

- 1Basic Science Program, Basic Research Laboratory, Frederick National Laboratory for Cancer Research, Frederick, MD, USA
- 2Chaire de Bioinformatique, Laboratoire Génomique Bioinformatique et Applications (EA 4627), Conservatoire National des Arts et Métiers, Paris, France
Very early after the identification of the human immunodeficiency virus (HIV), host genetics factors were anticipated to play a role in viral control and disease progression. As early as the mid-1990s, candidate gene studies demonstrated a central role for the chemokine co-receptor/ligand (e.g., CCR5) and human leukocyte antigen (HLA) systems. In the last decade, the advent of genome-wide arrays opened a new era for unbiased genetic exploration of the genome and brought big expectations for the identification of new unexpected genes and pathways involved in HIV/AIDS. More than 15 genome-wide association studies targeting various HIV-linked phenotypes have been published since 2007. Surprisingly, only the two HIV-chemokine co-receptors and HLA loci have exhibited consistent and reproducible statistically significant genetic associations. In this chapter, we will review the findings from the genome-wide studies focusing especially on non-progressive and HIV control phenotypes, and discuss the current perspectives.
Introduction
Shortly after the discovery of human immunodeficiency virus (HIV)-1 as the etiologic agent for AIDS, it became apparent that this infection was exhibiting a considerable phenotypic heterogeneity at different levels:
(i) Virus acquisition: some individuals, named highly exposed HIV-seronegatives, remain uninfected even after repeated exposure to the virus (e.g., discordant couples, persons exposed to HIV-contaminated blood during transfusion, or sex workers) (Ludlam et al., 1985; Langlade-Demoyen et al., 1994; Fowke et al., 1996).
(ii) Disease progression: some infected individuals, named long-term non-progressors (LTNP), are able to immunologically control the infection for a long period of time (≥8 years) without any antiretroviral therapy by maintaining high CD4+ T-cells levels; other individuals, named the elite controllers, are able to successfully suppress the virus to very low or even undetectable level; in contrast, rapid progressors succumb to AIDS in absence of treatment after only a few years of infection (Pantaleo and Fauci, 1996; Saksena et al., 2007).
(iii) Response to treatment: some individuals may develop severe side effects in response to certain antiretroviral drugs (Hetherington et al., 2002; Mallal et al., 2002).
This phenotypic heterogeneity may be attributed to a complex interplay between viral, environmental, and host genetic factors, and de facto, host genes have been extensively explored since the 1990s using candidate gene association studies. The first discovery involved a 32 bp deletion in CCR5 (CCR5-Δ32) coding for a truncated and non-functional protein version of the major HIV-1 entry co-receptor. Homozygous Δ32/Δ32 individuals no longer express the co-receptor on the cell surface, which affords these fortunate individuals near complete protection from HIV-1 infection, while Δ32/+ heterozygous individuals exhibit a decreased cell surface protein expression and a delayed AIDS progression (Dean et al., 1996; Liu et al., 1996; Samson et al., 1996). This critical finding has fostered the development of a new antiretroviral class of inhibitors, which are designed to interfere with the HIV-CCR5 interaction, and two of them are already included in current treatment regimens (Henrich and Kuritzkes, 2013). This successful story of translation to bedside reveals the power of leverage for host genetics in the fight against the AIDS epidemic.
Additional genetic associations were identified with HIV clinical course and highlighted notably the importance of the human leukocyte antigen (HLA) alleles (e.g., B*27, B*35, B*57), of several immunity-related genes (e.g., KIR, IL10, IFNγ), and of host genes encoding HIV restriction factors (e.g., CCL5, APOBEC3G, CUL5). While HLA associations were consistently replicated across studies (Kaslow et al., 1996; Carrington et al., 1999; Hendel et al., 1999), others are still controversial. The functional interpretation for most of these variants is yet to be discovered, but the detailed account of each candidate gene is beyond the scope of this article, and has been covered by previous enlightening reviews (Fellay, 2009; An and Winkler, 2010). Overall, most of the candidate gene associations displayed small to modest effect sizes, and combined all together account for a small fraction of the phenotype variability (O’Brien and Nelson, 2004).
Key Advance: Genome-Wide Association Studies
In this context, the advent of a series of technological and scientific progresses enabling the exploration of nearly the entire human genome without any a priori motivated the scientific community to pursue the discovery of host genes contributing to HIV-related outcomes. To a large extent, genome-wide association studies (GWAS) were a direct consequence of the achievement of International HapMap Consortium (2003), which compiled common human genetic variations across several continental populations. Due to the high correlation or linkage disequilibrium (LD) existing between genetic variants or SNPs, the complexity of the entire human genome can be recapitulated by a smaller subset of informative SNPs, called tagSNPs. Genotyping this subset of tagSNPs offered an unbiased exploration of most of the common genetic variation of the human genome, opening up opportunities for novel discoveries. At the same time, accurate high throughput chip technologies were developed to genotype cost-effectively, rapidly, and almost effortless hundreds of thousands to millions of SNPs in parallel (Gunderson et al., 2005), which combined with knowledge from the HapMap project, led to a highly prolific era for genetic interrogation of complex diseases.
