 Lander Egaña-Gorroño1
Lander Egaña-Gorroño1 Esteban Martínez2 Tuixent Escribà1 Marta Calvo2 José M. Gatell1,2
Esteban Martínez2 Tuixent Escribà1 Marta Calvo2 José M. Gatell1,2 Mireia Arnedo1*
Mireia Arnedo1*- 1Group of Genomics and Pharmacogenomics in HIV, Retrovirology and Viral Immunopathology Laboratory, Institut D’Investigacions Biomèdiques August Pi i Sunyer, Hospital Clínic Barcelona, Barcelona, Spain
- 2Department of Infectious Diseases, Hospital Clínic, Faculty of Medicine, University of Barcelona, Barcelona, Spain
Objectives: General population studies have shown associations between copy number variation (CNV) of the LPA gene Kringle-IV type-2 (KIV-2) coding region, single-nucleotide polymorphism (SNP) rs6415084 in LPA and coronary heart disease (CHD). Because risk factors for HIV-infected patients may differ from the general population, we aimed to assess whether these potential associations also occur in HIV-infected patients. Methods: A unicenter, retrospective, case–control (1:3) study. Eighteen HIV-patients with confirmed diagnosis of acute myocardial infarction (AMI) were adjusted for age, gender, and time since HIV diagnosis to 54 HIV-patients without CHD. After gDNA extraction from frozen blood, both CNV and SNP genotyping were performed using real-time quantitative PCR. All genetic and non-genetic variables for AMI were assessed in a logistic regression analysis. Results: Our results did not confirm any association in terms of lipoprotein(a) LPA structural genetic variants when comparing KIV-2 CNV (p = 0.67) and SNP genotypes (p = 0.44) between AMI cases and controls. However, traditional risk factors such as diabetes mellitus, hypertension, and CD4+ T cell count showed association (p < 0.05) with CHD. Conclusion: Although significant associations of AMI with diabetes, hypertension and CD4+ T cell count in HIV-patients were found, this study could not confirm the feasibility neither of KIV-2 CNV nor rs6415084 in LPA as genetic markers of CHD in HIV-infected patients.
Highlights:
● Individuals with HIV infection are at higher risk of coronary heart disease (CHD) than the non-infected population.
● Our results showed no evidence of LPA structural genetic variants associated with CHD in HIV-1-infected patients.
● Associations were found between diabetes mellitus, arterial hypertension, CD4+ T cell count, and CHD.
● The clinical usefulness of these biomarkers to predict CHD in HIV-1-infected population remains unproven.
● Further studies are needed to assess the contribution of common genetic variations to CHD in HIV-infected individuals.
Introduction
Since the first case reports of acute myocardial infarction (AMI) in HIV-1-infected patients on highly active antiretroviral therapy (HAART) were described, it has become increasingly evident that individuals with HIV infection are at higher risk of cardiovascular events than the general population (Triant et al., 2007). Besides, the contribution of classic risk factors, other factors such as viral replication, HIV-associated inflammation and/or immunodeficiency, and antiretroviral therapy have been associated with premature cardiovascular disease (CVD; Friis-Møller et al , 2003; Hansson, 2005). The study of genetic traits and their relation to complex diseases, e.g., CVD, still poses a major challenge.
Increased plasma concentrations of atherogenic lipoproteins play an important role in the development of atherosclerosis leading to premature AMI and ischemic stroke. One particular fraction, which is important in that respect, is lipoprotein(a) [Lp(a)]. Elevated plasma concentrations of Lp(a) have been associated with the risk of coronary heart disease (CHD) in the general population (Rhoads et al., 1986; Ridker et al., 1993; Ohira et al., 2006). The plasma concentration of Lp(a) varies over a wide range among individuals. Furthermore, the inter-individual variation in Lp(a) level is 90% genetically determined by the LPA locus on chromosome 6, although plasma Lp(a) in a particular individual remains stable over a lifetime (Boerwinkle et al., 1992).
