
Ca2+ signals are critical for T cell function. A number of ion channels regulate Ca2+ influx from the extracellular space in T cells, either by conducting Ca2+ ions or by modulating the membrane potential that provides the driving force for Ca2+ influx (Cahalan and Chandy, 2009; Feske et al., 2012). The best characterized Ca2+ channel in T cells is the Ca2+ release-activated Ca2+ (CRAC) channel, which mediates store-operated Ca2+ entry (SOCE) in response to T cell receptor (TCR) activation and is composed of ORAI and stromal interaction molecules (STIM) family proteins. Several other channels may also mediate Ca2+ influx directly in T cells including members of the transient receptor potential (TRP) family, P2X receptors, and voltage-gated Ca2+ (Cav) channels. Compared to CRAC channels, however, their contribution to TCR-induced Ca2+ influx and immunity is less well defined.
Ca2+ release-activated Ca2+ channels were first identified in T cells (and mast cells) over 20 years ago (Lewis and Cahalan, 1989; Hoth and Penner, 1992; Zweifach and Lewis, 1993). They mediate Ca2+ influx and have well defined electrophysiological properties (Parekh and Penner, 1997; Prakriya and Lewis, 2003). The long elusive molecular identity of the CRAC channel was solved with the discovery of ORAI1 by genome-wide RNAi screens and positional cloning in patients lacking CRAC channel function (Feske et al., 2006; Vig et al., 2006b; Zhang et al., 2006). ORAI1 and its two homologs, ORAI2 and ORAI3, are integral membrane proteins (Figure 1). Mutagenesis and structural analyses have showed that ORAI1 forms the pore of the CRAC channel through which Ca2+ ions enter the cell (Prakriya et al., 2006; Vig et al., 2006a; Yeromin et al., 2006; Hou et al., 2012; McNally et al., 2012). CRAC channels open after TCR-induced production of inositol 1,4,5-trisphosphate (InsP3) and release of Ca2+ from ER stores. Reduced Ca2+ levels in the ER trigger the activation of STIM 1 and 2 located in the ER membrane. After translocation to ER-plasma membrane junctions, STIM proteins bind to ORAI1 and open the CRAC channel pore, resulting in sustained Ca2+ influx. The molecular regulation of CRAC channel function has been described in detail elsewhere (Shaw et al., 2012).
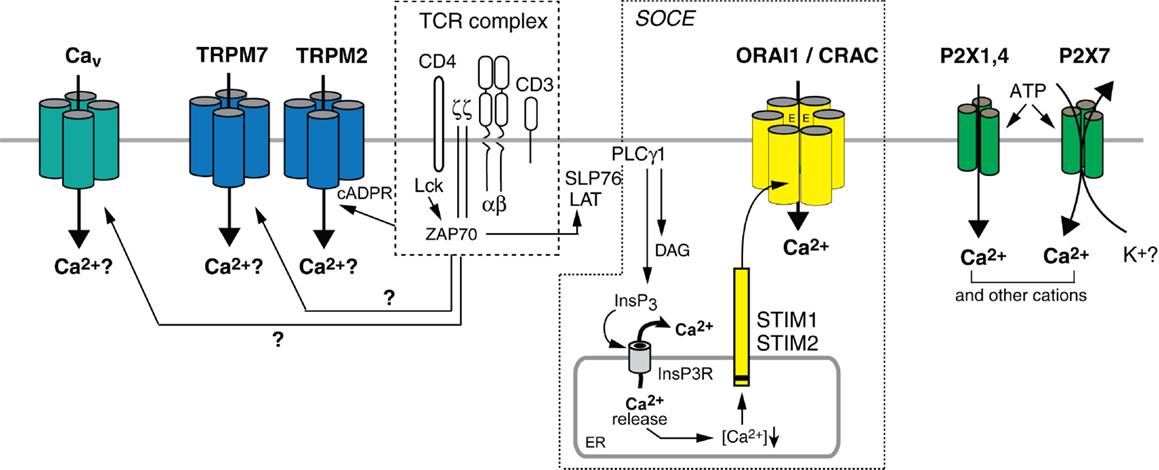
Figure 1. Ca2+ influx pathways in T cells. Stimulation of T cells through the TCR complex results in Ca2+ influx, which is involved in the regulation of many T cell functions. CRAC channels mediate store-operated Ca2+ entry (SOCE) following activation of PLCγ1 and production of InsP3. InsP3 binds to and opens Ca2+ permeable InsP3 receptors (InsP3R) in the ER, resulting in the release of Ca2+ from ER stores (Lewis, 2001; Feske, 2007). Ca2+ release from the ER causes the activation of STIM 1 and 2, which oligomerize and translocate to ER-plasma membrane junctions. STIM1 and STIM2 bind to ORAI1, the pore-forming subunit of the CRAC channel, thereby mediating its opening