

- Neuroscience Center of Excellence, Health Science Center, Louisiana State University, New Orleans, LA, USA
Inflammation, in conjunction with leukocytes, plays a key role in most acute kidney injury (AKI). Non-resolving renal inflammation leads to chronic fibrosis and renal failure. Resolvin D series (RvDs) and E series (RvEs), protectins, and maresins (MaRs) are endogenous omega-3 fatty acid-derived lipid mediators (LMs) that potently promote inflammation resolution by shortening neutrophil life span and promoting macrophage (Mf) non-phelogistic phagocytosis of apoptotic cells and the subsequent exit of Mfs from inflammatory tissue. 14S,21R-dihydroxy docosahexaenoic acid (14S,21R-diHDHA), a Mf-produced autacrine, reprograms Mfs to rescue vascular endothelia. RvD1, RvE1, or 14S,21R-diHDHA also switches Mfs to the phenotype that produces pro-resolving interleukin-10. RvDs or protectin/neuroprotectin D1 (PD1/NPD1) inhibits neutrophil infiltration into injured kidneys, blocks toll-like receptor -mediated inflammatory activation of Mfs and mitigates renal functions. RvDs also repress renal interstitial fibrosis, and PD1 promotes renoprotective heme-oxygenase-1 expression. These findings provide novel approaches for targeting inflammation resolution and LMs or modulation of LM-associated pathways for developing better clinical treatments for AKI.
Acute Kidney Injury: An Inflammatory Disease
Acute Kidney Injury: An Unmet Medical Challenge
Acute kidney injury (AKI), formerly known as “acute renal failure,” causes a decline of kidney function (Bonventre and Yang, 2011). AKI occurs in many conditions, and AKI mortality is quite significant (Bonventre and Yang, 2011). Patients with AKI have a high chance of developing chronic or end-stage renal disease if they survive. Pharmacologic treatment and renal replacement therapy are only preventive or supportive and have not reduced AKI mortality (Negi and Shigematsu, 2012). The current treatment for AKI is still only preventive or supportive (Bonventre and Yang, 2011). Kidney ischemia/reperfusion injury (KIR) is a common cause of AKI (Bonventre and Yang, 2011).
Inflammation, Leukocytes, and Inflammation Resolution: Crucial to Acute Kidney Injury and Chronic Fibrosis
Inflammation plays a critical role in pathogenesis and recovery of AKI (Bonventre and Yang, 2011). AKI is characterized by infiltration and activation of leukocytes neutrophils, macrophages (Mfs), dendritic cells (DCs), and lymphocytes as well as damage (apoptosis and necrosis) of vascular endothelia and tubular epithelia (Figure 1). The activated leukocytes produce reactive oxidative species (ROS) and inflammatory factors, both of which damage the surrounding tissue. Mfs and DCs participate in both the innate and adaptive immune responses. Mfs infiltrated into kidneys during the first 48 h after KIR are mainly inflammatory M1 type that injures the tissue, whereas non-inflammatory M2 Mfs predominate later and are correlated with kidney repair (Lee et al., 2011). Regulatory T-cells are protective in AKI (Ko et al., 2010). B-cell deficiency confers protection from KIR injury (Burne-Taney et al., 2003). This type of injury also stimulates expression of adhesion molecules by vascular endothelia, such as ICAM-1 and VCAM-1, promoting leukocyte accumulation around injured sites. The injury-enhanced interaction of endothelia and leukocytes produces inflammatory cytokines, prostaglandins, leukotrienes, and complements, compromising endothelial junctions due to swelling and loss of glycocalyx and actin cytoskeleton. AKI inflammation goes into a positive feedback amplification as more blood leukocytes infiltrate through the vascular endothelial barrier into other renal tissue and become activated until inflammation resolution dominates over the inflammation (Figure 1) (Borgeson and Godson, 2010; Bonventre and Yang, 2011). Tubular epithelia, mesangium, and pericytes also produce inflammatory factors after injury or interaction with leukocytes, such as TNF-α, IL-8, IL-6, and IL-1β, leading escalated kidney inflammation and damage (Figure 1) (Bonventre and Yang, 2011).
