 Stephanie Krieg1
Stephanie Krieg1 Evelyn Ullrich1,2,3*
Evelyn Ullrich1,2,3*- 1Hematology and Oncology, Department of Internal Medicine 5, University of Erlangen-Nuremberg, Erlangen, Germany
- 2Laboratory for Cellular Immunology, Department of Pediatric Hematology and Oncology, Children’s Hospital, Goethe University, Frankfurt, Germany
- 3Center for Cell and Gene Therapy, Goethe University, Frankfurt, Germany
There is a wide range of important pharmaceuticals used in treatment of cancer. Besides their known effects on tumor cells, there is growing evidence for modulation of the immune system. Immunomodulatory drugs (IMiDs®) play an important role in the treatment of patients with multiple myeloma or myelodysplastic syndrome and have already demonstrated antitumor, anti-angiogenic, and immunostimulating effects, in particular on natural killer (NK) cells. Tyrosine kinase inhibitors are directly targeting different kinases and are known to regulate effector NK cells and expression of NKG2D ligands (NKG2DLs) on tumor cells. Demethylating agents, histone deacetylases, and proteasome inhibitors interfere with the epigenetic regulation and protein degradation of malignant cells. There are first hints that these drugs also sensitize tumor cells to chemotherapy, radiation, and NK cell-mediated cytotoxicity by enhanced expression of TRAIL and NKG2DLs. However, these pharmaceuticals may also impair NK cell function in a dose- and time-dependent manner. In summary, this review provides an update on the effects of different novel molecules on the immune system focusing NK cells.
Introduction/Background
Immunomodulatory drugs (IMiDs®) have been introduced in the treatment of cancer with important clinical success. In addition to classical chemotherapeutic treatments, the novel classes of IMiDs® have been developed for the treatment of multiple myeloma (MM), myelodysplastic syndrome (MDS), chronic lymphatic leukemia (CLL), and B cell lymphoma (Bartlett et al., 2004a).
Tyrosine kinase inhibitors (TKIs) are administered in patients with tumors overexpressing kinases like BCR/ABL, stem cell factor receptor (c-KIT), platelet-derived growth factor receptor (PDGFR) and vascular endothelial growth factor receptor (VEGFR). TKIs are used in first-line for chronic and acute myeloid leukemia (CML and AML) with BCR/ABL overexpression as well as for gastrointestinal stromal tumors (GIST) with c-KIT mutations (Bennasroune et al., 2004). Furthermore, TKIs have been developed for the treatment of advanced metastatic renal cell carcinoma (RCC; Pirrotta et al., 2011).
Demethylating agents such as azacytidine and decitabine have been approved for treatment of AML, MDS, and other hematopoietic disorders (Issa et al., 2005; Garcia-Manero, 2008).
Histone deacetylases inhibitors (HDACis) have been designed as direct antitumor agents with major anti-proliferative effects. Some HDACis have been approved for the treatment of cutaneous T cell lymphoma (CTCL) and recently also for peripheral T cell lymphoma. HDACis are further tested in clinical phase II and III trials for multiple entities such as cervical, breast, and ovarian cancer, sarcoma, lymphoma, myeloma, and leukemia (Marks and Xu, 2009; Khan and La Thangue, 2012; Ning et al., 2012).
In addition, the proteasome inhibitor bortezomib has been approved for treatment of MM and mantle cell lymphoma (Voorhees and Orlowski, 2006; Orlowski and Kuhn, 2008).
Current studies addressing the use of immune modulating pharmaceutics in different other malignant diseases are in progress. Interestingly, these new pharmaceuticals do not only show direct antitumor effects, but also impact cellular immunity. In this review, we sum up the recent knowledge of the influence each of these drugs has on natural killer (NK) cells.
Natural killer cells are a heterogeneous lymphocyte population with cytotoxic antitumor capacity and multiple immunoregulatory properties. They express a wide range of activating receptors [e.g. natural cytotoxicity receptors (NCRs), some killer-cell immunoglobulin-like receptors (KIRs), and NKG2D], as well as inhibitory receptors (some KIRs, co-receptor NKG2A) that regulate NK cell activation and tolerance (Vivier et al., 2011). In the last years, exiting data revealed that modulation of the balance between activating and inhibiting NK cell signals, sensitization of malignant cells to NK cell-mediated cytotoxicity, optimization of the cross-talk of NK cells with other immune cells and the improvement of adoptive NK cell therapeutic protocols may provide novel immunotherapeutic concepts for treatment of cancer (Terme et al., 2008).
Immunomodulatory Drugs
Thalidomide was described as anti-angiogenic, antitumor, and immune modulatory agent affecting many cell types. To overcome the devastating side effects, including congenital defects when used during pregnancy, novel classes of IMiDs® have been developed (Bartlett et al., 2004a). The most important IMiDs® are lenalidomide (CC-5013, Revlimid®) and pomalidomide (CC-4047, Actimid®) but further thalidomide analogs are under clinical trials (Bartlett et al., 2004a; Reddy et al., 2008).
Thalidomide and IMiDs® were able to stimulate T cells to produce IFN-γ and IL-2 leading to NK cell activation (Davies et al., 2001; Hayashi et al., 2005). In addition, IMiDs® also showed direct effects on NK cells. Recent preclinical data revealed an augmentation of NK cell percentages in pomalidomide- and lenalidomide-treated peripheral blood mononuclear cells (PBMCs) from healthy donors that was mediated by the phosphoinositide (PI)-3 kinase signaling pathway causing enhanced IL-2 transcription and secretion in T cells (Hayashi et al., 2005). Pomalidomide induced expression of CD69 accompanied by a more than twofold increase of IFN-γ production and enhanced killing of K562 tumor cells (Payvandi et al., 2005). Interestingly, NK cells were required but not sufficient for lysis of K562, meaning that cytotoxicity was also dependent on other cell populations within the PBMCs (Zhu et al., 2008). In line with that, PBMCs treated with thalidomide, lenalidomide, or pomalidomide gained an increased killing capacity of MM tumor cells that was lost upon NK cell depletion (Davies et al., 2001). In addition to enhanced cytotoxicity, lenalidomide, and pomalidomide also increased antibody-dependent cell cytotoxicity (ADCC) toward MM cells (Hayashi et al., 2005). It has further been confirmed that the effect of pomalidomide on cytotoxicity and ADCC was induced by IL-2-producing T cells. Although signaling molecules that could directly mediate NK cell cytotoxicity and ADCC like ERK or p38 mitogen-activated protein kinase (MAPK) were not activated by pomalidomide, both IMiDs® were able to downregulate suppressor of cytokine signaling 1 (SOCS1) in NK, T, and NKT cells (Gorgun et al., 2010). Additionally, lenalidomide-treatment resulted in upregulation of CD16, CD40L, and LFA1 on NK cells thereby facilitating ADCC against MM cells (Tai et al., 2005). Recent work showed that lenalidomide does not affect NK cell proliferation among PBMCs but inhibits the production of IFN-γ in IL-12/IL-18 pre-activated, healthy donor-derived, purified NK cells. Furthermore, the NK cell phenotype was modified by enhancement of CD56 and decreased expression of KIRs and NKp46 (Dauguet et al., 2010). However, the reduced expression of receptors involved in NK cell-mediated killing was not sufficient to impair the cytolytic function of NK cells (Hayashi et al., 2005; Dauguet et al., 2010).
