
 Orso Maria Lucherini1
Orso Maria Lucherini1- 1Research Center of Systemic Autoimmune and Autoinflammatory Diseases, Unit of Rheumatology, Policlinico Le Scotte, University of Siena, Siena, Italy
- 2Institute of Pediatrics, Università Cattolica Sacro Cuore, Rome, Italy
Autoinflammatory disorders (AIDs) are a group of inherited diseases of innate immunity, characterized by seemingly unprovoked inflammation recurring with variable rhythmicity and involving skin, serosal membranes, synovial membranes, and gastrointestinal tube, with reactive amyloidosis as a potential severe long-term complication (Touitou and Koné-Paut, 2008). They can be categorized in hereditary monogenic disorders and multi-factorial polygenic disorders, encompassing an expanding number of conditions, as the well-known periodic fever, aphthosis, pharyngitis, and adenitis (PFAPA) syndrome (Masters et al., 2009).
Monogenic AIDs are caused by mutations in genes encoding proteins involved in the regulation or activation of the inflammatory response, and include familial Mediterranean fever (FMF), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), mevalonate kinase deficiency syndrome, the family of cryopyrin-associated periodic syndromes (CAPS), which in turn include familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome and neonatal onset multisystem inflammatory disease, NLRP12-associated autoinflammatory disorder (NLRP12AD), PAPA (pyogenic arthritis, pyoderma gangrenosum, acne) syndrome, Majeed’s syndrome, deficiency of interleukin-1 receptor antagonist, and – lastly – Blau’s syndrome. Recurrent multi-district inflammatory flares, which typically alternate with symptom-free intervals characterized by complete well-being and normalization of acute phase reactants, are the most striking marker of AIDs. The vast majority of these conditions are related to the activation of the interleukin-1 pathway, which results in a unifying common pathogenetic mechanism (Rigante, 2012). Table 1 summarizes their genetic characteristics, inheritance patterns, and the most peculiar clinical manifestations. The recognition of AIDs derives from the combination of clinical data, evaluation of acute phase reactants, clinical efficacy in response to specific drugs, and identification of specific mutations in the causative genes (Federici et al., 2006; Gattorno et al., 2008; Cantarini et al., 2010a, 2011, 2012a,b; Muscari et al., 2012). Genetic testing remains a feasible tool to corroborate the clinical diagnosis of AIDs.
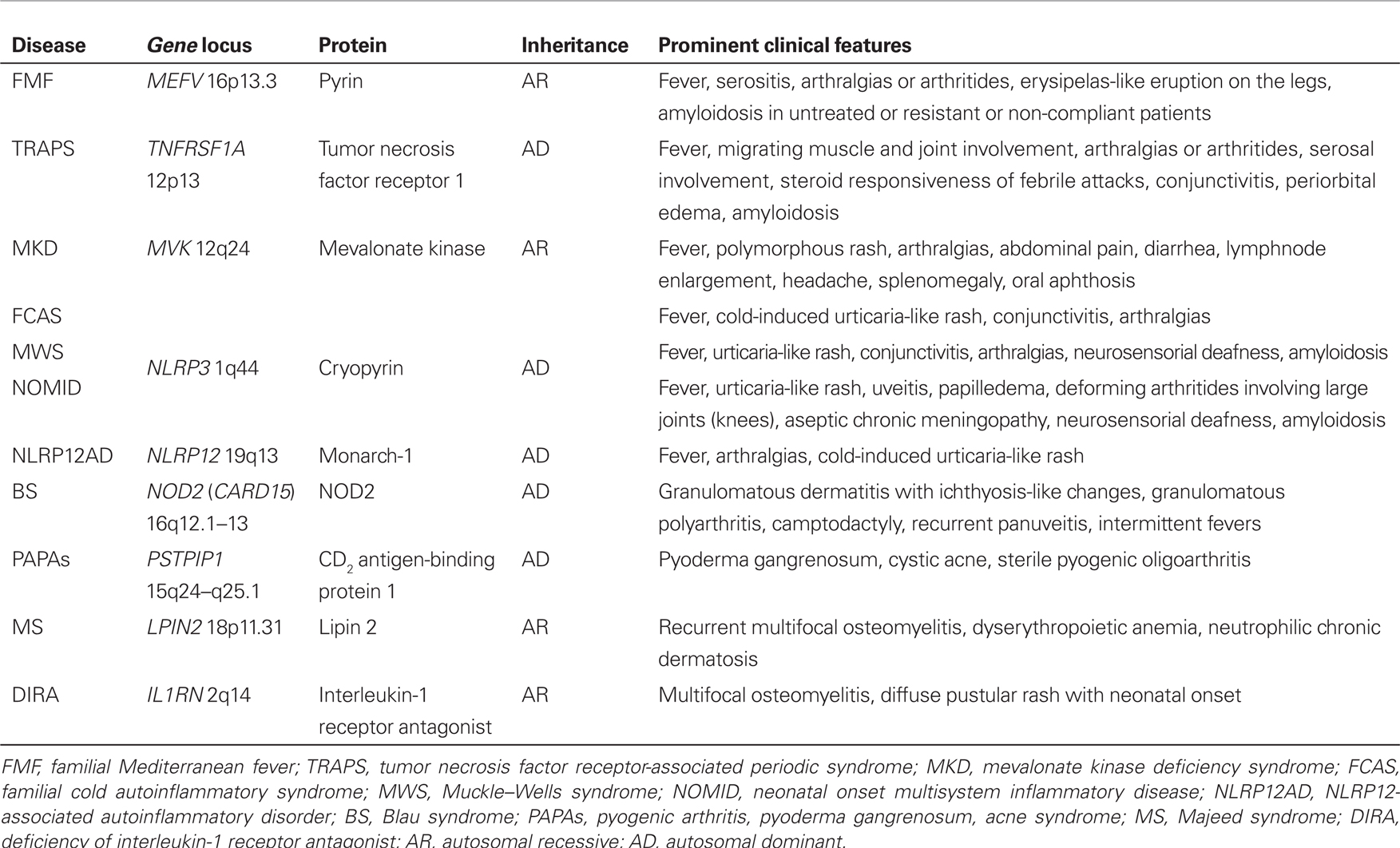
Table 1. Summary of the main genetic and clinical features of monogenic autoinflammatory disorders.
Most of these disorders are manifest in childhood, but a certain number of patients may experience disease onset during adulthood. To date, among monogenic AIDs a late disease onset has been described in FMF (Sohar et al., 1967; Sayarlioglu et al., 2005; Cantarini et al., 2010b), TRAPS (Aksentijevich et al., 2001; Dodé et al., 2002; Aganna et al., 2003; Cantarini et al., 2012c), CAPS (Vitale et al., 2012), and NLRP12AD (Borghini et al., 2011).
