.
Duarte, J. H., Zelenay, S., Bergman, M.-L., Martins, A. C., and Demengeot, J. (2009). Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopaenic conditions. Eur. J. Immunol. 39, 948–955.
Edinger, M., and Hoffmann, P. (2011). Regulatory T cells in stem cell transplantation: strategies and first clinical experiences. Curr. Opin. Immunol. 23, 679–684.
Fandrich, F., Dresske, B., Bader, M., and Schulze, M. (2002a). Embryonic stem cells share immune-privileged features relevant for tolerance induction. J. Mol. Med. (Berl) 80, 343–350.
Fandrich, F., Lin, X., Chai, G. X., Schulze, M., Ganten, D., Bader, M., Holle, J., Huang, D. S., Parwaresch, R., Zavazava, N., and Binas, B. (2002b). Preimplantation-stage stem cells induce long-term allogeneic graft acceptance without supplementary host conditioning. Nat. Med. 8, 171–178.
Fandrich, F., Zhou, X., Schlemminger, M., Lin, X., and Dresske, B. (2002c). Future strategies for tolerance induction: a comparative study between hematopoietic stem cells and macrophages. Hum. Immunol. 63, 805–812.
Fehr, T., Wang, S., Haspot, F., Kurtz, J., Blaha, P., Hogan, T., Chittenden, M., Wekerle, T., and Sykes, M. (2008). Rapid deletional peripheral CD8 T cell tolerance induced by allogeneic bone marrow: role of donor class II MHC and B cells. J. Immunol. 181, 4371–4380.
Fillatreau, S., Sweenie, C. H., Mcgeachy, M. J., Gray, D., and Anderton, S. M. (2002). B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 3, 944–950.
Fleming, B. D., and Mosser, D. M. (2011). Regulatory macrophages: setting the threshold for therapy. Eur. J. Immunol. 41, 2498–2502.
Ford, M. L., and Larsen, C. P. (2011). Transplantation tolerance: memories that haunt us. Sci. Transl. Med. 3, 86ps22.
Francis, R. S., Feng, G., Tha-In, T., Lyons, I. S., Wood, K. J., and Bushell, A. (2011). Induction of transplantation tolerance converts potential effector T cells into graft-protective regulatory T cells. Eur. J. Immunol. 41, 726–738.
Fudaba, Y., Spitzer, T. R., Shaffer, J., Kawai, T., Fehr, T., Delmonico, F., Preffer, F., Tolkoff-Rubin, N., Dey, B. R., Saidman, S. L., Kraus, A., Bonnefoix, T., Mcafee, S., Power, K., Kattleman, K., Colvin, R. B., Sachs, D. H., Cosimi, A. B., and Sykes, M. (2006). Myeloma responses and tolerance following combined kidney and nonmyeloablative marrow transplantation: in vivo and in vitro analyses. Am. J. Transplant. 6, 2121–2133.
Fugier-Vivier, I. J., Rezzoug, F., Huang, Y., Graul-Layman, A. J., Schanie, C. L., Xu, H., Chilton, P. M., and Ildstad, S. T. (2005). Plasmacytoid precursor dendritic cells facilitate allogeneic hematopoietic stem cell engraftment. J. Exp. Med. 201, 373–383.
Gao, W., Lu, Y., El Essawy, B., Oukka, M., Kuchroo, V. K., and Strom, T. B. (2007). Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am. J. Transplant. 7, 1722–1732.
Garcia, M. R., Ledgerwood, L., Yang, Y., Xu, J., Lal, G., Burrell, B., Ma, G., Hashimoto, D., Li, Y., Boros, P., Grisotto, M., Van Rooijen, N., Matesanz, R., Tacke, F., Ginhoux, F., Ding, Y., Chen, S. H., Randolph, G., Merad, M., Bromberg, J. S., and Ochando, J. C. (2010). Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J. Clin. Invest. 120, 2486–2496.
Gilliet, M., and Liu, Y. J. (2002). Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J. Exp. Med. 195, 695–704.
Gitelman, S. E., Bluestone, J., and Herold, K. C. (2012). T1DM Immunotherapy Using CD4+CD127lo/-CD25+ Polyclonal Tregs. Available at: http://clinicaltrials.gov/ct2/show/NCT01210664 (accessed August 6, 2012).
Golshayan, D., Jiang, S., Tsang, J., Garin, M. I., Mottet, C., and Lechler, R. I. (2007). In vitro-expanded donor alloantigen-specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood 109, 827–835.
Graca, L., Honey, K., Adams, E., Cobbold, S. P., and Waldmann, H. (2000). Cutting edge: anti-CD154 therapeutic antibodies induce infectious transplantation tolerance. J. Immunol. 165, 4783–4786.
Graca, L., Thompson, S., Lin, C. Y., Adams, E., Cobbold, S. P., and Waldmann, H. (2002). Both CD4(+)CD25(+) and CD4(+)CD25(-) regulatory cells mediate dominant transplantation tolerance. J. Immunol. 168, 5558–5565.
Granger, D. K., Briedenbach, W. C., Pidwell, D. J., Jones, J. W., Baxter-Lowe, L. A., and Kaufman, C. L. (2002). Lack of donor hyporesponsiveness and donor chimerism after clinical transplantation of the hand. Transplantation 74, 1624–1630.