Nevertheless, this global hypothesis-free approach also carries limitations:
(1) Genome-wide association studies requires a very large number of subjects with homogeneous ethnicity and phenotype in order to get enough power to overcome the cost of multiple comparisons and of very stringent genome-wide significance threshold. Power is the key in complex diseases: in contrast to Mendelian diseases caused by a single gene, complex diseases are often caused by a multitude of variants with small to medium effect size (odds-ratio, OR). To identify these variants while respecting the strict statistical expectations of GWAS, a very large number of samples is thus necessary. As an illustration of this limitation, a GWAS exploring adult height in about 200,000 Europeans identified over 180 loci with OR ranging from 1.02 to 1.3, which explain only 10% of the variance in adult height (Lango Allen et al., 2010).
(2) The first generation of genotyping arrays only covered common variations of the human genome (with a frequency ≥5%). As a consequence, less frequent SNPs, indels, and structural variants that may be involved in the pathogenesis could not be identified by this approach.
(3) Genotyping arrays targeting representative tagSNPs, the identification of the true causal variant is unlikely and often requires fine mapping.
(4) Africa contains the oldest populations in the world and has more generational time for recombination events to occur. African populations therefore exhibit a higher genetic diversity (larger number of polymorphisms) and a lower LD level. The genome-wide capture of common variation is thus currently less effective in African populations compared to European or Asian populations.
The scientific community embraced GWAS as a method to unravel the mysteries of genetics underlying complex diseases and traits. These studies proliferated to such a degree that a dedicated website listing all the GWAS findings was created (http://www.genome.gov/26525384). This enthusiasm extended to the HIV field with the publication of over 15 GWAS since the first article in 2007. In the next sections, we will review the main GWAS findings by emphasizing especially the differences in gene associations among different HIV-linked phenotypes: viral load control, AIDS progression, and HIV acquisition.
GWAS Focusing on Viral Load Control
The first GWAS focusing on HIV-related outcomes was published in 2007 for the Euro-CHAVI cohort (Fellay et al., 2007). This international consortium assessed genetic association with stable plasma RNA viral load during the asymptomatic stage of HIV infection for 486 European HIV-infected individuals (Figure 1). The exploration of 550,000 tagSNPs revealed only two independent signals respecting the genome-wide significance threshold (Figure 2): rs2395029 (P = 9.36 × 10−12) and rs9264942 (P = 3.77 × 10−9). Both polymorphisms are located in the highly polymorphic chromosome 6 HLA region: rs2395029 is located in the HCP5 gene and is in near complete LD with the previously known HLA-B*5701 HIV-1 protective allele; and rs9264942 is located 35 kb upstream HLA-C. For both associations, causal variants were sought and functional exploration was undertaken (see below).
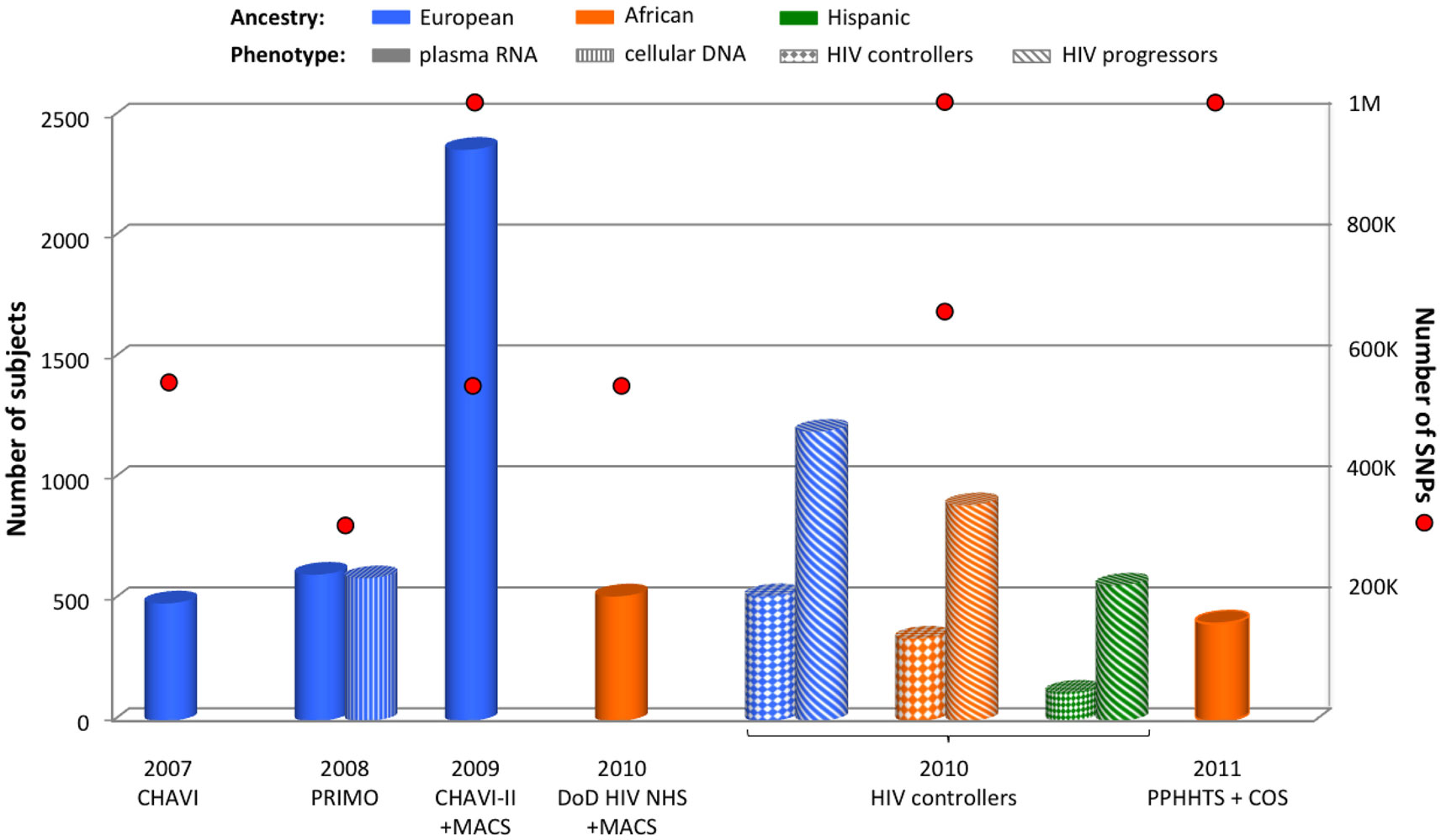
Figure 1. Summary of GWAS design focusing on viral load control. The histogram presents the number of subjects and red circles display the number of SNPs covered by the genotyping arrays. Two red circles are symbolized for one cohort when individuals were genotyped using two sets of arrays. Individual ancestry is color-coded: blue for European, orange for African, and green for Hispanic. The pattern of each cylinder illustrates the phenotype of interest: plain color for plasma RNA, vertical stripes for cellular DNA, solid diamonds for HIV controllers, and diagonal stripes for HIV progressors.