and sustained Ca2+ influx. The subunit composition of the CRAC channel awaits further studies; both tetrameric and hexameric assemblies of ORAI1 subunits were proposed (Ji et al., 2008; Mignen et al., 2008; Penna et al., 2008; Maruyama et al., 2009; Hou et al., 2012). TRPM2 is a Ca2+ permeable cation channel that can be activated by cADPR and NAADP in human T cells (Beck et al., 2006). Increased cADPR levels after TCR stimulation (Guse et al., 1999) activate SOCE by releasing Ca2+ from the ER through RyR channels and potentially activate TRPM2 channels directly. TRPM7 is a non-selective cation channel implicated in Mg2+ homeostasis in T cells; whether its ability to conduct Ca2+ contributes to T cell function and how it is activated by TCR stimulation is not understood. The L-type Cav channels Cav1.2, Cav1.3, and Cav1.4, which mediate depolarization-dependent Ca2+ influx in excitable cells including neurons may contribute to Ca2+ influx in T cells but their activation mechanism is unknown and their current properties are not well defined. P2X receptors are non-selective Ca2+ channels activated by extracellular ATP. Several homologs, P2X1, P2X4, and P2X7, were reported to mediate Ca2+ influx in T cells in vitro.
The essential role of CRAC channels for T cell function and adaptive immunity is best illustrated by patients with loss-of-function or null mutations in ORAI1 or STIM1 genes, whose T cells lack CRAC channel function and SOCE (Partiseti et al., 1994; Le Deist et al., 1995; Feske et al., 1996; McCarl et al., 2009; Picard et al., 2009; Feske, 2011; Fuchs et al., 2012). CRAC channel-deficient T cells proliferate poorly in vitro and have a profound defect in the production of cytokines such as IFNγ, TNFα, IL-2, and IL-17. Similar defects are found in CD4+ and CD8+ T cells from Stim1−/−, Orai1−/−, and Orai1R91W knock-in mice (Gwack et al., 2008; Beyersdorf et al., 2009; McCarl et al., 2010). SOCE-deficient T cells were found to be more resistant to apoptotic cell death and showed migration defects in vitro and in vivo (Ma et al., 2010; Kim et al., 2011; Greenberg et al., 2013) (and Stefan Feske unpublished data). Interestingly, SOCE is dispensable for the development and selection of conventional TCRαβ+ CD4+ and CD8+ T cells in SOCE-deficient patients and mice. However, their T cell function is severely compromised in vivo, apparent in absent delayed type hypersensitivity (DTH) responses to recall antigens in patients and mice (Le Deist et al., 1995; Feske et al., 1996; McCarl et al., 2010) and impaired skin allograft rejection in Orai1R93W knock-in mice (McCarl et al., 2010). Most importantly, impaired T cell function in ORAI1 and STIM1-deficient patients results in recurrent and chronic infections with a wide spectrum of viral, bacterial and fungal pathogens (Partiseti et al., 1994; Le Deist et al., 1995; Feske et al., 1996; McCarl et al., 2009; Picard et al., 2009; Byun et al., 2010; Feske, 2010; Fuchs et al., 2012).