Grimes, H. L., Schanie, C. L., Huang, Y., Cramer, D., Rezzoug, F., Fugier-Vivier, I., and Ildstad, S. T. (2004). Graft facilitating cells are derived from hematopoietic stem cells and functionally require CD3, but are distinct from T lymphocytes. Exp. Hematol. 32, 946–954.
Hara, M., Kingsley, C. I., Niimi, M., Read, S., Turvey, S. E., Bushell, A. R., Morris, P. J., Powrie, F., and Wood, K. J. (2001). IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J. Immunol. 166, 3789–3796.
Heidt, S., Hester, J., Shankar, S., Friend, P. J., and Wood, K. J. (2012). B-cell repopulation after alemtuzumab induction – transient increase in transitional B cells and long-term dominance of naive B cells. Am. J. Transplant. 12, 1784–1792.
Heidt, S., Segundo, D. S., Chadha, R., and Wood, K. J. (2010). The impact of Th17 cells on transplant rejection and the induction of tolerance. Curr. Opin. Organ Transplant. 15, 456–461.
Hendrikx, T. K., Velthuis, J. H., Klepper, M., Van Gurp, E., Geel, A., Schoordijk, W., Baan, C. C., and Weimar, W. (2009). Monotherapy rapamycin allows an increase of CD4 CD25 FoxP3 T cells in renal recipients. Transpl. Int. 22, 884-891.
Hequet, O., Morelon, E., Bourgeot, J. P., Dubois, V., Devauchelle, B., Dubernard, J. M., and Michallet, M. (2008). Allogeneic donor bone marrow cells recovery and infusion after allogeneic face transplantation from the same donor. Bone Marrow Transplant. 41, 1059–1061.
Hester, J., Mills, N., Shankar, S., Carvalho-Gaspar, M., Friend, P., and Wood, K. J. (2011). Th17 cells in alemtuzumab-treated patients: the effect of long-term maintenance immunosuppressive therapy. Transplantation 91, 744–750.
Hori, S., Nomura, T., and Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061.
Horibe, E. K., Sacks, J., Unadkat, J., Raimondi, G., Wang, Z., Ikeguchi, R., Marsteller, D., Ferreira, L. M., Thomson, A. W., Lee, W. P., and Feili-Hariri, M. (2008). Rapamycin-conditioned, alloantigen-pulsed dendritic cells promote indefinite survival of vascularized skin allografts in association with T regulatory cell expansion. Transpl. Immunol. 18, 307–318.
Huang, Y., Bozulic, L. D., Miller, T., Xu, H., Hussain, L. R., and Ildstad, S. T. (2011). CD8α+ plasmacytoid precursor DCs induce antigen-specific regulatory T cells that enhance HSC engraftment in vivo. Blood 117, 2494–2505.
Hutchinson, J. A., Brem-Exner, B. G., Riquelme, P., Roelen, D., Schulze, M., Ivens, K., Grabensee, B., Witzke, O., Philipp, T., Renders, L., Humpe, A., Sotnikova, A., Matthai, M., Heumann, A., Govert, F., Schulte, T., Kabelitz, D., Claas, F. H., Geissler, E. K., Kunzendorf, U., and Fandrich, F. (2008a). A cell-based approach to the minimization of immunosuppression in renal transplantation. Transpl. Int. 21, 742–754.
Hutchinson, J. A., Govert, F., Riquelme, P., Brasen, J. H., Brem-Exner, B. G., Matthai, M., Schulze, M., Renders, L., Kunzendorf, U., Geissler, E. K., and Fandrich, F. (2009). Administration of donor-derived transplant acceptance-inducing cells to the recipients of renal transplants from deceased donors is technically feasible. Clin. Transplant. 23, 140–145.
Hutchinson, J. A., Riquelme, P., Brem-Exner, B. G., Schulze, M., Matthai, M., Renders, L., Kunzendorf, U., Geissler, E. K., and Fandrich, F. (2008b). Transplant acceptance-inducing cells as an immune-conditioning therapy in renal transplantation. Transpl. Int. 21, 728–741.
Hutchinson, J. A., Riquelme, P., and Geissler, E. K. (2012). Human regulatory macrophages as a cell-based medicinal product. Curr. Opin. Organ Transplant. 17, 48–54.
Hutchinson, J. A., Riquelme, P., Sawitzki, B., Tomiuk, S., Miqueu, P., Zuhayra, M., Oberg, H. H., Pascher, A., Lutzen, U., Janssen, U., Broichhausen, C., Renders, L., Thaiss, F., Scheuermann, E., Henze, E., Volk, H. D., Chatenoud, L., Lechler, R. I., Wood, K. J., Kabelitz, D., Schlitt, H. J., Geissler, E. K., and Fandrich, F. (2011). Cutting Edge: Immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J. Immunol. 187, 2072–2078.
Issa, F., Hester, J., Goto, R., Nadig, S. N., Goodacre, T. E., and Wood, K. (2010). Ex vivo-expanded human regulatory T cells prevent the rejection of skin allografts in a humanized mouse model. Transplantation 90, 1321–1327.
