

- UCD Diabetes Research Centre, UCD Conway Institute, School of Medicine and Medical Sciences, University College Dublin, Dublin, Ireland
The role of inflammation in the pathogenesis of type 2 diabetes mellitus (T2DM) and its associated complications is increasingly recognized. The resolution of inflammation is actively regulated by endogenously produced lipid mediators such as lipoxins, resolvins, protectins, and maresins. Here we review the potential role of these lipid mediators in diabetes-associated pathologies, specifically focusing on adipose inflammation and diabetic kidney disease, i.e., diabetic nephropathy (DN). DN is one of the major complications of T2DM and we propose that pro-resolving lipid mediators may have therapeutic potential in this context. Adipose inflammation is also an important component of T2DM-associated insulin resistance and altered adipokine secretion. Promoting the resolution of adipose inflammation would therefore likely be a beneficial therapeutic approach in T2DM.
Inflammation and Counter-Regulatory Lipid Mediators
The inflammatory response is necessary for effective host defense, although it must eventually dissipate to ensure tissue homeostasis and avoid pathologic conditions such as abscess formation, scarring, fibrosis, and eventual organ failure (Lawrence and Gilroy, 2007). Indeed, compromised resolution has been proposed as an underlying mechanism in many prevalent chronic diseases such as arthritis, diabetes, and atherosclerosis (Serhan et al., 2008; Maderna and Godson, 2009). It is now recognized that the resolution of inflammation is a dynamically regulated process orchestrated by mediators that play important counter-regulatory roles including cytokines, chemokines, and lipid mediators such as the lipoxins (LXs), resolvins, and protectins (Serhan, 2009). These mediators reduce vascular permeability and inhibit polymorphonuclear cell (PMN) recruitment, while promoting recruitment of monocytes and stimulating efferocytosis (Serhan et al., 2008). It has also been proposed that pro-resolving lipids stimulate lymphatic drainage of leukocytes (Arita et al., 2005b). Interestingly, the signaling pathways initially inducing prostaglandin (PG)E2 and PGD2 formation and thus the onset of inflammation, may actively switch the production of lipid mediators from pro-inflammatory to pro-resolving by inducing 5-lipoxygenases (LO) necessary for production of LXs, protectins, and resolvins (Serhan and Savill, 2005). In this way physiological inflammation programs its own resolution and promotes tissue homeostasis (Levy et al., 2001).
Lipoxins
The LXs are produced endogenously at sites of inflammation as counter-regulatory lipid mediators with anti-inflammatory, pro-resolving, and anti-fibrotic bioactions (Serhan et al., 2008; Maderna and Godson, 2009). LXs are typically generated by transcellular metabolism between neutrophils, platelets, and resident tissue cells, such as epithelial cells (Lefer et al., 1988; Serhan, 2007), through the sequential action of 5-LO and either 12-LO or 15-LO (Serhan, 2005; Parkinson, 2006). LXs limit leukocyte chemotaxis (Lee et al., 1989) and activation of neutrophils and eosinophils (Bandeira-Melo et al., 2000), while stimulating Mφ efferocytosis of apoptotic cells (Godson et al., 2000; Mitchell et al., 2002; Reville et al., 2006). Lipoxin A4 (LXA4) and its positional isomer lipoxin B4 (LXB4) are the principal LX species found in mammals. Although the LXB4 receptor remains to be identified, the LXA4 receptor FPR2/ALX is expressed on cells of diverse lineage, including fibroblasts (Wu et al., 2006a), renal mesangial cells (McMahon et al., 2002; Mitchell et al., 2004), and epithelial cells (Nascimento-Silva et al., 2007). LXs are protective in several experimental models of disease, e.g., inflammatory bowel diseases (Fiorucci et al., 2004), periodontal disease (Serhan, 2004; Kantarci and Van Dyke, 2005; Kantarci et al., 2006), and cardiovascular disease (Serhan, 2005). LXs have also been reported to act as vasodilators (von der Weid et al., 2004) and may reprogram Mφs from a classically activated (M1) phenotype to a spectrum of alternative activation (Mitchell et al., 2002). The bioactions of LXs are summarized in Table 1. The impact of LXs in maintaining the exquisite equilibrium between effective host defense and homeostasis is remarkably illustrated by the fact that over production of LXs may compromise host defense to pathogens. In the case of Mycobacterium tuberculosis (M.tb), increased LXA4 production is associated with decreased TNF-α activity and results in an inadequate inflammatory response (Tobin et al., 2010). Conversely, LXA4 increases survival rate in Toxoplasma gondii infection where a compromised immune response due to diminished LO activity and LX biosynthesis is detrimental (Aliberti, 2005).
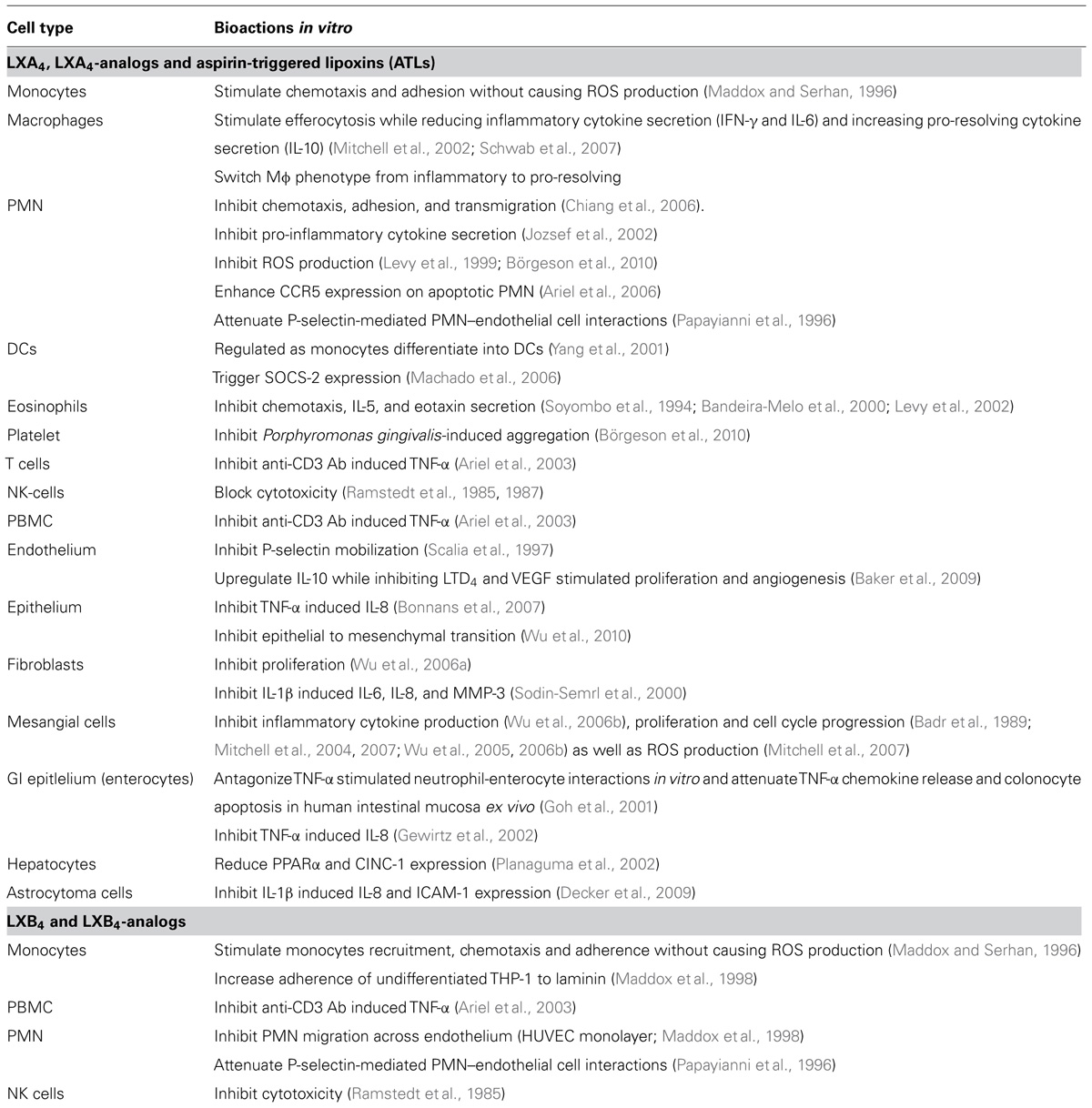
TABLE 1. Lipoxin induced bioactions.