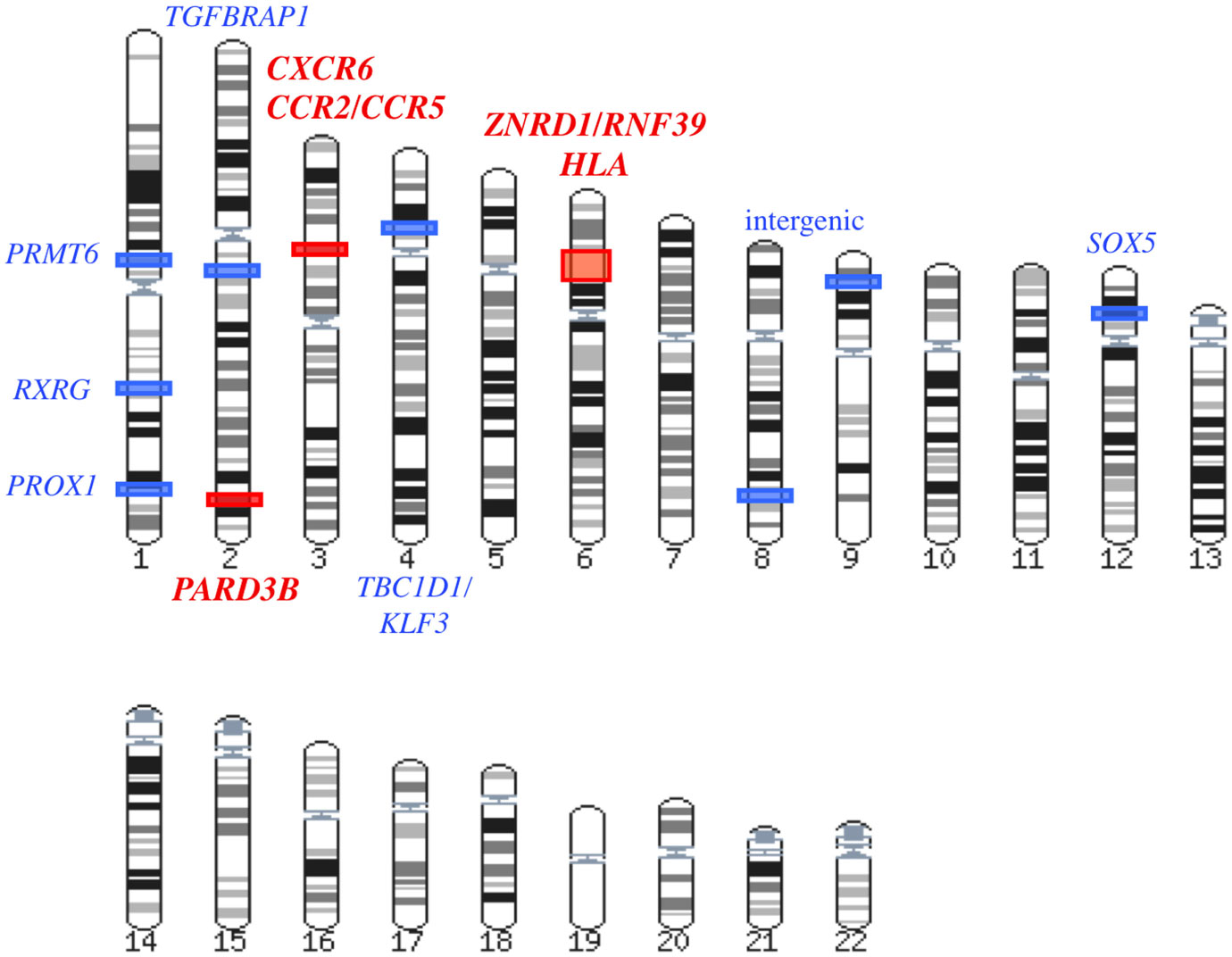
Figure 2. Summary of the GWAS results associated with AIDS progression and/or viral load control. In this karyotype, the statistical significance of the GWAS hits is color-coded: red for genome-wide significant loci and blue for candidate loci that did not reach genome-wide significance but were considered noteworthy by the authors of the study.