Issa, F., and Wood, K. J. (2010). CD4+ regulatory T cells in solid organ transplantation. Curr. Opin. Organ Transplant. 15, 757–764.
Jiang, S., Tsang, J., Game, D. S., Stevenson, S., Lombardi, G., and Lechler, R. I. (2006). Generation and expansion of human CD4+ CD25+ regulatory T cells with indirect allospecificity: potential reagents to promote donor-specific transplantation tolerance. Transplantation 82, 1738–1743.
Joffre, O., Santolaria, T., Calise, D., Al Saati, T., Hudrisier, D., Romagnoli, P., and van Meerwijk, J. P. (2008). Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat. Med. 14, 88–92.
Jones, N. D., Brook, M. O., Carvalho-Gaspar, M., Luo, S., and Wood, K. J. (2010). Regulatory T cells can prevent memory CD8+ T-cell-mediated rejection following polymorphonuclear cell depletion. Eur. J. Immunol. 40, 3107–3116.
Josefowicz, S. Z., and Rudensky, A. (2009). Control of regulatory T cell lineage commitment and maintenance. Immunity 30, 616–625.
Karim, M., Feng, G., Wood, K. J., and Bushell, A. R. (2005). CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood 105, 4871–4877.
Karim, M., Kingsley, C. I., Bushell, A. R., Sawitzki, B. S., and Wood, K. J. (2004). Alloantigen-induced CD25+CD4+ regulatory T cells can develop in vivo from CD25-CD4+ precursors in a thymus-independent process. J. Immunol. 172, 923–928.
Kaufman, C. L., Colson, Y. L., Wren, S. M., Watkins, S., Simmons, R. L., and Ildstad, S. T. (1994). Phenotypic characterization of a novel bone marrow-derived cell that facilitates engraftment of allogeneic bone marrow stem cells. Blood 84, 2436–2446.
Kawai, T., Cosimi, A. B., and Sachs, D. H. (2011). Preclinical and clinical studies on the induction of renal allograft tolerance through transient mixed chimerism. Curr. Opin. Organ Transplant. 16, 366–371.
Kawai, T., Cosimi, A. B., Spitzer, T. R., Tolkoff-Rubin, N., Suthanthiran, M., Saidman, S. L., Shaffer, J., Preffer, F. I., Ding, R., Sharma, V., Fishman, J. A., Dey, B., Ko, D. S., Hertl, M., Goes, N. B., Wong, W., Williams, W. W. Jr., Colvin, R. B., Sykes, M., and Sachs, D. H. (2008). HLA-mismatched renal transplantation without maintenance immunosuppression. N. Engl. J. Med. 358, 353–361.
Khan, A., Tomita, Y., and Sykes, M. (1996). Thymic dependence of loss of tolerance in mixed allogeneic bone marrow chimeras after depletion of donor antigen. Peripheral mechanisms do not contribute to maintenance of tolerance. Transplantation 62, 380–387.
Kingsley, C. I., Karim, M., Bushell, A. R., and Wood, K. J. (2002). CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4 and IL-10 dependent immunoregulation of alloresponses. J. Immunol. 168, 1080–1086.
Kingsley, C. I., Nadig, S. N., and Wood, K. J. (2007). Transplantation tolerance: lessons from experimental rodent models. Transpl. Int. 20, 828–841.
Kleinewietfeld, M., Starke, M., Di Mitri, D., Borsellino, G., Battistini, L., Rotzschke, O., and Falk, K. (2009). CD49d provides access to “untouched” human Foxp3+ Treg free of contaminating effector cells. Blood 113, 827–836.
Knechtle, S. J., Pascual, J., Bloom, D. D., Torrealba, J. R., Jankowska-Gan, E., Burlingham, W. J., Kwun, J., Colvin, R. B., Seyfert-Margolis, V., Bourcier, K., and Sollinger, H. W. (2009). Early and limited use of tacrolimus to avoid rejection in an alemtuzumab and sirolimus regimen for kidney transplantation: clinical results and immune monitoring. Am. J. Transplant. 9, 1087–1098.
Komatsu, N., Mariotti-Ferrandiz, M. E., Wang, Y., Malissen, B., Waldmann, H., and Hori, S. (2009). Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. U.S.A. 106, 1903–1908.
Kulahci, Y., Klimczak, A., Madajka, M., Altuntas, S., and Siemionow, M. (2010). Long-term survival of composite hemiface/mandible/tongue allografts correlates with multilineage chimerism development in the lymphoid and myeloid compartments of recipients. Transplantation 90, 843–852.
Lange, C. M., Tran, T. Y., Farnik, H., Jungblut, S., Born, T., Wagner, T. O., and Hirche, T. O. (2010). Increased frequency of regulatory T cells and selection of highly potent CD62L+ cells during treatment of human lung transplant recipients with rapamycin. Transpl. Int. 23, 266–276.
Lerut, J., and Sanchez-Fueyo, A. (2006). An appraisal of tolerance in liver transplantation. Am. J. Transplant. 6, 1774–1780.
Leventhal, J., Abecassis, M., Miller, J., Gallon, L., Ravindra, K., Tollerud, D. J., King, B., Elliott, M. J., Herzig, G., Herzig, R., and Ildstad, S. T. (2012). Chimerism and tolerance without GVHD or engraftment syndrome in HLA-mismatched combined kidney and hematopoietic stem cell transplantation. Sci. Transl. Med. 4, 124ra128.