Lipoxin Receptors and Synthetic Lipoxin Analogs
The principal LXA4 receptor is FPR2/ALX, which has been identified and cloned in numerous cell types, including monocytes and Mφs (Maddox et al., 1997), T cells (Ariel et al., 2003), synovial fibroblasts (Sodin-Semrl et al., 2000), renal mesangial cells (McMahon et al., 2002), and enterocytes (Gronert et al., 1998). In contrast to conventional GPCRs, which typically show very specific ligand binding, the FPR2/ALX receptor binds pleiotropic ligands, both lipids and small peptides, such as acute phase proteins (Chiang et al., 2000), and may elicit ligand-dependent pro-inflammatory or anti-inflammatory responses (Chiang et al., 2006; Maderna and Godson, 2009). Krishnamoorthy et al. (2010) recently found that LXA4 also interacts with another G-protein coupled receptor, namely GPR32.
LXA4 undergoes rapid inactivation in vivo, primarily by PG dehydrogenase-mediated oxidation and reduction (Serhan et al., 1995) and efforts have been made to design chemically stable LX analogs. Because the three-dimensional molecular structure of the FPR2/ALX receptor is as of yet unknown, designing LXA4 analogs is based on experimentally discovered structure/function relationship of LXA4. The LXA4 molecule can be considered in three regions; the lower chain, the upper chain, and the tetraene side chain (Duffy and Guiry, 2010). The first generation LXA4 analogs carry modifications in the lower alkyl chain, to increase metabolic stability and prevent oxidation (Clish et al., 1999). The second generation analogs are collectively referred to as 3-oxa-LXA4 and were constructed carrying modifications in the upper chain (Petasis et al., 2005), replacing the C3 methylene group with an oxygen molecule (Guilford and Parkinson, 2005). The third generation LXA4 analogs are characterized by replacement of the triene structure with a benzene ring (O’Sullivan et al., 2007; Petasis et al., 2008). Importantly, the o-[9, 12]-Benzo-15-epi-LXA4 has been shown to activate the FPR/ALX receptor in a similar manner to native LXA4, using an engineered β-arrestin system (Sun et al., 2009).
Resolvins, Protectins, and Marseins
Resolvins, protectins, and maresins are pro-resolving lipids discovered by Serhan et al. (2000) through sophisticated lipidomic analysis of resolution phase exudates in the murine dorsal air pouch model. Resolvins may be divided into the E series (RvEs) and D series (RvDs), which are generated from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), respectively, the most common forms of ω-3 PUFA. Similarly, protectins and maresins are generated from DHA. Like the LXs, resolvins are generated in a transcellular manner by the sequential action of LO. Protectins and maresins on the other hand are generated by single cells, but also through the action of LO. In neutrophils RvE1 has been shown to bind the GPCR LTB4 receptor BLT1 with a Kd of 45 nM (Arita et al., 2007), whereas in Mφ and dendritic cells RvE1 bind ChemR23 with a Kd of 11.3 ± 5.4 nM and Bmax indicating approximately 4,200 binding sites per cell (Arita et al., 2005b; Kohli and Levy, 2009). RvD1 has also been reported to interact both with FPR2/ALX and GPR32 in phagocytes (Krishnamoorthy et al., 2010). As of yet it is not entirely clear which receptor the protectins and maresins act through, although PD1 has a high affinity surface binding site on human PMN and retinal pigment epithelium cells (Bannenberg and Serhan, 2010). Resolvins, protectins, and maresins all display potent anti-inflammatory and pro-resolving effects inhibiting production of pro-inflammatory mediators, regulating neutrophil trafficking and promoting efferocytosis (Schwab et al., 2007; Serhan, 2009). The effects of these lipid mediators are summarized in Table 2.

TABLE 2. Resolvin, protectin, and maresin induced bioactions.
Inflammation and Type 2 Diabetes Mellitus
Diabetes mellitus (DM) is a serious metabolic disorder of glucose homeostasis reflecting destruction of the β-cells of the pancreas and subsequent lack of insulin production (type 1 DM, T1DM) or decreased target organ sensitivity to insulin and β-cell dysfunction (type 2 DM, T2DM). T2DM is defined as having a fasting plasma glucose ≥7.0 mmol/l and affects over 90% of diabetics, or an estimated 285 million people globally (Cusi, 2010). T2DM imposes significant socioeconomic burdens through its many diabetes-associated complications. These can be divided into microvascular complications [diabetic nephropathy (DN), neuropathy, and retinopathy] and macrovascular complications [atherosclerosis, ischemic heart disease, stroke, and peripheral vascular disease often resulting in amputations] (Wild et al., 2004). Risk factors of T2DM include genetic preposition, ethnicity, high blood pressure, and high cholesterol, but obesity is frequently cited as the primary cause.
The role of inflammation in diabetes is becoming more evident and elevated circulating interleukin (IL)-1β, IL-6, and C-reactive protein (CRP) are predictive of T2DM (Navarro and Mora, 2006; de Luca and Olefsky, 2008; Donath and Shoelson, 2011). These inflammatory markers are primarily derived from the adipose tissue and the liver. The hypothesis that the pathogenesis of T2DM reflects an inflammatory disorder is supported by preclinical studies and clinical trials using anti-inflammatory agents (Donath and Shoelson, 2011). Examples of these include IL-1β receptor blockers, anti-TNF-α and IL-6 therapies, as well as the use of salsalate. We will now briefly discuss current attempts to use anti-inflammatory therapeutics to attenuate the pathology of diabetes.
Interleukin-1β is a key regulator of inflammation both in T1DM and T2DM and has been shown to induce pancreatic β-cell apoptosis and exacerbate the systemic inflammation associated with diabetes, for instance by augmenting adipocyte TNF-α and IL-6 production (Akash et al., 2012). Patients with T2DM display increased IL-1β levels (Boni-Schnetzler et al., 2008), while its naturally occurring IL-1 receptor antagonist (IL-1Ra) is diminished (Maedler et al., 2004). Interest has been directed toward using IL-1Ra as a therapeutic in T2DM. Clinical trials show that the IL-1Ra anakinra improves glycemia and β-cell secretory functions, while attenuating systemic inflammation (Donath and Shoelson, 2011). For instance, anakinra administered over a 13-week period in T2DM patients increased insulin production, while glycosylated hemoglobin, i.e., HbA1c and the inflammatory marker CRP were significantly reduced (Larsen et al., 2007). The limitation with IL-1Ra lies in its short half-life, but successful attempts have been made to increase its stability by fusing IL-1Ra with peptides such as human serum albumin (HLA) or elastin-like polypeptides (ELPs), although these compounds remain to be tested in diabetic models (Akash et al., 2012).
TNF-α is also implicated in the pathogenesis of insulin resistance (IR) and its expression correlates with reduced insulin-stimulated glucose disposal (Kern et al., 2001). TNF-α is elevated both in obese rodents (Uysal et al., 1997) and obese humans (Hotamisligil et al., 1995; Kern et al., 2001) and furthermore decreases upon weight loss (Kern et al., 1995). TNF-α– / – ob/ob mice have significantly improved insulin sensitivity (Uysal et al., 1997) and obese mice lacking the TNF-α receptor are protected from high fat diet induced IR (Romanatto et al., 2009). However, in humans TNF-α neutralizing antibodies does not appear to improve insulin sensitivity in obese subjects (Ofei et al., 1996; Rosenvinge et al., 2007). Nevertheless, TNF-α blockers are often used to treat rheumatoid arthritis (RA) and it was recently reported that obese RA patients receiving TNF-α blockers displayed improved fasting glucose and increased circulating adiponectin levels (Stanley et al., 2010), possibly warranting more studies in the field. IL-6 is also an important inflammatory mediator in diabetes and increased levels correlate with IR (Pradhan et al., 2001), although it appears to have a dual role. Whereas IL-6 causes IR in adipocytes (Rotter et al., 2003) and anti-IL-6 therapy over a 6 month period diminished HbA1c in diabetic RA patients (Ogata et al., 2011), the IL-6 derived from skeletal muscle during exercise appears beneficial (Pedersen et al., 2003). The use of anti-IL-6 blockers as an anti-inflammatory therapeutic in diabetes has therefore been debated. Salsalate on the other hand is a very interesting drug in the context of diabetes and has been shown to reduce CRP, FFA, and triglycerides while increasing insulin sensitivity and adiponectin levels (Koska et al., 2009; Goldfine et al., 2010). Salsalate may, however, cause gastric irritation and should be used with caution in pregnancy (Torloni et al., 2006; Chyka et al., 2007). Collectively these studies indicate the potential of using anti-inflammatory therapeutics to attenuate T2DM.