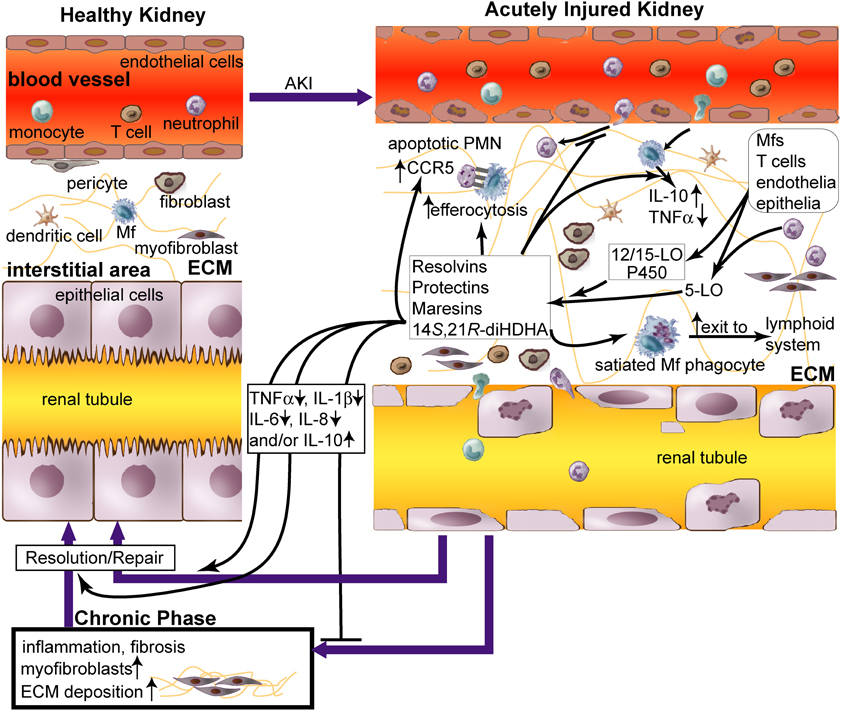
Figure 1. Role of lipid mediators, resolvins and protectins in inflammation resolution in acute renal injury (AKI). AKI causes (1) damage of renal vascular endothelium; (2) diapedesis of neutrophils and monocytes through endothelium; (3) accumulation of neutrophils, macrophages (Mfs), dendritic cells (DCs), T cells, fibroblasts, and myofibroblasts in renal tissue; and (4) apoptosis and necrosis of tubular epithelium. Impaired inflammation resolution that occurs during AKI leads to chronic fibrosis [i.e., excessive extracellular matrix (ECM, as light-green lines) produced by myofibroblasts and other cells in the kidneys] and ultimately chronic renal failure (Bonventre and Yang, 2011; Serhan and Petasis, 2011). Although leukocytes cause tissue damage when activated to inflammatory phenotypes by AKI in the initial phase, they could promote inflammation resolution and tissue repair when modulated by pro-resolving lipid mediators, including resolvins and protectins. Resolvins, protectins, maresins, and 14S,21R-diHDHA are produced by 12/15-lipoxygenase (LO), p450, and/or 5-LO, in trans-cellular or intracellular biosynthetic systems of leukocytes or leukocytes plus endothelia/epiothelia. These lipid mediators act as paracrines and autacrines of leukocytes to promote resolution of AKI-initiated inflammation and fibrosis and rescue of kidney functions. They also inhibit accumulation of neutrophils and Mfs in kidneys during acute inflammation in AKI (Duffield et al., 2006; Tian et al., 2012), promote Mf non-phlogistic efferocytosis of apoptotic neutrophils (Hong et al., 2008), enhance apoptotic leukocyte expression of CCR5 that scavenges critical inflammatory chemokines, and accelerate phagocyte exit from the inflammatory site via the lymphatics (Ariel et al., 2005; Ariel and Serhan, 2007). These lipid mediators increase the level of pro-resolving, anti-fibrotic IL-10 and reduce the levels of inflammatory cytokines TNFα, IL-1β, IL-6, and/or IL-8 in Mfs or renal tissue. Thus these lipid mediators act as effectors for inflammation resolution and injury repair, which restores AKI-damaged kidneys to homeostasis. Administration of these lipid mediators promotes resolution of inflammation and injury in AKI.
While leukocyte-led inflammation causes tissue injury; a natural force for inflammation resolution is gaining ground both in parallel and in series (Serhan et al., 2002; Kieran and Rabb, 2004; Kluth, 2007; Serhan and Petasis, 2011). Certain macrophage phenotypes promote inflammation resolution in AKI (Alikhan et al., 2011; Lee et al., 2011). Particularly, pro-resolving lipid mediators (LMs) are produced by leukocytes or interaction of leukocytes, endothelium, and epithelium (Hong et al., 2003; Serhan and Petasis, 2011). These mediators trigger signaling that reduces production of inflammatory factors, enhances non-phlogistic efferocytosis, and promotes the switch of inflammatory leukocytes to pro-resolution reparative phenotypes (Figure 1, Table 1) (Hong et al., 2008).
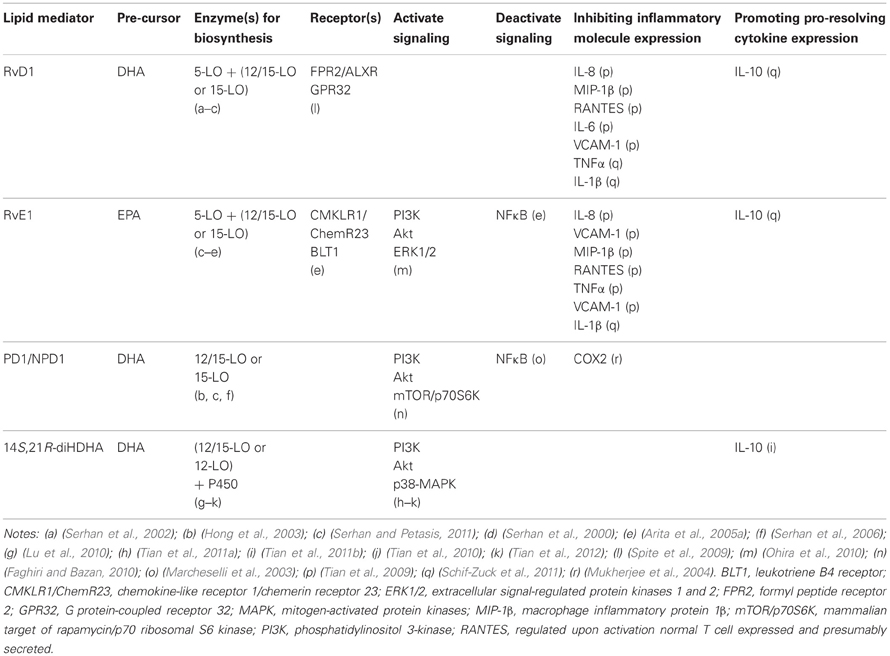
Table 1. Selected characteristics of n3-PUFAs-derived lipid mediators.
