



- 1Laboratory of Human Bacterial Pathogenesis, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Hamilton, MT, USA
- 2Department of Immunology and Infectious Diseases, Montana State University, Bozeman, MT, USA
Neutrophils constitute a critical part of innate immunity and are well known for their ability to phagocytose and kill invading microorganisms. The microbicidal processes employed by neutrophils are highly effective at killing most ingested bacteria and fungi. However, an alternative non-phagocytic antimicrobial mechanism of neutrophils has been proposed whereby microorganisms are eliminated by neutrophil extracellular traps (NETs). NETs are comprised of DNA, histones, and antimicrobial proteins extruded by neutrophils during NETosis, a cell death pathway reported to be distinct from apoptosis, phagocytosis-induced cell death, and necrosis. Although multiple laboratories have reported NETs using various stimuli in vitro, the molecular mechanisms involved in this process have yet to be definitively elucidated, and many questions regarding the formation and putative role or function of NETs in innate host defense remain unanswered. It is with these questions in mind that we provide some reflection and perspective on NETs and NETosis.
Neutrophil Turnover and Homeostasis
Neutrophils are short-lived granulocytes that mature in bone marrow for several days (Bainton et al., 1971; Weissman et al., 2001). During maturation, these cells acquire key functional attributes, including the ability to phagocytose and kill microorganisms (Bainton et al., 1971; Glasser and Fiederlein, 1987; Weissman et al., 2001; Rosenbauer and Tenen, 2007; Pillay et al., 2010). After maturation, neutrophils are released into the bloodstream and circulate and/or marginate for 10–24 h before migrating into tissues, where they may function for an additional 1–2 days before they undergo apoptosis and are cleared by macrophages or dendritic cells (Cartwright et al., 1964; Fliedner et al., 1964; Bainton et al., 1971; Savill et al., 1989; Voll et al., 1997; Fadok et al., 1998; Huynh et al., 2002; Martin et al., 2003; Rigby and DeLeo, 2012). In addition, neutrophils in the total blood granulocyte pool (circulating and marginating) can be removed by the liver, spleen, and bone marrow, although the precise mechanism for this turnover process remains incompletely determined (reviewed by Summers et al., 2010). The neutrophil lifespan is highly regulated, as it is critical to remove spent/effete neutrophils as a means to prevent accidental release of cytotoxic molecules and associated host tissue damage (Edwards et al., 2003; Duffin et al., 2010; Bratton and Henson, 2011; Milot and Filep, 2011). Neutrophil turnover in an adult human is typically on the order of 1011 cells per day (Athens et al., 1961; Dancey et al., 1976; Rankin, 2010). While the hematopoietic system is able to regulate steady-state levels of circulating neutrophils, it can also be switched to an emergency granulopoiesis response to accommodate the increased demand for neutrophils during infection (Hirai et al., 2006; Panopoulos and Watowich, 2008).
The neutrophil lifespan is regulated by a balance of pro- and anti-apoptotic factors present in the environment. Cytokines and other factors such as interleukin (IL)-1β, IL-2, IL-4, IL-15, interferon-γ, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and lipopolysaccharide (LPS) can prolong and/or enhance neutrophil function and delay apoptosis for several days (Colotta et al., 1992; Duffin et al., 2010). Although enhancing neutrophil function and survival presumably favors elimination of invading microbes, the persistence of these cytotoxic host cells increases the potential for prolonged inflammation and host tissue damage. Therefore, it is not surprising that neutrophil turnover is a highly regulated process.
Molecular control of neutrophil turnover or apoptosis is mediated by several mechanisms, including extrinsic pathways induced by extracellular signals and intrinsic pathways induced by intracellular signals. These signals include those triggered by death receptors, which bind ligands that activate caspases to promote apoptosis, mitochondrial release of cytochrome c, and processes mediated by the BCL-2 protein family (Edwards et al., 2003; Duffin et al., 2010). Spontaneous or constitutive apoptosis in neutrophils is an example of intrinsic apoptosis. Apoptosis elicited by FAS, tumor necrosis factor (TNF)-α, or TNF-related apoptosis inducing ligand (TRAIL), caused by the binding of these extracellular ligands to the cognate receptor anchored on the cell surface, is an example of extrinsic pathway apoptosis (Kennedy and DeLeo, 2009; Duffin et al., 2010). Phagocytosis may also lead to neutrophil apoptosis (Watson et al., 1996; Kobayashi et al., 2002; Zhang et al., 2003; Kennedy and DeLeo, 2009). Neutrophil phagocytosis-induced apoptosis or phagocytosis-induced cell death (PICD) promotes the resolution of infection by disposing spent or effete neutrophils containing dead or partially digested microbes in a non-inflammatory manner (Kennedy and DeLeo, 2009). This process is described below in the context of the resolution of inflammation.
Neutrophils and the Inflammatory Response
The importance of neutrophils in the immune response is underscored by human diseases caused by defects in neutrophil function, which result in increased risk of infection from bacteria and fungi (Nauseef and Clark, 2010). For example, neutropenia, which can be medically induced by cytotoxic drugs or cancer therapy, is associated with significant morbidity (Bodey et al., 1966; Dale et al., 1979; Frøland, 1984; Tobias and Schleien, 1991). In addition, the inflammatory response, which from a cellular perspective is largely comprised of neutrophils, is critical for defense against invading microorganisms. On the other hand, timely resolution of the inflammatory response is an important process that returns the host immune system to pre-infection homeostasis. Historically, neutrophils were considered to have a passive part in inflammation resolution; however, this view has changed over time, and it is now known that neutrophils actively help to resolve inflammation by blocking and scavenging chemokines and cytokines (Ariel et al., 2006), and also produce pro-resolving lipid mediators (Ariel et al., 2006; Serhan et al., 2008). Thus, given that neutrophils contain and produce a vast array of cytotoxic molecules and contribute to the regulation of inflammation, it should not be unexpected that these cells are involved in – or are the primary cause of – a variety of inflammatory disorders. For instance, in chronic obstructive pulmonary disease, the aminopeptidase activity of leukotriene A4 hydrolase (LTA4H) is inhibited, causing accumulation of proline-glycine-proline, which in turn, promotes neutrophil recruitment and chronic lung inflammation (Weathington et al., 2006). In mouse models, recruitment of neutrophils has been shown to be involved in arthritis (Chou et al., 2010) and multiple sclerosis (Carlson et al., 2008; Liu et al., 2010). More notably, recent studies have demonstrated that neutrophils and neutrophil responses (rather than bacterial pathogens per se) are the cause of severe pneumonia and tissue destruction in animal models of bacterial respiratory tract infection (Bartlett et al., 2008; Diep et al., 2010). Thus, it is clear that unchecked neutrophil activation and neutrophil lysis are phenomena that can have a significant negative impact on health of the host.