In vivo, administration of lenalidomide and pomalidomide resulted in a significant increase of NK cells in severe combined immunodeficient (SCID) mice with human lymphoma xenografts. The combination of IMiDs® together with rituximab prolonged the median survival and was abrogated by NK cell depletion (Hernandez-Ilizaliturri et al., 2005). In Raji-bearing SCID mice, application of IMiDs® led not only to NK cell expansion and activation but also to their migration into tumor beds. In this model, the effects of IMiDs® seemed to be related to dendritic cells (DCs) and their secreted cytokines (Reddy et al., 2008).
Remarkably, in patients responding to thalidomide-therapy an increased percentage of CD56+ cells has been observed (Davies et al., 2001). This observation is in line with other studies reporting an enhanced number of NK cells in patients during IMiD®-treatment (Bartlett et al., 2004b). In a clinical study addressing the effects of lenalidomide on MM patients with relapse after allogeneic stem cell transplantation, the monitoring of immune cell subpopulations revealed an increase of T, Treg, and activated NKp44+ NK cells (Lioznov et al., 2010).
To sum it up, IMiDs® mostly positively influence NK cell function, either direct or indirect via DC or T cell stimulation. Nevertheless, effects of IMiDs® on NK cells should be further investigated in clinical immunomonitoring studies.
Tyrosine Kinase Inhibitors
Tyrosine kinase receptors are involved in multiple cellular processes and have a crucial role in tumor development and progression. The inhibitors of those receptors are called TKIs and belong to a broad range of drugs (Bennasroune et al., 2004). Important TKIs are imatinib (Givec®, Gleevec®) and nilotinib (Tasigna®) that specifically inhibit BCR/ABL, PDGFR, and c-KIT, as well as dasatinib (Sprycel®) that additionally targets the SRC kinase (Krusch and Salih, 2011). Further multi-target TKIs like sunitinib (Sutent®) and sorafenib (Nexavar®) have been developed inhibiting various tyrosine kinases (Pirrotta et al., 2011).
Recent in vitro and in vivo studies indicated both direct inhibitory effects on immune cells including T and NK cells and indirect activatory or inhibitory effects on NK cell function via modification of markers on tumor cells caused by TKI-treatment (Seggewiss et al., 2005; Chen et al., 2008; Schade et al., 2008; Weichsel et al., 2008; Fraser et al., 2009). On side of the tumor, a direct control of the expression of the NKG2D ligands (NKG2DLs) MHC class I-related chain molecules (MIC)A/B by BCR/ABL has been shown and was reduced by different TKIs leading to decreased NK cell-mediated cytotoxicity and IFN-γ production (Boissel et al., 2006; Salih et al., 2010). A similar effect was shown after imatinib-treatment of a leukemic cell line transfected with high levels of BCR/ABL representing an ideal NK cell target. Imatinib led to diminished killing that was accompanied by decreased ICAM-1 expression on target cells and was most likely due to reduced formation of NK cell/target immunological synapses (Baron et al., 2002; Cebo et al., 2006). On the NK cell effector side, direct exposure of human NK cells with pharmacological doses of imatinib had no impact on NK cytotoxicity or cytokine production, whereas nilotinib negatively influenced cytokine production and dasatinib additionally abrogated cytotoxicity in vitro. The direct modulation of NK cells by dasatinib was apparently based on its impact on signaling cascades preventing phosphorylation of PI-3 kinase and ERK1/2 (Salih et al., 2010). Interestingly, inhibition by dasatinib seemed to be reversible as washing NK cells mainly restored cytotoxicity (Blake et al., 2008; Hassold et al., 2012).
Further, in a murine model NK cell activation could be induced by imatinib-treated DC in vitro and in vivo (Borg et al., 2004). The positive, most likely NK cell-dependent, antitumor effect of imatinib was further augmented by IL-2 in another murine model (Taieb et al., 2006). Other data showed, that frequencies of NK cells were not altered by imatinib-treatment in mice (Balachandran et al., 2011).
In contrary to the TKIs described so far, treatment of tumor cells with the multi-kinase inhibitors sorafenib and sunitinib increased their susceptibility for cytolysis by NK cells. Treatment of a hepatocellular carcinoma cell (HCC) line with sorafenib did not affect HLA class I expression but increased membrane-bound MICA and decreased soluble MICA resulting in enhanced NK cell-mediated cytotoxicity. Sorafenib led to a decline of the metalloprotease ADAM9 that is usually upregulated in human HCC resulting in MICA shedding (Kohga et al., 2010). Also, incubation of a nasopharyngeal carcinoma cell line with sunitinib increased the expression of NKG2DL better than sorafenib leading to a higher NK cell-mediated cytotoxicity (Huang et al., 2011). On the other side, in line with the other TKIs mentioned before, pharmacological concentrations of sorafenib but not sunitinib reduced cytotoxicity and cytokine production of resting and IL-2-activated NK cells in vitro by impaired granule mobilization apparently due to diminished phosphorylation of ERK1/2 and PI-3 kinase. Notably, sunitinib only altered cytotoxicity and cytokine production when added in high doses which were not reached in patients (Krusch et al., 2009).