Renal chronic fibrosis is the formation of excessive fibrous connective tissue in kidneys due to excessive accumulation in the extracellular matrix in response to chronic inflammation or repeated injury. Although appropriate local and transient renal fibrosis is needed for repair in the early phase of AKI, chronic fibrosis is a major detrimental feature in the later phases (Borgeson and Godson, 2010). Chronic fibrosis can eventually lead to end-stage renal failure. Leukocytes play a crucial role in renal fibrosis during AKI (Borgeson and Godson, 2010) (Figure 1). Lipoxin (LX) A4, protectin/neuroprotectin D1 (PD1/NPD1), or resolvin D1 (RvD1) suppresses chronic fibrosis in KIR-injured kidneys (Godson et al., 2000; Duffield et al., 2006; Borgeson and Godson, 2010; Borgeson et al., 2011). Mfs interact with fibroblasts and pericytes, the key cells that can transdifferentiate into fibrosis-forming myofibroblasts (Figure 1) (Duffield, 2010). Mfs under inflammatory activation produce pro-fibrotic factors—such as TGF-β1, IL-13, and platelet-derived growth factor (PDGF) (Ko et al., 2008)—and also contribute to renal fibrosis (Young et al., 1995). Moreover, Mf depletion reduces renal fibrosis, but Mfs also can produce anti-fibrotic factors, such as IL-10, specific matrix metalloproteinases, and Endo180, in addition to phogocytose fibrotic extracellular matrix, apoptotic myeofibroblasts, and tissue debris (Vernon et al., 2010). Adoptive transfer of Mfs into mice at the chronic inflammation phase ameliorates chronic renal fibrosis (Nishida et al., 2005). This demonstrates that certain Mf phenotypes contribute to the prevention or resolution of chronic renal fibrosis.
In the following sections, we will present a concise review on omega-3 polyunsaturated fatty acids (n3-PUFA)-derived LMs that promote the resolution of inflammation and chronic fibrosis as well as repair in AKI.
Specialized Anti-Inflammatory, Pro-Resolving Lipid Mediators Derived from n3-PUFAs: Resolvins, Protectins, and Maresins (Figure 1, Table 1)
Chemical Structures and Formation In vivo and In vitro
Docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), the major n3-PUFAs present in fish oils, have beneficial effects that could prove helpful in preventing and/or treating inflammatory diseases (Kelley et al., 1999; Simopoulos, 2002). As such, the molecular and cellular mechanisms behind these beneficial effects are of significant interest and have been explored (Bazan et al., 1984; Serhan et al., 2000, 2002; Hong et al., 2003; Marcheselli et al., 2003; Serhan, 2011). The structures and bioactivities of several families of novel n3-PUFAs-derived LMs that are both anti-inflammatory and pro-resolving have been discovered (Serhan, 2011). Some of these compounds were termed resolvins since they are formed in the resolution phase of inflammation and potently promote resolution (Serhan et al., 2002). Another DHA-derived LM, 10,17S-docosatriene, was discovered (Serhan, 2011). It was termed neuroprotectin D1 if generated in neural tissue for its protection in neurons, glial cells, and brain stroke; or protectin D1 for other tissue (Bazan, 2005; Serhan, 2011).
Resolvin D series (RvDs) are derived from DHA. During inflammation, endogenous DHA is converted to 17S hydroxyl-containing RvDs (RvD1–RvD6) and docosa-conjugated triene-containing PD1/NPD1 via 15-lipoxygenase (LO) (15S-lipoxygenation)-initiated biochemical pathways (Serhan et al., 2002; Hong et al., 2003; Marcheselli et al., 2003) or to 14S hydroxyl-containing maresins (MaRs) via 12-LO (12S-lipoxygenation)-initiated biochemical pathways. 5-LO catalyzes sequentially with 15-LO or 12/15-LO, generating RvDs (Hong et al., 2003) and some MaRs (Serhan et al., 2009). PD1, in isolated human cells and murine cells, was found to be 10R,17S-dihydroxy-docosa-4Z, 7Z, 11E, 13E, 15Z, 19Z-hexaenoic acid (Serhan et al., 2006). RvD1, RvD2, PD1/NPD1, and/or their biosynthetical pathway marker 17S-hydroxyl DHA (17S-HDHA) have been found in blood (Hong et al., 2003), ischemia-injured brains, retinal pigment epithelial cells, and/or AKI kidneys (Marcheselli et al., 2003; Mukherjee et al., 2004; Duffield et al., 2006; Bazan et al., 2011), demonstrating the existence of these compounds and/or pathways in injured tissue or cells. The interaction of endothelial cells and leukocytes promotes their biosynthesis (Tian et al., 2009) (Figure 1). RvE1 and 17R-hydroxyl epimers of RvDs and PD1, on the other hand, are generated through 15R-lipoxygenation pathways catalyzed by aspirin-acetylated cyclocygenase-2 (COX-2) or cytochrome P450 (in contrast to the typical 15-LO-catalyzed 15S-lipoxygenation of arachidonic acid) (Serhan et al., 2000, 2006). They were found in exudates, blood, and brains of humans and animals treated with aspirin (Serhan, 2011; Bazan et al., 2012). This provides new molecular insights for aspirin-based anti-inflammatory medication besides inhibiting COXs to produce inflammatory prostaglandins and thromboxins.
Recently we found several additional new pro-healing LMs: 14S,21R-dihydroxy-docosa-4Z, 7Z, 10Z, 12E, 16Z, 19Z-hexaenoic acid (14S,21R-diHDHA) and its epimers (Lu et al., 2010; Tian et al., 2011a,b). 14S,21R-diHDHA, as a positional isomer of maresin-1 (Serhan et al., 2009), is generated from DHA in Mfs, neutrophils, and cutaneous wounds. 12-LO and P450 catalyze sequentially to convert DHA to 14S,21R-diHDHA and 14S,21S-diHDHA through the intermediacy of 14S-HDHA (formed via 12S-lipoxygenation from DHA) (Lu et al., 2010; Tian et al., 2011a).