Response to Infection
Neutrophils are recruited rapidly to the site of infection in response to chemotactic stimuli released by the host and/or invading microorganism. Inasmuch as neutrophils are the most abundant leukocyte in humans, there can be a tremendous influx of neutrophils to the site of infection. At such sites, neutrophils bind and ingest microorganisms through a process known as phagocytosis (reviewed in Rigby and DeLeo, 2012). Ingested microbes are typically destroyed by the combined effects of NADPH oxidase-derived reactive oxygen species (ROS) and cytotoxic molecules delivered from cytoplasmic granules into the phagosome. Neutrophil granules contain numerous antimicrobial peptides (AMPs) and proteins, and matrix protein-degrading proteases, including alpha-defensins, cathelicidins, azurocidin, cathepsins, lactoferrin, lysozyme, proteinase-3, gelatinase, collagenase, and elastase (Faurschou and Borregaard, 2003; Nauseef and Clark, 2010; Rigby and DeLeo, 2012). Of note, these cytotoxic agents are normally targeted into the formed phagosome, thereby limiting inadvertent extracellular release and potential damage to host tissues (Nauseef and Clark, 2010).
It is well documented that neutrophil PICD occurs following ingestion of numerous microorganisms in vitro (Watson et al., 1996; Colamussi et al., 1999; Engelich et al., 2001; Kobayashi et al., 2003a,b, 2012; Kennedy and DeLeo, 2009), and in vivo this phenomenon likely promotes clearance of effete neutrophils containing dead or dying microbes (Figure 1; Kobayashi et al., 2012). Importantly, this process would prevent local host tissue damage that can occur if these spent host cells are not removed and undergo lysis, and thus ultimately promotes the resolution of inflammation (Whyte et al., 1993; Savill, 1997; Kobayashi et al., 2002, 2003a, 2012; Kim et al., 2004; Iyoda et al., 2005; Ariel et al., 2006; Kobayashi and DeLeo, 2009; Rigby and DeLeo, 2012). Such a process is considered normal for neutrophils during infection and healthy for the host. On the other hand, pathogenic microorganisms circumvent killing by neutrophils, and in doing so ultimately alter the normal process of neutrophil turnover during infection, by either delaying apoptosis or causing neutrophil lysis (Kobayashi et al., 2003a, 2010; DeLeo, 2004; Voyich et al., 2005). The resulting neutrophil lysis releases tissue-damaging molecules, not only allowing pathogen survival but also exacerbating the inflammatory response (Figure 1). This process can lead to disease and can be considered unhealthy for the host. As one example, some strains of Staphylococcus aureus are known to cause lysis of human neutrophils after phagocytosis (Rogers and Tompsett, 1952; Voyich et al., 2005, 2006; Kobayashi et al., 2010). Indeed, the possibility that S. aureus survive after phagocytosis and ultimately disseminate to cause disease (which can be explained at least in part by neutrophil lysis after trafficking) has been reviewed recently (Thwaites and Gant, 2011). These authors describe neutrophils as “Trojan horses” for the dissemination or metastasis of S. aureus (Thwaites and Gant, 2011). In accordance with the observations in vitro, S. aureus is an abundant cause of pyogenic infections in humans. Therefore, the ability of S. aureus to cause neutrophil lysis is likely a component of virulence.
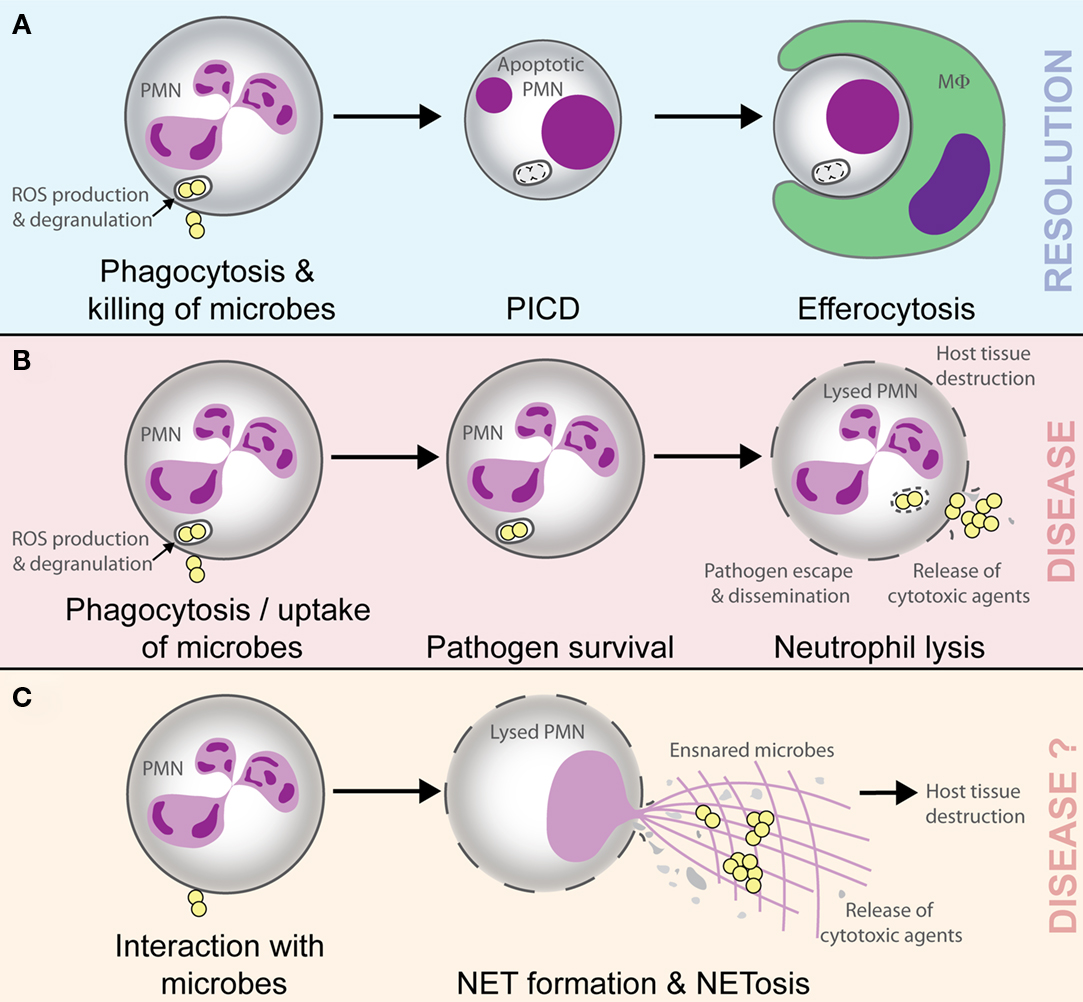
Figure 1. Possible outcomes of the interaction of microbes with neutrophils. Phagocytosis and killing of microorganisms by neutrophils (polymorphonuclear leukocyte, PMN) triggers host cell apoptosis and ultimate removal by macrophages (MΦ) or dendritic cells. This process promotes resolution of the inflammatory response (A). Pathogenic microbes such as Staphylococcus aureus can cause lysis of PMN after phagocytosis, thereby facilitating escape/dissemination of the invading pathogen and release of cytotoxic molecules that cause host tissue damage and disease (B). NETs ensnare and may kill microbes, but there is accompanying lysis of neutrophils and release of cytotoxic molecules that are known to cause host tissue damage and promote inflammatory disease. In this regard, the outcome of NETosis and the formation of NETs should be similar to that in (B; i.e., disease; C).