Besides immunity to infection, CRAC channels in T cells regulate immunological tolerance and inflammation. CD4+ T cells from mice lacking ORAI1 or STIM1 function showed strongly impaired expression of proinflammatory cytokines such as IFN-γ and IL-17 (Ma et al., 2010; McCarl et al., 2010). Importantly, these mice were resistant to T cell-mediated intestinal and CNS inflammation in animal models of colitis and multiple sclerosis. Complete absence of CRAC channel function in mice with T cell-specific deletion of Stim1 and Stim2 genes, in addition, results in impaired development and function of Foxp3+ regulatory T (Treg) cells (Oh-Hora et al., 2008). As a result, STIM1/2-deficient mice over time develop severe myelo-lymphoproliferative disease with lymphadenopathy, splenomegaly, and pulmonary inflammation (Oh-Hora et al., 2008). Intriguingly, these mice show an exocrine gland autoimmune disease resembling Sjogren’s syndrome in humans (Cheng et al., 2012). Reduced numbers of Treg cells are also found in ORAI1- and STIM1-deficient patients (Picard et al., 2009) (and unpublished data), most of which suffer from autoimmune thrombocytopenia and hemolytic anemia due to autoantibodies against erythrocytes and platelets (Feske, 2011). The complete lack of SOCE in STIM1/2-deficient mice not only impaired the development of Treg cells but also that of natural killer T (NKT) cells and TCRαβ+ CD8αα+ intraepithelial lymphocytes (IEL) in the gut (Oh-Hora et al., 2013). These findings indicate that low to moderate SOCE is sufficient for the postselection maturation of agonist-selected T cells (Treg cells, NKT cells, IEL), whereas strong SOCE is required for the proinflammatory function of Th1 and Th17 cells.
Transient receptor potential channels belong to a large family of ion channels, which conduct monovalent and divalent cations including Ca2+ (Nilius and Owsianik, 2011). Before the discovery of ORAI1 as the CRAC channel, several TRPC channels were proposed to mediate Ca2+ influx in T cells. However, a significant role of TRPC channels in Ca2+ influx and T cell mediated immune function has not been established. By contrast, TRPM7 is essential for T cell development as mice with T cell-specific deletion of Trpm7 had a severe block in T cell development at the CD4−CD8− double negative stage (Jin et al., 2008). This is the most profound effect of any ion channel on lymphocyte development demonstrated so far. TRPM7 is Mg2+ permeable and widely considered to regulate cellular Mg2+ homeostasis. However, T cells from Trpm7−/− mice had normal Mg2+ influx and total Mg2+ levels, raising the question whether impaired T cell development is caused by dysregulated Mg2+ homeostasis or rather by impaired influx of other cations including Ca2+ which TRPM7 is able to conduct as well. Another TRP channel, TRPM2 is a non-selective, Ca2+ permeable cation channel and in human T cells, TRPM2 can be activated by a variety of intracellular agonists including adenosine diphosphate ribose (ADPR), cyclic ADPR (cADPR), and Nicotinic acid adenine dinucleotide phosphate (NAADP) (Beck et al., 2006). TCR stimulation was reported to result in increased intracellular cADPR levels and Ca2+ release from the ER through ryanodine receptors (RyR) (Guse et al., 1999), thereby initiating SOCE; alternatively, elevated cADPR levels could directly activate TRPM2 channels. However, the physiological function of TRPM2 channels in T cells is unknown. It is intriguing to speculate that TRPM2 may be involved in inflammatory T cell responses similar to their role in CXCL2 chemokine expression and NADPH oxidase function in monocytes (Yamamoto et al., 2008) and phagocytes (Di et al., 2011).