Bioactions
Resolvins, protectins, and MaRs recapitulate beneficial bioactions of DHA or EPA with several order-of-magnitudes higher potency (in nanomolar and picomolar range) compared to their precursors (DHA or EPA) (Serhan and Petasis, 2011). These LMs have potent anti-inflammatory and pro-resolving effects, since they inhibit inflammatory factor expression and neutrophil infiltration, and since they promote non-phlogistic Mf phagocytosis of apoptotic cells (Serhan and Petasis, 2011). Such actions have been revealed in many in vivo models of inflammatory diseases, as well as in vitro experiments on diverse types of cells critical to these diseases. These actions include dermal inflammation, peritonitis, periodontitis, colitis and intestinal inflammation, asthma and airway inflammation, cystic fibrosis, acute lung or kidney injury, glomerulonephritis, and brain stroke (Marcheselli et al., 2003; Serhan and Petasis, 2011). RvE1 and its analogs are currently undergoing clinic trials for diseases of the eye, lung, kidney, skin, and intestines (Serhan and Petasis, 2011). Bazan et al. discovered that PD1/NPD1 resolves inflammation in brain and eye (Marcheselli et al., 2003; Mukherjee et al., 2004; Lukiw et al., 2005). PD1 or LXA4 blocks inflammatory cytokine secretion from human T-cells and enhances CCR5 expression on apoptotic PMN (Figure 1), which accelerates clearance of inflammatory CCR5 ligands (Ariel et al., 2003, 2005). PD1 also promotes T-cell apoptosis (Ariel et al., 2005), as well as reduces the neutrophil lifespan in peritonitis (Bannenberg et al., 2005) and neutrophil-survival signaling for IL-1β (Hong et al., 2003). RvE1 promotes phagocytosis-induced neutrophil apoptosis and resolution of pulmonary inflammation (El Kebir et al., 2012). Several comprehensive reviews on these mediators are already available (Borgeson and Godson, 2010; Serhan and Petasis, 2011; Bazan, 2012).
14S,21-diHDHA and 14R,21-diHDHA promote or restore wound healing (Lu et al., 2010) impaired by alcohol intoxication (Tian et al., 2010) or diabetes (Tian et al., 2011a). Also, 14S,21R-diHDHA enhances VEGF release, vascularization, and migration of endothelial cells in diabetic mice. It also remedies angiogenic and pro-healing functions of mesenchymal stem cells (MSCs) and Mfs attenuated by diabetes, including their production of VEGF and/or IL-10 (Tian et al., 2011b). 14S,21R-diHDHA reduces hyperglycemia-induced ROS generation by inflammatorily-activated Mfs (Tian et al., 2011b). Thus, 14S,21R-diHDHA is a specific pro-resolving LM that may promote the protection or repair of the renal endothelium and epithelium during AKI.
Receptors (Table 1)
Two G-protein-coupled receptors have been identified for RvE1: (1) BLT1 in neutrophils; and (2) CMKLR1/ChemR23 in Mfs and DCs (Arita et al., 2005a,b). RvD1 has also been reported to interact with both FPR2 or LXA4 receptor (ALXR) and GPR32 in phagocytes (Spite et al., 2009). ALXR is expressed in neutrophils (Fiore et al., 1994) and monocytes (Maddox et al., 1997), and it activates T-cells (Ariel et al., 2003), intestinal or bronchial epithelial cells (Bonnans et al., 2003; Kucharzik et al., 2003), and renal mesangial cells (McMahon et al., 2000; Maderna and Godson, 2009), implying ALXR existence in renal podocytes and tubular epithelium. PD1/NPD1 stereoselectively and specifically binds with retinal pigment epithelial cells and neutrophils, suggesting specific receptors for NPD1 in both the immune and visual systems (Marcheselli et al., 2010). However, the exact NPD1 receptor(s) needs to be identified. The receptors of other resolvins, protectins, and marsins are likely to exist based on their structure-activity association, but have not been discovered yet.
Cell Signaling (Table 1)
Through CMKLR1 or BLT1 receptors, RvE1 represses the activation of NFκB (Arita et al., 2005a, 2007), a crucial regulator of innate immune responses in kidneys (Mulay et al., 2012). NPD1 or RvE1-CMKLR1 interactions activate PI3K and Akt, which involves mTOR signaling; RvE1 also activates ERK1/2 (Faghiri and Bazan, 2010) 14S,21R-diHDHA activates PI3K, Akt, and P38-MAPK, but not ERK1/2 (Tian et al., 2010, 2011a, 2012). PI3K-Akt signaling regulates cell survival, and activation of MAPK pathways is essential in wound healing and associated angiogenesis (Tian et al., 2010, 2011a). These signaling systems are relevant to AKI (Borgeson and Godson, 2010; Tian et al., 2012).
Metabolic Deactivation
RvD1 is converted by eicosanoid oxidoreductases (EORs) to 17-oxo-RvD1 and 8-oxo-RvD1. The former is an inactivation metabolite, while the latter is still effective in suppressing neutrophil infiltration (Sun et al., 2007). RvE1 is metabolized to 12-oxo-RvE, 18-oxo-RvE1, 10,11-dihydroxy RvE, 19-hydroxy RvE1, 20-hydroxy RvE1 in tissue or cells, of which the first four metabolites are inactive partially or completely in inflammation resolution, and thus are representative for RvE1 metabolic deactivation (Arita et al., 2006; Hong et al., 2008). Human neutrophils convert PD1 to its omega-22 hydroxy product (Serhan and Petasis, 2011). The metabolic deactivation of resolvins could be excessively up-regulated in pathological conditions, resulting in their deficiency, or diminishing the pharmacological efficacy of administered resolvins. Molecular engineering has been used to overcome this problem; for example, A p-fluorophenoxyl added to RvE1 ω-terminal blocks the critical metabolic inactivation of RvE1 without attenuating the anti-inflammatory pro-resolving activities (Arita et al., 2005a; Hong et al., 2008).