Resolution of Adipose Inflammation in Type 2 Diabetes Mellitus
There is a growing appreciation that adipose tissue is not merely an insulating energy store but is actually an endocrine organ regulating appetite, glucose and lipid metabolism, blood pressure, inflammation, and immune function (Kershaw and Flier, 2004). Adipose tissue has been shown to play a particularly important role in the systemic inflammation associated with obesity, IR, and diabetes. Factors such as prolonged obesity or aging cause a state of systemic low-grade inflammation, which induces monocyte recruitment to the adipose tissue. Adipose tissue is a source of pro-inflammatory cytokines and adipose tissue Mφ (ATM) derived TNF-α, IL6, and IL-1β contribute to adipose IR and exacerbates systemic inflammation (Lumeng et al., 2007b). Promoting resolution of adipose inflammation would likely be a beneficial therapeutic approach, reducing the risk of developing obesity-associated complications, such as IR and T2DM (Donath and Shoelson, 2011).
Given the spectrum of anti-inflammatory and pro-resolution bioactions of LXs and other counter-regulatory lipid mediators, these may provide a potential intervention to attenuate adipose inflammation (Gonzalez-Periz and Claria, 2010). We recently reported a role of LXA4 in adipose inflammation, culturing adipose explants of aging mice as an ex vivo model of adipose inflammation (Börgeson et al., 2012). We confirmed that LXA4 increased expression of critical components of insulin sensitivity, including the glucose transporter GLUT-4 and IRS-1, consistent with restoring insulin sensitivity in the tissue. Furthermore, LXA4 decreased IL-6 secretion while increasing production of the pro-resolving IL-10, suggesting that LXA4 acted in a pro-resolving manner (Börgeson et al., 2012). Indeed, IL-10 inversely correlates with T2DM and has been shown to inhibit IL-6-induced IR, attenuate MCP-1 secretion, and promote GLUT-4 and IRS-1 expression (Lumeng et al., 2007a; Gonzalez-Periz and Claria, 2010). The study also demonstrated that LXA4 partially rescued MΦ-inhibited adipose glucose uptake in vitro (Börgeson et al., 2012). Inflammatory MΦs are a key component of augmented adipose IR (Lumeng et al., 2007b; Cusi, 2010; Spencer et al., 2010). Importantly, LXA4-mediated reversal of insulin desensitization correlated with restored adipose Akt activation, which is necessary for translocation of the glucose sensitizing GLUT-4 receptor from the cytosol to the plasma membrane (Börgeson et al., 2012). Interestingly, RvD1 also increased insulin-stimulated pAkt in adipose tissue of obese db/db mice (Hellmann et al., 2011). Furthermore, LXA4 inhibited MΦ TNF-α production, which is an important cytokine previously demonstrated to inhibit adipose glucose uptake in vitro (Gao et al., 2003). LXA4 also inhibited MCP-1 secretion, though the importance of MCP-1 in adipose inflammation has been debated (Chen et al., 2005; Inouye et al., 2007). The reduction of inflammatory cytokines may suggest that LXA4 promoted restoration of insulin sensitivity by altering MΦ phenotype toward resolution. Finally, LXA4 also appeared to have a direct impact on adipocytes as it rescued TNF-α-induced desensitization to insulin-stimulated Akt activation, which also correlated with increased GLUT-4 translocation.
The beneficial effects of ω-3 PUFA, RvE1, and PD1 have also been shown in ob/ob mice (Gonzalez-Periz et al., 2009). Both ω-3 PUFA enriched diet and intraperitoneal injections of RvE1 increased expression of genes involved in glucose transport (GLUT-4) and insulin signaling (IRS-1), as well as genes involved in insulin sensitivity (PPARγ). Similar to ω-3 PUFA, RvE1 increased adiponectin levels, as did PD1 when incubated with adipose explants from ob/ob mice (Gonzalez-Periz et al., 2009). Additional studies show that RvD1 decrease accumulation of ATMs and improve insulin sensitivity while reducing fasting blood glucose in db/db diabetic mice (Hellmann et al., 2011). Interestingly, the total number of ATMs remained unaltered with RvD1 treatment, but the ratio of M2:M1 increased. The number of adipose crown like structures (CLS) in obese animals was also reduced by 50–60% (Hellmann et al., 2011) and RvD1 significantly increased circulating adiponectin and adipose phosphorylation of AMPK. The study also reports diminished IL-6 secretion (Hellmann et al., 2011), which has previously been shown to suppress adiponectin in 3T3-L1 adipocytes (Fasshauer et al., 2003) and may explain the restored adiponectin levels, which in turn have been shown to increase insulin sensitivity (Kristiansen and Mandrup-Poulsen, 2005; Kadowaki et al., 2006).
Inflammation and Diabetic Nephropathy
Diabetic nephropathy presents a particularly important problem as it develops in 25–40% of diabetic patients and is the major cause of end-stage kidney disease (Ritz et al., 1999). DN is a type of chronic kidney disease (CKD) rising in prevalence in concert with chronic DM in susceptible individuals. In addition to being the leading cause of renal failure, T2DM is also an independent risk factor in the development of cardiovascular disease (Syed and Khan, 2011). DN reflects the convergence of inflammatory, metabolic, and hemodynamic factors. Inflammation causes glomerulosclerosis, tubular atrophy, damage to renal vasculature, and fibrosis (Ferenbach et al., 2007). Renal matrix accumulation arises in response to paracrine and autocrine mediators produced by resident and infiltrating cells, such as mesangial cells and Mφs.
Promoting inflammatory resolution is likely an attractive approach when trying to prevent renal fibrosis and CKD (Börgeson and Godson, 2010). The mechanisms by which resolution of renal inflammation occurs naturally and how they are subverted in disease are only beginning to be understood. Important components include efferocytosis of apoptotic cells and a change of the cytokine milieu from pro-inflammatory to anti-inflammatory/pro-resolving (Ferenbach et al., 2007; Börgeson and Godson, 2010). Biphasic regulation of renal inflammation and NF-κB also appears important, where the first peak mediated through p65/p50 heterodimers induces inflammation through pro-inflammatory mediators such as MCP-1 and RANTES. The second peak on the other hand (p50/p50 homodimers) promotes resolution by downregulating MCP-1/CCL1, RANTES/CCL5, and TNF-α (Panzer et al., 2009), while inducing expression of pro-resolving IL-10 (Cao et al., 2006). Similarly, to other pathologies it also appears that the phenotype of Mφs is important in CKD (Wada et al., 2004; Sung et al., 2007). Whereas M1 Mφs are detrimental, the M2a and perhaps even more so the M2c phenotype is beneficial (Wang and Harris, 2011).
Mφs play an important role in DN as previously reported by Tesch (2008), 2010. Mφs increase production of ROS, pro-inflammatory cytokines, and pro-fibrotic growth factors that contribute to the formation of myo-fibroblasts. Mφs also appear to directly activate fibroblasts to a pro-fibrotic (myo-fibroblast) phenotype through secretion of galectin-3 (Henderson et al., 2008). Inhibition of Mφ recruitment has been suggested to attenuate disease in several models of renal fibrosis with varying efficacy (Wada et al., 2004; Sung et al., 2007). For instance, MCP-1– / – mice are protected against renal injury in a model of T1DM (Chow et al., 2006) and furthermore urinary levels of MCP-1 are predictive of renal injury in humans and have been proposed as a diagnostic marker of progressive diabetic kidney disease (Tesch, 2008). There is a growing appreciation that the plasticity of Mφs is an important factor in disease progression (Duffield, 2011; Wang and Harris, 2011) and that Mφs also contribute to the resolution of renal inflammation. For instance, Mφ efferocytosis of apoptotic cells is coupled to the generation of anti-inflammatory mediators such as IL-10 (Ricardo et al., 2008). To this effect, re-programming Mφs ex vivo toward a M2 phenotype (IL-4/IL-13 stimulation) provides protection in models of renal disease, whereas the M1 phenotype (LPS stimulation) is detrimental (Wang et al., 2007). Additional research suggests that M2a and M2c phenotypes are both renoprotective, but that the latter appears to be the more effective (Wang and Harris, 2011).