Voltage-gated Ca2+ (Cav) channels are highly Ca2+ selective channels that play an important role in Ca2+ influx and the function of electrically excitable cells such as neurons following cell depolarization (Tsien et al., 1987). In T cells, several members of the L-type family of CaV channels (CaV1) were reported to be expressed but their contribution to Ca2+ influx has remained controversial (Hogan et al., 2010). Recent studies showed that genetic deletion of CaV1.4 in mouse T cells and knockdown of Cav1.2 and Cav1.3 in human T cells attenuates TCR-induced Ca2+ influx (Cabral et al., 2010; Omilusik et al., 2011). Similarly, mutation of the regulatory β3 and β4 subunits of CaV1 channels in mice results in reduced Ca2+ influx and impaired IL-4, IFNγ, and TNFα production in CD4+ and CD8+ T cells following TCR stimulation (Badou et al., 2006; Jha et al., 2009). CD8+ T cells lacking functional β3 regulatory subunits or Cav1.4 channels were more susceptible to apoptosis (Jha et al., 2009; Omilusik et al., 2011). Cav1.4-deficient mice also showed reduced cytotoxic function of CD8+ T cells in vitro and impaired CD8+ T cell responses to infection with Listeria monocytogenes in vivo (Omilusik et al., 2011). Despite these intriguing findings, the pathways by which TCR signaling activates CaV1 channels are unknown. In contrast to excitable cells, depolarization of T cells fails to open CaV1 channels and mediate Ca2+ influx. It has been speculated that CaV1 channels in T cells are activated by an alternative, voltage-independent mechanism, but the nature of this mechanism remains to be elucidated. In addition, native CaV1 channel-like Ca2+ currents have so far been reported only once despite efforts by numerous labs and appear to be restricted to naïve CD4+ and CD8+ T cells (Omilusik et al., 2011). Intriguingly, these currents were abolished in T cells from Cav1.4−/− mice. Future studies will need to investigate the electrophysiological properties of CaV1 channels in T cells, clarify the molecular mechanisms that regulate their activation, and investigate the role of CaV1 channels in adaptive immunity.
P2X receptors are not, unlike the Ca2+ channels discussed above, activated by TCR stimulation but by extracellular ATP. Three P2X receptors, P2X1, P2X4 and P2X7 (Yip et al., 2009; Woehrle et al., 2010), were reported to mediate Ca2+ influx in T cells. The opening of P2X7 causes Ca2+ influx and activation of enzymes such as calcineurin, resulting in T cell proliferation (Baricordi et al., 1996) and IL-2 production (Adinolfi et al., 2005; Woehrle et al., 2010). Inhibition of P2X1, P2X4, and P2X7 function by RNAi or chemical antagonists attenuated Ca2+ influx and IL-2 production in Jurkat cells and human CD4+ T cells in vitro (Yip et al., 2009; Woehrle et al., 2010). A Ca2+ dependent role for individual P2X receptors in T cell immunity in vivo, however, is not well established. Using P2X7−/− mice, one study found that P2X7 is proinflammatory in T cells by promoting the differentiation and function of Th17 cells and inhibiting the stability of Treg cells (Schenk et al., 2011). The role of P2X7 in T cell-mediated autoimmunity, however, is ambiguous as both increased (Chen and Brosnan, 2006) and decreased (Sharp et al., 2008) CNS inflammation were observed in P2X7−/− mice when analyzed in animal models of multiple sclerosis. In addition, it is unclear whether the main function of P2X7 receptors in T cells is to mediate Ca2+ influx. At the high ATP concentrations (∼1 mM) used to measure Ca2+ influx in most studies, P2X7 channels form a large pore (Junger, 2011) that is permeable to a variety of anorganic and organic cations (Chused et al., 1996; Adriouch et al., 2002). Similarly high ATP concentrations are used to activate inflammasomes in innate immune cells, in which P2X7 channels are thought to mediate K+ efflux and thereby production of IL-1β (Ferrari et al., 2006; Tschopp and Schroder, 2010). Future studies need to evaluate if P2X7 receptors modulate T cell function through Ca2+ influx or other mechanisms. P2X1 and P2X4 conduct Ca2+ more selectively and open at lower (micromolar) ATP concentrations (Junger, 2011). However, P2X1 and P2X4-deficient mice have no reported immunological phenotype (Mulryan et al., 2000; Yamamoto et al., 2006) and their role in T cell immunity in vivo remains poorly understood.
Ca2+ signals have long been recognized as essential for T cell function and several channels may contribute to Ca2+ influx in T cells. Whereas the role of CRAC channels to T cell function and adaptive immunity is well documented by findings in ORAI1 and STIM1-deficient patients and mice, the contributions of TRP, CaV1, and P2X receptor channels remain to be more clearly defined. These channels could contribute to Ca2+ influx in specific T cell subsets, at distinct stages of T cell development or following stimuli other than TCR engagement. A better understanding of the contributions of different Ca2+ influx pathways in T cells will be essential to define potential drug targets for the modulation of T cell function in a variety of diseases caused by aberrant T cell function.
Acknowledgments
This work was funded by NIH grant AI097302.
References
Adinolfi, E., Callegari, M. G., Ferrari, D., Bolognesi, C., Minelli, M., Wieckowski, M. R., et al. (2005). Basal activation of the P2×7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol. Biol. Cell 16, 3260–3272.