Lipoprotein(a) is composed of an apolipoprotein(a) [Apo(a)] molecule encoded by LPA, which is connected via a disulphide bond to the apolipoprotein B-100 of a proatherogenic LDL-cholesterol particle (McLean et al., 1987; Berglund and Ramakrishnan, 2004). The Apo(a) molecule is composed of a signal peptide region, 10 types of kringles that differ in sequence but are homologous to plasminogen kringle IV (KIV1–10), a kringle homologous to plasminogen kringle V (KV) and an inactive protease-like domain (McLean et al., 1987; Gavish et al., 1989). The size of Apo(a) is determined by a copy number variation (CNV) of the kringle-IV type 2 (KIV-2) coding region (encoded by exons 4 and 5 in LPA gene), and this being negatively correlated with Lp(a) levels (Lanktree et al., 2009). The genetically determined KIV-2 repeat number affects the final size of the Apo(a) protein, with larger isoforms being compromised with respect to protein folding, transport, and secretion (Lanktree et al., 2010). Furthermore, the LPA single-nucleotide polymorphism (SNP) rs6415084 (C → T), within the same haplotype block as the KIV-2 CNV, has been reported to be significantly associated with both Lp(a) concentrations and the KIV-2 copy number (CN; Paultre et al., 2000; Clarke et al., 2009). Several studies have identified increased atherogenesis and CHD in individuals with fewer apo(a) KIV-2 repeats (Kraft et al., 1992, 1996; Sandholzer et al., 1992). The number of KIV-2 repeats varies among subjects and ranges from 11 to >50 (Kannel et al., 1976; Lackner et al., 1993). Contrary to KIV-2 with CN > 25, a CN ≥22 leads to significantly higher Lp(a) levels, which are more frequent in CHD patients (Sandholzer et al., 1992; Kraft et al., 1996). The aim of this study was to test whatever association between KIV-2 CNV and CHD could be confirmed in HIV-infected patients, as well as association between classical clinical risk factors and CHD.
Materials and Methods
The study population consisted of 72 HIV-1-infected individuals under HAART who were followed-up at the Hospital Clínic of Barcelona during the study period (January 1, 1997 to December 31, 2008). They had all signed the ethical informed consent for genetic testing. The retrospective case–control study (1:3) was based on CHD patients from whom genetic testing could be performed (n = 18). CHD was defined according to the criteria of the Joint European Society of Cardiology and the American College of Cardiology Committee for the Redefinition of Myocardial Infarction (The Joint European Society of Cardiology/American College of Cardiology Committee, 2000). Patients with CHD, had either an ST-segment elevation myocardial infarction (n = 11, 61%) or a non-ST-segment elevation myocardial infarction (n = 7, 39%) if there was or not an evolving ST-segment elevation >0.1 mV in two contiguous leads, respectively. Each case was matched by age, gender, and time since HIV diagnosis to three controls without CHD (n = 54). Besides age and gender, relevant data on HIV infection and on clinical risk factors for CVD were collected. DNA was isolated from frozen blood according to manufacturer’s instructions using the QIAamp DNA Blood Mini Kit, automated by QIAcube (QIAGEN). For the CNV analysis a multiplex qPCR was carried out using custom TaqMan probes for exons 4 and 5 in LPA and single-copy reference gene RNaseP (part number 4316844) in the Applied Biosystems 7900HT Fast Real-time PCR System. Reaction volumes contained 4 µl of water, 1 µl of 20× TaqMan primer/probe mix for LPA, 1 µl of 20× TaqMan primer/probe mix for RNaseP, 10 µl of 2× Genotyping Master Mix (Applied Biosystems), and 4 µl of genomic DNA at a final concentration of 5–10 ng. Six replicates were run in all qPCRs. Thermocycler conditions were as follows: 95°C hot-start for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Absolute quantification (AQ) files were then exported to the Copy Caller Software (Applied Biosystems) for relative quantifications (RQ) and final CN analysis.
Intronic SNP rs6415084 in LPA gene was genotyped by TaqMan Allelic Discrimination, using TaqMan SNP genotyping assays predesigned by Applied Biosystems (part number C_27422575_10). Reaction volumes contained 1.25 µl of 20× TaqMan SNP Assay, 12.5 µl of 2× Universal Master Mix (Applied Biosystems), and 11.25 µl of genomic DNA at a final concentration of 1–20 ng. Thermocycler conditions were as follows: 95°C hot-start for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. 15% of the total samples were re-genotyped in order to check reproducibility.
A basic case/control association test, based on a Fisher’s exact test, was performed by comparison of calculated CN variants and SNP genotypes between AMI cases and controls using SNPator software (CEGEN, Barcelona, Spain). A multivariate analysis through logistic regression model was performed in order to assess the influence of genetic and non-genetic variables using SPSS v.15. With the sample size available, any difference above 20% would have been detected with a power of 80%.