Resolvin D Series and Protectin D1 Resolve Inflammation and Mitigate AKI (Figure 1, Table 1)
Based on the findings that DHA-derived RvDs and PD1 promote inflammation resolution (Serhan, 2011) and DHA supplementation reduces KIR injury in dogs and rats (Neumayer et al., 1992; Kielar et al., 2003), Duffield and colleagues studied the treatment of murine KIR with RvDs and PD1. The study showed that administration of RvDs (RvD1:RvD2:RvD3 = 1:2:1), RvD1, or PD1 attenuates functional and morphological kidney injury, reduces accumulation of inflammatory neutrophils and Mfs, and suppresses TLR-mediated activation of Mfs. TLR signaling in Mfs and lymphocytes is involved in sustained chronic inflammation (Foell et al., 2007; Kato et al., 2008). RvDs treatment until 72 h after ischemia inhibits renal interstitial chronic fibrosis (Duffield et al., 2006). Interstitial chronic fibrosis and persistent leukocyte infiltration (chronic inflammation), resulting from AKI, leads to scarring and chronic renal failure (Morgera et al., 2002; Duffield and Bonventre, 2004). RvDs and PD1 likely have additional cellular sites of action in the kidney, e.g., on the endothelium and vascular tone, interstitial fibroblasts, mesangial cells, pericytes, DCs, and T-cells because of their pro-resolving and anti-fibrotic ability (Duffield et al., 2006). They may modulate the actions of monocytes/Mfs and neutrophils in the kidney. Hassan and Gronert found that PD1 amplified renoprotective heme-oxygenase-1 (HO-1) expression in ischemia-injured and non-injured kidneys, while PD1 inhibited neutrophil infiltration in murine KIR (Hassan and Gronert, 2009). These results support their notion that the interaction of the 12/15-LO and HO-1 systems provides a positive feedback loop that amplifies anti-inflammatory, pro-resolving signals.
Godson and colleagues found that arachidonic acid-derived LXs are pro-resolving in several types of renal injury; LXs play a reparative role in glomerulonephritis, and reduce proteinuria, glomerular inflammation, and mesangial cell proliferation (Kieran et al., 2004; Wu et al., 2006; Borgeson and Godson, 2010). LXs are protective against murine KIR, where a LX-stable analogue gives functional and morphological protection and attenuates inflammatory cytokine responses (Leonard et al., 2002). LXs also up-regulate genes of tight-junction proteins claudin 1, 3, and 7, which likely reduce inflammatory leukocyte infiltration; Moreover, they found that LXs attenuate renal chronic fibrosis and related gene expression in mesangial cells (Borgeson and Godson, 2010). LXA4 analog or RvE1 remarkably prolong renal allograft survival in mice, which is consistent with LXA4 inhibition of calcineurin activity and inflammatory cytokine release by human neutrophils. Also, RvE1 counter-regulates leukocytes partially via increased LXA4 biosynthesis (Levy et al., 2011). Since AKI is the major complication of renal allograft transplantation (Bellomo et al., 2012), these results further demonstrate the effectiveness of LXA4 or RvE1 in reducing AKI. LX actions converge with the pro-resolving characteristics of RvD1, as LXA4 and RvD1 both activate the same G-protein coupled receptors ALXR/FPR2 and GPR32.
14S,21R-diHDHA Promotes Mesenchymal Stem Cells in Resolution of Inflammation and Prevention of AKI
MSCs have shown potential to resolve inflammation and repair injury in renal failure (Togel et al., 2005). MSCs treated with 14S,21R-diHDHA more efficiently inhibit KIR-induced elevation of serum creatinine levels and reduce renal tubular cell death, as well as infiltration of neutrophils, Mfs, and DCs to renal tissue. Conditioned media from 14S,21R-diHDHA-treated MSCs reduce the generation of TNF-α and ROS by Mfs under KIR conditions. Infusion of 14S,21R-diHDHA-treated MSCs more efficiently reduce KIR-renal damage compared to untreated MSCs. Treated MSCs are resistant to apoptosis in vivo (when transplanted under capsules of AKI-injured kidneys) and in vitro (when cultured under simulated KIR conditions). This enhancement of MSC viability involves PI3K-Akt signaling. Additionally, treatment of MSCs with 14S,21R-diHDHA promotes secretion of renotrophic hepatocyte growth factor and insulin growth factor-1. In brief, 14S,21R-diHDHA promotes MSC amelioration of AKI (Tian et al., 2012).
Resolvins, Protectins, and Maresins Act on Leukocytes Related to Fibrosis in AKI
Although the mechanisms that resolvins and PD1 use to reduce renal chronic fibrosis in AKI (Duffield et al., 2006) remain to be further delineated, the following findings provide hints for future research on this subject. PD1, RvD1, or RvE1 switches Mfs to pro-resolving phenotypes, including CD11blow Mfs, which are more capable in efferocytosis and emigration to lymphoid organs for inflammation resolution (Figure 1) (Schwab et al., 2007; Schif-Zuck et al., 2011; Ariel and Serhan, 2012). RvD1, RvE1, or 14S,21R-diHDHA induces Mfs to produce more anti-fibrotic IL-10 (Schif-Zuck et al., 2011; Tian et al., 2011b). These pro-resolving LMs, acting in concert in AKI, not only inhibit inflammation, but also shift the macrophage roles from pro-inflammatory (M1) or pro-fibrotic phenotypes to phenotypes that promote resolution as well as anti-fibrotic, regulatory functions (Figure 1, Table 1) (Duffield et al., 2006; Serhan and Petasis, 2011; Ariel and Serhan, 2012).