Experimental Therapeutics and DN
Diabetic nephropathy is a chronic disease and current therapeutics primarily focus on glycemic and blood-pressure control through drugs targeting the renin–angiotensin system (RAS), such as angiotensin-converting-enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs). However, these treatment regimes only slow the progression of the disease, but do not halt or reverse it. Furthermore, prolonged use of RAS inhibitors may induce hyperkalemia, reduction in systemic blood pressure and decreased renal blood flow. Therefore, there is a profound need for novel therapeutic strategies in this field and the search is ongoing (Decleves and Sharma, 2010; Shepler et al., 2012). Examples of experimental therapeutics that show potential include bardoxolone methyl, which in clinical trials increases estimated glomerular filtration rate (eGFR) and creatinine clearance, while inhibiting inflammation in diabetic patients with stage 3b-4 CKD (Pergola et al., 2011a,b; Thomas and Cooper, 2011). Pirfenidone is an oral anti-fibrotic and anti-inflammatory agent which shows therapeutic potential in DN, although it was initially developed for treatment of idiopathic pulmonary fibrosis. In a randomized, double blind study pirfenidone increased eGFR and decreased markers of inflammation (TNF, INF-γ, and IL-1; Sharma et al., 2011) and has also demonstrated anti-fibrotic potential in both in vitro (Hewitson et al., 2001) and in vivo (RamachandraRao et al., 2009; Takakuta et al., 2010) models of renal disease. Vitamin D analogs, e.g., paricalcitol, may also be renoprotective agents through negatively regulating the RAS system and attenuation of renal fibrosis in rodent unilateral ureteric obstruction (UUO) models inhibiting accumulation of ECM as well as TGF-β1 and MCP-1 gene expression signaling (Li and Batuman, 2009; Li, 2010). Vitamin D analogs have also been suggested to prevent podocyte injury by promoting expression of slit diaphragm proteins (Li, 2011) and shows promising potential in emerging clinical trials reducing proteinuria in CKD patients (Li, 2010).
As inflammation is a common denominator in CKD and a hallmark of DN, pro-resolving therapeutics may have potential benefit. We recently reported that LXs are protective in CKD, as pre-treatment with LXA4 and benzo-LXA4 modulates inflammation and fibrosis in early UUO-induced injury (Börgeson et al., 2011a). UUO is an established model of progressive tubulointerstitial fibrosis and inflammation, relevant to CKD of diverse etiologies, including DN. UUO induces marked Mφ infiltration, tubular cell death, fibroblast activation, and possible phenotypic transition of resident renal cells characteristic of progressive renal fibrosis (Higgins et al., 2007; Chevalier et al., 2009). Benzo-LXA4 and LXA4 attenuated UUO-induced fibrotic responses such as collagen accumulation by inhibiting collagen-1α2 gene expression, expression of collagen chaperone HSP47 and TGF-β1 signaling pathways (Börgeson et al., 2011a). Interestingly, RvD1 has also been demonstrated to attenuate collagen deposition in a murine model of renal ischemia reperfusion (Duffield et al., 2006). Specifically, LXs inhibited UUO-induced TGF-β1 canonical (Smad2) and non-canonical (Akt, Erk, and p38MAPK) signaling pathways, translating to reduced pro-fibrotic signaling (Börgeson et al., 2011a). Although LXA4 did not alter the expression of TGF-β1, it did inhibit expression of MMP2 and CTGF. This is indeed noteworthy since MMP2 activates latent TGF-β1 and is a major driver of TGF-β1-mediated fibrosis. The LXA4 mediated reduction of CTGF, both at mRNA and protein levels, would likely result in reduced fibrotic responses. The anti-fibrotic effect of LXs has been demonstrated in several in vitro systems, inhibiting proliferation and cell cycle progression in mesangial cells (Börgeson and Godson, 2010). Recent data also demonstrate protection by RvE and RvD in murine UUO (Qu et al., 2012). LXs also appeared to shift Mφ phenotype and displayed significant pro-resolving actions in UUO-induced CKD. Whereas the total number of Mφ and MCP-1 remained unaltered, LX treated animals displayed decreased pro-inflammatory IFN-γ and TNF-α cytokines and increased pro-resolving IL-10 levels (Börgeson et al., 2011a). Indeed, it appeared that the LXs induced a shift the Mφ phenotype toward an early stage M2c reparative phenotype, based on the high IL-10 expression induced by benzo-LXA4, although TGF-β1 remained unaffected (Börgeson et al., 2011a).
Micro RNA (miRNA) may also prove an important therapeutic target in DN, as they have demonstrated importance in CKD pathogenesis (Kato et al., 2007; Wang et al., 2008; Long et al., 2010). We recently reported that whereas TGF-β1 downregulates expression of the miRNA let-7c in renal epithelia, LXA4 enhances let-7c expression, and attenuates TGF-β1 fibrotic responses as let-7c targets expression of the TGF-βR1 (Brennan et al., in revision). Importantly, LXs inhibit ROS production (Börgeson and Godson, 2010; Börgeson et al., 2011b; Wu et al., 2012), which may be analogous to the antioxidant effect of bardoxolone methyl (Rojas-Rivera et al., 2012). Indeed, bardoxolone methyl is an antioxidant inflammation modulator (AIM) compound, targeting Nrf2 which is a master regulator of the antioxidant response. Interestingly, LXA4 has been shown to inhibit LPS-mediated ROS production and to downregulate Nrf2 protein levels in human umbilical vein endothelial cells (HUVECs; Pang et al., 2011).
Conclusion
Increasing evidence supports the role of chronic inflammation in the pathogenesis of T2DM and associated complications such as DN. Pro-resolving mediators, such as LXs, resolvins, and protectins, attenuate diabetes-related pathologies, including kidney disease and adipose inflammation. Thus promoting the resolution of inflammation through use of these lipids may provide a novel therapeutic strategy in the fight against diabetes-related pathologies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in Professor Godson’s laboratory is supported by Science Foundation Ireland, the Health Research Board and the Government of Ireland Programme for Research in Third Level Institutions. Emma Börgeson was supported by an Embark IRCSET PhD scholarship.
Abbreviations
ACE inhibitors, angiotensin-converting-enzyme inhibitors; AIM, antioxidant inflammation modulator; ARBs, angiotensin receptor blockers; ATMs, adipose tissue Mφs; CKD, chronic kidney disease; CLS, crown like structures; CRP, C-reactive protein; DHA, docosahexaenoic acid; DN, diabetic nephropathy; eGFR, estimated glomerular filtration rate; EPA, eicosapentaenoic acid; HUVECs, human umbilical vein endothelial cells; IL, interleukin; LO, lipoxygenase; LXA4, lipoxin A4 (S),6(R),15,Trihydroxyeicosa-7E,9E,11Z,13E-tetraenoic acid; M.tb, Mycobacterium tuberculosis; maresins, MΦ mediators in resolving inflammation; miRNA, micro RNA; Mφ, macrophages; PG, prostaglandin; PMN, polymorphonuclear cell; RA, rheumatoid arthritis; RAS, renin–angiotensin system; Rvs, resolvins; T2DM, type 2 diabetes mellitus; UUO, unilateral ureteric obstruction.
References
Akash, M. S., Shen, Q., Rehman, K., and Chen, S. (2012). Interleukin-1 receptor antagonist: a new therapy for type 2 diabetes mellitus. J. Pharm. Sci. 101, 1647–1658.
Aliberti, J. (2005). Host persistence: exploitation of anti-inflammatory pathways by Toxoplasma gondii. Nat. Rev. Immunol. 5, 162–170.
Ariel, A., Chiang, N., Arita, M., Petasis, N. A., and Serhan, C. N. (2003). Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J. Immunol. 170, 6266–6272.