Neutrophil Extracellular Traps and NETosis
Until fairly recently, phagocyte biologists were content with a model of neutrophil function in which these phagocytes bind, ingest, and subsequently kill microorganisms. The idea that neutrophils would extrude DNA in a cytolytic process that captures microorganisms was unheard of – until Brinkmann et al. (2004) reported the formation of structures known as neutrophil extracellular traps (NETs). These unique structures, which are discussed in detail in this issue of Frontiers in Immunology, are composed of DNA, histones, and antimicrobial proteins, and can ensnare pathogens. Since the report by Brinkmann et al. (2004), extracellular traps have been shown to be produced in vitro by a number of different cell types, including neutrophils, mast cells, eosinophils, and endothelial cells (Palić et al., 2007; von Köckritz-Blickwede et al., 2008; Yousefi et al., 2008; Chuammitri et al., 2009; Katzenback and Belosevic, 2009; Aulik et al., 2010; Gupta et al., 2010; Wardini et al., 2010; Webster et al., 2010; Lin et al., 2011; Scapinello et al., 2011). Moreover, recent studies have investigated possible mechanisms for the induction of NETs. For example, it has been reported that formation of NETs requires activation of the Raf-MEK-ERK pathway through protein kinase C (Hakkim et al., 2011) and histone citrullination (Neeli et al., 2008; Li et al., 2010; Hemmers et al., 2011; Leshner et al., 2012). These findings suggest that NETosis and formation of NETs involves specific signal transduction events. Thus, it is tempting to advocate the importance of these structures in host defense due to the apparent simplicity and elegance of the phenomenon by which they occur. However, many questions remain about the role of NETs in host defense and the molecular mechanisms underlying their formation are incompletely characterized. In addition, the evidence for formation of NETs in vivo is not very compelling, and whether the formation of NETs is of benefit to the host remains an open question. Indeed, it was suggested early on that NETs form only under extreme circumstances and can injure host tissues (Clark et al., 2007).
While use of NETs appears as an alternative mechanism for pathogen control and elimination, production of NETs and NETosis (as a cytolytic process) seems at variance with the highly regulated control of neutrophil turnover (including PICD) and homeostasis, as discussed above. That is, utilization of NETs for host defense contrasts with the considerable effort made by the host to prevent inadvertent neutrophil lysis, release of cytotoxic agents, and post-lysis sequelae, such as inflammatory disorders. Notably, NETs have been implicated in a number of pathologic processes consistent with inflammatory disorders involving lysed neutrophils and cytotoxic molecules from neutrophils. For example, NETs can cause collateral damage in the form of endothelial and tissue damage (Clark et al., 2007; Ma and Kubes, 2008; Marin-Esteban et al., 2012) and may be partially responsible for sputum viscosity and tissue damage in cystic fibrosis patients (Papayannopoulos et al., 2011). NETs have been implicated in systemic lupus erythematosus and systemic vasculitis (Hakkim et al., 2010; Amulic and Hayes, 2011; Bosch, 2011; Garcia-Romo et al., 2011; Lande et al., 2011; Villanueva et al., 2011; Knight and Kaplan, 2012; Liu et al., 2012), gout, asthma, keratinocyte damage, and lupus nephritis (Mitroulis et al., 2011; Marin-Esteban et al., 2012), and may also be involved in the hyper reaction of the immune system by triggering physiological signals and causing pre-eclampsia (Gupta et al., 2005, 2006; Brinkmann and Zychlinsky, 2007). NETs are present in transfusion-related acute lung injury (Thomas et al., 2012), atherosclerotic carotid arteries (Döring, 2012), are toxic to vasculature (Clark et al., 2007; Gupta et al., 2010; Villanueva et al., 2011; Saffarzadeh et al., 2012), and facilitate thrombosis where they could provide a scaffold for red blood cell adhesion (Fuchs et al., 2010, 2012; Van Den Berg and Reitsma, 2011; Brill et al., 2012). NETs may also contribute to cancer-associated thrombosis, since neutrophils from mice with experimentally induced cancers are more likely to form NETs than those from control mice (Demers et al., 2012). It is also of note that extracellular histones, a signature component of NETs, contribute to host death during sepsis (Xu et al., 2009). Whether the structures reported as NETs in these aforementioned inflammatory syndromes are distinct from the remains of necrotic neutrophils is unclear, but in any case the process or phenomenon is associated with a negative outcome for the host – similar to the prediction in the model described in Figure 1.
Given the association of NETs and NETosis with inflammatory disorders, and coupled with a highly regulated neutrophil turnover process, the frequency with which formation of NETs occurs should be fairly low. Indeed, even under optimal NET-inducing conditions in vitro, only one-third of activated neutrophils, and perhaps as few as 10%, make NETs (Brinkmann and Zychlinsky, 2007; Fuchs et al., 2007; Munafo et al., 2009). The kinetics of NETosis vary depending on type and concentration of stimulus, isolation procedure of neutrophils, and the sensitivity of the detection method (Fuchs et al., 2012). Despite the fact that NET formation is stimulated by pathogens, it is still not clear whether NETosis that occurs during host-pathogen interactions is a programmed mechanism, a hijacking of host pathways by pathogen-produced factors, or simply an incidental component of neutrophil lysis. For instance, S. aureus is well known to cause lysis of neutrophils in vitro and in vivo, but the pathogen has also been reported to induce NETs (Brinkmann et al., 2004; Jann et al., 2009; Yipp et al., 2012). It is also not clear whether the release of NETs always leads to cell death (and the possibility of host tissue damage) or if it is an extrusion of DNA by intact cells (Yousefi et al., 2009; Remijsen et al., 2011; Guimarães-Costa et al., 2012; Yipp et al., 2012). Although it is difficult to understand how neutrophils can remain intact and viable after release of nuclear DNA, the question of whether NET formation always causes cytolysis or can occur with intact cells is important and must be resolved by the field.