To increase the power of this first study, additional European subjects were recruited from the Euro-CHAVI consortium and from the USA-based MACS cohort (n = 2,362 including the previous 486 participants) (Fellay et al., 2009). These individuals were genotyped on the Illumina 550K or 1 M platforms (Figure 1), and the analysis of RNA viral load at set point confirmed the two previous HLA signals. Numerous significant variants were also identified in the HLA region, but after correcting the analysis for rs2395029 and rs9264942 effects, only four independent SNPs remained: C6orf12 rs259919 upstream ZNRD1, rs9468692 correlated with a missense TRIM10 variant, rs9266409 downstream HLA-B, and the missense rs8192591 NOTCH4 SNP. In addition, genotyping of SNPs previously associated with viral load control in candidate gene studies confirmed the role of the chemokine receptors variants: CCR5-Δ32, CCR5-P1, and CCR2-64I.
Plasma HIV-RNA was assessed in 605 seroconverters from the French PRIMO cohort during the first 6 months after HIV infection, as well as cellular HIV-DNA (reflecting the HIV reservoir) in a subset of 590 subjects (Dalmasso et al., 2008) (Figure 1). Although no signal passed the Bonferroni threshold for genome-wide significance, the HIV-RNA GWAS still pointed out the importance of the HLA region and HCP5 rs2395029 for viral load control. It also suggested a potential chromosome 4 signal for rs11725412, located between TBC1D1 and KLF3 genes, which was supported by an enrichment of the protective allele in a group of 45 long-term HIV controllers (HIV-RNA <400 copies/mL after 10 years of infection). In the GWAS focusing on cellular HIV-DNA phenotype, the top signal was again HCP5 rs2395029 (P = 6.72 × 10−7, not reaching genome-wide significance), thus revealing the importance of the HLA region for both HIV replication and HIV reservoir.
Similarly to the Euro-CHAVI cohort design, 515 HIV-infected African-Americans with stable plasma RNA viral load at set point were recruited from the Department of Defense HIV Natural History Study (DoD HIV NHS) and the MACS (Figure 1) (Pelak et al., 2010). The GWAS did not reveal any genome-wide significant signal, but supplemental HLA typing revealed LD between the top SNP, rs2523608, and HLA-B*5703. This HLA allele was significantly associated with viral load control (P = 5.6 × 10−10), and explained 10% of viral load variance at set point in this cohort. Therefore, in a similar fashion to what has been observed for individuals of European ancestry, this first GWAS in an African ancestry group emphasized the main role played by HLA-B*57 alleles for viral load control.
The international HIV controllers study explored the genetic determinants of HIV-1 control by comparing HIV controllers (cases defined by a stable plasma HIV-RNA <2,000 copies/mL during at least 1 year without any treatment) with untreated chronically infected individuals (controls) with different ancestries: 516 cases vs. 1,196 controls for Europeans, 341 cases vs. 892 controls for African-Americans, and 117 cases vs. 560 controls for Hispanics (Figure 1) (International HIV Controllers Study et al., 2010). The GWAS in European individuals confirmed the two previously known HLA SNPs and revealed two additional independent HLA SNPs: the −35 kb HLA-C rs9264942 SNP, the HCP5 rs2395029, rs4418214 close to MICA, and the intronic PSORS1C3 rs3131018. The GWAS in African-Americans identified four different independent HLA SNPs: the previously known HLA-B*5703/rs2523608, rs2255221, rs2523590, and rs9262632. The Hispanic GWAS was performed on a much smaller sample size and no signal reached genome-wide significance. However, the strongest signals were also located within the HLA region.
In a study focusing on HIV-1 serodiscordant heterosexual couples from East and Southern Africa (the Partners in Prevention HSV/HIV Transmission Study, PPHHTS) and from South Africa and Uganda (the Couples Observational Study, COS), a subset of 403 African seropositive individuals was available for analysis with stable plasma HIV-RNA at set point (Figure 1) (Lingappa et al., 2011). This GWAS did not reveal any genome-wide significant SNP.
Overall, these various GWAS identified the HLA region as the primary driver of viral control in individuals with European or African ancestry.
GWAS Focusing on AIDS Disease Progression
The Euro-CHAVI GWAS described above also explored host genetic factors associated with AIDS progression on a subset of 337 individuals from the Euro-CHAVI cohort (Figure 3) (Fellay et al., 2007). The progression phenotype was defined as the time to antiretroviral therapy initiation or to the drop of CD4+ T-cell count below 350/μL. A set of SNPs in the chromosome 6 ZNRD1/RNF39 locus approached the genome-wide significance threshold (Figure 2). Their effect on progression and viral load set point was demonstrated to be independent from both the HCP5 and −35 kb HLA-C SNPs.