Ariel, A., Fredman, G., Sun, Y. P., Kantarci, A., Van Dyke, T. E., Luster, A. D., et al. (2006). Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat. Immunol. 7, 1209–1216.
Ariel, A., Li, P. L., Wang, W., Tang, W. X., Fredman, G., Hong, S., et al. (2005). The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J. Biol. Chem. 280, 43079–43086.
Arita, M., Bianchini, F., Aliberti, J., Sher, A., Chiang, N., Hong, S., et al. (2005a). Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201, 713–722.
Arita, M., Yoshida, M., Hong, S., Tjonahen, E., Glickman, J. N., Petasis, N. A., et al. (2005b). Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. U.S.A. 102, 7671–7676.
Arita, M., Ohira, T., Sun, Y. P., Elangovan, S., Chiang, N., and Serhan, C. N. (2007). Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 178, 3912–3917.
Badr, K. F., Deboer, D. K., Schwartzberg, M., and Serhan, C. N. (1989). Lipoxin A4 antagonizes cellular and in vivo actions of leukotriene D4 in rat glomerular mesangial cells: evidence for competition at a common receptor. Proc. Natl. Acad. Sci. U.S.A. 86, 3438–3442.
Baker, N., O’Meara, S. J., Scannell, M., Maderna, P., and Godson, C. (2009). Lipoxin A4: anti-inflammatory and anti-angiogenic impact on endothelial cells. J. Immunol. 182, 3819–3826.
Bandeira-Melo, C., Bozza, P. T., Diaz, B. L., Cordeiro, R. S., Jose, P. J., Martins, M. A., et al. (2000). Cutting edge: lipoxin (LX) A4 and aspirin-triggered 15-epi-LXA4 block allergen-induced eosinophil trafficking. J. Immunol. 164, 2267–2271.
Bannenberg, G., and Serhan, C. N. (2010). Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochim. Biophys. Acta 1801, 1260–1273.
Boni-Schnetzler, M., Thorne, J., Parnaud, G., Marselli, L., Ehses, J. A., Kerr-Conte, J., et al. (2008). Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta-cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 93, 4065–4074.
Bonnans, C., Gras, D., Chavis, C., Mainprice, B., Vachier, I., Godard, P., et al. (2007). Synthesis and anti-inflammatory effect of lipoxins in human airway epithelial cells. Biomed. Pharmacother. 61, 261–267.
Börgeson, E., Docherty, N. G., Murphy, M., Rodgers, K., Ryan, A., O’Sullivan, T.P., et al. (2011a). Lipoxin A(4) and benzo-lipoxin A(4) attenuate experimental renal fibrosis. FASEB J. 25, 2967–2979.
Börgeson, E., Lonn, J., Bergstrom, I., Brodin, V. P., Ramstrom, S., Nayeri, F., et al. (2011b). Lipoxin A(4) inhibits Porphyromonas gingivalis-induced aggregation and reactive oxygen species production by modulating neutrophil-platelet interaction and CD11b expression. Infect. Immun. 79, 1489–1497.
Börgeson, E., and Godson, C. (2010). Molecular circuits of resolution in renal disease. Sci. World J. 10, 1370–1385.
Börgeson, E., Lonn, J., Bergstrom, I., Brodin, V. P., Ramstrom, S., Nayeri, F., et al. (2010). Lipoxin A4 inhibits Porphyromonas gingivalis-induced aggregation and reactive oxygen species production by modulating neutrophil-platelet interaction and CD11b expression. Infect. Immun. 79, 1489–1497.
Börgeson, E., Mcgillicuddy, F. C., Harford, K. A., Corrigan, N., Higgins, D. F., Maderna, P., et al. (2012). Lipoxin A4 attenuates adipose inflammation. FASEB J. 26, 4287–4294.
Campbell, E. L., Louis, N. A., Tomassetti, S. E., Canny, G. O., Arita, M., Serhan, C. N., et al. (2007). Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J. 21, 3162–3170.
Cao, S., Zhang, X., Edwards, J. P., and Mosser, D. M. (2006). NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J. Biol. Chem. 281, 26041–26050.
Chen, A., Mumick, S., Zhang, C., Lamb, J., Dai, H., Weingarth, D., et al. (2005). Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes. Res. 13, 1311–1320.
Chevalier, R. L., Forbes, M. S., and Thornhill, B. A. (2009). Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 75, 1145–1152.
Chiang, N., Fierro, I. M., Gronert, K., and Serhan, C. N. (2000). Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 191, 1197–1208.
Chiang, N., Serhan, C. N., Dahlen, S. E., Drazen, J. M., Hay, D. W., Rovati, G. E., et al. (2006). The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 58, 463–487.
Chow, F. Y., Nikolic-Paterson, D. J., Ozols, E., Atkins, R. C., Rollin, B. J., and Tesch, G. H. (2006). Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 69, 73–80.
Chyka, P. A., Erdman, A. R., Christianson, G., Wax, P. M., Booze, L. L., Manoguerra, A. S., et al. (2007). Salicylate poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin. Toxicol. (Phila.) 45, 95–131.
Clish, C. B., O’Brien, J. A., Gronert, K., Stahl, G. L., Petasis, N. A., and Serhan, C. N. (1999). Local and systemic delivery of a stable aspirin-triggered lipoxin prevents neutrophil recruitment in vivo. Proc. Natl. Acad. Sci. U.S.A. 96, 8247–8252.
Cusi, K. (2010). The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr. Diab. Rep. 10, 306–315.
Decker, Y., Mcbean, G., and Godson, C. (2009). Lipoxin A4 inhibits IL-1beta-induced IL-8 and ICAM-1 expression in 1321N1 human astrocytoma cells. Am. J. Physiol. Cell Physiol. 296, C1420–C1427.
Decleves, A. E., and Sharma, K. (2010). New pharmacological treatments for improving renal outcomes in diabetes. Nat. Rev. Nephrol. 6, 371–380.
de Luca, C., and Olefsky, J. M. (2008). Inflammation and insulin resistance. FEBS Lett. 582, 97–105.
Dona, M., Fredman, G., Schwab, J. M., Chiang, N., Arita, M., Goodarzi, A., et al. (2008). Resolvin E1, an EPA-derived mediator in whole blood, selectively counterregulates leukocytes and platelets. Blood 112, 848–855.
Donath, M. Y., and Shoelson, S. E. (2011). Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107.
Duffield, J. S. (2011). Macrophages in kidney repair and regeneration. J. Am. Soc. Nephrol. 22, 199–201.
Duffield, J. S., Hong, S., Vaidya, V. S., Lu, Y., Fredman, G., Serhan, C. N., et al. (2006). Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 177, 5902–5911.
Duffy, C. D., and Guiry, P. J. (2010). Recent advances in the chemistry and biology of stable synthetic Lipoxin analogues. Med. Chem. Comm. 1, 249–265.
Fasshauer, M., Kralisch, S., Klier, M., Lossner, U., Bluher, M., Klein, J., et al. (2003). Adiponectin gene expression and secretion is inhibited by interleukin-6 in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 301, 1045–1050.
Ferenbach, D., Kluth, D. C., and Hughes, J. (2007). Inflammatory cells in renal injury and repair. Semin. Nephrol. 27, 250–259.
Fiorucci, S., Wallace, J. L., Mencarelli, A., Distrutti, E., Rizzo, G., Farneti, S., et al. (2004). A beta-oxidation-resistant lipoxin A4 analog treats hapten-induced colitis by attenuating inflammation and immune dysfunction. Proc. Natl. Acad. Sci. U.S.A. 101, 15736–15741.
Fredman, G., Van Dyke, T. E., and Serhan, C. N. (2010). Resolvin E1 regulates adenosine diphosphate activation of human platelets. Arterioscler. Thromb. Vasc. Biol. 30, 2005–2013.
Gao, Z., Zuberi, A., Quon, M. J., Dong, Z., and Ye, J. (2003). Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. J. Biol. Chem. 278, 24944–24950.
Gewirtz, A. T., Collier-Hyams, L. S., Young, A. N., Kucharzik, T., Guilford, W. J., Parkinson, J. F., et al. (2002). Lipoxin a4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. J. Immunol. 168, 5260–5267.
Godson, C., Mitchell, S., Harvey, K., Petasis, N. A., Hogg, N., and Brady, H. R. (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 164, 1663–1667.