In immunomonitoring analysis, NK cell percentages did not differ between imatinib-treated Philadelphia chromosome positive ALL patients and healthy donors (Maggio et al., 2011). In CML patients, the NK cell percentages were decreased at diagnosis and did not recover during imatinib therapy. This was accompanied by reduced degranulation response to tumor cells (Chen et al., 2012). Another study compared NK cell numbers of patients who received imatinib with complete molecular response for more than 2 years, patients that stopped therapy, and healthy donors. Interestingly, NK cell numbers were significantly increased in patients that stopped therapy. Of note, increasing cell numbers correlated with increased NK cell activity (Ohyashiki et al., 2012). During imatinib therapy of GIST patients an increase of INF-γ production by NK cells was observed and correlated with a positive therapy response (Borg et al., 2004). Although GIST patients displayed less NKp30+ NK cells and fewer NKp30-dependent lytic potential, both were at least partially restored during imatinib therapy. On the other hand, NKG2D showed a normal expression on NK cells in GIST patients, but nevertheless imatinib increased NKG2D-dependent cytotoxicity. Additionally, after 2 months of therapy, imatinib led to increased IFN-γ production of patient-derived NK cells after restimulation with IL-2 or DCs in vitro (Menard et al., 2009). In contrast to the observation of NK cell suppression by dasatinib in vitro as well as in murine models, where dasatinib-treatment led to reduced lysis of tumor cells, some dasatinib-treated patients showed an increased number of NK large granular lymphocytes associated with improved leukemic control and prolonged survival (Fraser et al., 2009; Kim et al., 2009; Mustjoki et al., 2009; Kreutzman et al., 2010; Tanaka et al., 2012). Regarding the effects of sunitinib in clinical settings, there was no significant change concerning NK cell numbers measured in 43 metastatic clear cell renal cancer patients (Powles et al., 2011). Recently, a link between KIR genotypes and therapy response to TKIs has been assessed. Interestingly, the lower BCR/ABL1 transcript levels in peripheral blood of 86 first-line dasatinib-treated CML patients after 12 months were associated with the absence of three inhibitory KIRs and a similar trend was visible for two activating KIRs including KIR2DS1 (Kreutzman et al., 2012). KIR genotyping in 174 CML patients on first-line therapy with imatinib showed that the expression of KIR2DS1 was accompanied by a significantly lower 2-year probability to achieve cytogenetic response (Marin et al., 2012). In contrast, another study showed no significant impact of the KIR genotype on clinical outcome in 130 CML patients (Ali et al., 2012). However, missing KIR ligand KIR/HLA genotype combination might be a prognostic factor as in the case of increased survival in neuroblastoma patients treated with anti-GD2 antibody (Tarek et al., 2012).
Altogether, TKIs display various direct and indirect effects on NK cells and function. Dasatinib, imatinib, and nilotinib showed a negative influence on NK cell ligands on tumor cells whereas the effect of sorafenib and sunitinib revealed an obverse effect. With exception of imatinib the direct effects on NK cells seem to be mostly inhibitory and should be further investigated.
Demethylating Agents
DNA hypermethylation followed by silencing of tumor-suppressor genes is supposed to play an important role in leukemogenesis. Demethylating agents like azacytidine (5-azacytidine, Vidaza®) and decitabine (5-aza-2′-deoxycytidine, 5-aza-2′-dc, Dacogen®) are cytidine analogs irreversibly inhibiting the DNA methyltransferase and in addition induce genome-wide damages in a dose-dependent manner. Demethylating agents have already revealed their direct effects on malignant cells (Christman, 2002; Issa et al., 2005; Stone et al., 2009).
It was shown that azacytidine and decitabine alone or combined with other drugs induced expression of the NKG2DLs MICA/B or UL16-binding proteins (ULBP) on various tumor cell lines (Rohner et al., 2007; Tang et al., 2008; Wu et al., 2009; Schmiedel et al., 2011). Interactions of NKG2D with chemically induced MICB significantly enhanced the killing capacity of a NK cell line upon pre-treatment with decitabine, most likely due to DNA damage and promotor DNA demethylation (Gasser et al., 2005; Tang et al., 2008). NK cell-dependent antitumor activity was also increased upon treatment of patient-derived primary AML blasts with a combination of demethylating agents and other differentiation-promoting drugs (Rohner et al., 2007). Demethylating agents do not only affect tumor cells, they additionally influence gene expression in effector cells. NK cell lines, clones and developing NK cells all exhibiting different KIR genotypes, displayed a stable transcription of KIRs after treatment with decitabine (Santourlidis et al., 2002; Chan et al., 2005). In contrast to the augmented tumor susceptibility to lysis upon azanucleoside-treatment, it has been reported that azacytidine induced expression of inhibitory KIRs and led to impaired granzyme B and perforin release resulting in limited cytolytic function (Gao et al., 2009).
An important comparative analysis of azacytidine and decitabine revealed different impacts on NK cell cytotoxicity. While azacytidine-treated PBMC-derived NK cells displayed a diminished cytotoxicity, decitabine-treated cells showed the opposite and strongly dose-dependent effect. In line with the impact on cytotoxicity, azacytidine led to decreased and decitabine to increased IFN-γ production by NK cells upon stimulation with tumor cells or IL-2. The expression of activating receptors was only influenced by azacytidine resulting in slight downregulation of CD16, NKG2D, and NKp30 on NK cells. Of note, the relatively short exposure time to demethylating agents in the described in vitro setting did neither alter the KIR profile of NK cells nor the NKG2DL expression of target cells that has been described previously. Altogether, the inhibitory effect of azacytidine seemed to be caused by impaired mRNA synthesis, whereas decitabine increased the responsiveness to activation stimuli resulting in enhanced gene transcription (Schmiedel et al., 2011).
However, one should be aware that these studies have mainly been performed in vitro and so far analysis of NK cell phenotype and function from patients treated with demethylating agents are pending.
Histone Deacetylases Inhibitors
Histone deacetylases (HDACs) play an important role in the epigenetic modulation of gene expression affecting cell-cycle arrest, differentiation and apoptosis in all nucleated cells. Mutations in genes encoding HDACs and alterations in HDAC expression have been found in many types of cancer. Therefore, inhibition of HDACs seems to be a good point of attack in tumor therapy (Glozak and Seto, 2007). Mentionable HDACis are sodium valproate (VPA), trichostatin A (TSA), sodium butyrate, vorinostat (Zolinza®), romidepsin (Istodax®), and chidamide (CS055/HBI-8000).
Microarray analysis revealed that VPA augmented MICA/B expression on HCC cells leading to elevated lysis by NK cells (Armeanu et al., 2005). A similar effect could be shown in vitro in treated leukemic blasts from AML patients that enhanced MICA/B and ULBP1 expression, thereby increasing cytolysis by KIR–HLA class I-mismatched cells (Diermayr et al., 2008). Recently, a similar effect was shown on a B-ALL cell line and a proportion of primary B-ALL samples. Those tumor cells upregulated NKG2DL and displayed an enhanced, NKG2D-dependent degranulation of NK cells (Jardine et al., 2012). TSA-treated leukemic cell lines and patient-derived leukemic cells also increased MICA/B expression via histone acetylation of promotors and mediated NK cell cytotoxicity (Kato et al., 2007). These NK cell-activating effects could also be demonstrated for vorinostat and sodium butyrate. This study further showed a cell-dependent involvement of cytotoxicity mediated via DNAX accessory molecule-1 (DNAM-1) and NCRs (Schmudde et al., 2008). Noteworthy, romidepsin-dependent upregulation of MICA/B on cancer cells showed an essential involvement of the glycogen synthase kinase-3, whereas the sodium butyrate-induced upregulation was influenced by increased binding of Sp1 together with heat shock transcription factor 1 to the MICA/B promotor (Skov et al., 2005; Zhang et al., 2009). On the effector side, one of the newest HDACi, chidamide, led to enhanced lysis of K562 tumor cells in vitro by increased expression of proteins involved in NK cell functions like CD16, NKG2D, and granzyme A. Gene expression studies were performed and a time-dependent induction of NK cell receptors (CD16, NKG2D, and KLRG1), cytotoxic enzymes (granzyme and perforin), and molecules important for apoptosis (FASLG) was observed (Ning et al., 2012). On the other hand, there is increasing evidence that HDACis can also inhibit cytotoxicity even of activated NK cells. In one study, VPA- and vorinostat-induced inhibition was accompanied by diminished expression of NKp46 and NKp30 and reduced granule exocytosis (Ogbomo et al., 2007). Recent data on the effect of vorinostat on NK cells from healthy donors and patients with CTCL also demonstrated a suppression of cytotoxicity in vitro (Anshelevich et al., 2010). A further study distinguished between activated and resting NK cells and revealed a downregulation of NKG2D and NKp46 on resting, VPA-, TSA-, or sodium butyrate-treated, and an additional downregulation of NKp44 and CD25 on activated, TSA-treated NK cells. These HDACis also significantly impaired IFN-γ production of NK cells (Rossi et al., 2012). VPA decreased IFN-γ expression of cytokine-pre-treated NK cells on mRNA and protein levels that was associated by decreased phosphorylation of STAT5 and attenuated expression of T-bet. Valproic acids further reduced expression of perforin and granzyme B, whereas only the transcript levels and not the protein levels of granzyme A and Fas ligand were affected (Alvarez-Breckenridge et al., 2012).