Concluding Remarks and Perspectives
The discoveries of n3-PUFA-derived resolvins, protectins, and MaRs in the last two decades have provided unconventional knowledge and opened new frontiers for understanding the mechanisms involved in inflammation resolution. These LMs are produced endogenously by enzymes in leukocytes and tissue and act as paracrines and autacrines of leukocytes. Experiments have already shown that selected LMs promote resolution of AKI-caused inflammation and chronic fibrosis and rescue kidney function. LMs inhibit recruitment of neutrophils and monocytes to kidneys during acute inflammation, and they likely switch Mfs and T-cells toward anti-inflammatory pro-resolving phenotypes in AKI, as observed in other inflammatory conditions (Figure 1, Table 1). Mechanisms behind the actions of these LMs and their regulatory roles on leukocytes provide the basis for developing leukocyte-related modalities for efficient AKI treatment. These LMs or their mimics may be of therapeutic importance for treating AKI. More studies need to be conducted to further delineate the kinetic process for these LMs in reprogramming the phenotypes of leukocytes, which regulate the resolution of renal inflammation and chronic fibrosis and recover renal functions in AKI. Additional up-stream or down-stream signaling pathways involved should also be studied, as they may yield novel mechanistic targets and insights for AKI treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by NIH grant R01DK087800 (Song Hong) and LSUHSC Research Enhancement Fund (Song Hong). We appreciate Mr. Ryan R. Labadens for his editing services and Yue-Liang Brewerton for graphic assistance. We apologize for omitting many relevant reports due to space limitations.
References
Alikhan, M. A., Jones, C. V., Williams, T. M., Beckhouse, A. G., Fletcher, A. L., Kett, M. M., et al. (2011). Colony-stimulating factor-1 promotes kidney growth and repair via alteration of macrophage responses. Am. J. Pathol. 179, 1243–1256.
Ariel, A., Chiang, N., Arita, M., Petasis, N. A., and Serhan, C. N. (2003). Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J. Immunol. 170, 6266–6272.
Ariel, A., Li, P. L., Wang, W., Tang, W. X., Fredman, G., Hong, S., et al. (2005). The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J. Biol. Chem. 280, 43079–43086.
Ariel, A., and Serhan, C. N. (2007). Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 28, 176–183.
Ariel, A., and Serhan, C. N. (2012). New lives given by cell death: macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Front. Immunol. 3:4. doi: 10.3389/fimmu.2012.00004
Arita, M., Bianchini, F., Aliberti, J., Sher, A., Chiang, N., Hong, S., et al. (2005a). Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201, 713–722.
Arita, M., Yoshida, M., Hong, S., Tjonahen, E., Glickman, J. N., Petasis, N. A., et al. (2005b). Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2, 4, 6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. U.S.A. 102, 7671–7676.
Arita, M., Oh, S. F., Chonan, T., Hong, S., Elangovan, S., Sun, Y. P., et al. (2006). Metabolic inactivation of resolvin E1 and stabilization of its anti-inflammatory actions. J. Biol. Chem. 281, 22847–22854.
Arita, M., Ohira, T., Sun, Y. P., Elangovan, S., Chiang, N., and Serhan, C. N. (2007). Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 178, 3912–3917.
Bannenberg, G. L., Chiang, N., Ariel, A., Arita, M., Tjonahen, E., Gotlinger, K. H., et al. (2005). Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355.
Bazan, N. G. (2005). Neuroprotectin D1 (NPD1): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 15, 159–166.
Bazan, N. G. (2012). The docosanoid neuroprotectin D1 induces homeostatic regulation of neuroinflammation and cell survival. Prostaglandins Leukot. Essent. Fatty Acids 88, 127–129.
Bazan, N. G., Birkle, D. L., and Reddy, T. S. (1984). Docosahexaenoic acid (22:6, n-3) is metabolized to lipoxygenase reaction products in the retina. Biochem. Biophys. Res. Commun. 125, 741–747.
Bazan, N. G., Eady, T. N., Khoutorova, L., Atkins, K. D., Hong, S., Lu, Y., et al. (2012). Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp. Neurol. 236, 122–130.
Bazan, N. G., Musto, A. E., and Knott, E. J. (2011). Endogenous signaling by omega-3 docosahexaenoic acid-derived mediators sustains homeostatic synaptic and circuitry integrity. Mol. Neurobiol. 44, 216–222.
Bonnans, C., Mainprice, B., Chanez, P., Bousquet, J., and Urbach, V. (2003). Lipoxin A4 stimulates a cytosolic Ca2+ increase in human bronchial epithelium. J. Biol. Chem. 278, 10879–10884.
Bonventre, J. V., and Yang, L. (2011). Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 121, 4210–4221.
Borgeson, E., Docherty, N. G., Murphy, M., Rodgers, K., Ryan, A., O'Sullivan, T. P., et al. (2011). Lipoxin A(4) and benzo-lipoxin A(4) attenuate experimental renal fibrosis. FASEB J. 25, 2967–2979.
Borgeson, E., and Godson, C. (2010). Molecular circuits of resolution in renal disease. Sci. World J. 10, 1370–1385.
Burne-Taney, M. J., Ascon, D. B., Daniels, F., Racusen, L., Baldwin, W., and Rabb, H. (2003). B cell deficiency confers protection from renal ischemia reperfusion injury. J. Immunol. 171, 3210–3215.
Duffield, J. S. (2010). Macrophages and immunologic inflammation of the kidney. Semin. Nephrol. 30, 234–254.
Duffield, J. S., and Bonventre, J. V. (2004). “Acute renal failure from Bench to Bedside,” in Chronic Kidney Disease, Dialysis and Transplant 42, 2nd Edn. eds B. J. G. Pereira, M. H. Sayegh, and P. Blake (Philadelphia, PA: Elsevier Saunders), 765–786.
Duffield, J. S., Hong, S., Vaidya, V. S., Lu, Y., Fredman, G., Serhan, C. N., et al. (2006). Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 177, 5902–5911.
El Kebir, D., Gjorstrup, P., and Filep, J. G. (2012). Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. U.S.A. 109, 14983–14988.