Adriouch, S., Dox, C., Welge, V., Seman, M., Koch-Nolte, F., and Haag, F. (2002). Cutting edge: a natural P451L mutation in the cytoplasmic domain impairs the function of the mouse P2×7 receptor. J. Immunol. 169, 4108–4112.
Badou, A., Jha, M. K., Matza, D., Mehal, W. Z., Freichel, M., Flockerzi, V., et al. (2006). Critical role for the beta regulatory subunits of Cav channels in T lymphocyte function. Proc. Natl. Acad. Sci. U.S.A. 103, 15529–15534.
Baricordi, O. R., Ferrari, D., Melchiorri, L., Chiozzi, P., Hanau, S., Chiari, E., et al. (1996). An ATP-activated channel is involved in mitogenic stimulation of human T lymphocytes. Blood 87, 682–690.
Beck, A., Kolisek, M., Bagley, L. A., Fleig, A., and Penner, R. (2006). Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J. 20, 962–964.
Beyersdorf, N., Braun, A., Vogtle, T., Varga-Szabo, D., Galdos, R. R., Kissler, S., et al. (2009). STIM1-independent T cell development and effector function in vivo. J. Immunol. 182, 3390–3397.
Byun, M., Abhyankar, A., Lelarge, V., Plancoulaine, S., Palanduz, A., Telhan, L., et al. (2010). Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 207, 2307–2312.
Cabral, M. D., Paulet, P. E., Robert, V., Gomes, B., Renoud, M. L., Savignac, M., et al. (2010). Knocking down Cav1 calcium channels implicated in Th2 cell activation prevents experimental asthma. Am. J. Respir. Crit. Care Med. 181, 1310–1317.
Cahalan, M. D., and Chandy, K. G. (2009). The functional network of ion channels in T lymphocytes. Immunol. Rev. 231, 59–87.
Chen, L., and Brosnan, C. F. (2006). Exacerbation of experimental autoimmune encephalomyelitis in P2×7R-/- mice: evidence for loss of apoptotic activity in lymphocytes. J. Immunol. 176, 3115–3126.
Cheng, K. T., Alevizos, I., Liu, X., Swaim, W. D., Yin, H., Feske, S., et al. (2012). STIM1 and STIM2 protein deficiency in T lymphocytes underlies development of the exocrine gland autoimmune disease, Sjogren’s syndrome. Proc. Natl. Acad. Sci. U.S.A. 109, 14544–14549.
Chused, T. M., Apasov, S., and Sitkovsky, M. (1996). Murine T lymphocytes modulate activity of an ATP-activated P2Z-type purinoceptor during differentiation. J. Immunol. 157, 1371–1380.
Di, A., Gao, X. P., Qian, F., Kawamura, T., Han, J., Hecquet, C., et al. (2011). The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 13, 29–34.
Ferrari, D., Pizzirani, C., Adinolfi, E., Lemoli, R. M., Curti, A., Idzko, M., et al. (2006). The P2×7 receptor: a key player in IL-1 processing and release. J. Immunol. 176, 3877–3883.
Feske, S. (2007). Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 7, 690–702.
Feske, S. (2011). Immunodeficiency due to defects in store-operated calcium entry. Ann. N. Y. Acad. Sci. 1238, 74–90.
Feske, S., Gwack, Y., Prakriya, M., Srikanth, S., Puppel, S. H., Tanasa, B., et al. (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185.
Feske, S., Muller, J. M., Graf, D., Kroczek, R. A., Drager, R., Niemeyer, C., et al. (1996). Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. Eur. J. Immunol. 26, 2119–2126.
Feske, S., Skolnik, E. Y., and Prakriya, M. (2012). Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 12, 532–547.
Fuchs, S., Rensing-Ehl, A., Speckmann, C., Bengsch, B., Schmitt-Graeff, A., Bondzio, I., et al. (2012). Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J. Immunol. 188, 1523–1533.
Greenberg, M. L., Yu, Y., Leverrier, S., Zhang, S. L., Parker, I., and Cahalan, M. D. (2013). Orai1 function is essential for T cell homing to lymph nodes. J. Immunol. 190, 3197–3206.