Li, J., Zhang, K., Ye, P., Wang, S., and Xia, J. (2009). CCR5 blockade in combination with rapamycin prolongs cardiac allograft survival in mice. Clin. Exp. Immunol. 157, 437–445.
Li, Z., Benghiat, F. S., Charbonnier, L. M., Kubjak, C., Rivas, M. N., Cobbold, S. P., Waldmann, H., De Wilde, V., Petein, M., Schuind, F., Goldman, M., and Le Moine, A. (2008). CD8+ T-Cell depletion and rapamycin synergize with combined coreceptor/stimulation blockade to induce robust limb allograft tolerance in mice. Am. J. Transplant. 8, 2527–2536.
Li, Z., Benghiat, F. S., Kubjak, C., Schuind, F., Goldman, M., and Le Moine, A. (2007). Donor T-cell development in host thymus after heterotopic limb transplantation in mice. Transplantation 83, 815–818.
Liu, W., Putnam, A. L., Xu-Yu, Z., Szot, G. L., Lee, M. R., Zhu, S., Gottlieb, P. A., Kapranov, P., Gingeras, T. R., Fazekas de St Groth, B., Clayberger, C., Soper, D. M., Ziegler, S. F., and Bluestone, J. A. (2006). CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 203, 1701–1711.
LoCascio, S. A., Morokata, T., Chittenden, M., Preffer, F. I., Dombkowski, D. M., Andreola, G., Crisalli, K., Kawai, T., Saidman, S. L., Spitzer, T. R., Tolkoff-Rubin, N., Cosimi, A. B., Sachs, D. H., and Sykes, M. (2010). Mixed chimerism, lymphocyte recovery, and evidence for early donor-specific unresponsiveness in patients receiving combined kidney and bone marrow transplantation to induce tolerance. Transplantation 90, 1607–1615.
Long, E., and Wood, K. J. (2009). Regulatory T cells in transplantation: transferring mouse studies to the clinic. Transplantation 88, 1050–1056.
Lopez, M., Clarkson, M. R., Albin, M., Sayegh, M. H., and Najafian, N. (2006). A novel mechanism of action for anti-thymocyte globulin: induction of CD4+CD25+Foxp3+ regulatory T cells. J. Am. Soc. Nephrol. 17, 2844–2853.
Lu, L., Li, W., Fu, F., Chambers, F. G., Qian, S., Fung, J. J., and Thomson, A. W. (1997). Blockade of the CD40-CD40 ligand pathway potentiates the capacity of donor-derived dendritic cell progenitors to induce long-term cardiac allograft survival. Transplantation 64, 1808–1815.
Lutz, M. B., Suri, R. M., Niimi, M., Ogilvie, A. L., Kukutsch, N. A., Rossner, S., Schuler, G., and Austyn, J. M. (2000). Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur. J. Immunol. 30, 1813–1822.
Ma, A., Qi, S., Wang, Z., Massicotte, E., Dupuis, M., Daloze, P., and Chen, H. (2009). Combined therapy of CD4(+)CD25(+) regulatory T cells with low-dose sirolimus, but not calcineurin inhibitors, preserves suppressive function of regulatory T cells and prolongs allograft survival in mice. Int. Immunopharmacol. 9, 553–563.
Mathes, D. W., Solari, M. G., Randolph, M. A., Gazelle, G. S., Yamada, K., Huang, C. A., Sachs, D. H., and Lee, W. P. (2005). Long-term acceptance of renal allografts following prenatal inoculation with adult bone marrow. Transplantation 80, 1300–1308.
Mauri, C., and Blair, P. A. (2010). Regulatory B cells in autoimmunity: developments and controversies. Nat. Rev. Rheumatol. 6, 636–643.
Mauri, C., Gray, D., Mushtaq, N., and Londei, M. (2003). Prevention of arthritis by interleukin 10-producing B cells. J. Exp. Med. 197, 489–501.
Menges, M., Rossner, S., Voigtlander, C., Schindler, H., Kukutsch, N. A., Bogdan, C., Erb, K., Schuler, G., and Lutz, M. B. (2002). Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J. Exp. Med. 195, 15–21.
Miyao, T., Floess, S., Setoguchi, R., Luche, H., Fehling, H. J., Waldmann, H., Huehn, J., and Hori, S. (2012). Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 36, 262–275.
Miyara, M., Yoshioka, Y., Kitoh, A., Shima, T., Wing, K., Niwa, A., Parizot, C., Taflin, C., Heike, T., Valeyre, D., Mathian, A., Nakahata, T., Yamaguchi, T., Nomura, T., Ono, M., Amoura, Z., Gorochov, G., and Sakaguchi, S. (2009). Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 30, 899–911.
Morelli, A. E., and Thomson, A. W. (2007). Tolerogenic dendritic cells and the quest for transplant tolerance. Nat. Rev. Immunol. 7, 610–621.
Nasir, S., Bozkurt, M., Klimczak, A., and Siemionow, M. (2008). Large antigenic skin load in total abdominal wall transplants permits chimerism induction. Ann. Plast. Surg. 61, 572–579.