Adult-onset FMF is fairly rare, however it has been reported as late as age 65 (Cantarini et al., 2010b), and advanced age is not an exclusion criterion (Livneh et al., 1997): this picture is mainly related to low-penetrance mutations, giving rise to a milder disease, generally similar to that of younger patients (Sayarlioglu et al., 2005).
On the contrary, a more significant variability in terms of clinical phenotype may be observed in TRAPS (Aksentijevich et al., 2001; Dodé et al., 2002; Aganna et al., 2003; Ravet et al., 2006; Cantarini et al., 2009, 2010c,d,e,f,g; Rigante et al., 2011; Brizi et al., 2012): this heterogeneity is largely related to the wide spectrum of known TNFRSF1A mutations, which can be distinguished into high-penetrance and low-penetrance variants (Touitou et al., 2004). As in FMF, adult-onset of symptoms is usually related to low-penetrance mutations, which are associated with feeble clinical signs and a lower risk of amyloidosis (Aksentijevich et al., 2001; Dodé et al., 2002; Aganna et al., 2003). In addition, TRAPS patients carrying low-penetrance TNFRSF1A variants may show atypical clinical manifestations and symptoms that mimic other AIDs and/or autoimmune diseases, such as idiopathic recurrent acute pericarditis, thus hindering differential diagnosis (Cantarini et al., 2009, 2010d,e,g, 2012a; Rigante et al., 2011). These alleles have also been described in patients with recurrent inflammatory attacks who lack the most typical TRAPS manifestations, even when the duration of fever episodes is short and might resemble FMF, and even in healthy controls (Dodé et al., 2002; Ravet et al., 2006; Cantarini et al., 2010a, 2011; Muscari et al., 2012). Their pathogenetic role and the genotype-phenotype correlation are still a huge matter of debate. Moreover, the absence of segregation in some families suggests the existence of other genes with a non-permissive power on AIDs expression (D’Osualdo et al., 2006).
On the other hand, CAPS are primarily characterized by the onset of symptoms during early-infancy: nevertheless, we have recently described a case series of patients presenting FCAS-like symptoms, carrying the low-penetrance Q703K mutation in the NLRP3 gene, all characterized by disease onset during adulthood, and with clinical manifestations triggered or worsened by cold exposure (Vitale et al., 2012). Other NLRP3 mutations of unknown pathogenetic significance can also be found both in patients with recurrent inflammatory attacks and in healthy controls: thus, their causal role remains doubtful and the question of whether these variants are low-penetrance disease-associated mutations or asymptomatic polymorphisms has been often raised (Aróstegui et al., 2004; Aksentijevich et al., 2007; Verma et al., 2008). As in TRAPS, it has been suggested that these alleles might exert a possible proinflammatory effect, causing an inflammatory phenotype in concomitance with other eventual environmental and/or genetic factors.