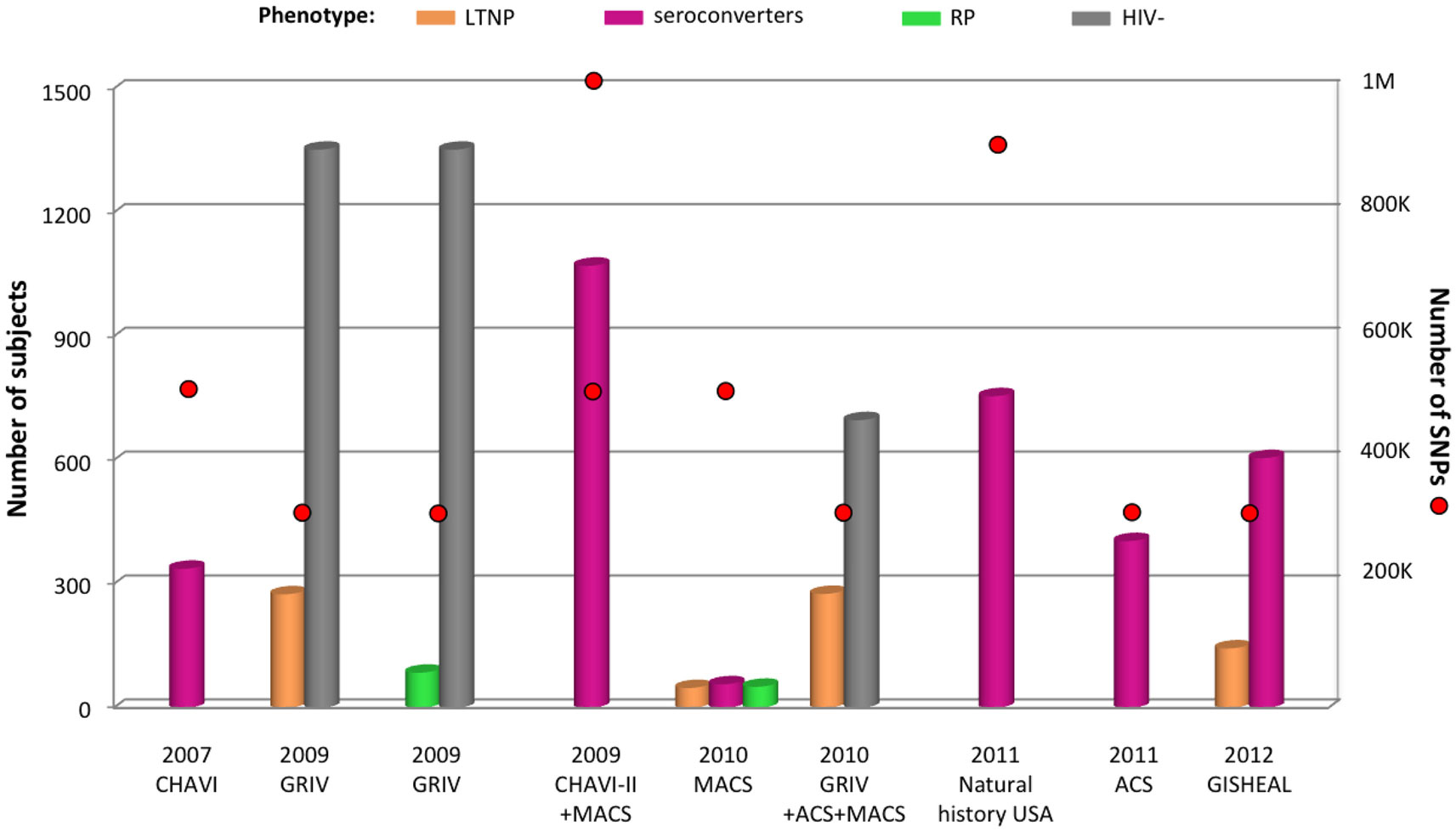
Figure 3. Summary of GWAS design focusing on disease progression. The histogram presents the number of subjects and red circles display the number of SNPs covered by the genotyping arrays. Two red circles are symbolized for one cohort when individuals were genotyped using two sets of arrays. All subjects are from European ancestry. The different phenotypes are color-coded: gold for long-term non-progressors (LTNP), pink for medium seroconverters, green for rapid progressors (RP), and gray for uninfected individuals. The natural history USA study comprises seroconverters from the following cohorts: ALIVE, DCG, HGDS, MACS, MHCS, and SFCC.
The first genome-wide explorations of cohorts specifically designed to concentrate on AIDS progression rather than on viral load control were performed in the French GRIV cohort of participants with extreme progression profiles (Figure 3) (Le Clerc et al., 2009; Limou et al., 2009). GRIV patients were enrolled based on plasma CD4+ T-cell count, a major AIDS progression predictor in untreated patients. The 275 GRIV LTNP, defined by a CD4+ T-cell count above 500/μL for more than 8 years after HIV infection in absence of antiretroviral therapy, were compared to 1,352 HIV-1 seronegative individuals. The strongest signal associated with long-term non-progression was obtained for HCP5 rs2395029 (P = 6.79 × 10−10). As illustrated by this result, host factors controlling viral load are likely to be correlated with stable high CD4+ T-cell count. Likewise, this explains the abundance of HLA SNPs (46 out of the 50 top signals) in the meta-analysis between the GRIV LTNP and the Euro-CHAVI HIV-RNA GWAS. These results emphasize the critical role of HLA both for early viral control and long-term non-progression. In addition, comparable to the Euro-CHAVI study, signals approaching genome-wide significance were identified independently from HCP5 rs2395029 for ZNRD1/RNF39 SNPs.
The only GWAS addressing rapid progression to date was performed by comparing the GRIV rapid progressors, defined by a drop of CD4+ T-cell count below 300/μL within 3 years after the last seronegative test, with 1,352 seronegative controls (Figure 3) (Le Clerc et al., 2009). The limited sample size for rapid progressors (n = 85) was not powered to detect variants respecting the genome-wide significance threshold. However, the use of an alternative statistical strategy, termed the false-discovery rate (Benjamini and Hochberg, 1995), revealed six independent loci potentially associated with rapid progression: PRMT6 and RXRG (chr.1), SOX5 (chr.12), TGFBRAP1 (chr.2), and two intergenic loci (chr.8 and chr.9). These four candidate genes were biologically relevant for HIV/AIDS pathogenesis and notably emphasized a potential key role for the TGFβ pathway and for HIV replication control for progression. Nonetheless, the absence of a replication cohort enriched in rapid progressors is a severe limitation for further investigation of this unique progression phenotype.