In a murine tumor model, treatment with TSA showed reduced expression of NK1.1, NKG2D, and NKp46 accompanied with diminished IFN-γ production after ex vivo cytokine stimulation (Rossi et al., 2012). Furthermore, immunomonitoring performed on romidepsin-treated CTCL patients showed suppression of cellular immune functions, including NK cell cytotoxicity (Kelly-Sell et al., 2012).
In sum, these data show not only the promising effect on tumor sensitization to NK cell cytotoxicity, but also a potential negative impact on antitumor NK cell activity by different HDACis with exception of new agents like chidamide. However, further evaluation in clinical studies is needed.
Proteasome Inhibitors
Proteasomes belong to the intracellular machinery responsible for protein degradation. Inhibitors of proteasomes can directly inhibit tumor growth or sensitize cells to apoptotic effects of other agents. The mechanisms by which bortezomib (Velcade®), an important proteasome inhibitor, acts, may differ dependent on tumor targets and still have to be further examined (Voorhees and Orlowski, 2006).
In general, tumor-treatment with proteasome inhibitors seemed to sensitize various tumor cells and cell lines such as breast cancer, melanoma, RCC, CML, and others to NK cell-mediated lysis (Lundqvist et al., 2006, 2010; Ames et al., 2009; Yong et al., 2009). First studies showed that bortezomib led to expression of death receptor DR5 on RCC cells that correlated with susceptibility toward NK cells (Lundqvist et al., 2006). Further in vitro analysis indicated that bortezomib-treated tumors had higher TRAIL-induced caspase 8 activity (Lundqvist et al., 2009). Additionally, bortezomib and other tested proteasome inhibitors with distinct mechanisms also strongly increased NKG2DL expression such as ULBP in head and neck squamous cell carcinoma cells and other cell lines (Vales-Gomez et al., 2008; Butler et al., 2009). This could be recently confirmed as bortezomib augmented NKG2DL on a B-ALL cell line and a proportion of primary B-ALL samples. All treated cells displayed an enhanced degranulation of NK cells mediated by NKG2D signaling (Jardine et al., 2012). In line with that in vitro treated primary plasma cells upregulated DNAM-1 and NKG2DL accompanied with increased degranulation of NK cells (Soriani et al., 2009). On the contrary, other studies described a bortezomib-dependent downregulation of HLA class I on MM cell lines and patient-derived MM cells in a dose- and time-dependent manner. This effect was also observed on MM cells derived from patients treated with bortezomib (Shi et al., 2008). However, besides this augmentation of cytotoxicity, pro-apoptotic effects of bortezomib on PBMC-derived, resting NK cells have been reported. This effect might be due to oxidative stress as glutathione significantly abolished total NK cell apoptosis by preventing loss of mitochondrial membrane potential. Furthermore, bortezomib-treated NK cells downregulated NKp46 leading to diminished NKp46-mediated NK cell activity (Wang et al., 2009). Besides, NK cytotoxicity could also be diminished by downregulation of TRAIL via inhibition of NFκB on IL-2-activated NK cells upon bortezomib-treatment in vitro (Feng et al., 2010).
Murine studies showed a NK cell-sensitizing effect of bortezomib-treated renal tumors to both perforin/granzyme and TRAIL-mediated lysis in vitro. In addition, bortezomib-treatment combined with Treg depletion significantly enhanced tumor lytic capacity of adoptively transferred autologous NK cells in vivo (Lundqvist et al., 2009). Of note, additional work in this mouse model revealed that bortezomib-treated tumors underlying enhanced NK cell killing paradoxically developed resistance to antigen-specific T cells that was probably due to changes in proteasomal processing and presentation of antigens (Lundqvist et al., 2010). In another in vitro mouse model, sensitization of tumor cells with bortezomib led to lysis that was mediated by enhanced DR5 and Fas expression accompanied with downregulation of MHC class I. The authors could show in a murine tumor model that bortezomib-treatment in bone marrow transplantation together with NK cell immunotherapy increased survival (Hallett et al., 2008).
Natural killer cells from RCC patients showed an increased killing against autologous tumor cell lines after ex vivo sensitization with bortezomib (Lundqvist et al., 2006). A first phase I trial showed safety in adoptive transfer of activated NK cells. This protocol was combined with Treg depletion and bortezomib-treatment for tumor sensitization prior to adoptive NK cell transfer into patients with advanced stage of cancer (Lundqvist et al., 2011).
Although tumor cells displayed an increased NK cell susceptibility upon stimulation with proteasome inhibitors, the effect on NK cells seemed to be contrary and should be closer examined. In addition, further clinical phase II and III studies are needed to address the specific tumor entity and combination therapy that might promise best efficacy of the combination of tumor sensitization with proteasome inhibitors and donor lymphocyte infusion of NK cells (NK-DLI).
Concluding Remarks and Outlook
In summary, the heterogeneous impact on NK cell function reported for the different immune modulating substances, referred in this review, is mediated by either indirect or direct effects on NK and tumor cells and is further depending on the tumor entity, timing, and dosing of the different pharmaceutics (Table 1).