Also NLRP12AD has been shown to be characterized by neonatal or early-infancy onset (Goldbach-Mansky, 2012). However, Borghini et al. (2011) have recently described a 32-year-old woman found to be a carrier of the D294E NLRP12 mutation: this patient experienced FCAS-like symptoms since the age of 20. In agreement with these findings, we recently diagnosed a 27-year-old Caucasian woman with NLRP12AD and this patient carried the F402L mutation presenting a daily low-grade fever (<38°C) since the age 22 (unpublished data). Hence, we underscore that patients carrying NLRP12 mutations might undoubtedly display a disease onset during adulthood.
The differential diagnosis of AIDs can be complicated by PFAPA syndrome, which frequently occurs in pediatric patients. This syndrome does not have a documented genetic basis, and spontaneous resolution of fever episodes is commonly observed a few years after symptom onset (Marshall et al., 1987, 1989; Thomas et al., 1999). Recent medical literature has included dozens of suspected cases in adults as well, suggesting that it should be taken into consideration also in adults (Cavuoto and Bonagura, 2008; Padeh et al., 2008; Colotto et al., 2011; Cantarini et al., 2012d,e; Cazzato et al., 2013).
In conclusion, although little data is available in the literature in comparison with the medical amount of clinical notes related to the pediatric population, the increasingly frequent reports of adult patients with AIDs is gradually allowing a more extensive and detailed information regarding their potential belated onset, genotype-phenotype correlations, overall prognosis, and management of therapy. Both a delayed diagnosis during adulthood and adult-onset of symptoms are more and more observed: in these cases the presence of low-penetrance mutations, giving nuanced clinical pictures in comparison with children, can be advocated. Low-penetrance mutations may also be responsible for oligosymptomatic diseases in some cases, and for the appearance of atypical clinical manifestations in others, but may even function as susceptibility alleles to inflammation, rather than disease-associated mutations (Dodé et al., 2002; Ravet et al., 2006; Cantarini et al., 2010d, 2012a). Indeed, we suggest caution in the interpretation of low-penetrance mutations in probands with suspected AIDs in order to avoid false positive diagnoses and overtreatment, given the high frequency of healthy carriers and the influence of additional, still unknown, genetic and/or environmental modifying factors.
References
Aganna, E., Hammond, L., Hawkins, P. N., Aldea, A., McKee, S. A., van Amstel, H. K., et al. (2003). Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum. 48, 2632–2644.
Aksentijevich, D. I., Putnam, C., Remmers, E. F., Mueller, J. L., Le, J., Kolodner, R. D., et al. (2007). The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 56, 1273–1285.
Aksentijevich, I., Galon, J., Soares, M., Mansfield, E., Hull, K., Oh, H. H., et al. (2001). The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am. J. Hum. Genet. 69, 301–314.
Aróstegui, J. I., Aldea, A., Modesto, C., Rua, M. J., Argüelles, F., González-Enseñat, M. A., et al. (2004). Clinical and genetic heterogeneity among Spanish patients with recurrent autoinflammatory syndromes associated with the CIAS1/PYPAF1/NALP3 gene. Arthritis Rheum. 50, 4045–4050.
Borghini, S., Tassi, S., Chiesa, S., Caroli, F., Carta, S., Caorsi, R., et al. (2011). Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum. 63, 830–839.
Brizi, M. G., Galeazzi, M., Lucherini, O. M., Cantarini, L., and Cimaz, R. (2012). Successful treatment of tumor necrosis factor receptor-associated periodic syndrome with canakinumab. Ann. Intern. Med. 156, 907–908.
Cantarini, L., Iacoponi, F., Lucherini, O. M., Obici, L., Brizi, M. G., Cimaz, R., et al. (2011). Validation of a diagnostic score for the diagnosis of autoinflammatory diseases in adults. Int. J. Immunopathol. Pharmacol. 24, 695–702.
Cantarini, L., Lucherini, O. M., Brucato, A., Barone, L., Cumetti, D., Iacoponi, F., et al. (2012a). Clues to detect tumor necrosis factor receptor-associated periodic syndrome (TRAPS) among patients with idiopathic recurrent acute pericarditis: results of a multicentre study. Clin. Res. Cardiol. 101, 525–531.