One could hypothesize that the formation of NETs represents a directed host defense mechanism. If the process is host-directed, does this suggest there is an advantage to the use of NETs for removal of microbes versus traditional phagocytosis-based uptake and subsequent killing of microbial invaders? The antimicrobial activities of NETs have been ascribed to the histones, AMPs, and other cytoplasmic components associated with extracellular DNA. However, it is important to note that NETs provide a low concentration of AMPs compared to that present in the phagosome, and NETs lack the ability to produce microbicidal ROS. Published studies to date suggest that the formation of NETs does not lead to the universal killing of all microorganisms, although NETs can reduce the burden of selected of microorganisms in vitro (Table 1). This finding is perhaps not surprising, since solubilized azurophilic granule components isolated from disrupted neutrophils have varied capacity to kill different bacterial species (Bertram et al., 1986; Joiner et al., 1989; Levy et al., 1999; Palazzolo-Ballance et al., 2008; Nordenfelt et al., 2009). Moreover, several microorganisms are known to circumvent killing by NETs using a variety of strategies, including altering bacterial surface affinity to NETs (Wartha et al., 2007; Carlin et al., 2009; Juneau et al., 2011) and secreting NET-degrading DNases (Beiter et al., 2006; Buchanan et al., 2006; Midon et al., 2011; Palmer et al., 2011). As an alternative hypothesis, the formation of NETs (especially if it requires lysis of neutrophils) could be considered an incidental event rather than something intended by the host innate immune system. An incidental process seems more consistent with our understanding of the regulation of neutrophil turnover and homeostasis.
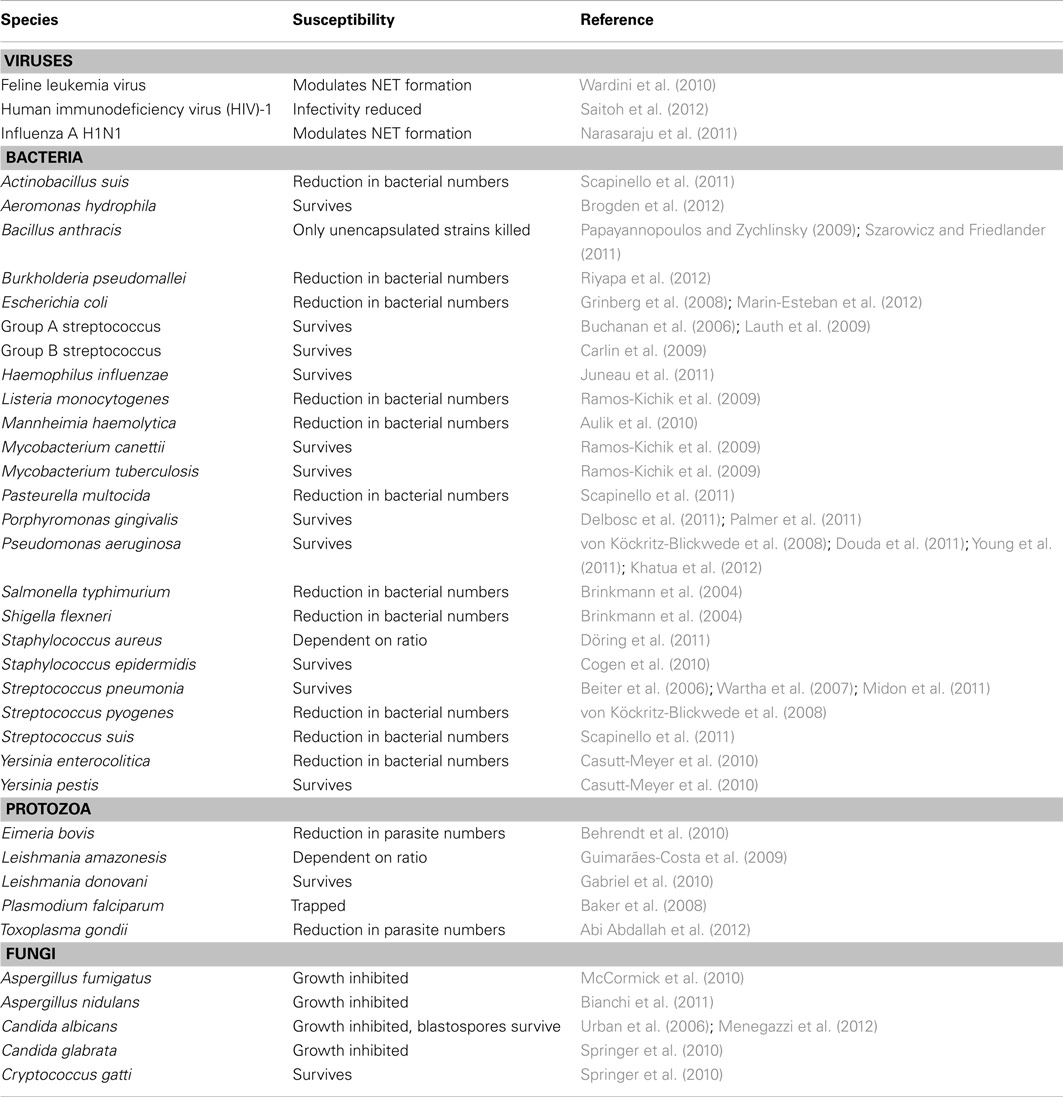
Table 1. Microbial susceptibility to NETs.
Concluding Perspective
Neutrophil extracellular traps have been suggested as an alternative or additional component of the innate host defense against microorganisms. Although progress has been made, many questions related to NET formation and function remain unanswered. Do NETs commonly occur in vivo? Compelling evidence is lacking. Are NETs formed by live neutrophils or does the process (i.e., NETosis) always result in cytolysis? If it is accompanied by neutrophil lysis, how does this phenomenon fit with what we know about the control of neutrophil turnover and the host efforts to prevent inflammatory syndromes? Importantly, is the pathway that leads to the formation of NETs a host-directed mechanism or simply an incidental phenomenon in neutrophils? These and other questions can only be answered by continued investigation into the biology and function of NETs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Thea Lu, Scott D. Kobayashi, and Frank R. DeLeo are supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. Mark T. Quinn is supported by a grant (GM103500) from the National Institute of General Medical Sciences, National Institutes of Health.
References
Abi Abdallah, D., Lin, C., Ball, C., King, M., Duhamel, G., and Denkers, E. (2012). Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect. Immun. 80, 768–777.
Ariel, A., Fredman, G., Sun, Y.-P., Kantarci, A., Van Dyke, T. E., Luster, A. D., et al. (2006). Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat. Immunol. 7, 1209–1216.
Athens, J. W., Haab, O. P., Raab, S. O., Mauer, A. M., Ashenbrucker, H., Cartwright, G. E., et al. (1961). Leukokinetic studies. IV. The total blood, circulating and marginal granulocyte pools and the granulocyte turnover rate in normal subjects. J. Clin. Invest. 40, 989–995.
Aulik, N., Hellenbrand, K., Klos, H., and Czuprynski, C. (2010). Mannheimia haemolytica and its leukotoxin cause neutrophil extracellular trap formation by bovine neutrophils. Infect. Immun. 78, 4454–4466.