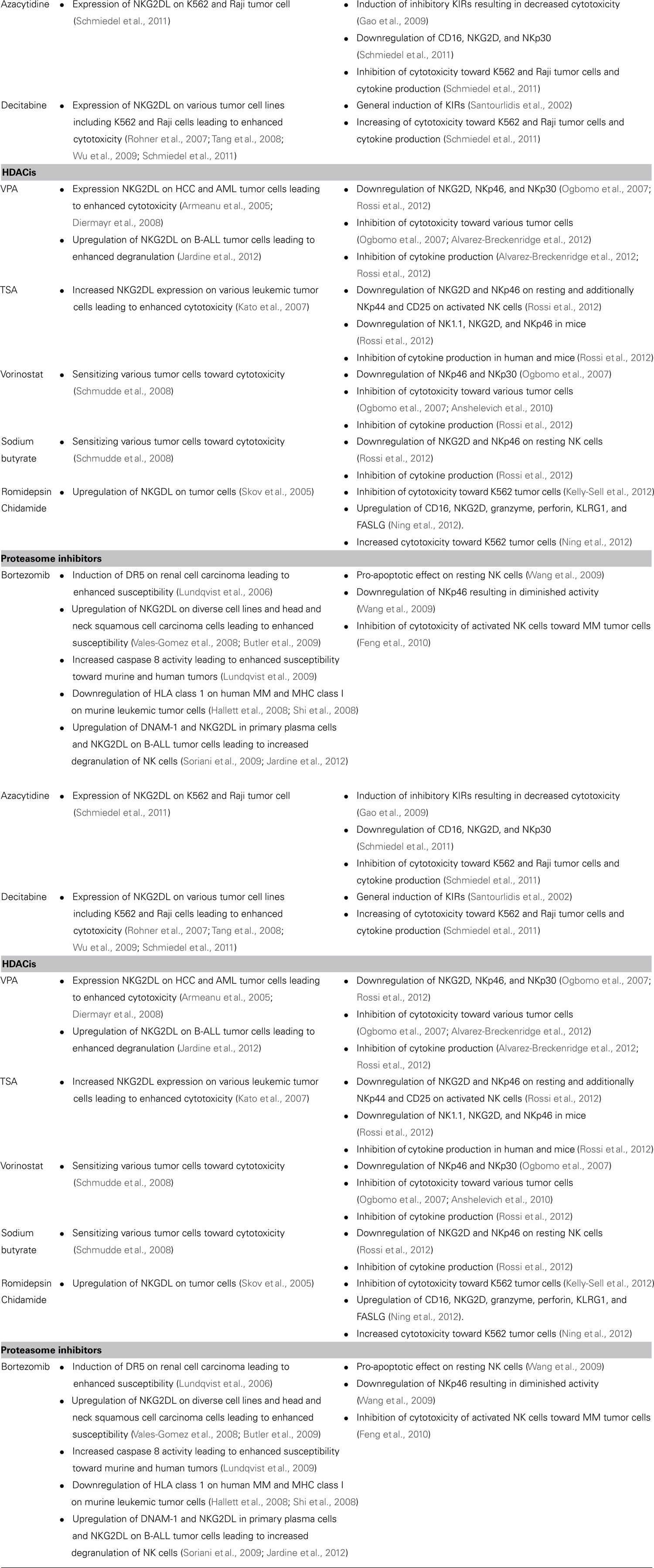
TABLE 1. Overview on the impact of immunomodulating agents on NK cells.
The majority of IMiDs® predominantly mediates immune stimulation positively influencing NK cell-mediated antitumor capacity. TKIs, HDACis, and proteasome inhibitors exhibit various effects. They display mostly negative impacts on NK cell function although treatment of tumor cells may result in contradictory results by sensitization to apoptosis-induction. Exceptions were the new HDACi chidamide and the TKI imatinib that showed a positive effect on NK cell activation. Both of the demethylating agents described exhibit opposite effects on NK cell function.
For better understanding of the underlying mechanisms, unambiguous data on NK cell function from preclinical and clinical studies are still needed. In addition to the complex balance of both tumor and NK cells, there are multiple effects on the other players of the immune system such as antigen presenting cells and cytolytic T cells that regulate the outcoming immune effects.
Finally, an ideal therapeutic concept for cancer patients might combine inhibitors of immunosuppression with tumor sensitizers and cellular therapy protocols such as adoptive NK cell transfer. This review underlines the urgent need of further immunomonitoring studies that should address functional aspects of immune cells on autologous patient-derived tumor cells under therapy with immune modulating agents. Growing knowledge from such studies will allow the right choice of the ideal immune modulators at the optimal dosing and timing to obtain the best possible outcome for the patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to all investigators whose works were not cited in this article due to space limitations. We thank Alexander Steinle for critically reading the manuscript. Evelyn Ullrich has been supported by the LOEWE Center for Cell and Gene Therapy, Frankfurt, funded by the Hessian Ministry of Higher Education, Research and the Arts, Germany (III L 4- 518/17.004). Stephanie Krieg and Evelyn Ullrich were supported by the IZKF Erlangen, Germany.
Abbreviations
ADCC, antibody-dependent cell cytotoxicity; AML, acute myeloid leukemia; CLL, chronic lymphatic leukemia; CML, chronic myeloid leukemia; CTCL, cutaneous T cell lymphoma; DC, dendritic cell; DLI, donor lymphocyte infusion; DNAM-1, DNAX accessory molecule-1; GIST, gastrointestinal stromal tumors; HCC, hepatocellular carcinoma cell; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; IMiD®, immunomodulatory drug; KIR, killer-cell immunoglobulin-like receptors; MAPK, mitogen-activated protein kinase; MDS, myelodysplastic syndrome; MIC, MHC class I-related chain molecules; MM, multiple myeloma; NCR, natural cytotoxicity receptor; NK, natural killer; NKG2DL, NKG2D ligand; PBMC, peripheral blood mononuclear cells; PDGFR, platelet-derived growth factor receptor; PI, phosphoinositide; RCC, renal cell carcinoma; SCID, severe combined immunodeficient; SOCS1, suppressor of cytokine signaling 1; TCL, T cell lymphoma; TKI, tyrosine kinase inhibitor; TSA, trichostatin A; VEGFR, vascular endothelial growth factor receptor; VPA, sodium valproate.
References
Ali, S., Sergeant, R., O’Brien, S. G., Foroni, L., Hedgley, C., Gerrard, G., et al. (2012). Dasatinib may overcome the negative prognostic impact of KIR2DS1 in newly diagnosed patients with chronic myeloid leukemia. Blood 120, 697–698.
Alvarez-Breckenridge, C. A., Yu, J., Price, R., Wei, M., Wang, Y., Nowicki, M. O., et al. (2012). The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J. Virol. 86, 4566–4577.
Ames, E., Hallett, W. H., and Murphy, W. J. (2009). Sensitization of human breast cancer cells to natural killer cell-mediated cytotoxicity by proteasome inhibition. Clin. Exp. Immunol. 155, 504–513.
Anshelevich, A., Wysocka, M., Benoit, B. M., and Rook, A. H. (2010). “Minimizing the immunosuppressive effects of histone deacetylase inhibitors (HDACi): implications for therapy of cutaneous T-cell lymphoma (CTCL),” in First World Congress of Cutaneous Lymphomas, September 22–25, Chicago, IL.
Armeanu, S., Bitzer, M., Lauer, U. M., Venturelli, S., Pathil, A., Krusch, M., et al. (2005). Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 65, 6321–6329.