Results
Baseline characteristics of the study participants are shown in Table 1. The median age was 44 years (interquartile range 12). Men represented 88.9% in both groups. Groups of risk for HIV acquisition were 50% homosexuals, 25% heterosexuals, and 25% injection drugs users in both groups. The median CD4+ T cell count (cell/µl) was 107 (interquartile range 261) in the cases and 210 (interquartile range 275) in the controls. HIV viral load (log) was 4.81 (interquartile range 5.92) in the cases and 4.62 (interquartile range 5.34) in the controls. A total of 88.9% of cases and 64.8% of controls were exposed to protease inhibitor containing regimens. Diabetes mellitus and usual smokers were present in 22.2 and 55.5% of cases and 14.8 and 37.03% of controls, respectively. A total of 22.2% of cases and 9.25% of controls had hypertension, and 22.2% of cases and 3.7% of controls had family antecedents of CVD. At least, one AIDS event was suffered by 2 (11.1%) cases and 12 (22.2%) controls. Both groups had a median Framingham risk score (FRS) of 2% (interquartile range 14 in cases and 5.5 in controls).
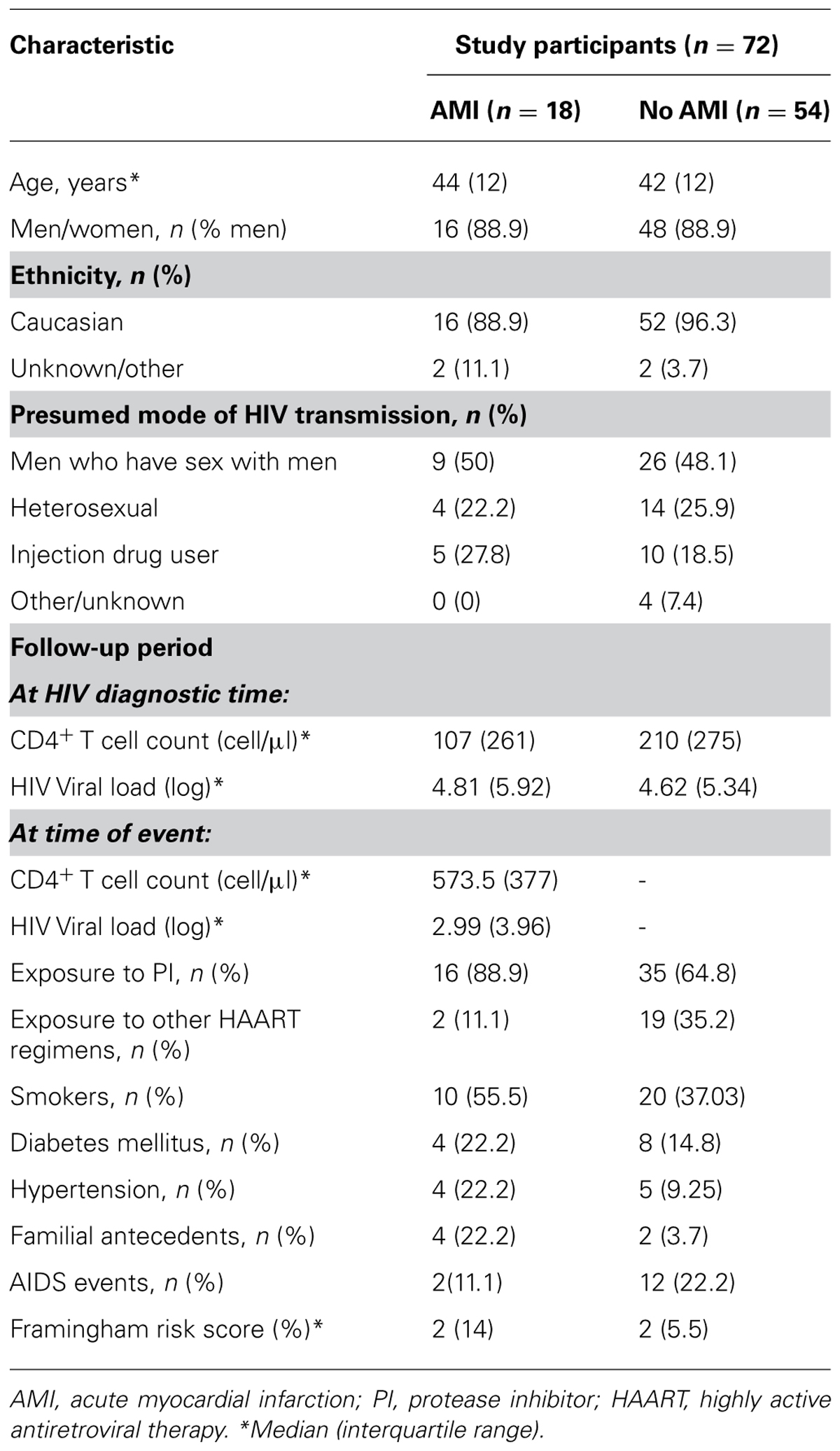
TABLE 1. Baseline characteristics of the study participants.