Goh, J., Baird, A. W., O’Keane, C., Watson, R. W., Cottell, D., Bernasconi, G., et al. (2001). Lipoxin A(4) and aspirin-triggered 15-epi-lipoxin A(4) antagonize TNF-alpha-stimulated neutrophil-enterocyte interactions in vitro and attenuate TNF-alpha-induced chemokine release and colonocyte apoptosis in human intestinal mucosa ex vivo. J. Immunol. 167, 2772–2780.
Goldfine, A. B., Fonseca, V., Jablonski, K. A., Pyle, L., Staten, M. A., and Shoelson, S. E. (2010). The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann. Intern. Med. 152, 346–357.
Gonzalez-Periz, A., and Claria, J. (2010). Resolution of adipose tissue inflammation. Sci. World J. 10, 832–856.
Gonzalez-Periz, A., Horrillo, R., Ferre, N., Gronert, K., Dong, B., Moran-Salvador, E., et al. (2009). Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 23, 1946–1957.
Gronert, K., Gewirtz, A., Madara, J. L., and Serhan, C. N. (1998). Identification of a human enterocyte lipoxin A4 receptor that is regulated by interleukin (IL)-13 and interferon gamma and inhibits tumor necrosis factor alpha-induced IL-8 release. J. Exp. Med. 187, 1285–1294.
Guilford, W. J., and Parkinson, J. F. (2005). Second-generation beta-oxidation resistant 3-oxa-lipoxin A4 analogs. Prostaglandins Leukot. Essent. Fatty Acids 73, 245–250.
Hellmann, J., Tang, Y., Kosuri, M., Bhatnagar, A., and Spite, M. (2011). Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J. 25, 2399–2407.
Henderson, N. C., Mackinnon, A. C., Farnworth, S. L., Kipari, T., Haslett, C., Iredale, J. P., et al. (2008). Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 172, 288–298.
Hewitson, T. D., Kelynack, K. J., Tait, M. G., Martic, M., Jones, C. L., Margolin, S. B., et al. (2001). Pirfenidone reduces in vitro rat renal fibroblast activation and mitogenesis. J. Nephrol. 14, 453–460.
Higgins, D. F., Kimura, K., Bernhardt, W. M., Shrimanker, N., Akai, Y., Hohenstein, B., et al. (2007). Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest. 117, 3810–3820.
Hotamisligil, G. S., Arner, P., Caro, J. F., Atkinson, R. L., and Spiegelman, B. M. (1995). Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Invest. 95, 2409–2415.
Inouye, K. E., Shi, H., Howard, J. K., Daly, C. H., Lord, G. M., Rollins, B. J., et al. (2007). Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes 56, 2242–2250.
Jozsef, L., Zouki, C., Petasis, N. A., Serhan, C. N., and Filep, J. G. (2002). Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-kappa B and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc. Natl. Acad. Sci. U.S.A. 99, 13266–13271.
Kadowaki, T., Yamauchi, T., Kubota, N., Hara, K., Ueki, K., and Tobe, K. (2006). Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 116, 1784–1792.
Kantarci, A., Hasturk, H., and Van Dyke, T. E. (2006). Host-mediated resolution of inflammation in periodontal diseases. Periodontology 2000 40, 144–163.
Kantarci, A., and Van Dyke, T. E. (2005). Lipoxin signaling in neutrophils and their role in periodontal disease. Prostaglandins Leukot. Essent. Fatty Acids 73, 289–299.
Kato, M., Zhang, J., Wang, M., Lanting, L., Yuan, H., Rossi, J. J., et al. (2007). MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. U.S.A. 104, 3432–3437.
Kern, P. A., Ranganathan, S., Li, C., Wood, L., and Ranganathan, G. (2001). Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 280, E745–E751.
Kern, P. A., Saghizadeh, M., Ong, J. M., Bosch, R. J., Deem, R., and Simsolo, R. B. (1995). The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J. Clin. Invest. 95, 2111–2119.
Kershaw, E. E., and Flier, J. S. (2004). Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 89, 2548–2556.
Kohli, P., and Levy, B. D. (2009). Resolvins and protectins: mediating solutions to inflammation. Br. J. Pharmacol. 158, 960–971.
Koska, J., Ortega, E., Bunt, J. C., Gasser, A., Impson, J., Hanson, R. L., et al. (2009). The effect of salsalate on insulin action and glucose tolerance in obese non-diabetic patients: results of a randomised double-blind placebo-controlled study. Diabetologia 52, 385–393.
Krishnamoorthy, S., Recchiuti, A., Chiang, N., Yacoubian, S., Lee, C. H., Yang, R., et al. (2010). Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 1660–1665.
Kristiansen, O. P., and Mandrup-Poulsen, T. (2005). Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabetes 54(Suppl. 2), S114–S124.
Larsen, C. M., Faulenbach, M., Vaag, A., Volund, A., Ehses, J. A., Seifert, B., et al. (2007). Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 356, 1517–1526.
Lawrence, T., and Gilroy, D. W. (2007). Chronic inflammation: a failure of resolution? Int. J. Exp. Pathol. 88, 85–94.
Lee, T. H., Horton, C. E., Kyan-Aung, U., Haskard, D., Crea, A. E., and Spur, B. W. (1989). Lipoxin A4 and lipoxin B4 inhibit chemotactic responses of human neutrophils stimulated by leukotriene B4 and N-formyl-L-methionyl-L-leucyl-L-phenylalanine. Clin. Sci. (Lond.) 77, 195–203.
Lefer, A. M., Stahl, G. L., Lefer, D. J., Brezinski, M. E., Nicolaou, K. C., Veale, C. A., et al. (1988). Lipoxins A4 and B4: comparison of icosanoids having bronchoconstrictor and vasodilator actions but lacking platelet aggregatory activity. Proc. Natl. Acad. Sci. U.S.A. 85, 8340–8344.
Levy, B. D., Clish, C. B., Schmidt, B., Gronert, K., and Serhan, C. N. (2001). Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2, 612–619.
Levy, B. D., De Sanctis, G. T., Devchand, P. R., Kim, E., Ackerman, K., Schmidt, B. A., et al. (2002). Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4). Nat. Med. 8, 1018–1023.
Levy, B. D., Fokin, V. V., Clark, J. M., Wakelam, M. J., Petasis, N. A., and Serhan, C. N. (1999). Polyisoprenyl phosphate (PIPP) signaling regulates phospholipase D activity: a ‘stop’ signaling switch for aspirin-triggered lipoxin A4. FASEB J. 13, 903–911.
Li, M., and Batuman, V. (2009). Vitamin D: a new hope for chronic kidney disease? Kidney Int. 76, 1219–1221.
Long, J., Wang, Y., Wang, W., Chang, B. H., and Danesh, F. R. (2010). Identification of microRNA-93 as a novel regulator of vascular endothelial growth factor in hyperglycemic conditions. J. Biol. Chem. 285, 23457–23465.
Lukiw, W. J., Cui, J. G., Marcheselli, V. L., Bodker, M., Botkjaer, A., Gotlinger, K., et al. (2005). A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 115, 2774–2783.
Lumeng, C. N., Bodzin, J. L., and Saltiel, A. R. (2007a). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184.
Lumeng, C. N., Deyoung, S. M., Bodzin, J. L., and Saltiel, A. R. (2007b). Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 56, 16–23.
Machado, F. S., Johndrow, J. E., Esper, L., Dias, A., Bafica, A., Serhan, C. N., et al. (2006). Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat. Med. 12, 330–334.
Maddox, J. F., Colgan, S. P., Clish, C. B., Petasis, N. A., Fokin, V. V., and Serhan, C. N. (1998). Lipoxin B4 regulates human monocyte/neutrophil adherence and motility: design of stable lipoxin B4 analogs with increased biologic activity. FASEB J. 12, 487–494.
Maddox, J. F., Hachicha, M., Takano, T., Petasis, N. A., Fokin, V. V., and Serhan, C. N. (1997). Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J. Biol. Chem. 272, 6972–6978.
Maddox, J. F., and Serhan, C. N. (1996). Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: selective inactivation by dehydrogenation and reduction. J. Exp. Med. 183, 137–146.