Similarly to the first GWAS on Euro-CHAVI, the extended Euro-CHAVI and MACS cohort study assessed the progression phenotype for a subset of 1,071 subjects (Figure 3) (Fellay et al., 2009). This analysis confirmed the major role of HCP5 rs2395029 and −35 kb HLA-C rs9264942 both for viral load control and disease progression. Due to increased power, the ZNRD1/RNF39 candidate locus was for the first time significantly associated with AIDS progression (P = 1.8 × 10−8).
The HCP5 and −35 kb HLA-C associations with disease progression are mainly driven by their influence on early viral load control: both P-values are much weaker if viral load is considered as a covariate in the statistical analysis of the extended Euro-CHAVI and MACS cohort (P < 10−3). On the other hand, ZNRD1/RNF39 association is largely independent of viremia, suggesting a different mechanism of protection (Fellay et al., 2009). Similarly in the GRIV LTNP cohort, viral load was significantly lower in LTNP carrying the protective HCP5 rs2395029-G allele, while none of the ZNRD1/RNF39 polymorphisms seemed to correlate with viral load (Limou et al., 2009). In order to identify additional host factors impacting long-term non-progression without necessarily controlling viral load at very low level, the GRIV LTNP GWAS data were re-analyzed after excluding the elite controllers (with a viral load <100 copies/mL, Figure 3) (Limou et al., 2010). Individuals from the ACS and MACS cohorts respecting this specific non-progression phenotype were also included to increase power. The comparison of these 276 unique LTNP with 697 HIV-uninfected controls revealed a significant association for CXCR6 rs2234358 (P = 2.1 × 10−8, Figure 2). This newly identified chromosome 3 signal was replicated in three independent European studies and shown to be independent from the CCR2/CCR5 and HLA associations. CXCR6 is a minor HIV co-receptor (Deng et al., 1997) and is involved in inflammation (Landro et al., 2009) through the trafficking of effector T-cells (Kim et al., 2001) and the activation of NKT cells (Germanov et al., 2008). Interestingly, hepatic NK mice cells were shown to develop a specific adaptive immunity to HIV-1 antigens through a mechanism requiring the expression of CXCR6 (Paust et al., 2010).
Another GWAS was performed on a subset of subjects enriched with extreme progression profiles from the European-American MACS cohort: 51 rapid progressors vs. 57 medium progressors vs. 48 LTNP (Figure 3) (Herbeck et al., 2010). This GWAS was the first published study using the Affymetrix genotyping arrays, which carry a different set of SNPs (especially for the HLA region) compared to Illumina arrays. The top 25 signals were assessed for replication in an independent cohort of 590 seroconverters. No SNP reached genome-wide significance, but a potential signal was suggested for three related SNPs located 36 kb upstream the PROX1 gene, encoding for a negative regulator of IFNγ (Wang et al., 2008).
Patients enrolled from six USA natural history cohorts (MACS, MHCS, DCG, HGDS, SFCC, and ALIVE) were genotyped using large coverage Affymetrix 6.0 arrays (Figure 3) (Troyer et al., 2011). Collectively, 755 European-American seroconverters were evaluated for association with progression to AIDS 1987. An intronic SNP in PARD3B reached genome-wide significance (P = 3.37 × 10−9), and several other PARD3B polymorphisms were among the top signals (Figure 2). One of these variants was replicated by comparing rapid progressors from the GRIV and ACS cohorts with uninfected controls (P = 0.025). PARD3B interacts with members of the SMAD family, which are involved in multiple signaling pathways and biological activities, and directly interact with HIV-1 (Ptak et al., 2008).
The Dutch seroconverters from ACS (n = 404) were tested for association with survival time to AIDS 1993-diagnosis or to AIDS-related death (Figure 3) (van Manen et al., 2011). The top 10 signals were assessed for replication in the independent GRIV cohort, and no genome-wide significant result was revealed. Potential associations were highlighted, but to date, none of them have been replicated. The HCP5 rs2395029 SNP trended to association with time to AIDS 1993 or to AIDS-related death, but as previously reported, this effect was only marginal when the analysis was corrected for the effect of viral load at set point (van Manen et al., 2009).
Finally, the French-Italian GISHEAL cohort recruited 144 LTNP defined by a CD4+ T-cell count above 500/μL for more than 7 years after HIV infection in the absence of antiretroviral therapy (Figure 3) (Guergnon et al., 2012). The GWAS comparing these LTNP with the 605 seroconverters from the PRIMO cohort confirmed the importance of the HLA region for long-term non-progression (rs2395029, P = 1.61 × 10−11).
Surprisingly, GWAS exploring host factors involved in AIDS disease progression only focused on population of European ancestry. These studies confirmed that the HLA region is also strongly associated with long-term non-progression. The chromosome 6 ZNRD1/RNF39 locus, the chemokine receptor region (CCR2/CCR5 and CXCR6), and the PARD3B gene were also identified as key regulators of disease progression. Lastly, some biologically relevant candidates genes were suggested (such as PRMT6, TGFBRAP1, or PROX1), but they still require replication in an independent genetic study.