Newell, K. A., Asare, A., Kirk, A. D., Gisler, T. D., Bourcier, K., Suthanthiran, M., Burlingham, W. J., Marks, W. H., Sanz, I., Lechler, R. I., Hernandez-Fuentes, M. P., Turka, L. A., and Seyfert-Margolis, V. L. (2010). Identification of a B cell signature associated with renal transplant tolerance in humans. J. Clin. Invest. 120, 1836–1847.
Orlando, G., Hematti, P., Stratta, R. J., Burke, G. W. III, Cocco, P. D., Pisani, F., Soker, S., and Wood, K. (2010). Clinical operational tolerance after renal transplantation: current status and future challenges. Ann. Surg. 252, 915–928.
Ozer, K., Izycki, D., Zielinski, M., and Siemionow, M. (2004). Development of donor-specific chimerism and tolerance in composite tissue allografts under alphabeta-T-cell receptor monoclonal antibody and cyclosporine a treatment protocols. Microsurgery 24, 248–254.
Ozer, K., Oke, R., Gurunluoglu, R., Zielinski, M., Izycki, D., Prajapati, R., and Siemionow, M. (2003). Induction of tolerance to hind limb allografts in rats receiving cyclosporine A and antilymphocyte serum: effect of duration of the treatment. Transplantation 75, 31–36.
Petit, F., Minns, A. B., Nazzal, J. A., Hettiaratchy, S. P., Lantieri, L. A., Randolph, M. A., and Lee, W. P. (2004). Prolongation of skin allograft survival after neonatal injection of donor bone marrow and epidermal cells. Plast. Reconstr. Surg. 113, 270–276.
Petruzzo, P., Revillard, J. P., Kanitakis, J., Lanzetta, M., Hakim, N. S., Lefrancois, N., Owen, E., and Dubernard, J. M. (2003). First human double hand transplantation: efficacy of a conventional immunosuppressive protocol. Clin. Transplant. 17, 455–460.
Pilat, N., Baranyi, U., Klaus, C., Jaeckel, E., Mpofu, N., Wrba, F., Golshayan, D., Muehlbacher, F., and Wekerle, T. (2010). Treg-therapy allows mixed chimerism and transplantation tolerance without cytoreductive conditioning. Am. J. Transplant. 10, 751–762.
Pilat, N., and Wekerle, T. (2010). Mechanistic and therapeutic role of regulatory T cells in tolerance through mixed chimerism. Curr. Opin. Organ Transplant. 15, 725–30.
Poirier, N., Azimzadeh, A. M., Zhang, T., Dilek, N., Mary, C., Nguyen, B., Tillou, X., Wu, G., Reneaudin, K., Hervouet, J., Martinet, B., Coulon, F., Allain-Launay, E., Karam, G., Soulillou, J. P., Pierson, R. N. III, Blancho, G., and Vanhove, B. (2010). Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci. Transl. Med. 2, 17ra10.
Posselt, A. M., Bellin, M. D., Tavakol, M., Szot, G. L., Frassetto, L. A., Masharani, U., Kerlan, R. K., Fong, L., Vincenti, F. G., Hering, B. J., Bluestone, J. A., and Stock, P. G. (2010). Islet transplantation in type 1 diabetics using an immunosuppressive protocol based on the anti-LFA-1 antibody efalizumab. Am. J. Transplant. 10, 1870–1880.
Presser, D., Sester, U., Mohrbach, J., Janssen, M., Kohler, H., and Sester, M. (2009). Differential kinetics of effector and regulatory T cells in patients on calcineurin inhibitor-based drug regimens. Kidney Int. 76, 557–566.
Putnam, A. L., Brusko, T. M., Lee, M. R., Liu, W., Szot, G. L., Ghosh, T., Atkinson, M. A., and Bluestone, J. A. (2009). Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes 58, 652–662.
Qin, S., Cobbold, S. P., Pope, H., Elliott, J., Kioussis, D., Davies, J., and Waldmann, H. (1993). “Infectious” transplantation tolerance. Science 259, 974–977.
Qin, S. X., Wise, M., Cobbold, S. P., Leong, L., Kong, Y. C., Parnes, J. R., and Waldmann, H. (1990). Induction of tolerance in peripheral T cells with monoclonal antibodies. Eur. J. Immunol. 20, 2737–2745.
Quatra, F., Lowenberg, D. W., Buncke, H. J., Romeo, O. M., Brooks, D., Buntic, R. F., and Baxter-Lowe, L. A. (2006). Induction of tolerance to composite tissue allograft in a rat model. Microsurgery 26, 573–578.
Raimondi, G., Sumpter, T. L., Matta, B. M., Pillai, M., Corbitt, N., Vodovotz, Y., Wang, Z., and Thomson, A. W. (2010). Mammalian target of rapamycin inhibition and alloantigen-specific regulatory T cells synergize to promote long-term graft survival in immunocompetent recipients. J. Immunol. 184, 624–636.
Riley, J. L., June, C. H., and Blazar, B. R. (2009). Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity 30, 656–665.
Roncarolo, M. G., and Battaglia, M. (2007). Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat. Rev. Immunol. 7, 585–598.