All participants were successfully genotyped. As it was expected, the LPA KIV-2 repeat analysis ranged between 17 and 48 copies. The case/control analysis showed CN ≥22 in seven patients (one case vs. six controls) and CN > 25 in 65 patients [17 cases vs. 48 controls; p = 0.46; OR = 0.44 (0.049–3.99)]. SNP rs6415084 was checked to confirm Hardy–Weinberg equilibrium. Genotyping assay showed common homozygosity (C/C) in 17 patients (3 cases vs. 14 controls), rare homozygosity (T/T) in 20 patients (7 cases vs. 13 controls), and heterozygosity (C/T) in 35 patients [8 cases vs. 27 controls; p = 0.42; OR = 0.57 (0.144, 2.274)].
Logistic regression analysis showed significant associations (p < 0.05) between traditional risk factors such as diabetes mellitus, hypertension, and CD4+ T cell count at HIV-infected diagnosis and CHD (Table 2). The strength of the association was quantified using the odds ratios and the 95% confidence intervals.
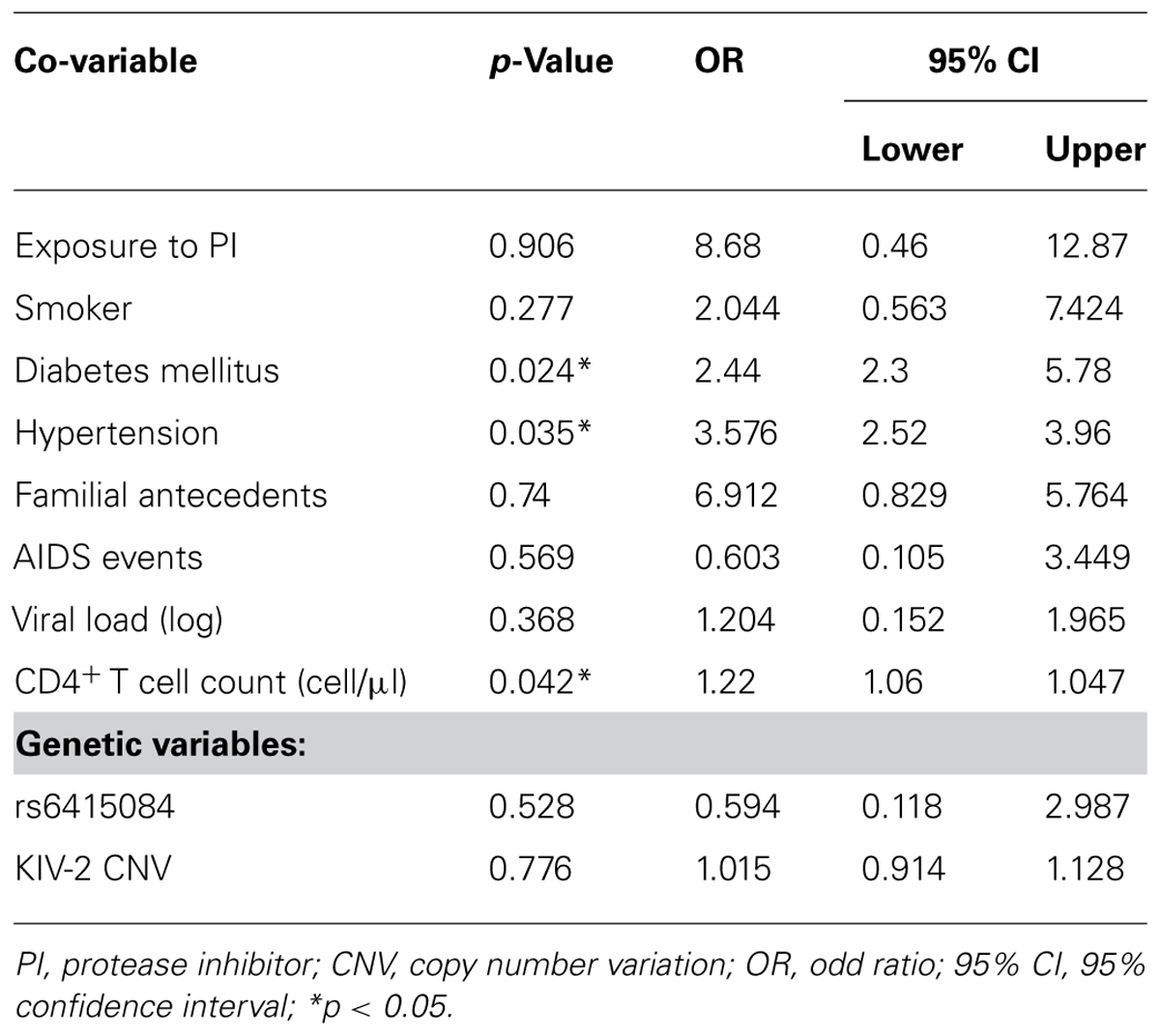
TABLE 2. Multivariate analysis through logistic regression including all genetic and non-genetic co-variables.