GWAS Focusing on HIV Acquisition
The first GWAS exploring HIV acquisition focused on mother-to-child transmission in Malawi (Joubert et al., 2010). In this underpowered study, 100 HIV+ infants and 126 HIV− infants from HIV+ mothers enrolled in the Malaria and HIV in Pregnancy prospective study were compared (Figure 4), but no genetic variant reached genome-wide significance.
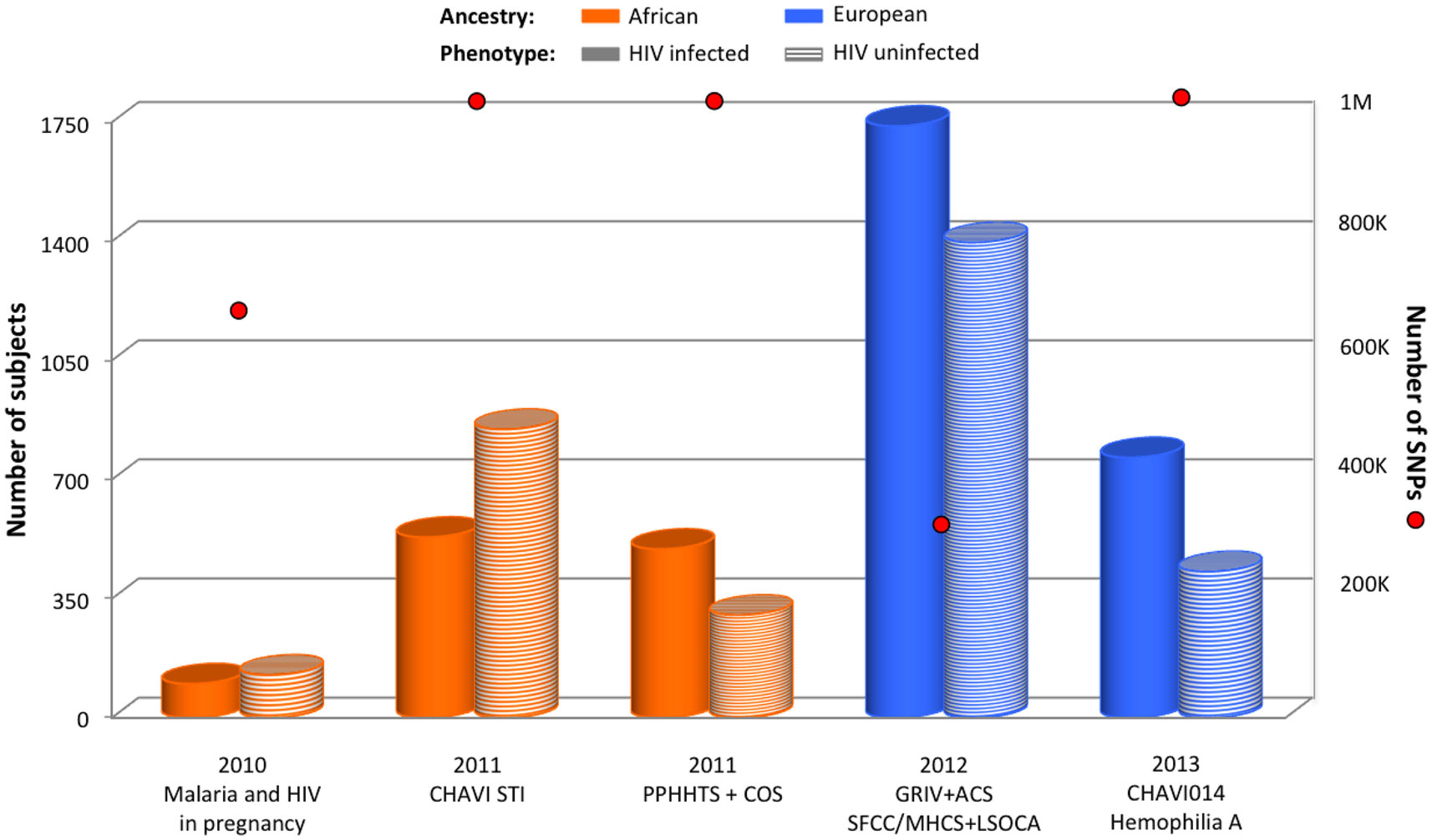
Figure 4. Summary of GWAS design focusing on HIV acquisition. The histogram presents the number of subjects and red circles display the number of SNPs covered by the genotyping arrays. Individual ancestry is color-coded: orange for African and blue for European. The pattern of each cylinder illustrates the phenotype of interest: plain color for HIV-infected, and horizontal stripes for HIV-uninfected.
A second larger GWAS performed on individuals from Malawi compared 531 HIV+ with 848 sexually active HIV− recruited by CHAVI in sexually transmitted infection (STI) clinics (Figure 4) (Petrovski et al., 2011). It was assumed that the latter group was exposed to HIV-1 because of the very high prevalence of the infection among sexually active adults in Malawi. No genome-wide significant SNP was identified.
A third study performed on individuals from Africa explored the HIV acquisition in serodiscordant heterosexual couples (PPHHTS/COS study) (Lingappa et al., 2011). In this study, the risk of exposure to HIV was carefully scored, and 496 HIV+ individuals were compared to 302 HIV− individuals (Figure 4). Nevertheless, this GWAS also failed to reveal any significant signal.