Bainton, D. F., Ullyot, J. L., and Farquhar, M. G. (1971). The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J. Exp. Med. 134, 907–934.
Baker, V. S., Imade, G. E., Molta, N. B., Tawde, P., Pam, S. D., Obadofin, M. O., et al. (2008). Cytokine-associated neutrophil extracellular traps and antinuclear antibodies in Plasmodium falciparum infected children under six years of age. Malar. J. 7, 41.
Bartlett, A. H., Foster, T. J., Hayashida, A., and Park, P. W. (2008). α-Toxin facilitates the generation of CXC chemokine gradients and stimulates neutrophil homing in Staphylococcus aureus pneumonia. J. Infect. Dis. 198, 1529–1535.
Behrendt, J. H., Ruiz, A., Zahner, H., Taubert, A., and Hermosilla, C. (2010). Neutrophil extracellular trap formation as innate immune reactions against the apicomplexan parasite Eimeria bovis. Vet. Immunol. Immunopathol. 133, 1–8.
Beiter, K., Wartha, F., Albiger, B., Normark, S., Zychlinsky, A., and Henriques-Normark, B. (2006). An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 16, 401–407.
Bertram, T. A., Canning, P. C., and Roth, J. A. (1986). Preferential inhibition of primary granule release from bovine neutrophils by a Brucella abortus extract. Infect. Immun. 52, 285–292.
Bianchi, M., Niemiec, M. J., Siler, U., Urban, C. F., and Reichenbach, J. (2011). Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J. Allergy Clin. Immunol. 127, 1243–1252.e1247.
Bodey, G. P., Buckley, M., Sathe, Y. S., and Freireich, E. J. (1966). Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann. Intern. Med. 64, 328–340.
Bratton, D., and Henson, P. (2011). Neutrophil clearance: when the party is over, clean-up begins. Trends Immunol. 32, 350–357.
Brill, A., Fuchs, T. A., Savchenko, A. S., Thomas, G. M., Martinod, K., De Meyer, S. F., et al. (2012). Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 10, 136–144.
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535.
Brinkmann, V., and Zychlinsky, A. (2007). Beneficial suicide: why neutrophils die to make NETs. Nat. Rev. Microbiol. 5, 577–582.
Brogden, G., Von Köckritz-Blickwede, M., Adamek, M., Reuner, F., Jung-Schroers, V., Naim, H. Y., et al. (2012). β-Glucan protects neutrophil extracellular traps against degradation by Aeromonas hydrophila in carp (Cyprinus carpio). Fish Shellfish Immunol. 33, 1060–1064.
Buchanan, J. T., Simpson, A. J., Aziz, R. K., Liu, G. Y., Kristian, S. A., Kotb, M., et al. (2006). DNase expression allows the pathogen Group A Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. 16, 396–400.
Carlin, A., Uchiyama, S., Chang, Y.-C., Lewis, A., Nizet, V., and Varki, A. (2009). Molecular mimicry of host sialylated glycans allows a bacterial pathogen to engage neutrophil Siglec-9 and dampen the innate immune response. Blood 113, 3333–3336.
Carlson, T., Kroenke, M., Rao, P., Lane, T., and Segal, B. (2008). The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 205, 811–823.
Cartwright, G. E., Athens, J. W., and Wintrobe, M. M. (1964). The kinetics of granulopoiesis in normal man. Blood 24, 780–803.
Casutt-Meyer, S., Renzi, F., Schmaler, M., Jann, N. J., Amstutz, M., and Cornelis, G. R. (2010). Oligomeric coiled-coil adhesin YadA is a double-edged sword. PLoS ONE 5:e15159. doi:10.1371/journal.pone.0015159
Chou, R. C., Kim, N. D., Sadik, C. D., Seung, E., Lan, Y., Byrne, M. H., et al. (2010). Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 33, 266–278.
Chuammitri, P., Ostojić, J., Andreasen, C. B., Redmond, S. B., Lamont, S. J., and Palić, D. (2009). Chicken heterophil extracellular traps (HETs): novel defense mechanism of chicken heterophils. Vet. Immunol. Immunopathol. 129, 126–131.
Clark, S. R., Ma, A. C., Tavener, S. A., McDonald, B., Goodarzi, Z., Kelly, M. M., et al. (2007). Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13, 463–469.
Cogen, A. L., Yamasaki, K., Muto, J., Sanchez, K. M., Crotty Alexander, L., Tanios, J., et al. (2010). Staphylococcus epidermidis antimicrobial (δ-toxin (phenol-soluble modulin-(γ) cooperates with host antimicrobial peptides to kill Group A Streptococcus. PLoS ONE 5:e8557. doi:10.1371/journal.pone.0008557
Colamussi, M. L., White, M. R., Crouch, E., and Hartshorn, K. L. (1999). Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood 93, 2395–2403.
Colotta, F., Re, F., Polentarutti, N., Sozzani, S., and Mantovani, A. (1992). Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80, 2012–2020.
Dale, D. C., Guerry, D., Wewerka, J. R., Bull, J. M., and Chusid, M. J. (1979). Chronic neutropenia. Medicine (Baltimore) 58, 128–144.
Dancey, J. T., Deubelbeiss, K. A., Harker, L. A., and Finch, C. A. (1976). Neutrophil kinetics in man. J. Clin. Invest. 58, 705–715.
Delbosc, S., Alsac, J.-M., Journe, C., Louedec, L., Castier, Y., Bonnaure-Mallet, M., et al. (2011). Porphyromonas gingivalis participates in pathogenesis of human abdominal aortic aneurysm by neutrophil activation. Proof of concept in rats. PLoS ONE 6:e18679. doi:10.1371/journal.pone.0018679
DeLeo, F. R. (2004). Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 9, 399–413.
Demers, M., Krause, D. S., Schatzberg, D., Martinod, K., Voorhees, J. R., Fuchs, T. A., et al. (2012). Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. U.S.A. 109, 13076–13081.
Diep, B., Chan, L., Tattevin, P., Kajikawa, O., Martin, T., Basuino, L., et al. (2010). Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc. Natl. Acad. Sci. U.S.A. 107, 5587–5592.
Döring, P., Grumann, D., Kolata, J., Goosmann, C., Löffler, B., Völker, U., et al. (2011). Neutrophil cell death induction by Staphylococcus aureus from nasal colonization and chronic furunculosis. Int. J. Med. Microbiol. 301, 40.
Döring, Y. (2012). Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 125, 1673–1683.
Douda, D., Jackson, R., Grasemann, H., and Palaniyar, N. (2011). Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J. Immunol. 187, 1856–1865.
Duffin, R., Leitch, A. E., Fox, S., Haslett, C., and Rossi, A. G. (2010). Targeting granulocyte apoptosis: mechanisms, models, and therapies. Immunol. Rev. 236, 28–40.