Balachandran, V. P., Cavnar, M. J., Zeng, S., Bamboat, Z. M., Ocuin, L. M., Obaid, H., et al. (2011). Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 17, 1094–1100.
Baron, F., Turhan, A. G., Giron-Michel, J., Azzarone, B., Bentires-Alj, M., Bours, V., et al. (2002). Leukemic target susceptibility to natural killer cytotoxicity: relationship with BCR-ABL expression. Blood 99, 2107–2113.
Bartlett, J. B., Dredge, K., and Dalgleish, A. G. (2004a). The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat. Rev. Cancer 4, 314–322.
Bartlett, J. B., Michael, A., Clarke, I. A., Dredge, K., Nicholson, S., Kristeleit, H., et al. (2004b). Phase I study to determine the safety, tolerability and immunostimulatory activity of thalidomide analogue CC-5013 in patients with metastatic malignant melanoma and other advanced cancers. Br. J. Cancer 90, 955–961.
Bennasroune, A., Gardin, A., Aunis, D., Crémel, G., and Hubert, P. (2004). Tyrosine kinase receptors as attractive targets of cancer therapy. Crit. Rev. Oncol. Hematol. 50, 23–38.
Blake, S. J., Bruce Lyons, A., Fraser, C. K., Hayball, J. D., and Hughes, T. P. (2008). Dasatinib suppresses in vitro natural killer cell cytotoxicity. Blood 111, 4415–4416.
Boissel, N., Rea, D., Tieng, V., Dulphy, N., Brun, M., Cayuela, J. M., et al. (2006). BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J. Immunol. 176, 5108–5116.
Borg, C., Terme, M., Taïeb, J., Ménard, C., Flament, C., Robert, C., et al. (2004). Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J. Clin. Invest. 114, 379–388.
Butler, J. E., Moore, M. B., Presnell, S. R., Chan, H. W., Chalupny, N. J., and Lutz, C. T. (2009). Proteasome regulation of ULBP1 transcription. J. Immunol. 182, 6600–6609.
Cebo, C., Da Rocha, S., Wittnebel, S., Turhan, A. G., Abdelali, J., Caillat-Zucman, S., et al. (2006). The decreased susceptibility of Bcr/Abl targets to NK cell-mediated lysis in response to imatinib mesylate involves modulation of NKG2D ligands, GM1 expression, and synapse formation. J. Immunol. 176, 864–872.
Chan, H. W., Miller, J. S., Moore, M. B., and Lutz, C. T. (2005). Epigenetic control of highly homologous killer Ig-like receptor gene alleles. J. Immunol. 175, 5966–5974.
Chen, C. I., Koschmieder, S., Kerstiens, L., Schemionek, M., Altvater, B., Pscherer, S., et al. (2012). NK cells are dysfunctional in human chronic myelogenous leukemia before and on imatinib treatment and in BCR-ABL-positive mice. Leukemia 26, 465–474.
Chen, J., Schmitt, A., Chen, B., Rojewski, M., Rübeler, V., Fei, F., et al. (2008). Nilotinib hampers the proliferation and function of CD8+ T lymphocytes through inhibition of T cell receptor signalling. J. Cell. Mol. Med. 12, 2107–2118.
Christman, J. K. (2002). 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21, 5483–5495.
Dauguet, N., Fournie, J., Poupot, R., and Poupot, M. (2010). Lenalidomide down regulates the production of interferon-gamma and the expression of inhibitory cytotoxic receptors of human natural killer cells. Cell. Immunol. 264, 163–170.
Davies, F. E., Raje, N., Hideshima, T., Lentzsch, S., Young, G., Tai, Y. T., et al. (2001). Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 98, 210–216.
Diermayr, S., Himmelreich, H., Durovic, B., Mathys-Schneeberger, A., Siegler, U., Langenkamp, U., et al. (2008). NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR–HLA class I specificities. Blood 111, 1428–1436.
Feng, X., Yan, J., Wang, Y., Zierath, J. R., Nordenskjöld, M., Henter, J. I., et al. (2010). The proteasome inhibitor bortezomib disrupts tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression and natural killer (NK) cell killing of TRAIL receptor-positive multiple myeloma cells. Mol. Immunol. 47, 2388–2396.
Fraser, C. K., Blake, S. J., Diener, K. R., Lyons, A. B., Brown, M. P., Hughes, T. P., et al. (2009). Dasatinib inhibits recombinant viral antigen-specific murine CD4+ and CD8+ T-cell responses and NK-cell cytolytic activity in vitro and in vivo. Exp. Hematol. 37, 256–265.
Gao, X. N., Lin, J., Wang, L. L., and Yu, L. (2009). Demethylating treatment suppresses natural killer cell cytolytic activity. Mol. Immunol. 46, 2064–2070.
Garcia-Manero, G. (2008). Demethylating agents in myeloid malignancies. Curr. Opin. Oncol. 20, 705–710.
Gasser, S., Orsulic, S., Brown, E. J., and Raulet, D. H. (2005). The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436, 1186–1190.
Gorgun, G., Calabrese, E., Soydan, E., Hideshima, T., Perrone, G., Bandi, M., et al. (2010). Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood 116, 3227–3237.
Hallett, W. H., Ames, E., Motarjemi, M., Barao, I., Shanker, A., Tamang, D. L., et al. (2008). Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J. Immunol. 180, 163–170.
Hassold, N., Seystahl, K., Kempf, K., Urlaub, D., Zekl, M., Einsele, H., et al. (2012). Enhancement of natural killer cell effector functions against selected lymphoma and leukemia cell lines by dasatinib. Int. J. Cancer 131, E916–E927.
Hayashi, T., Hideshima, T., Akiyama, M., Podar, K., Yasui, H., Raje, N., et al. (2005). Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br. J. Haematol. 128, 192–203.
Hernandez-Ilizaliturri, F. J., Reddy, N., Holkova, B., Ottman, E., and Czuczman, M. S. (2005). Immunomodulatory drug CC-5013 or CC-4047 and rituximab enhance antitumor activity in a severe combined immunodeficient mouse lymphoma model. Clin. Cancer Res. 11, 5984–5992.
Huang, Y., Wang, Y., Li, Y., Guo, K., and He, Y. (2011). Role of sorafenib and sunitinib in the induction of expressions of NKG2D ligands in nasopharyngeal carcinoma with high expression of ABCG2. J. Cancer Res. Clin. Oncol. 137, 829–837.
Issa, J. P., Kantarjian, H., and Kirkpatrick, P. (2005). Azacitidine. Nat. Rev. Drug Discov. 4, 275–276.
Jardine, L., Hambleton, S., Bigley, V., Pagan, S., Wang, X. N., and Collin, M. (2012). Sensitizing primary acute lymphoblastic leukemia to natural killer cell recognition by induction of NKG2D ligands. Leuk. Lymphoma. doi: 10.3109/10428194.2012.708026 [Epub ahead of print].