Faghiri, Z., and Bazan, N. G. (2010). PI3K/Akt and mTOR/p70S6K pathways mediate neuroprotectin D1-induced retinal pigment epithelial cell survival during oxidative stress-induced apoptosis. Exp. Eye Res. 90, 718–725.
Fiore, S., Maddox, J. F., Perez, H. D., and Serhan, C. N. (1994). Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med. 150, 253–260.
Foell, D., Wittkowski, H., Vogl, T., and Roth, J. (2007). S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 81, 28–37.
Godson, C., Mitchell, S., Harvey, K., Petasis, N. A., Hogg, N., and Brady, H. R. (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 164, 1663–1667.
Hassan, I. R., and Gronert, K. (2009). Acute changes in dietary omega-3 and omega-6 polyunsaturated fatty acids have a pronounced impact on survival following ischemic renal injury and formation of renoprotective docosahexaenoic acid-derived protectin D1. J. Immunol. 182, 3223–3232.
Hong, S., Gronert, K., Devchand, P. R., Moussignac, R. L., and Serhan, C. N. (2003). Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J. Biol. Chem. 278, 14677–14687.
Hong, S., Porter, T. F., Lu, Y., Oh, S. F., Pillai, P. S., and Serhan, C. N. (2008). Resolvin E1 metabolome in local inactivation during inflammation-resolution. J. Immunol. 180, 3512–3519.
Kato, S., Chmielewski, M., Honda, H., Pecoits-Filho, R., Matsuo, S., Yuzawa, Y., et al. (2008). Aspects of immune dysfunction in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 3, 1526–1533.
Kelley, D. S., Taylor, P. C., Nelson, G. J., Schmidt, P. C., Ferretti, A., Erickson, K. L., et al. (1999). Docosahexaenoic acid ingestion inhibits natural killer cell activity and production of inflammatory mediators in young healthy men. Lipids 34, 317–324.
Kielar, M. L., Jeyarajah, D. R., Zhou, X. J., and Lu, C. Y. (2003). Docosahexaenoic acid ameliorates murine ischemic acute renal failure and prevents increases in mRNA abundance for both TNF-alpha and inducible nitric oxide synthase. J. Am. Soc. Nephrol. 14, 389–396.
Kieran, N. E., Maderna, P., and Godson, C. (2004). Lipoxins: potential anti-inflammatory, proresolution, and antifibrotic mediators in renal disease. Kidney Int. 65, 1145–1154.
Kieran, N. E., and Rabb, H. (2004). Immune responses in kidney preservation and reperfusion injury. J. Investig. Med. 52, 310–314.
Kluth, D. C. (2007). Pro-resolution properties of macrophages in renal injury. Kidney Int. 72, 234–236.
Ko, G. J., Boo, C. S., Jo, S. K., Cho, W. Y., and Kim, H. K. (2008). Macrophages contribute to the development of renal fibrosis following ischaemia/reperfusion-induced acute kidney injury. Nephrol. Dial. Transplant. 23, 842–852.
Ko, G. J., Zakaria, A., Womer, K. L., and Rabb, H. (2010). Immunologic research in kidney ischemia/reperfusion injury at Johns Hopkins University. Immunol. Res. 47, 78–85.
Kucharzik, T., Gewirtz, A. T., Merlin, D., Madara, J. L., and Williams, I. R. (2003). Lateral membrane LXA4 receptors mediate LXA4's anti-inflammatory actions on intestinal epithelium. Am. J. Physiol. Cell Physiol. 284, C888–C896.
Lee, S., Huen, S., Nishio, H., Nishio, S., Lee, H. K., Choi, B. S., et al. (2011). Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 22, 317–326.
Leonard, M. O., Hannan, K., Burne, M. J., Lappin, D. W., Doran, P., Coleman, P., et al. (2002). 15-Epi-16-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure. J. Am. Soc. Nephrol. 13, 1657–1662.
Levy, B. D., Zhang, Q. Y., Bonnans, C., Primo, V., Reilly, J. J., Perkins, D. L., et al. (2011). The endogenous pro-resolving mediators lipoxin A4 and resolvin E1 preserve organ function in allograft rejection. Prostaglandins Leukot. Essent. Fatty Acids 84, 43–50.
Lu, Y., Tian, H., and Hong, S. (2010). Novel 14, 21-dihydroxy-docosahexaenoic acids: structures, formation pathways, and enhancement of wound healing. J. Lipid Res. 51, 923–932.
Lukiw, W. J., Cui, J. G., Marcheselli, V. L., Bodker, M., Botkjaer, A., Gotlinger, K., et al. (2005). A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 115, 2774–2783.
Maddox, J. F., Hachicha, M., Takano, T., Petasis, N. A., Fokin, V. V., and Serhan, C. N. (1997). Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J. Biol. Chem. 272, 6972–6978.
Marcheselli, V. L., Hong, S., Lukiw, W. J., Tian, X. H., Gronert, K., Musto, A., et al. (2003). Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 278, 43807–43817.
Marcheselli, V. L., Mukherjee, P. K., Arita, M., Hong, S., Antony, R., Sheets, K., et al. (2010). Neuroprotectin D1/protectin D1 stereoselective and specific binding with human retinal pigment epithelial cells and neutrophils. Prostaglandins Leukot. Essent. Fatty Acids 82, 27–34.
McMahon, B., Stenson, C., Mcphillips, F., Fanning, A., Brady, H. R., and Godson, C. (2000). Lipoxin A4 antagonizes the mitogenic effects of leukotriene D4 in human renal mesangial cells. Differential activation of MAP kinases through distinct receptors. J. Biol. Chem. 275, 27566–27575.
Morgera, S., Kraft, A. K., Siebert, G., Luft, F. C., and Neumayer, H. H. (2002). Long-term outcomes in acute renal failure patients treated with continuous renal replacement therapies. Am. J. Kidney Dis. 40, 275–279.
Mukherjee, P. K., Marcheselli, V. L., Serhan, C. N., and Bazan, N. G. (2004). Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 101, 8491–8496.