Maedler, K., Sergeev, P., Ehses, J. A., Mathe, Z., Bosco, D., Berney, T., et al. (2004). Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc. Natl. Acad. Sci. U.S.A. 101, 8138–8143.
Marcheselli, V. L., Hong, S., Lukiw, W. J., Tian, X. H., Gronert, K., Musto, A., et al. (2003). Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 278, 43807–43817.
McMahon, B., Mitchell, D., Shattock, R., Martin, F., Brady, H. R., and Godson, C. (2002). Lipoxin, leukotriene, and PDGF receptors cross-talk to regulate mesangial cell proliferation. FASEB J. 16, 1817–1819.
Mitchell, D., O’Meara, S. J., Gaffney, A., Crean, J. K., Kinsella, B. T., and Godson, C. (2007). The Lipoxin A4 receptor is coupled to SHP-2 activation: implications for regulation of receptor tyrosine kinases. J. Biol. Chem. 282, 15606–15618.
Mitchell, D., Rodgers, K., Hanly, J., McMahon, B., Brady, H. R., Martin, F., et al. (2004). Lipoxins inhibit Akt/PKB activation and cell cycle progression in human mesangial cells. Am. J. Pathol. 164, 937–946.
Mitchell, S., Thomas, G., Harvey, K., Cottell, D., Reville, K., Berlasconi, G., et al. (2002). Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 13, 2497–2507.
Mukherjee, P. K., Marcheselli, V. L., Serhan, C. N., and Bazan, N. G. (2004). Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 101, 8491–8496.
Nascimento-Silva, V., Arruda, M. A., Barja-Fidalgo, C., and Fierro, I. M. (2007). Aspirin-triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: a novel antioxidative mechanism. Thromb. Haemost. 97, 88–98.
Navarro, J. F., and Mora, C. (2006). Diabetes, inflammation, proinflammatory cytokines, and diabetic nephropathy. Sci. World J. 6, 908–917.
Ofei, F., Hurel, S., Newkirk, J., Sopwith, M., and Taylor, R. (1996). Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45, 881–885.
Ogata, A., Morishima, A., Hirano, T., Hishitani, Y., Hagihara, K., Shima, Y., et al. (2011). Improvement of HbA1c during treatment with humanised anti-interleukin 6 receptor antibody, tocilizumab. Ann. Rheum. Dis. 70, 1164–1165.
O’Sullivan, T. P., Vallin, K. S., Shah, S. T., Fakhry, J., Maderna, P., Scannell, M., et al. (2007). Aromatic lipoxin A4 and lipoxin B4 analogues display potent biological activities. J. Med. Chem. 50, 5894–5902.
Pang, H., Yi, P., Wu, P., Liu, Z., Gong, J., Hao, H., et al. (2011). Effect of lipoxin A4 on lipopolysaccharide-induced endothelial hyperpermeability. Sci. World J. 11, 1056–1067.
Panzer, U., Steinmetz, O. M., Turner, J. E., Meyer-Schwesinger, C., Von Ruffer, C., Meyer, T. N., et al. (2009). Resolution of renal inflammation: a new role for NF-kappaB1 (p50) in inflammatory kidney diseases. Am. J. Physiol. Renal Physiol. 297, F429–F439.
Papayianni, A., Serhan, C. N., and Brady, H. R. (1996). Lipoxin A4 and B4 inhibit leukotriene-stimulated interactions of human neutrophils and endothelial cells. J. Immunol. 156, 2264–2272.
Parkinson, J. F. (2006). Lipoxin and synthetic lipoxin analogs: an overview of anti-inflammatory functions and new concepts in immunomodulation. Inflamm. Allergy Drug Targets 5, 91–106.
Pedersen, B. K., Steensberg, A., Keller, P., Keller, C., Fischer, C., Hiscock, N., et al. (2003). Muscle-derived interleukin-6: lipolytic, anti-inflammatory and immune regulatory effects. Pflugers Arch. 446, 9–16.
Pergola, P. E., Krauth, M., Huff, J. W., Ferguson, D. A., Ruiz, S., Meyer, C. J., et al. (2011a). Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am. J. Nephrol. 33, 469–476.
Pergola, P. E., Raskin, P., Toto, R. D., Meyer, C. J., Huff, J. W., Grossman, E. B., et al. (2011b). Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 365, 327–336.
Petasis, N. A., Akritopoulou-Zanze, I., Fokin, V. V., Bernasconi, G., Keledjian, R., Yang, R., et al. (2005). Design, synthesis and bioactions of novel stable mimetics of lipoxins and aspirin-triggered lipoxins. Prostaglandins Leukot. Essent. Fatty Acids 73, 301–321.
Petasis, N. A., Keledjian, R., Sun, Y. P., Nagulapalli, K. C., Tjonahen, E., Yang, R., et al. (2008). Design and synthesis of benzo-lipoxin A4 analogs with enhanced stability and potent anti-inflammatory properties. Bioorg. Med. Chem. Lett. 18, 1382–1387.
Planaguma, A., Titos, E., Lopez-Parra, M., Gaya, J., Pueyo, G., Arroyo, V., et al. (2002). Aspirin (ASA) regulates 5-lipoxygenase activity and peroxisome proliferator-activated receptor alpha-mediated CINC-1 release in rat liver cells: novel actions of lipoxin A4 (LXA4) and ASA-triggered 15-epi-LXA4. FASEB J. 16, 1937–1939.
Pradhan, A. D., Manson, J. E., Rifai, N., Buring, J. E., and Ridker, P. M. (2001). C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286, 327–334.
Qu, X., Zhang, X., Yao, J., Song, J., Nikolic-Paterson, D. J., and Li, J. (2012). Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J. Pathol. doi: 10.1002/path.4050 [Epub ahead of print].
RamachandraRao, S. P., Zhu, Y., Ravasi, T., Mcgowan, T. A., Toh, I., Dunn, S. R., et al. (2009). Pirfenidone is renoprotective in diabetic kidney disease. J. Am. Soc. Nephrol. 20, 1765–1775.
Ramstedt, U., Ng, J., Wigzell, H., Serhan, C. N., and Samuelsson, B. (1985). Action of novel eicosanoids lipoxin A and B on human natural killer cell cytotoxicity: effects on intracellular cAMP and target cell binding. J. Immunol. 135, 3434–3438.
Ramstedt, U., Serhan, C. N., Nicolaou, K. C., Webber, S. E., Wigzell, H., and Samuelsson, B. (1987). Lipoxin A-induced inhibition of human natural killer cell cytotoxicity: studies on stereospecificity of inhibition and mode of action. J. Immunol. 138, 266–270.
Reville, K., Crean, J. K., Vivers, S., Dransfield, I., and Godson, C. (2006). Lipoxin A4 redistributes myosin IIA and Cdc42 in macrophages: implications for phagocytosis of apoptotic leukocytes. J. Immunol. 176, 1878–1888.
Ricardo, S. D., Van Goor, H., and Eddy, A. A. (2008). Macrophage diversity in renal injury and repair. J. Clin. Invest. 118, 3522–3530.
Ritz, E., Rychlik, I., Locatelli, F., and Halimi, S. (1999). End-stage renal failure in type 2 diabetes: a medical catastrophe of worldwide dimensions. Am. J. Kidney Dis. 34, 795–808.
Rojas-Rivera, J., Ortiz, A., and Egido, J. (2012). Antioxidants in kidney diseases: the impact of bardoxolone methyl. Int. J. Nephrol. 2012, 321714.
Romanatto, T., Roman, E. A., Arruda, A. P., Denis, R. G., Solon, C., Milanski, M., et al. (2009). Deletion of tumor necrosis factor-alpha receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J. Biol. Chem. 284, 36213–36222.
Rosenvinge, A., Krogh-Madsen, R., Baslund, B., and Pedersen, B. K. (2007). Insulin resistance in patients with rheumatoid arthritis: effect of anti-TNFalpha therapy. Scand. J. Rheumatol. 36, 91–96.
Rotter, V., Nagaev, I., and Smith, U. (2003). Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J. Biol. Chem. 278, 45777–45784.
Scalia, R., Gefen, J., Petasis, N. A., Serhan, C. N., and Lefer, A. M. (1997). Lipoxin A4 stable analogs inhibit leukocyte rolling and adherence in the rat mesenteric microvasculature: role of P-selectin. Proc. Natl. Acad. Sci. U.S.A. 94, 9967–9972.