Kato, N., Tanaka, J., Sugita, J., Toubai, T., Miura, Y., Ibata, M., et al. (2007). Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia 21, 2103–2108.
Kelly-Sell, M. J., Kim, Y. H., Straus, S., Benoit, B., Harrison, C., Sutherland, K., et al. (2012). The histone deacetylase inhibitor, romidepsin, suppresses cellular immune functions of cutaneous T-cell lymphoma patients. Am. J. Hematol. 87, 354–360.
Khan, O., and La Thangue, N. B. (2012). HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol. Cell Biol. 90, 85–94.
Kim, D. H., Kamel-Reid, S., Chang, H., Sutherland, R., Jung, C. W., Kim, H. J., et al. (2009). Natural killer or natural killer/T cell lineage large granular lymphocytosis associated with dasatinib therapy for Philadelphia chromosome positive leukemia. Haematologica 94, 135–139.
Kohga, K., Takehara, T., Tatsumi, T., Ishida, H., Miyagi, T., Hosui, A., et al. (2010). Sorafenib inhibits the shedding of major histocompatibility complex class I-related chain A on hepatocellular carcinoma cells by down-regulating a disintegrin and metalloproteinase 9. Hepatology 51, 1264–1273.
Kreutzman, A., Jaatinen, T., Greco, D., Vakkila, E., Richter, J., Ekblom, M., et al. (2012). Killer-cell immunoglobulin-like receptor gene profile predicts good molecular response to dasatinib therapy in chronic myeloid leukemia. Exp. Hematol. 40, 906–913.e1.
Kreutzman, A., Juvonen, V., Kairisto, V., Ekblom, M., Stenke, L., Seggewiss, R., et al. (2010). Mono/oligoclonal T and NK cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood 116, 772–782.
Krusch, M., and Salih, H. R. (2011). Effects of BCR-ABL inhibitors on anti-tumor immunity. Curr. Med. Chem. 18, 5174–5184.
Krusch, M., Salih, J., Schlicke, M., Baessler, T., Kampa, K. M., Mayer, F., et al. (2009). The kinase inhibitors sunitinib and sorafenib differentially affect NK cell antitumor reactivity in vitro. J. Immunol. 183, 8286–8294.
Lioznov, M., El-Cheikh, J. Jr., Hoffmann, F., Hildebrandt, Y., Ayuk, F., Wolschke, C., et al. (2010). Lenalidomide as salvage therapy after allo-SCT for multiple myeloma is effective and leads to an increase of activated NK (NKp44+) and T (HLA-DR+) cells. Bone Marrow Transplant. 45, 349–353.
Lundqvist, A., Abrams, S. I., Schrump, D. S., Alvarez, G., Suffredini, D., Berg, M., et al. (2006). Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 66, 7317–7325.
Lundqvist, A., Berg, M., Smith, A., and Childs, R. W. (2011). Bortezomib treatment to potentiate the anti-tumor immunity of ex-vivo expanded adoptively infused autologous natural killer cells. J. Cancer 2, 383–385.
Lundqvist, A., Su, S., Rao, S., and Childs, R. (2010). Cutting edge: bortezomib-treated tumors sensitized to NK cell apoptosis paradoxically acquire resistance to antigen-specific T cells. J. Immunol. 184, 1139–1142.
Lundqvist, A., Yokoyama, H., Smith, A., Berg, M., and Childs, R. (2009). Bortezomib treatment and regulatory T-cell depletion enhance the antitumor effects of adoptively infused NK cells. Blood 113, 6120–6127.
Maggio, R., Peragine, N., De Propris, M. S., Vitale, A., Elia, L., Calabrese, E., et al. (2011). Immunocompetent cell functions in Ph+ acute lymphoblastic leukemia patients on prolonged imatinib maintenance treatment. Cancer Immunol. Immunother. 60, 599–607.
Marin, D., Gabriel, I. H., Ahmad, S., Foroni, L., de Lavallade, H., Clark, R., et al. (2012). KIR2DS1 genotype predicts for complete cytogenetic response and survival in newly diagnosed chronic myeloid leukemia patients treated with imatinib. Leukemia 26, 296–302.
Marks, P. A., and Xu, W. S. (2009). Histone deacetylase inhibitors: potential in cancer therapy. J. Cell. Biochem. 107, 600–608.
Menard, C., Blay, J. Y., Borg, C., Michiels, S., Ghiringhelli, F., Robert, C., et al. (2009). Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 69, 3563–3569.
Mustjoki, S., Ekblom, M., Dybedal, I., Epling-Burnette, P. K., Guilhot, F., et al. (2009). Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia 23, 1398–1405.
Ning, Z. Q., Li, Z. B., Newman, M. J., Shan, S., Wang, X. H., Pan, D. S., et al. (2012). Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 69, 901–909.
Ogbomo, H., Michaelis, M., Kreuter, J., Doerr, H. W., and Cinatl, J. Jr. (2007). Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett. 581, 1317–1322.
Ohyashiki, K., Katagiri, S., Tauchi, T., Ohyashiki, J. H., Maeda, Y., Matsumura, I., et al. (2012). Increased natural killer cells and decreased CD3+ CD8+ CD62L+ T cells in CML patients who sustained complete molecular remission after discontinuation of imatinib. Br. J. Haematol. 157, 254–256.
Orlowski, R. Z., and Kuhn, D. J. (2008). Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin. Cancer Res. 14, 1649–1657.
Payvandi, F., Wu, L., Naziruddin, S. D., Haley, M., Parton, A., Schafer, P. H., et al. (2005). Immunomodulatory drugs (IMiDs®) increase the production of IL-2 from stimulated T cells by increasing PKC-theta activation and enhancing the DNA-binding activity of AP-1 but not NF-kappaB, OCT-1, or NF-AT. J. Interferon Cytokine Res. 25, 604–616.
Pirrotta, M. T., Bernardeschi, P., and Fiorentini, G. (2011). Targeted-therapy in advanced renal cell carcinoma. Curr. Med. Chem. 18, 1651–1657.
Powles, T., Chowdhury, S., Bower, M., Saunders, N., Lim, L., Shamash, J., et al. (2011). The effect of sunitinib on immune subsets in metastatic clear cell renal cancer. Urol. Int. 86, 53–59.
Reddy, N., Hernandez-Ilizaliturri, F. J., Deeb, G., Roth, M., Vaughn, M., Knight, J., et al. (2008). Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br. J. Haematol. 140, 36–45.
Rohner, A., Langenkamp, U., Siegler, U., Kalberer, C. P., and Wodnar-Filipowicz, A. (2007). Differentiation-promoting drugs up-regulate NKG2D ligand expression and enhance the susceptibility of acute myeloid leukemia cells to natural killer cell-mediated lysis. Leuk. Res. 31, 1393–1402.