Cantarini, L., Rigante, D., Brizi, M. G., Lucherini, O. M., Sebastiani, G. D., Vitale, A., et al. (2012b). Clinical and biochemical landmarks in systemic autoinflammatory diseases. Ann. Med. 44, 664–673.
Cantarini, L., Lucherini, O. M., Muscari, I., Frediani, B., Galeazzi, M., Brizi, M. G., et al. (2012c). Tumour necrosis factor receptor-associated periodic syndrome (TRAPS): state of the art and future perspectives. Autoimmun. Rev. 12, 38–43.
Cantarini, L., Vitale, A., Bartolomei, B., Galeazzi, M., and Rigante, D. (2012d). Diagnosis of PFAPA syndrome applied to a cohort of 17 adults with unexplained recurrent fevers. Clin. Exp. Rheumatol. 30, 269–271.
Cantarini, L., Vitale, A., Galeazzi, M., and Frediani, B. (2012e). A case of resistant adult-onset periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome responsive to anakinra. Clin. Exp. Rheumatol. 30, 593.
Cantarini, L., Lucherini, O. M., Cimaz, R., Baldari, C. T., Bellisai, F., Rossi Paccani, S., et al. (2009). Idiopathic recurrent pericarditis refractory to colchicine treatment can reveal tumor necrosis factor receptor-associated periodic syndrome. Int. J. Immunopathol. Pharmacol. 22, 1051–1058.
Cantarini, L., Lucherini, O. M., Iacoponi, F., Cimaz, R., Simonini, G., Rigante, D., et al. (2010a). Development and preliminary validation of a diagnostic score for identifying patients affected with adult-onset autoinflammatory disorders. Int. J. Immunopathol. Pharmacol. 23, 1133–1141.
Cantarini, L., Capecchi, P. L., Lucherini, O. M., Laghi Pasini, F., and Galeazzi, M. (2010b). Familial Mediterranean fever diagnosed in an elderly patient. Clin. Exp. Rheumatol. 28(Suppl. 60), S91.
Cantarini, L., Rigante, D., Lucherini, O. M., Cimaz, R., Laghi Pasini, F., Baldari, C. T., et al. (2010c). Role of etanercept in the treatment of tumor necrosis factor receptor-associated periodic syndrome: personal experience and review of the literature. Int. J. Immunopathol. Pharmacol. 23, 701–707.
Cantarini, L., Lucherini, O. M., Baldari, C. T., Laghi Pasini, F., and Galeazzi, M. (2010d). Familial clustering of recurrent pericarditis may disclose tumour necrosis factor receptor-associated periodic syndrome. Clin. Exp. Rheumatol. 28, 405–407.
Cantarini, L., Lucherini, O. M., Cimaz, R., and Galeazzi, M. (2010e). Recurrent pericarditis caused by a rare mutation in the TNFRSF1A gene and with excellent response to anakinra treatment. Clin. Exp. Rheumatol. 28, 802.
Cantarini, L., Lucherini, O. M., Cimaz, R., Baldari, C. T., Laghi Pasini, F., and Galeazzi, M. (2010f). Sacroileitis and pericarditis: atypical presentation of tumor necrosis factor receptor-associated periodic syndrome and response to etanercept therapy. Clin. Exp. Rheumatol. 28, 290–291.
Cantarini, L., Imazio, M., Brucato, A., Lucherini, O. M., and Galeazzi, M. (2010g). Innate versus acquired immune response in the pathogenesis of recurrent idiopathic pericarditis. Autoimmun. Rev. 9, 436–440.
Cavuoto, M., and Bonagura, V. R. (2008). Adult-onset periodic fever, aphthous stomatitis, pharyngitis, and adenitis. Ann. Allergy Asthma Immunol. 100, 170.
Cazzato, M., Neri, R., Possemato, N., Puccini, R., and Bombardieri, S. (2013). A case of adult periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome associated with endocapillary proliferative glomerulonephritis. Clin. Rheumatol. 32(Suppl. 1), 33–36.
Colotto, M., Maranghi, M., Durante, C., Rossetti, M., Renzi, A., and Anatra, M. G. (2011). PFAPA syndrome in a young adult with a history of tonsillectomy. Intern. Med. 50, 223–225.
Dodé, C., André, M., Bienvenu, T., Hausfater, P., Pêcheux, C., Bienvenu, J., et al. (2002). The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 46, 2181–2188.
D’Osualdo, A., Ferlito, F., Prigione, I., Obici, L., Meini, A., Zulian, F., et al. (2006). Neutrophils from patients with TNFRSF1A mutations display resistance to tumor necrosis factor-induced apoptosis: pathogenetic and clinical implications. Arthritis Rheum. 54, 998–1008.