Guse, A. H., Da Silva, C. P., Berg, I., Skapenko, A. L., Weber, K., Heyer, P., et al. (1999). Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature 398, 70–73.
Gwack, Y., Srikanth, S., Oh-Hora, M., Hogan, P. G., Lamperti, E. D., Yamashita, M., et al. (2008). Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol. Cell. Biol. 28, 5209–5222.
Hogan, P. G., Lewis, R. S., and Rao, A. (2010). Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28, 491–533.
Hoth, M., and Penner, R. (1992). Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355, 353–356.
Hou, X., Pedi, L., Diver, M. M., and Long, S. B. (2012). Crystal structure of the calcium release-activated calcium channel Orai. Science 338, 1308–1313.
Jha, M. K., Badou, A., Meissner, M., McRory, J. E., Freichel, M., Flockerzi, V., et al. (2009). Defective survival of naive CD8+ T lymphocytes in the absence of the beta3 regulatory subunit of voltage-gated calcium channels. Nat. Immunol. 10, 1275–1282.
Ji, W., Xu, P., Li, Z., Lu, J., Liu, L., Zhan, Y., et al. (2008). Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc. Natl. Acad. Sci. U.S.A. 105, 13668–13673.
Jin, J., Desai, B. N., Navarro, B., Donovan, A., Andrews, N. C., and Clapham, D. E. (2008). Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 322, 756–760.
Junger, W. G. (2011). Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 11, 201–212.
Kim, K. D., Srikanth, S., Yee, M. K., Mock, D. C., Lawson, G. W., and Gwack, Y. (2011). ORAI1 deficiency impairs activated T cell death and enhances T cell survival. J. Immunol. 187, 3620–3630.
Le Deist, F., Hivroz, C., Partiseti, M., Thomas, C., Buc, H. A., Oleastro, M., et al. (1995). A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood 85, 1053–1062.
Lewis, R. S. (2001). Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 19, 497–521.
Lewis, R. S., and Cahalan, M. D. (1989). Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1, 99–112.
Ma, J., McCarl, C. A., Khalil, S., Luthy, K., and Feske, S. (2010). T-cell-specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur. J. Immunol. 2010, 9.
Maruyama, Y., Ogura, T., Mio, K., Kato, K., Kaneko, T., Kiyonaka, S., et al. (2009). Tetrameric Orai1 is a teardrop-shaped molecule with a long, tapered cytoplasmic domain. J. Biol. Chem. 284, 13676–13685.
McCarl, C. A., Khalil, S., Ma, J., Oh-Hora, M., Yamashita, M., Roether, J., et al. (2010). Store-operated Ca2+ entry through ORAI1 is critical for T cell-mediated autoimmunity and allograft rejection. J. Immunol. 185, 5845–5858.
McCarl, C. A., Picard, C., Khalil, S., Kawasaki, T., Rother, J., Papolos, A., et al. (2009). ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. 124, e1317.
McNally, B. A., Somasundaram, A., Yamashita, M., and Prakriya, M. (2012). Gated regulation of CRAC channel ion selectivity by STIM1. Nature 482, 241–245.
Mignen, O., Thompson, J. L., and Shuttleworth, T. J. (2008). Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J. Physiol. (Lond.) 586, 419–425.
Mulryan, K., Gitterman, D. P., Lewis, C. J., Vial, C., Leckie, B. J., Cobb, A. L., et al. (2000). Reduced vas deferens contraction and male infertility in mice lacking P2×1 receptors. Nature 403, 86–89.
Nilius, B., and Owsianik, G. (2011). The transient receptor potential family of ion channels. Genome Biol. 12, 218.
Oh-Hora, M., Komatsu, N., Pishyareh, M., Feske, S., Hori, S., Taniguchi, M., et al. (2013). Agonist-selected T cell development requires strong T cell receptor signaling and store-operated calcium entry. Immunity 38, 1–15.
Oh-Hora, M., Yamashita, M., Hogan, P. G., Sharma, S., Lamperti, E., Chung, W., et al. (2008). Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 9, 432–443.
Omilusik, K., Priatel, J. J., Chen, X., Wang, Y. T., Xu, H., Choi, K. B., et al. (2011). The Ca(v)1.4 calcium channel is a critical regulator of T cell receptor signaling and naive T cell homeostasis. Immunity 35, 349–360.