Edwards, S. W., Moulding, D. A., Derouet, M., and Moots, R. J. (2003). Regulation of neutrophil apoptosis. Chem. Immunol. Allergy 83, 204–224.
Engelich, G., White, M., and Hartshorn, K. L. (2001). Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J. Leukoc. Biol. 69, 50–56.
Fadok, V. A., Bratton, D. L., Konowal, A., Freed, P. W., Westcott, J. Y., and Henson, P. M. (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898.
Faurschou, M., and Borregaard, N. (2003). Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 5, 1317–1327.
Fliedner, T. M., Cronkite, E. P., and Robertson, J. S. (1964). Granulocytopoiesis. I. Senescence and random loss of neutrophilic granulocytes in human beings. Blood 24, 402–414.
Frøland, S. S. (1984). Bacterial infections in the compromised host. Scand. J. Infect. Dis. Suppl. 43, 7–16.
Fuchs, T., Abed, U., Goosmann, C., Hurwitz, R., Schulze, I., Wahn, V., et al. (2007). Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176, 231–241.
Fuchs, T., Brill, A., Duerschmied, D., Schatzberg, D., Monestier, M., Myers, D., et al. (2010). Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. U.S.A. 107, 15880–15885.
Fuchs, T. A., Brill, A., and Wagner, D. D. (2012). Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler. Thromb. Vasc. Biol. 32, 1777–1783.
Gabriel, C., McMaster, W. R., Girard, D., and Descoteaux, A. (2010). Leishmania donovani promastigotes evade the antimicrobial activity of neutrophil extracellular traps. J. Immunol. 185, 4319–4327.
Garcia-Romo, G. S., Caielli, S., Vega, B., Connolly, J., Allantaz, F., Xu, Z., et al. (2011). Netting neutrophils are major inducers of Type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra20.
Glasser, L., and Fiederlein, R. L. (1987). Functional differentiation of normal human neutrophils. Blood 69, 937–944.
Grinberg, N., Elazar, S., Rosenshine, I., and Shpigel, N. (2008). Beta-hydroxybutyrate abrogates formation of bovine neutrophil extracellular traps and bactericidal activity against mammary pathogenic Escherichia coli. Infect. Immun. 76, 2802–2807.
Guimarães-Costa, A., Nascimento, M. T. C., Froment, G. S., Soares, R. P. P., Morgado, F. N., Conceição-Silva, F., et al. (2009). Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. U.S.A. 106, 6748–6753.
Guimarães-Costa, A. B., Nascimento, M. T. C., Wardini, A. B., Pinto-Da-Silva, L. H., and Saraiva, E. M. (2012). ETosis: a microbicidal mechanism beyond cell death. J. Parasitol. Res. 2012, 929743.
Gupta, A., Hasler, P., Gebhardt, S., Holzgreve, W., and Hahn, S. (2006). Occurrence of neutrophil extracellular DNA traps (NETs) in pre-eclampsia. Ann. N. Y. Acad. Sci. 1075, 118–122.
Gupta, A. K., Hasler, P., Holzgreve, W., Gebhardt, S., and Hahn, S. (2005). Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum. Immunol. 66, 1146–1154.
Gupta, A. K., Joshi, M. B., Philippova, M., Erne, P., Hasler, P., Hahn, S., et al. (2010). Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 584, 3193–3197.
Hakkim, A., Fuchs, T. A., Martinez, N. E., Hess, S., Prinz, H., Zychlinsky, A., et al. (2011). Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 7, 75–77.
Hakkim, A., Fürnrohr, B., Amann, K., Laube, B., Abed, U., Brinkmann, V., et al. (2010). Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. U.S.A. 107, 9813–9818.
Hemmers, S., Teijaro, J. R., Arandjelovic, S., and Mowen, K. A. (2011). PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE 6:e22043. doi:10.1371/journal.pone.0022043
Hirai, H., Zhang, P., Dayaram, T., Hetherington, C., Mizuno, S.-I., Imanishi, J., et al. (2006). C/EBPbeta is required for “emergency” granulopoiesis. Nat. Immunol. 7, 732–739.
Huynh, M.-L. N., Fadok, V. A., and Henson, P. M. (2002). Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J. Clin. Invest. 109, 41–50.
Iyoda, T., Nagata, K., Akashi, M., and Kobayashi, Y. (2005). Neutrophils accelerate macrophage-mediated digestion of apoptotic cells in vivo as well as in vitro. J. Immunol. 175, 3475–3483.
Jann, N., Schmaler, M., Kristian, S., Radek, K., Gallo, R., Nizet, V., et al. (2009). Neutrophil antimicrobial defense against Staphylococcus aureus is mediated by phagolysosomal but not extracellular trap-associated cathelicidin. J. Leukoc. Biol. 86, 1159–1169.
Joiner, K. A., Ganz, T., Albert, J., and Rotrosen, D. (1989). The opsonizing ligand on Salmonella typhimurium influences incorporation of specific, but not azurophil, granule constituents into neutrophil phagosomes. J. Cell Biol. 109, 2771–2782.
Juneau, R., Pang, B., Weimer, K. E. D., Armbruster, C., and Swords, W. E. (2011). Nontypeable Haemophilus influenzae initiates formation of neutrophil extracellular traps. Infect. Immun. 79, 431–438.
Katzenback, B. A., and Belosevic, M. (2009). Isolation and functional characterization of neutrophil-like cells, from goldfish (Carassius auratus L.) kidney. Dev. Comp. Immunol. 33, 601–611.
Kennedy, A., and DeLeo, F. (2009). Neutrophil apoptosis and the resolution of infection. Immunol. Res. 43, 25–61.
Khatua, B., Bhattacharya, K., and Mandal, C. (2012). Sialoglycoproteins adsorbed by Pseudomonas aeruginosa facilitate their survival by impeding neutrophil extracellular trap through siglec-9. J. Leukoc. Biol. 91, 641–655.
Kim, S., Elkon, K. B., and Ma, X. (2004). Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity 21, 643–653.
Knight, J. S., and Kaplan, M. J. (2012). Lupus neutrophils: “NET” gain in understanding lupus pathogenesis. Curr. Opin. Rheumatol. 24, 441–450.
Kobayashi, S., Braughton, K., Palazzolo-Ballance, A., Kennedy, A., Sampaio, E., Kristosturyan, E., et al. (2010). Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J. Innate Immun. 2, 560–575.
Kobayashi, S., Braughton, K., Whitney, A., Voyich, J., Schwan, T., Musser, J., et al. (2003a). Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc. Natl. Acad. Sci. U.S.A. 100, 10948–10953.
Kobayashi, S., Voyich, J., Somerville, G., Braughton, K., Malech, H., Musser, J., et al. (2003b). An apoptosis-differentiation program in human polymorphonuclear leukocytes facilitates resolution of inflammation. J. Leukoc. Biol. 73, 315–322.
Kobayashi, S., and DeLeo, F. (2009). Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip. Rev. Syst. Biol. Med. 1, 309–333.