Sachs, D. H., Sykes, M., Kawai, T., and Cosimi, A. B. (2011). Immuno-intervention for the induction of transplantation tolerance through mixed chimerism. Semin. Immunol. 23, 165–173.
Sagoo, P., Ali, N., Garg, G., Nestle, F. O., Lechler, R. I., and Lombardi, G. (2011). Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci. Transl. Med. 3, 83ra42.
Sagoo, P., Lombardi, G., and Lechler, R. I. (2008). Regulatory T cells as therapeutic cells. Curr. Opin. Organ Transplant. 13, 645–653.
Sagoo, P., Perucha, E., Sawitzki, B., Tomiuk, S., Stephens, D. A., Miqueu, P., Chapman, S., Craciun, L., Sergeant, R., Brouard, S., Rovis, F., Jimenez, E., Ballow, A., Giral, M., Rebollo-Mesa, I., Le Moine, A., Braudeau, C., Hilton, R., Gerstmayer, B., Bourcier, K., Sharif, A., Krajewska, M., Lord, G. M., Roberts, I., Goldman, M., Wood, K. J., Newell, K., Seyfert-Margolis, V., Warrens, A. N., Janssen, U., Volk, H. D., Soulillou, J. P., Hernandez-Fuentes, M. P., and Lechler, R. I. (2010). Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans. J. Clin. Invest. 120, 1848–1861.
Saitovitch, D., Bushell, A., Mabbs, D. W., Morris, P. J., and Wood, K. J. (1996). Kinetics of induction of transplantation tolerance with a nondepleting anti-Cd4 monoclonal antibody and donor-specific transfusion before transplantation. A critical period of time is required for development of immunological unresponsiveness. Transplantation 61, 1642–1647.
Saitovitch, D., Bushell, A., Morris, P. J., and Wood, K. J. (1997). Modulation of the CD4 molecule is a major event in the induction of transplantation tolerance with anti-CD4 monoclonal antibodies. Transplant. Proc. 29, 1159.
Saitovitch, D., Bushell, A. R., Morris, P. J., and Wood, K. J. (1995). Induction of transplantation tolerance using a nondepleting anti-CD4 MAb and donor-specific transfusion before transplantation: evidence that a critical period of time is required for the development of immunological unresponsiveness. Transplant. Proc. 27, 117–118.
Sato, K., Yamashita, N., Baba, M., and Matsuyama, T. (2003). Regulatory dendritic cells protect mice from murine acute graft-versus-host disease and leukemia relapse. Immunity 18, 367–379.
Sawyer, G. J., Dalchau, R., and Fabre, J. W. (1993). Indirect T cell allorecognition: a cyclosporine A resistant pathway for T cell help for antibody production to donor MHC antigens. Transpl. Immunol. 1, 77–81.
Scandling, J. D., Busque, S., Dejbakhsh-Jones, S., Benike, C., Millan, M. T., Shizuru, J. A., Hoppe, R. T., Lowsky, R., Engleman, E. G., and Strober, S. (2008). Tolerance and chimerism after renal and hematopoietic-cell transplantation. N. Engl. J. Med. 358, 362–368.
Scandling, J. D., Busque, S., Shizuru, J. A., Engleman, E. G., and Strober, S. (2011). Induced immune tolerance for kidney transplantation. N. Engl. J. Med. 365, 1359–1360.
Seddiki, N., Santner-Nanan, B., Martinson, J., Zaunders, J., Sasson, S., Landay, A., Solomon, M., Selby, W., Alexander, S. I., Nanan, R., Kelleher, A., and Fazekas De St Groth, B. (2006). Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J. Exp. Med. 203, 1693–1700.
Setoguchi, K., Schenk, A. D., Ishii, D., Hattori, Y., Baldwin, W. M. III, Tanabe, K., and Fairchild, R. L. (2011). LFA-1 antagonism inhibits early infiltration of endogenous memory CD8 T cells into cardiac allografts and donor-reactive T cell priming. Am. J. Transplant. 11, 923–935.
Seung, E., Mordes, J. P., Rossini, A. A., and Greiner, D. L. (2003). Hematopoietic chimerism and central tolerance created by peripheral-tolerance induction without myeloablative conditioning. J. Clin. Invest. 112, 795–808.
Siemionow, M. (2011). New minimal immunosuppression strategies for composite tissue allograft transplantation: the cleveland clinic experience. J. Am. Acad. Orthop. Surg. 19(Suppl. 1), S38–S39.
Siemionow, M., and Klimczak, A. (2009). Immunodepletive anti-alpha/beta-TCR antibody in transplantation of composite tissue allografts: Cleveland Clinic research experience. Immunotherapy 1, 585–598.
Siemionow, M., Oke, R., Ozer, K., Izycki, D., and Prajapati, R. (2002a). Induction of donor-specific tolerance in rat hind-limb allografts under antilymphocyte serum and cyclosporine A protocol. J. Hand Surg. Am. 27, 1095–1103.
Siemionow, M., Ortak, T., Izycki, D., Oke, R., Cunningham, B., Prajapati, R., and Zins, J. E. (2002b). Induction of tolerance in composite-tissue allografts. Transplantation 74, 1211–1217.