Parekh, A. B., and Penner, R. (1997). Store depletion and calcium influx. Physiol. Rev. 77, 901–930.
Partiseti, M., Le Deist, F., Hivroz, C., Fischer, A., Korn, H., and Choquet, D. (1994). The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J. Biol. Chem. 269, 32327–32335.
Penna, A., Demuro, A., Yeromin, A. V., Zhang, S. L., Safrina, O., Parker, I., et al. (2008). The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 456, 116–120.
Picard, C., McCarl, C. A., Papolos, A., Khalil, S., Luthy, K., Hivroz, C., et al. (2009). STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N. Engl. J. Med. 360, 1971–1980.
Prakriya, M., Feske, S., Gwack, Y., Srikanth, S., Rao, A., and Hogan, P. G. (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233.
Prakriya, M., and Lewis, R. S. (2003). CRAC channels: activation, permeation, and the search for a molecular identity. Cell Calcium 33, 311–321.
Schenk, U., Frascoli, M., Proietti, M., Geffers, R., Traggiai, E., Buer, J., et al. (2011). ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci. Signal. 4, ra12.
Sharp, A. J., Polak, P. E., Simonini, V., Lin, S. X., Richardson, J. C., Bongarzone, E. R., et al. (2008). P2×7 deficiency suppresses development of experimental autoimmune encephalomyelitis. J. Neuroinflammation 5, 33.
Shaw, P. J., Qu, B., Hoth, M., and Feske, S. (2012). Molecular regulation of CRAC channels and their role in lymphocyte function. Cell. Mol. Life Sci. PMID: 23052215. [Epub ahead of print].
Tschopp, J., and Schroder, K. (2010). NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10, 210–215.
Tsien, R. W., Hess, P., McCleskey, E. W., and Rosenberg, R. L. (1987). Calcium channels: mechanisms of selectivity, permeation, and block. Annu. Rev. Biophys. Biophys. Chem. 16, 265–290.
Vig, M., Beck, A., Billingsley, J. M., Lis, A., Parvez, S., Peinelt, C., et al. (2006a). CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 16, 2073–2079.
Vig, M., Peinelt, C., Beck, A., Koomoa, D. L., Rabah, D., Koblan-Huberson, M., et al. (2006b). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223.
Woehrle, T., Yip, L., Elkhal, A., Sumi, Y., Chen, Y., Yao, Y., et al. (2010). Pannexin-1 hemichannel-mediated ATP release together with P2×1 and P2×4 receptors regulate T-cell activation at the immune synapse. Blood 116, 3475–3484.
Yamamoto, K., Sokabe, T., Matsumoto, T., Yoshimura, K., Shibata, M., Ohura, N., et al. (2006). Impaired flow-dependent control of vascular tone and remodeling in P2×4-deficient mice. Nat. Med. 12, 133–137.
Yamamoto, S., Shimizu, S., Kiyonaka, S., Takahashi, N., Wajima, T., Hara, Y., et al. (2008). TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 14, 738–747.
Yeromin, A. V., Zhang, S. L., Jiang, W., Yu, Y., Safrina, O., and Cahalan, M. D. (2006). Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443, 226–229.
Yip, L., Woehrle, T., Corriden, R., Hirsh, M., Chen, Y., Inoue, Y., et al. (2009). Autocrine regulation of T-cell activation by ATP release and P2×7 receptors. FASEB J. 23, 1685–1693.
Zhang, S. L., Yeromin, A. V., Zhang, X. H., Yu, Y., Safrina, O., Penna, A., et al. (2006). Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. U.S.A. 103, 9357–9362.
Citation: Feske S (2013) Ca2+ influx in T cells: how many Ca2+ channels? Front. Immunol. 4:99. doi: 10.3389/fimmu.2013.00099
Received: 18 March 2013; Accepted: 12 April 2013;
Published online: 24 April 2013.
Edited by:
Gergely Toldi, Semmelweis University, HungaryReviewed by:
Gergely Toldi, Semmelweis University, HungaryCopyright: © 2013 Feske. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence:ZmVza2VzMDFAbnl1bWMub3Jn