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874.
Serhan, C. N. (2004). Clues for new therapeutics in osteoporosis and periodontal disease: new roles for lipoxygenases? Expert Opin. Ther. Targets 8, 643–652.
Serhan, C. N. (2005). Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fatty Acids 73, 141–162.
Serhan, C. N. (2007). Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 25, 101–137.
Serhan, C. N. (2009). Systems approach to inflammation resolution: identification of novel anti-inflammatory and pro-resolving mediators. J. Thromb. Haemost. 7(Suppl. 1), 44–48.
Serhan, C. N., Clish, C. B., Brannon, J., Colgan, S. P., Chiang, N., and Gronert, K. (2000). Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204.
Serhan, C. N., Hong, S., Gronert, K., Colgan, S. P., Devchand, P. R., Mirick, G., et al. (2002). Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037.
Serhan, C. N., Maddox, J. F., Petasis, N. A., Akritopoulou-Zanze, I., Papayianni, A., Brady, H. R., et al. (1995). Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry 34, 14609–14615.
Serhan, C. N., and Savill, J. (2005). Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197.
Serhan, C. N., Yacoubian, S., and Yang, R. (2008). Anti-inflammatory and proresolving lipid mediators. Annu. Rev. Pathol. 3, 279–312.
Serhan, C. N., Yang, R., Martinod, K., Kasuga, K., Pillai, P. S., Porter, T. F., et al. (2009). Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 206, 15–23.
Sharma, K., Ix, J. H., Mathew, A. V., Cho, M., Pflueger, A., Dunn, S. R., et al. (2011). Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 22, 1144–1151.
Shepler, B., Nash, C., Smith, C., Dimarco, A., Petty, J., and Szewciw, S. (2012). Update on potential drugs for the treatment of diabetic kidney disease. Clin. Ther. 34, 1237–1246.
Sodin-Semrl, S., Taddeo, B., Tseng, D., Varga, J., and Fiore, S. (2000). Lipoxin A4 inhibits IL-1 beta-induced IL-6, IL-8, and matrix metalloproteinase-3 production in human synovial fibroblasts and enhances synthesis of tissue inhibitors of metalloproteinases. J. Immunol. 164, 2660–2666.
Soyombo, O., Spur, B. W., and Lee, T. H. (1994). Effects of lipoxin A4 on chemotaxis and degranulation of human eosinophils stimulated by platelet-activating factor and N-formyl-L-methionyl-L-leucyl-L-phenylalanine. Allergy 49, 230–234.
Spencer, M., Yao-Borengasser, A., Unal, R., Rasouli, N., Gurley, C. M., Zhu, B., et al. (2010). Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am. J. Physiol. Endocrinol. Metab. 299, E1016–E1027.
Stanley, T. L., Zanni, M. V., Johnsen, S., Rasheed, S., Makimura, H., Lee, H., et al. (2010). TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J. Clin. Endocrinol. Metab. 96, E146–E150.
Sun, Y. P., Tjonahen, E., Keledjian, R., Zhu, M., Yang, R., Recchiuti, A., et al. (2009). Anti-inflammatory and pro-resolving properties of benzo-lipoxin A(4) analogs. Prostaglandins Leukot. Essent. Fatty Acids 81, 357–366.
Sung, S. A., Jo, S. K., Cho, W. Y., Won, N. H., and Kim, H. K. (2007). Reduction of renal fibrosis as a result of liposome encapsulated clodronate induced macrophage depletion after unilateral ureteral obstruction in rats. Nephron Exp. Nephrol. 105, e1–e9.
Syed, I. A., and Khan, W. A. (2011). Glycated haemoglobin – a marker and predictor of cardiovascular disease. J. Pak. Med. Assoc. 61, 690–695.
Takakuta, K., Fujimori, A., Chikanishi, T., Tanokura, A., Iwatsuki, Y., Yamamoto, M., et al. (2010). Renoprotective properties of pirfenidone in subtotally nephrectomized rats. Eur. J. Pharmacol. 629, 118–124.
Tesch, G. H. (2008). MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 294, F697–F701.
Thomas, M. C., and Cooper, M. E. (2011). Diabetes: bardoxolone improves kidney function in type 2 diabetes. Nat. Rev. Nephrol. 7, 552–553.
Tobin, D. M., Vary, J. C. Jr., Ray, J. P., Walsh, G. S., Dunstan, S. J., Bang, N. D., et al. (2010). The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell 140, 717–730.
Torloni, M. R., Cordioli, E., Zamith, M. M., Hisaba, W. J., Nardozza, L. M., Santana, R. M., et al. (2006). Reversible constriction of the fetal ductus arteriosus after maternal use of topical diclofenac and methyl salicylate. Ultrasound Obstet. Gynecol. 27, 227–229.
Uysal, K. T., Wiesbrock, S. M., Marino, M. W., and Hotamisligil, G. S. (1997). Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389, 610–614.
von der Weid, P.Y., Hollenberg, M. D., Fiorucci, S., and Wallace, J. L. (2004). Aspirin-triggered, cyclooxygenase-2-dependent lipoxin synthesis modulates vascular tone. Circulation 110, 1320–1325.
Wada, T., Furuichi, K., Sakai, N., Iwata, Y., Kitagawa, K., Ishida, Y., et al. (2004). Gene therapy via blockade of monocyte chemoattractant protein-1 for renal fibrosis. J. Am. Soc. Nephrol. 15, 940–948.
Wang, Q., Wang, Y., Minto, A. W., Wang, J., Shi, Q., Li, X., et al. (2008). MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J. 22, 4126–4135.
Wang, Y., Wang, Y. P., Zheng, G., Lee, V. W., Ouyang, L., Chang, D. H., et al. (2007). Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 72, 290–299.
Wild, S., Roglic, G., Green, A., Sicree, R., and King, H. (2004). Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27, 1047–1053.
Wu, S. H., Lu, C., Dong, L., Zhou, G. P., He, Z. G., and Chen, Z. Q. (2005). Lipoxin A4 inhibits TNF-alpha-induced production of interleukins and proliferation of rat mesangial cells. Kidney Int. 68, 35–46.
Wu, S. H., Wu, X. H., Lu, C., Dong, L., and Chen, Z. Q. (2006a). Lipoxin A4 inhibits proliferation of human lung fibroblasts induced by connective tissue growth factor. Am. J. Respir. Cell Mol. Biol. 34, 65–72.
Wu, S. H., Wu, X. H., Lu, C., Dong, L., Zhou, G. P., and Chen, Z. Q. (2006b). Lipoxin A4 inhibits connective tissue growth factor-induced production of chemokines in rat mesangial cells. Kidney Int. 69, 248–256.
Wu, S. H., Zhang, Y. M., Tao, H. X., and Dong, L. (2010). Lipoxin A(4) inhibits transition of epithelial to mesenchymal cells in proximal tubules. Am. J. Nephrol. 32, 122–136.
Wu, Y., Zhai, H., Wang, Y., Li, L., Wu, J., Wang, F., et al. (2012). Aspirin-Triggered Lipoxin A(4) Attenuates lipopolysaccharide-induced intracellular ROS in BV2 microglia cells by inhibiting the function of NADPH oxidase. Neurochem. Res. 37, 1690–1696.
Keywords: inflammation, resolution, lipxoins, resolvins, protectins, renal inflammation
Citation: Börgeson E and Godson C (2012) Resolution of inflammation: therapeutic potential of pro-resolving lipids in type 2 diabetes mellitus and associated renal complications. Front. Immun. 3:318. doi: 10.3389/fimmu.2012.00318
Received: 31 July 2012; Accepted: 29 September 2012;
Published online: 18 October 2012.
Edited by:
Janos G. Filep, University of Montreal, CanadaReviewed by:
Pablo Pelegrin, Fundacion Formacion Investigacion Sanitaria Region Murcia - Hospital Universitario Virgen Arrixaca, SpainMasato Kubo, Research Institute for Biological Science, Tokyo University of Science, Japan
Copyright: © 2012 Börgeson and Godson. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Catherine Godson, UCD Diabetes Research Centre, UCD Conway Institute, School of Medicine and Medical Sciences, University College Dublin, Dublin, Ireland. e-mail:Y2F0aGVyaW5lLmdvZHNvbkB1Y2QuaWU=