Conclusion
The most important finding in our study is that we were able to detect associations between clinical risk factors and CHD. Non-genetic variables such us diabetes mellitus, hypertension, and CD4+ T cell count at HIV-infected diagnosis reached significance in the logistic regression analysis. The influence of CD4+ T cell level on CVD is supported by previously reported studies (Paultre et al., 2000). However, any previously described associations between LPA genetics biomarkers (KIV-2 CNV nor rs6415084) and CHD were found. The association between SNPs in LPA gene and circulating Lp(a) levels, and consequently with CHD, may be mediated by various mechanisms: (i) some of the SNPs may be in linkage disequilibrium with the KIV-2 repeat polymorphism which has been shown to explain approximately 50% of the genetic variation in Lp(a) concentrations; (ii) certain SNPs may directly influence the transcriptional and/or translational processes of the LPA gene (Paultre et al., 2000); and (iii) some non-causal SNPs may be in linkage disequilibrium with SNPs having causal effect on Lp(a) concentrations. The main important limitation of our study was the low absolute number of HIV-patients with documented AMI. Furthermore, larger number of individuals could not be included in the study because of the unavailability of data or blood sample for the purpose of this study. Lp(a) concentrations from frozen blood could not be measured. Although the power of the study was limited due to the small sample size analyzed, there is no other study regarding CNV in LPA and CHD in HIV-infected patients.
In summary, our results did not confirm evidence of LPA structural genetic variants associated with CHD in HIV-1-infected patients. Although the power of the study was limited, traditional risk factors are contributing to the progression of CHD amongst HIV-infected population as well as they do in non-HIV population. Moreover, there are factors in HIV-infected population that are not present in non-HIV-infected individuals and are unrelated to genetics: HIV and HAART. The pathogenesis of HIV-1-infection and/or exposure to HAART might contribute to CHD in a stronger way than LPA genetic variants do. The clinical utility of these biomarkers to predict CHD in HIV-1-infected population therefore remains unproven. Further collaborative studies with a larger number of HIV-infected patients are needed.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Retrovirology and Viral Immunopathology Laboratory of the Institut D’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). Mireia Arnedo is a recipient of a grant from the Miguel Servet program 06/0164 IDIBAPS-ISCIII and Lander Egaña-Gorroño is a recipient of a pre-doctoral grant (FI10/00174, ISCIII). This work was partly financed by a grant from Fondo de Investigación Sanitaria (09/00369), Instituto de Salud Carlos III, and Fondo Europeo para el Desarrollo Regional (FEDER).
References
Berglund, L., and Ramakrishnan, R. (2004). Lipoprotein(a): an elusive cardiovascular risk factor. Arterioscler. Thromb. Vasc. Biol. 24, 2219–2226.
Boerwinkle, E., Leffert, C. C., Lin, J., Lackner, C., Chiesa, G., and Hobbs, H. H. (1992). Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J. Clin. Invest. 90, 52–60.
Clarke, R., Peden, J. F., Hopewell, J. C., Kyriakou, T., Goel, A., Heath, S. C., et al. for the PROCARDIS Consortium. (2009). Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Eng. J. Med. 361, 2518–2528.
Friis-Møller, N., Sabin, C. A., Weber, R., d’Arminio Monforte, A., El-Sadr, W. M., Reiss, P., et al. (2003). Combination antiretroviral therapy and the risk of myocardial infarction. N. Engl. J. Med. 349, 1993–2003.
Gavish, D., Azrolan, N., and Breslow, J. L. (1989). Plasma lp(a) concentration is inversely correlated with the ratio of Kringle IV/Kringle V encoding domains in the apo(a) gene. J. Clin. Invest. 84, 2021–2027.