Siemionow, M., Zielinski, M., Ozmen, S., and Izycki, D. (2005). Intraosseus transplantation of donor-derived hematopoietic stem and progenitor cells induces donor-specific chimerism and extends composite tissue allograft survival. Transplant. Proc. 37, 2303–2308.
Siemionow, M. Z., Izycki, D. M., and Zielinski, M. (2003). Donor-specific tolerance in fully major histocompatibility major histocompatibility complex-mismatched limb allograft transplants under an anti-alphabeta T-cell receptor monoclonal antibody and cyclosporine A protocol. Transplantation 76, 1662–1668.
Spitzer, T. R., Sykes, M., Tolkoff-Rubin, N., Kawai, T., Mcafee, S. L., Dey, B. R., Ballen, K., Delmonico, F., Saidman, S., Sachs, D. H., and Cosimi, A. B. (2011). Long-term follow-up of recipients of combined human leukocyte antigen-matched bone marrow and kidney transplantation for multiple myeloma with end-stage renal disease. Transplantation 91, 672–676.
Stary, G., Klein, I., Bauer, W., Koszik, F., Reininger, B., Kohlhofer, S., Gruber, K., Skvara, H., Jung, T., and Stingl, G. (2011). Glucocorticosteroids modify Langerhans cells to produce TGF-beta and expand regulatory T cells. J. Immunol. 186, 103–112.
Tang, Q., Henriksen, K. J., Boden, E. K., Tooley, A. J., Ye, J., Subudhi, S. K., Zheng, X. X., Strom, T. B., and Bluestone, J. A. (2003). Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J. Immunol. 171, 3348–3352.
Tavazzi, E., Ferrante, P., and Khalili, K. (2011). Progressive multifocal leukoencephalopathy: an unexpected complication of modern therapeutic monoclonal antibody therapies. Clin. Microbiol. Infect. 17, 1776–1780.
Taylor, K. N., Shinde-Patil, V. R., Cohick, E., and Colson, Y. L. (2007). Induction of FoxP3+CD4+25+ regulatory T cells following hemopoietic stem cell transplantation: role of bone marrow-derived facilitating cells. J. Immunol. 179, 2153–2162.
Tran, D. Q., Andersson, J., Hardwick, D., Bebris, L., Illei, G. G., and Shevach, E. M. (2009). Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human FOXP3+ regulatory T cells allows for their purification from expansion cultures. Blood 113, 5125–5133.
Trzonkowski, P., Bieniaszewska, M., Juscinska, J., Dobyszuk, A., Krzystyniak, A., Marek, N., Mysliwska, J., and Hellmann, A. (2011). Expanded T regulatory cells in graft versus host disease in humans – when safe, when efficacious? [E-letter]. Blood (April 6, 2011).
Trzonkowski, P., Bieniaszewska, M., Juscinska, J., Dobyszuk, A., Krzystyniak, A., Marek, N., Mysliwska, J., and Hellmann, A. (2009). First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin. Immunol. 133, 22–26.
Trzonkowski, P., Zilvetti, M., Chapman, S., Wieckiewicz, J., Sutherland, A., Friend, P., and Wood, K. J. (2008). Homeostatic repopulation by CD28-CD8+ T cells in alemtuzumab-depleted kidney transplant recipients treated with reduced immunosuppression. Am. J. Transplant. 8, 338–347.
Tsang, J. Y., Tanriver, Y., Jiang, S., Xue, S. A., Ratnasothy, K., Chen, D., Stauss, H. J., Bucy, R. P., Lombardi, G., and Lechler, R. (2008). Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J. Clin. Invest. 118, 3619–3628.
van Kooten, C., Lombardi, G., Gelderman, K. A., Sagoo, P., Buckland, M., Lechler, R., and Cuturi, M. C. (2011). Dendritic cells as a tool to induce transplantation tolerance: obstacles and opportunities. Transplantation 91, 2–7.
van Maurik, A., Herber, M., Wood, K. J., and Jones, N. D. (2002). Cutting edge: CD4+CD25+ alloantigen-specific immunoregulatory cells that can prevent CD8+ T cell-mediated graft rejection: implications for anti-CD154 immunotherapy. J. Immunol. 169, 5401–5404.
Vella, J. P., Spadafora-Ferreira, M., Murhpy, B., Alexander, S. I., Harmon, W., Carpenter, C. B., and Sayegh, M. H. (1997). Indirect allorecognition of major histocompatibility complex allopeptides in human renal transplant recipients with chronic graft dysfunction. Transplantation 64, 795–800.
Verhasselt, V., Vosters, O., Beuneu, C., Nicaise, C., Stordeur, P., and Goldman, M. (2004). Induction of FOXP3-expressing regulatory CD4pos T cells by human mature autologous dendritic cells. Eur. J. Immunol. 34, 762–772.
Vincenti, F. (2008). Costimulation blockade in autoimmunity and transplantation. J. Allergy Clin. Immunol. 121, 299–306; quiz 307–308.
Vlad, G., Ho, E. K., Vasilescu, E. R., Fan, J., Liu, Z., Cai, J. W., Jin, Z., Burke, E., Deng, M., Cadeiras, M., Cortesini, R., Itescu, S., Marboe, C., Mancini, D., and Suciu-Foca, N. (2007). Anti-CD25 treatment and FOXP3-positive regulatory T cells in heart transplantation. Transpl. Immunol. 18, 13–21.