Federici, L., Rittore-Domingo, C., Kone-Paut, I., Jorgensen, C., Rodière, M., Le Quellec, A., et al. (2006). A decision tree for genetic diagnosis of hereditary periodic fever in unselected patients. Ann. Rheum. Dis. 65, 1427–1432.
Gattorno, M., Sormani, M. P., D’Osualdo, A., Pelagatti, M. A., Caroli, F., Federici, S., et al. (2008). A diagnostic score for molecular analysis of hereditary autoinflammatory syndromes with periodic fever in children. Arthritis Rheum. 58, 1823–1832.
Goldbach-Mansky, R. (2012). Immunology in clinic review series; focus on autoinflammatory diseases: update on monogenic autoinflammatory diseases: the role of interleukin (IL)-1 and an emerging role for cytokines beyond IL-1. Clin. Exp. Immunol. 167, 391–404.
Livneh, A., Langevit, P., Zemer, D., Zaks, N., Kees, S., Lidar, T., et al. (1997). Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 10, 1879–1885.
Marshall, G. S., Edwards, K. M., Butler, J., and Lawton, A. R. (1987). Syndrome of periodic fever, pharyngitis, and aphthous stomatitis. J. Pediatr. 110, 43–46.
Marshall, G. S., Edwards, K. M., and Lawton, A. R. (1989). PFAPA syndrome. Pediatr. Infect. Dis. J. 8, 658–659.
Masters, S. L., Simon, A., Aksentijevich, I., and Kastner, D. L. (2009). Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu. Rev. Immunol. 27, 621–668.
Muscari, I., Iacoponi, F., Cantarini, L., Lucherini, O. M., Simonini, G., Brizi, M. G., et al. (2012). The diagnostic evaluation of patients with potential adult-onset autoinflammatory disorders: our experience and review of the literature. Autoimmun. Rev. 12, 10–13.
Padeh, S., Stoffman, N., and Berkun, Y. (2008). Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr. Med. Assoc. J. 10, 358–360.
Ravet, N., Rouaghe, S., Dodé, C., Bienvenu, J., Stirnemann, J., Lévy, P., et al. (2006). Clinical significance of P46L and R92Q substitutions in the tumor necrosis factor superfamily 1A gene. Ann. Rheum. Dis. 65, 1158–1162.
Rigante, D. (2012). The fresco of autoinflammatory diseases from the pediatric perspective. Autoimmun. Rev. 11, 348–356.
Rigante, D., Cantarini, L., Imazio, M., Lucherini, O. M., Sacco, E., Galeazzi, M., et al. (2011). Autoinflammatory diseases and cardiovascular manifestations. Ann. Med. 43, 341–346.
Sayarlioglu, M., Cefle, A., Inanc, M., Kamali, S., Dalkilic, E., Gul, A., et al. (2005). Characteristics of patients with adult-onset familial Mediterranean fever in Turkey: analysis of 401 cases. Int. J. Clin. Pract. 59, 202–205.
Sohar, E., Gafni, J., Pras, M., and Heller, H. (1967). Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am. J. Med. 43, 227–253.
Thomas, K. T., Feder, H. M. Jr., Lawton, A. R., and Edwards, K. M. (1999). Periodic fever syndrome in children. J. Pediatr. 135, 15–21.
Touitou, I., and Koné-Paut, I. (2008). Autoinflammatory diseases. Best Pract. Res. Clin. Rheumatol. 22, 811–829.
Touitou, I., Lesage, S., McDermott, M., Cuisset, L., Hoffman, H., Dode, C., et al. (2004). Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum. Mutat. 24, 194–198.
Verma, D., Lerm, M., Blomgran Julinder, R., Eriksson, P., Söderkvist, P., and Särndahl, E. (2008). Gene polymorphisms in the NALP3 inflammasome are associated with interleukin-1 production and severe inflammation: relation to common inflammatory diseases? Arthritis Rheum. 58, 888–894.
Citation: Cantarini L, Lucherini OM and Rigante D (2013) Caution should be used in the recognition of adult-onset autoinflammatory disorders: facts or fiction? Front. Immunol. 4:96. doi: 10.3389/fimmu.2013.00096
Received: 05 March 2013; Accepted: 10 April 2013;
Published online: 23 April 2013.
Edited by:
Uday Kishore, Brunel University, UKReviewed by:
Uday Kishore, Brunel University, UKCopyright: © 2013 Cantarini, Lucherini and Rigante. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence:Y2FudGFyaW5pbHVjYUBob3RtYWlsLmNvbQ==