Rossi, L. E., Avila, D. E., Spallanzani, R. G., Ziblat, A., Fuertes, M, B., Lapyckyj, L., et al. (2012). Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J. Leukoc. Biol. 91, 321–331.
Salih, J., Hilpert, J., Placke, T., Grünebach, F., Steinle, A., Salih, H. R., et al. (2010). The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int. J. Cancer 127, 2119–2128.
Santourlidis, S., Trompeter, H. I., Weinhold, S., Eisermann, B., Meyer, K. L., Wernet, P., et al. (2002). Crucial role of DNA methylation in determination of clonally distributed killer cell Ig-like receptor expression patterns in NK cells. J. Immunol. 169, 4253–4261.
Schade, A. E., Schieven, G. L., Townsend, R., Jankowska, A. M., Susulic, V., Zhang, R., et al. (2008). Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood 111, 1366–1377.
Schmiedel, B. J., Arélin, V., Gruenebach, F., Krusch, M., Schmidt, S. M., Salih, H. R., et al. (2011). Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int. J. Cancer 128, 2911–2922.
Schmudde, M., Braun, A., Pende, D., Sonnemann, J., Klier, U., Beck, J. F., et al. (2008). Histone deacetylase inhibitors sensitize tumour cells for cytotoxic effects of natural killer cells. Cancer Lett. 272, 110–121.
Seggewiss, R., Loré, K., Greiner, E., Magnusson, M. K., Price, D. A., Douek, D. C., et al. (2005). Imatinib inhibits T-cell receptor-mediated T-cell proliferation and activation in a dose-dependent manner. Blood 105, 2473–2479.
Shi, J., Tricot, G. J., Malaviarachchi, P. A., Szmania, S. M., Kellum, R. E., Storrie, B., et al. (2008). Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood 111, 1309–1317.
Skov, S., Pedersen, M. T., Andresen, L., Straten, P. T., Woetmann, A., Odum, N., et al. (2005). Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 65, 11136–11145.
Soriani, A., Zingoni, A., Cerboni, C., Iannitto, M. L., Ricciardi, M. R., Di Gialleonardo, V., et al. (2009). ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 113, 3503–3511.
Stone, R., Sekeres, M., and Garcia-Manero, G. (2009). Evolving strategies in the treatment of MDS and AML. Clin. Adv. Hematol. Oncol. 7, 1–14.
Tai, Y. T., Li, X. F., Catley, L., Coffey, R., Breitkreutz, I., Bae, J., et al. (2005). Immunomodulatory drug lenalidomide (CC-5013, IMiD3) augments anti-CD40 SGN-40-induced cytotoxicity in human multiple myeloma: clinical implications. Cancer Res. 65, 11712–11720.
Taieb, J., Chaput, N., Ménard, C., Apetoh, L., Ullrich, E., Bonmort, M., et al. (2006). A novel dendritic cell subset involved in tumor immunosurveillance. Nat. Med. 12, 214–219.
Tanaka, H., Nakashima, S., and Usuda, M. (2012). Rapid and sustained increase of large granular lymphocytes and rare cytomegalovirus reactivation during dasatinib treatment in chronic myelogenous leukemia patients. Int. J. Hematol. 308–319.
Tang, K. F., He, C. X., Zeng, G. L., Wu, J., Song, G. B., Shi, Y, S., et al. (2008). Induction of MHC class I-related chain B (MICB) by 5-aza-2′-deoxycytidine. Biochem. Biophys. Res. Commun. 370, 578–583.
Tarek, N., Le Luduec, J. B., Gallagher, M. M., Zheng, J., Venstrom, J. M., Chamberlain, E., et al. (2012). Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J. Clin. Invest. 122, 3260–3270.
Terme, M., Ullrich, E., Delahaye, N. F., Chaput, N., and Zitvogel, L. (2008). Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat. Immunol. 9, 486–494.
Vales-Gomez, M., Chisholm, S. E., Cassady-Cain, R. L., Roda-Navarro, P., and Reyburn, H. T. (2008). Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res. 68, 1546–1554.
Vivier, E., Raulet, D. H., Moretta, A., Caligiuri, M. A., Zitvogel, L., Lanier, L. L., et al. (2011). Innate or adaptive immunity? The example of natural killer cells. Science 331, 44–49.
Voorhees, P. M., and Orlowski, R. Z. (2006). The proteasome and proteasome inhibitors in cancer therapy. Annu. Rev. Pharmacol. Toxicol. 46,189–213.
Wang, X., Ottosson, A., Ji, C., Feng, X., Nordenskjöld, M., Henter, J. I., et al. (2009). Proteasome inhibition induces apoptosis in primary human natural killer cells and suppresses NKp46-mediated cytotoxicity. Haematologica 94, 470–478.
Weichsel, R., Dix, C., Wooldridge, L., Clement, M., Fenton-May, A., Sewell, A. K., et al. (2008). Profound inhibition of antigen-specific T-cell effector functions by dasatinib. Clin. Cancer Res. 14, 2484–2491.
Wu, J. F., Zeng, G. L., Shen, W., Yang, M., Wang, F., Tian, L., et al. (2009). Up-regulation of major histocompatibility complex class I-related molecules A (MICA) induced by 5-aza-2′-deoxycytidine. Zhonghua Gan Zang Bing Za Zhi 17, 675–678.
Yong, A. S., Keyvanfar, K., Hensel, N., Eniafe, R., Savani, B. N., Berg, M., et al. (2009). Primitive quiescent CD34+ cells in chronic myeloid leukemia are targeted by in vitro expanded natural killer cells, which are functionally enhanced by bortezomib. Blood 113, 875–882.
Zhang, C., Wang, Y., Zhou, Z., Zhang, J., and Tian, Z. (2009). Sodium butyrate upregulates expression of NKG2D ligand MICA/B in HeLa and HepG2 cell lines and increases their susceptibility to NK lysis. Cancer Immunol. Immunother. 58, 1275–1285.
Keywords: NK cell, IMiD®, TKI, Glivec®, bortezomib, thalidomide, lenalidomide, HDACi
Citation: Krieg S and Ullrich E (2013) Novel immune modulators used in hematology: impact on NK cells. Front. Immun. 3:388. doi: 10.3389/fimmu.2012.00388
Received: 30 September 2012; Paper pending published: 21 October 2012;
Accepted: 04 December 2012; Published online: 03 January 2013.
Edited by:
Eric Vivier, Centre d’Immunologie de Marseille-Luminy, FranceReviewed by:
Stephan Gasser, National University of Singapore, SingaporeMichael G. Brown, University of Virginia School of Medicine, USA
Copyright: © 2013 Krieg and Ullrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Evelyn Ullrich, Laboratory for Cellular Immunology, Department of Pediatric Hematology and Oncology, Children’s Hospital, Goethe University, Theodor-Stern-Kai 7, 60590 Frankfurt, Germany. e-mail:ZXZlbHluLnVsbHJpY2hAa2d1LmRl