Mulay, S. R., Thomasova, D., Ryu, M., and Anders, H. J. (2012). MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int. 81, 1199–1211.
Negi, S., and Shigematsu, T. (2012). Current therapeutic strategies for acute kidney injury. Clin. Exp. Nephrol. 16, 672–678.
Neumayer, H. H., Heinrich, M., Schmissas, M., Haller, H., Wagner, K., and Luft, F. C. (1992). Amelioration of ischemic acute renal failure by dietary fish oil administration in conscious dogs. J. Am. Soc. Nephrol. 3, 1312–1320.
Nishida, M., Okumura, Y., Fujimoto, S., Shiraishi, I., Itoi, T., and Hamaoka, K. (2005). Adoptive transfer of macrophages ameliorates renal fibrosis in mice. Biochem. Biophys. Res. Commun. 332, 11–16.
Ohira, T., Arita, M., Omori, K., Recchiuti, A., Van Dyke, T. E., and Serhan, C. N. (2010). Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J. Biol. Chem. 285, 3451–3461.
Schif-Zuck, S., Gross, N., Assi, S., Rostoker, R., Serhan, C. N., and Ariel, A. (2011). Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur. J. Immunol. 41, 366–379.
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874.
Serhan, C. N. (2011). The resolution of inflammation: the devil in the flask and in the details. FASEB J. 25, 1441–1448.
Serhan, C. N., Clish, C. B., Brannon, J., Colgan, S. P., Chiang, N., and Gronert, K. (2000). Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 192, 1197–1204.
Serhan, C. N., Gotlinger, K., Hong, S., Lu, Y., Siegelman, J., Baer, T., et al. (2006). Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J. Immunol. 176, 1848–1859.
Serhan, C. N., Hong, S., Gronert, K., Colgan, S. P., Devchand, P. R., Mirick, G., et al. (2002). Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037.
Serhan, C. N., and Petasis, N. A. (2011). Resolvins and protectins in inflammation resolution. Chem. Rev. 111, 5922–5943.
Serhan, C. N., Yang, R., Martinod, K., Kasuga, K., Pillai, P. S., Porter, T. F., et al. (2009). Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 206, 15–23.
Simopoulos, A. P. (2002). Omega-3 fatty acids in inflammation and autoimmune diseases. J. Am. Coll. Nutr. 21, 495–505.
Spite, M., Norling, L. V., Summers, L., Yang, R., Cooper, D., Petasis, N. A., et al. (2009). Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291.
Sun, Y. P., Oh, S. F., Uddin, J., Yang, R., Gotlinger, K., Campbell, E., et al. (2007). Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem. 282, 9323–9334.
Tian, H., Lu, Y., Shah, S. P., and Hong, S. (2010). Novel 14S, 21-dihydroxy-docosahexaenoic acid rescues wound healing and associated angiogenesis impaired by acute ethanol intoxication/exposure. J. Cell. Biochem. 111, 266–273.
Tian, H., Lu, Y., Shah, S. P., and Hong, S. (2011a). 14S, 21R-dihydroxydocosahexaenoic acid remedies impaired healing and mesenchymal stem cell functions in diabetic wounds. J. Biol. Chem. 286, 4443–4453.
Tian, H., Lu, Y., Shah, S. P., and Hong, S. (2011b). Autacoid 14S, 21R-dihydroxy-docosahexaenoic acid counteracts diabetic impairment of macrophage prohealing functions. Am. J. Pathol. 179, 1780–1791.
Tian, H., Lu, Y., Shah, S. P., Wang, Q., and Hong, S. (2012). 14S, 21R-dihydroxy-docosahexaenoic acid treatment enhances mesenchymal stem cell amelioration of renal ischemia/reperfusion injury. Stem Cells Dev. 21, 1187–1199.
Tian, H., Lu, Y., Sherwood, A. M., Hongqian, D., and Hong, S. (2009). Resolvins E1 and D1 in choroid-retinal endothelial cells and leukocytes: biosynthesis and mechanisms of anti-inflammatory actions. Invest. Ophthalmol. Vis. Sci. 50, 3613–3620.
Togel, F., Hu, Z., Weiss, K., Isaac, J., Lange, C., and Westenfelder, C. (2005). Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am. J. Physiol. Renal Physiol. 289, F31–F42.
Vernon, M. A., Mylonas, K. J., and Hughes, J. (2010). Macrophages and renal fibrosis. Semin. Nephrol. 30, 302–317.
Wu, S. H., Liao, P. Y., Dong, L., and Jiang, X. Y. (2006). [Protective effects of 15-methyl-lipoxin A4 on mesangioproliferative nephritis in rats]. Zhongguo Dang Dai Er Ke Za Zhi 8, 225–230.
Keywords: resolvins, protectins/neuroprotectins, maresins, 14S,21R-diHDHA, inflammation-resolution, kidney-injury, fibrosis, leukocytes
Citation: Hong S and Lu Y (2013) Omega-3 fatty acid-derived resolvins and protectins in inflammation resolution and leukocyte functions: targeting novel lipid mediator pathways in mitigation of acute kidney injury. Front. Immun. 4:13. doi: 10.3389/fimmu.2013.00013
Received: 26 November 2012; Accepted: 07 January 2013;
Published online: 30 January 2013.
Edited by:
Janos G. Filep, University of Montreal, CanadaReviewed by:
Hiroki Yoshida, Saga University Faculty of Medicine, JapanJunji Yodoi, Kyoto University, Japan
Yasunobu Arima, Osaka University, Japan
Copyright © 2013 Hong and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Song Hong, Neuroscience Center of Excellence, Louisiana State University, Health Science Center, Lions Building, 2020 Gravier St., Suite D, New Orleans, LA 70112, USA. e-mail:c2hvbmdAbHN1aHNjLmVkdQ==