Kobayashi, S., Voyich, J., Buhl, C., Stahl, R., and DeLeo, F. (2002). Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc. Natl. Acad. Sci. U.S.A. 99, 6901–6906.
Kobayashi, S. D., Rigby, K. M., and DeLeo, F. R. (2012). “Bacteria-induced host cell death,” in Bacterial Pathogenesis: Molecular and Cellular Mechanisms, eds C. Locht and M. Simonet (Norfolk: Caister Academic Press), 317–362.
Lande, R., Ganguly, D., Facchinetti, V., Frasca, L., Conrad, C., Gregorio, J., et al. (2011). Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra19.
Lauth, X., Von Köckritz-Blickwede, M., McNamara, C., Myskowski, S., Zinkernagel, A., Beall, B., et al. (2009). M1 protein allows Group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J. Innate Immun. 1, 202–214.
Leshner, M., Wang, S., Lewis, C., Zheng, H., Chen, X. A., Santy, L., et al. (2012). PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 3:307. doi:10.3389/fimmu.2012.00307
Levy, O., Martin, S., Eichenwald, E., Ganz, T., Valore, E., Carroll, S. F., et al. (1999). Impaired innate immunity in the newborn: newborn neutrophils are deficient in bactericidal/permeability-increasing protein. Pediatrics 104, 1327–1333.
Li, P., Li, M., Lindberg, M., Kennett, M., Xiong, N., and Wang, Y. (2010). PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862.
Lin, A., Rubin, C., Khandpur, R., Wang, J., Riblett, M., Yalavarthi, S., et al. (2011). Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 187, 490–500.
Liu, C., Tangsombatvisit, S., Rosenberg, J., Mandelbaum, G., Gillespie, E., Gozani, O., et al. (2012). Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 14, R25–R25.
Liu, L., Belkadi, A., Darnall, L., Hu, T., Drescher, C., Cotleur, A. C., et al. (2010). CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat. Neurosci. 13, 319–326.
Ma, A. C., and Kubes, P. (2008). Platelets, neutrophils, and neutrophil extracellular traps (NETs) in sepsis. J. Thromb. Haemost. 6, 415–420.
Marin-Esteban, V., Turbica, I., Dufour, G., Semiramoth, N., Gleizes, A., Gorges, R., et al. (2012). Afa/Dr diffusely adhering Escherichia coli strain C1845 induces neutrophil extracellular traps that kill bacteria and damage human enterocyte-like cells. Infect. Immun. 80, 1891–1899.
Martin, C., Burdon, P. C. E., Bridger, G., Gutierrez-Ramos, J.-C., Williams, T. J., and Rankin, S. M. (2003). Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 19, 583–593.
McCormick, A., Heesemann, L., Wagener, J., Marcos, V., Hartl, D., Loeffler, J., et al. (2010). NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect. 12, 928–936.
Menegazzi, R., Decleva, E., and Dri, P. (2012). Killing by neutrophil extracellular traps: fact or folklore? Blood 119, 1214–1216.
Midon, M., Schäfer, P., Pingoud, A., Ghosh, M., Moon, A., Cuneo, M., et al. (2011). Mutational and biochemical analysis of the DNA-entry nuclease EndA from Streptococcus pneumoniae. Nucleic Acids Res. 39, 623–634.
Milot, E., and Filep, J. G. (2011). Regulation of neutrophil survival/apoptosis by Mcl-1. ScientificWorldJournal 11, 1948–1962.
Mitroulis, I., Kambas, K., Chrysanthopoulou, A., Skendros, P., Apostolidou, E., Kourtzelis, I., et al. (2011). Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS ONE 6:e29318. doi:10.1371/journal.pone.0029318
Munafo, D., Johnson, J., Brzezinska, A., Ellis, B., Wood, M., and Catz, S. (2009). DNase I inhibits a late phase of reactive oxygen species production in neutrophils. J. Innate Immun. 1, 527–542.
Narasaraju, T., Yang, E., Samy, R. P., Ng, H. H., Poh, W. P., Liew, A.-A., et al. (2011). Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 179, 199–210.
Nauseef, W. M., and Clark, R. A. (2010). Granulocytic Phagocytes. New York: Elsevier/Churchill Livingstone.
Neeli, I., Khan, S., and Radic, M. (2008). Histone deimination as a response to inflammatory stimuli in neutrophils. J. Immunol. 180, 1895–1902.
Nordenfelt, P., Bauer, S., Lönnbro, P., and Tapper, H. (2009). Phagocytosis of Streptococcus pyogenes by all-trans retinoic acid-differentiated HL-60 cells: roles of azurophilic granules and NADPH oxidase. PLoS ONE 4:e7363. doi:10.1371/journal.pone.0007363
Palazzolo-Ballance, A., Reniere, M., Braughton, K., Sturdevant, D., Otto, M., Kreiswirth, B., et al. (2008). Neutrophil microbicides induce a pathogen survival response in community-associated methicillin-resistant Staphylococcus aureus. J. Immunol. 180, 500–509.
Palić, D., Ostojić, J., Andreasen, C. B., and Roth, J. A. (2007). Fish cast NETs: neutrophil extracellular traps are released from fish neutrophils. Dev. Comp. Immunol. 31, 805–816.
Palmer, L. J., Chapple, I. L., Wright, H. J., Roberts, A., and Cooper, P. R. (2011). Extracellular deoxyribonuclease production by periodontal bacteria. J. Periodontal Res. 47, 439–445.
Panopoulos, A. D., and Watowich, S. S. (2008). Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and “emergency” hematopoiesis. Cytokine 42, 277–288.
Papayannopoulos, V., Staab, D., and Zychlinsky, A. (2011). Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS ONE 6:e28526. doi:10.1371/journal.pone.0028526
Papayannopoulos, V., and Zychlinsky, A. (2009). NETs: a new strategy for using old weapons. Trends Immunol. 30, 513–521.
Pillay, J., Den Braber, I., Vrisekoop, N., Kwast, L., De Boer, R., Borghans, J. A. M., et al. (2010). In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 116, 625–627.
Ramos-Kichik, V., Mondragón-Flores, R., Mondragón-Castelán, M., Gonzalez-Pozos, S., Muñiz-Hernandez, S., Rojas-Espinosa, O., et al. (2009). Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb.) 89, 29–37.
Remijsen, Q., Kuijpers, T. W., Wirawan, E., Lippens, S., Vandenabeele, P., and Vanden Berghe, T. (2011). Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 18, 581–588.
Rigby, K., and DeLeo, F. (2012). Neutrophils in innate host defense against Staphylococcus aureus infections. Semin. Immunopathol. 34, 237–259.
Riyapa, D., Buddhisa, S., Korbsrisate, S., Cuccui, J., and Wren, B. W. (2012). Neutrophil extracellular traps exhibit antibacterial activity against Burkholderia pseudomallei and are influenced by bacterial and host factors. Infect. Immun. 80, 3921–3929.