Hansson, G. K. (2005). Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695.
Kannel, W. B., McGee, D., and Gordon, T. (1976). Cardiovascular risk factors. Insights from Framingham Heart Study. Am. J. Cardiol. 38, 46–51.
Kraft, H. G., Köchl, S., Menzel, H. J., Sandholzer, C., and Utermann, G. (1992). The apolipoprotein(a) gene: a transcribed hypervariable locus controlling plasma lipoprotein (a) concentration. Hum. Genet. 90, 220–230.
Kraft, H. G., Lingenhel, A., Köchl, S., Hoppichler, F., Kronenberg, F., Abe, A., et al. (1996). Apolipoprotein(a) kringle IV repeat number predicts risk for coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 16, 713–719.
Lackner, C., Cohen, J. C., and Hobbs, H. H. (1993). Molecular definition of the extreme size polymorphism in apolipoprotein (a). Hum. Mol. Genet. 2, 933–940.
Lanktree, M. B., Anand, S. S., Yusuf, S., and Hegele, R. A. (2010). Comprehensive analysis of genomic variation in the LPA locus and its relationship to plasma lipoprotein (a) in south Asians, Chinese and European Caucasians. Circ. Cardiovasc. Genet. 3, 39–46.
Lanktree, M. B., Rajakumar, C., Brunt, J. H., Koschinsky, M. L., Connelly, P. W., and Hegele, R. A. (2009). Determination of lipoprotein(a) kringle repeat number from genomic DNA: copy number variation genotyping using qPCR. J. Lipid Res. 50, 768–772.
McLean, J. W., Tomlinson, J. E., Kuang, W. J., Eaton, D. L., Chen, E. Y., Fless, G. M., et al. (1987). cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 330, 132–137.
Ohira, T., Schreiner, P. J., Morrisett, J. D., Chambless, L. E., Rosamond, W. D., and Folsom, A. R. (2006). Lipoprotein (a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke 37, 1407–1412.
Paultre, F., Pearson, T. A., Weil, H. F., Tuck, C. H., Myerson, M., Rubin, J., et al. (2000). High levels of Lp(a) with a small apo(a) isoform are associated with coronary artery disease in African American and White men. Arterioscler. Thromb. Vasc. Biol. 20, 2619–2624.
Rhoads, G. G., Dahlen, G., Berg, K., Morton, N. E., and Dannenberg, A. L. (1986). Lp(a) lipoprotein as a risk factor for myocardial infarction. JAMA 256, 2540–2544.
Ridker, P. M., Hennekens, C. H., and Stampfer, M. J. (1993). A prospective study of lipoprotein (a) and the risk of myocardial infarction. JAMA 270, 2195–2199.
Sandholzer, C., Saha, N., Kark, J. D., Rees, A., Jaross, W., Dieplinger, H., et al. (1992). Apo(a) isoforms predict risk for coronary heart disease. A study in six populations. Arterioscler. Thromb. 12, 1214–1226.
The Joint European Society of Cardiology/American College of Cardiology Committee. (2000). Myocardial Infarction redefined: a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. Eur. Heart J. 21, 1502–1513.
Keywords: HIV, coronary heart disease, host genetics, copy number variation, LPA gene
Citation: Egaña-Gorroño L, Martínez E, Escribà T, Calvo M, Gatell JM and Arnedo M (2012) Association study of lipoprotein(a) genetic markers, traditional risk factors, and coronary heart disease in HIV-1-infected patients. Front. Immun. 3:367. doi: 10.3389/fimmu.2012.00367
Received: 28 August 2012; Accepted: 18 November 2012;
Published online: 06 December 2012.
Edited by:
Guido Poli, Vita-Salute San Raffaele University, ItalyReviewed by:
Angelo A. Manfredi, Vita-Salute San Raffaele University, ItalyGiuseppe Tambussi, Istituto di Ricovero e Cura a Carattere Scientifico Ospedale San Raffaele, Italy
Copyright: © 2012 Egaña-Gorroño, Martínez, Escribà, Calvo, Gatell and Arnedo. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Mireia Arnedo, Group of Genomics and Pharmacogenomics in HIV, Retrovirology and Viral Immunopathology Laboratory, Institut D’Investigacions Biomèdiques August Pi i Sunyer, Hospital Clínic Barcelona, Casanovas 143, Barcelona 08036, Spain. e-mail:bWFybmVkb0BjbGluaWMudWIuZXM=