Vogtenhuber, C., Bucher, C., Highfill, S. L., Koch, L. K., Goren, E., Panoskaltsis-Mortari, A., Taylor, P. A., Farrar, M. A., and Blazar, B. R. (2010). Constitutively active Stat5b in CD4+ T-cells inhibits graft-versus-host disease (GVHD) lethality associated with increased regulatory T-cell (Treg) potency and decreased T effector cell (Teff) responses. Blood 116, 466–474.
Voo, K. S., Wang, Y. H., Santori, F. R., Boggiano, C., Wang, Y. H., Arima, K., Bover, L., Hanabuchi, S., Khalili, J., Marinova, E., Zheng, B., Littman, D. R., and Liu, Y. J. (2009). Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. U.S.A. 106, 4793–4798.
Vukmanovic-Stejic, M., Agius, E., Booth, N., Dunne, P. J., Lacy, K. E., Reed, J. R., Sobande, T. O., Kissane, S., Salmon, M., Rustin, M. H., and Akbar, A. N. (2008). The kinetics of CD4+Foxp3+ T cell accumulation during a human cutaneous antigen-specific memory response in vivo. J. Clin. Invest. 118, 3639–3650.
Waldmann, H., Adams, E., Fairchild, P., and Cobbold, S. (2006). Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol. Rev. 212, 301–313.
Warnecke, G., Bushell, A., Nadig, S. N., and Wood, K. J. (2007). Regulation of transplant arteriosclerosis by CD25+CD4+ T cells generated to alloantigen in vivo. Transplantation 83, 1459–1465.
Weaver, T. A., Charafeddine, A. H., Agarwal, A., Turner, A. P., Russell, M., Leopardi, F. V., Kampen, R. L., Stempora, L., Song, M., Larsen, C. P., and Kirk, A. D. (2009). Alefacept promotes co-stimulation blockade based allograft survival in nonhuman primates. Nat. Med. 15, 746–749.
Webster, K. E., Walters, S., Kohler, R. E., Mrkvan, T., Boyman, O., Surh, C. D., Grey, S. T., and Sprent, J. (2009). In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 206, 751–760.
Wekerle, T., Kurtz, J., Ito, H., Ronquillo, J. V., Dong, V., Zhao, G., Shaffer, J., Sayegh, M. H., and Sykes, M. (2000). Allogeneic bone marrow transplantation with co-stimulatory blockade induces macrochimerism and tolerance without cytoreductive host treatment. Nat. Med. 6, 464–469.
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., Nomura, T., and Sakaguchi, S. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275.
Wood, K. J. (2003). Passenger leukocytes and microchimerism: what role in tolerance induction? Transplantation 75, 17S–20S.
Wood, K. J., Bushell, A., and Hester, J. (2012). Regulatory immune cells in transplantation. Nat. Rev. Immunol. 12, 417–430.
Wood, K. J., Bushell, A. R., Darby, C. R., Pearson, T. C., West, L., and Morris, P. J. (1991). Mechanism of induction of transplantation tolerance using donor antigen and anti-CD4 monoclonal antibody. Transplant. Proc. 23, 133–134.
Wood, K. J., and Goto, R. (2012). Mechanisms of rejection: current perspectives. Transplantation 93, 1–10.
Yamaguchi, T., Hirota, K., Nagahama, K., Ohkawa, K., Takahashi, T., Nomura, T., and Sakaguchi, S. (2007). Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity 27, 145–159.
Yang, X. O., Nurieva, R., Martinez, G. J., Kang, H. S., Chung, Y., Pappu, B. P., Shah, B., Chang, S. H., Schluns, K. S., Watowich, S. S., Feng, X. H., Jetten, A. M., and Dong, C. (2008). Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56.
Zeiser, R., Leveson-Gower, D. B., Zambricki, E. A., Kambham, N., Beilhack, A., Loh, J., Hou, J. Z., and Negrin, R. S. (2008). Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood 111, 453–462.
Zhang, L., Yang, X. Q., Cheng, J., Hui, R. S., and Gao, T. W. (2010). Increased Th17 cells are accompanied by FoxP3(+) Treg cell accumulation and correlated with psoriasis disease severity. Clin. Immunol. 135, 108–117.
Keywords: tolerance, immune regulation, cellular therapy, chimerism, regulatory T cell, clinical trials, transplantation
Citation: Issa F and Wood KJ (2012) Translating tolerogenic therapies to the clinic – where do we stand? Front. Immun. 3:254. doi: 10.3389/fimmu.2012.00254
Received: 24 April 2012; Accepted: 30 July 2012;
Published online: 20 August 2012.
Edited by:
Stephen Paul Cobbold, University of Oxford, UKReviewed by:
Ani Chandraker, Brigham and Women’s Hospital, USAAlf Hamann, Charite University Medicine and German Rheumatism Research Center Berlin, Germany
Copyright: © 2012 Issa and Wood. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Kathryn J. Wood, Transplantation Research Immunology Group, Nuffield Department of Surgical Sciences, Level 6, John Radcliffe Hospital, University of Oxford, Oxford OX3 9DU, UK. e-mail:a2F0aHJ5bi53b29kQG5kcy5veC5hYy51aw==