Rogers, D. E., and Tompsett, R. (1952). The survival of staphylococci within human leukocytes. J. Exp. Med. 95, 209–230.
Rosenbauer, F., and Tenen, D. G. (2007). Transcription factors in myeloid development: balancing differentiation with transformation. Nat. Rev. Immunol. 7, 105–117.
Saffarzadeh, M., Juenemann, C., Queisser, M. A., Lochnit, G., Barreto, G., Galuska, S. P., et al. (2012). Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE 7:e32366. doi:10.1371/journal.pone.0032366
Saitoh, T., Komano, J., Saitoh, Y., Misawa, T., Takahama, M., Kozaki, T., et al. (2012). Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12, 109–116.
Savill, J. S., Wyllie, A. H., Henson, J. E., Walport, M. J., Henson, P. M., and Haslett, C. (1989). Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest. 83, 865–875.
Scapinello, S., Brooks, A. S., MacInnes, J. I., Hammermueller, J., Clark, M. E., and Caswell, J. L. (2011). Bactericidal activity of porcine neutrophil secretions. Vet. Immunol. Immunopathol. 139, 113–118.
Serhan, C. N., Chiang, N., and Van Dyke, T. E. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361.
Springer, D. J., Ren, P., Raina, R., Dong, Y., Behr, M. J., McEwen, B. F., et al. (2010). Extracellular fibrils of pathogenic yeast Cryptococcus gattii are important for ecological niche, murine virulence and human neutrophil interactions. PLoS ONE 5:e10978. doi:10.1371/journal.pone.0010978
Summers, C., Rankin, S. M., Condliffe, A. M., Singh, N., Peters, A. M., and Chilvers, E. R. (2010). Neutrophil kinetics in health and disease. Trends Immunol. 31, 318–324.
Szarowicz, S. E., and Friedlander, A. (2011). Bacillus anthracis stimulates the formation of neutrophil extracellular traps. Mol. Biol. Cell 22, 4705.
Thomas, G. M., Carbo, C., Curtis, B. R., Martinod, K., Mazo, I. B., Schatzberg, D., et al. (2012). Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood 119, 6335–6345.
Thwaites, G. E., and Gant, V. (2011). Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat. Rev. Microbiol. 9, 215–222.
Tobias, J. D., and Schleien, C. (1991). Granulocyte transfusions – a review for the intensive care physician. Anaesth. Intensive Care 19, 512–520.
Urban, C. F., Reichard, U., Brinkmann, V., and Zychlinsky, A. (2006). Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 8, 668–676.
Van Den Berg, Y. W., and Reitsma, P. H. (2011). Not exclusively tissue factor: neutrophil extracellular traps provide another link between chemotherapy and thrombosis. J. Thromb. Haemost. 9, 2311–2312.
Villanueva, E., Yalavarthi, S., Berthier, C., Hodgin, J., Khandpur, R., Lin, A., et al. (2011). Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 187, 538–552.
Voll, R. E., Herrmann, M., Roth, E. A., Stach, C., Kalden, J. R., and Girkontaite, I. (1997). Immunosuppressive effects of apoptotic cells. Nature 390, 350–351.
von Köckritz-Blickwede, M., Goldmann, O., Thulin, P., Heinemann, K., Norrby Teglund, A., Rohde, M., et al. (2008). Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111, 3070–3080.
Voyich, J., Braughton, K., Sturdevant, D., Whitney, A., Sad-Salim, B., Porcella, S., et al. (2005). Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol. 175, 3907–3919.
Voyich, J., Otto, M., Mathema, B., Braughton, K., Whitney, A., Welty, D., et al. (2006). Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J. Infect. Dis. 194, 1761–1770.
Wardini, A., Guimarães-Costa, A., Nascimento, M. T. C., Nadaes, N., Danelli, M. G. M., Mazur, C., et al. (2010). Characterization of neutrophil extracellular traps in cats naturally infected with feline leukemia virus. J. Gen. Virol. 91, 259–264.
Wartha, F., Beiter, K., Albiger, B., Fernebro, J., Zychlinsky, A., Normark, S., et al. (2007). Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell. Microbiol. 9, 1162–1171.
Watson, R. W., Redmond, H. P., Wang, J. H., and Bouchier Hayes, D. (1996). Bacterial ingestion, tumor necrosis factor-alpha, and heat induce programmed cell death in activated neutrophils. Shock 5, 47–51.
Weathington, N. M., Van Houwelingen, A. H., Noerager, B. D., Jackson, P. L., Kraneveld, A. D., Galin, F. S., et al. (2006). A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat. Med. 12, 317–323.
Webster, S., Daigneault, M., Bewley, M., Preston, J., and Marriott, H. (2010). Distinct cell death programs in monocytes regulate innate responses following challenge with common causes of invasive bacterial disease. J. Immunol. 185, 2968–2979.
Weissman, I. L., Anderson, D. J., and Gage, F. (2001). Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 17, 387–403
Whyte, M. K., Meagher, L. C., MacDermot, J., and Haslett, C. (1993). Impairment of function in aging neutrophils is associated with apoptosis. J. Immunol. 150, 5124–5134.
Xu, J., Zhang, X., Pelayo, R., Monestier, M., Ammollo, C. T., Semeraro, F., et al. (2009). Extracellular histones are major mediators of death in sepsis. Nat. Med. 15, 1318–1321.
Yipp, B. G., Petri, B., Salina, D., Jenne, C. N., Scott, B. N. V., Zbytnuik, L. D., et al. (2012). Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 18, 1386–1393.
Young, R. L., Malcolm, K. C., Kret, J. E., Caceres, S. M., Poch, K. R., Nichols, D. P., et al. (2011). Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE 6:e23637. doi:10.1371/journal.pone.0023637
Yousefi, S., Gold, J. A., Andina, N., Lee, J. J., Kelly, A. M., Kozlowski, E., et al. (2008). Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 14, 949–953.
Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., and Simon, H. U. (2009). Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 16, 1438–1444.
Keywords: neutrophil, apoptosis, necrosis, phagocytosis, inflammation
Citation: Lu T, Kobayashi SD, Quinn MT and DeLeo FR (2012) A NET outcome. Front. Immun. 3:365. doi: 10.3389/fimmu.2012.00365
Received: 10 October 2012; Accepted: 16 November 2012;
Published online: 05 December 2012.
Edited by:
Marko Radic, University of Tennessee, USAReviewed by:
Robert A. Harris, Karolinska Institutet, SwedenMartin Herrmann, Universitätsklinikum Erlangen, Germany
Copyright: © 2012 Lu, Kobayashi, Quinn and DeLeo. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Frank R. DeLeo, Laboratory of Human Bacterial Pathogenesis, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health, 903 South 4th Street, Hamilton, MT 59840, USA. e-mail:ZmRlbGVvQG5pYWlkLm5paC5nb3Y=