A first GWAS exploring HIV acquisition in individuals from European ancestry compared 764 HIV+ individuals from the GRIV and ACS cohorts with 1,073 HIV− individuals (Figure 4) (Limou et al., 2012). The top signals were assessed for replication in two independent groups of European descent (MHCS-SFCC and LSOCA). The rs6996198-T allele (chr.8) was significantly associated with a protection from HIV-1 infection (P = 7.76 × 10−8). This protective allele was associated with a higher CYP7B1 expression, a gene located 45 kb upstream the variant. CYP7B1 is involved in the steroid and oxysterol metabolism (Stiles et al., 2009), but also in immune control and regulation – notably in the IgA (Bauman et al., 2009) and proinflammatory cytokines (Dulos et al., 2005) production, and in the induction of programed cell death (Rusinol et al., 2004).
Recently, European hemophiliacs exposed to potentially HIV-contaminated blood through transfusion (CHAVI014) were investigated: 765 HIV+ individuals were compared to 430 HIV− individuals who were not CCR5-Δ32 homozygous (Figure 4) (Lane et al., 2013). The extensive screening of the genetic data did not reveal any significant association.
In summary, the GWAS focusing on HIV acquisition did not reveal any signal in African ancestry populations, and only one potential association with CYP7B1 was identified in a European ancestry study.
Causal Variant and Functional Exploration
Amongst the genetic variants highlighted through the GWAS (Figure 2), several were further explored to unravel the biological mechanism underlying the association, especially for the HLA region. The complex LD pattern in this region makes it very difficult to discriminate the causal variant(s), in particular in the context of a locus rich in immune-related genes that are all candidates of interest for the control of HIV replication and pathogenesis, and that have almost all been previously associated with various immune-related diseases.
For example, HCP5 rs2395029 is in LD with HLA-B*5701, a securely identified HLA protective allele, but is also in LD with polymorphisms from MICB, BAT1, TNF, and LTB. MICB is a ligand for CD8+ T-cells and NK cells, which are both key players in the anti-HIV immune response. BAT1 is involved in splicing and RNA export (Reed and Hurt, 2002), and is a negative regulator of proinflammatory cytokines (Allcock et al., 2001). TNF is a key proinflammatory cytokine that has been widely investigated in HIV-1 infection (Kedzierska and Crowe, 2001). LTB is an inflammation modulator involved in the development of immune cells (Rennert et al., 1996). HCP5 itself was considered as a candidate of interest: since it encodes a human endogenous retrovirus with sequence homology to HIV-1 pol (Kulski and Dawkins, 1999), it was hypothesized that it may act as an antisense RNA interfering with HIV replication. However, the two allelic forms do not impact HIV-1 replication in vitro, suggesting that HCP5 rs2395029 is probably not the causal variant (Yoon et al., 2010). HLA-B*5701 was previously associated with low viremia and delayed onset of AIDS through candidate gene studies (Stephens, 2005). The potential function of this allele has been widely explored: notably, the presentation of specific HIV-1 epitopes able to initiate a strong cytotoxic T-lymphocyte response, and the TCR clonotype recognizing the HLA-peptide complex were largely emphasized (Klein et al., 1998; Migueles et al., 2000; Gillespie et al., 2002, 2006; Miura et al., 2009; Elahi et al., 2011; Chen et al., 2012; Kloverpris et al., 2012). Following this path, the international HIV controllers study confirmed the overwhelming importance of HLA alleles for viral control (B*5701, B*2705, B*14/C*0802, B*52, and A*25 as protective alleles, and B*35 and C*07 as risk alleles) and identified putative causal variants in the HLA-B peptide-binding groove (including residues at position 67, 70, and 97) in Europeans (International HIV Controllers Study et al., 2010). Similarly in African-Americans, they confirmed the importance of HLA-B*5703 and HLA-B*8101 alleles for viral control and identified the HLA-B groove residues 63, 97, and 116 as critical for the association (International HIV Controllers Study et al., 2010; McLaren et al., 2012a). Even though the mechanisms underpinning HIV-1 control and disease non-progression remain unclear, these coding variants are predicted to impact the conformation of the HLA-B groove and the HIV peptides that are presented, as well as the subsequent engagement of TCR or other receptors (such as the NK cell receptors). As suggested by the recent study by McLaren et al. (2012a), the role played by HLA-B on viral control does not rule out the implication of other polymorphisms in the HLA region, outside the HLA-B binding groove and possibly outside the class I HLA genes.
The −35 kb (HLA-C)-C allele, associated with viral control, was correlated with higher HLA-C gene expression (Stranger et al., 2005) and higher HLA-C cell surface expression (Thomas et al., 2009). The signal was proposed to be the result of LD between the −35 kb-T allele and the HLA-C*07 allele, known to be associated with very low cell surface expression and more rapid disease progression (Corrah et al., 2011). Interestingly, the −35 kb-T allele was also demonstrated to be in LD with a polymorphism of the HLA-C 3′UTR (ins263 or rs67384697-G): this polymorphism results in the creation of a binding site for miR-148a causing the silencing of the HLA-C mRNA expression and a low HLA-C cell surface expression (Figure 5) (Kulkarni et al., 2011). This mechanism offers a highly plausible functional basis for the high HLA-C cell surface expression observed amongst HIV-infected individuals controlling the virus replication. However, since HLA-C expression occurs as a continuous gradient rather than a bimodal pattern that would be expected if miR-148a were the sole regulation pathway, additional unknown factors are likely to affect HLA-C expression.
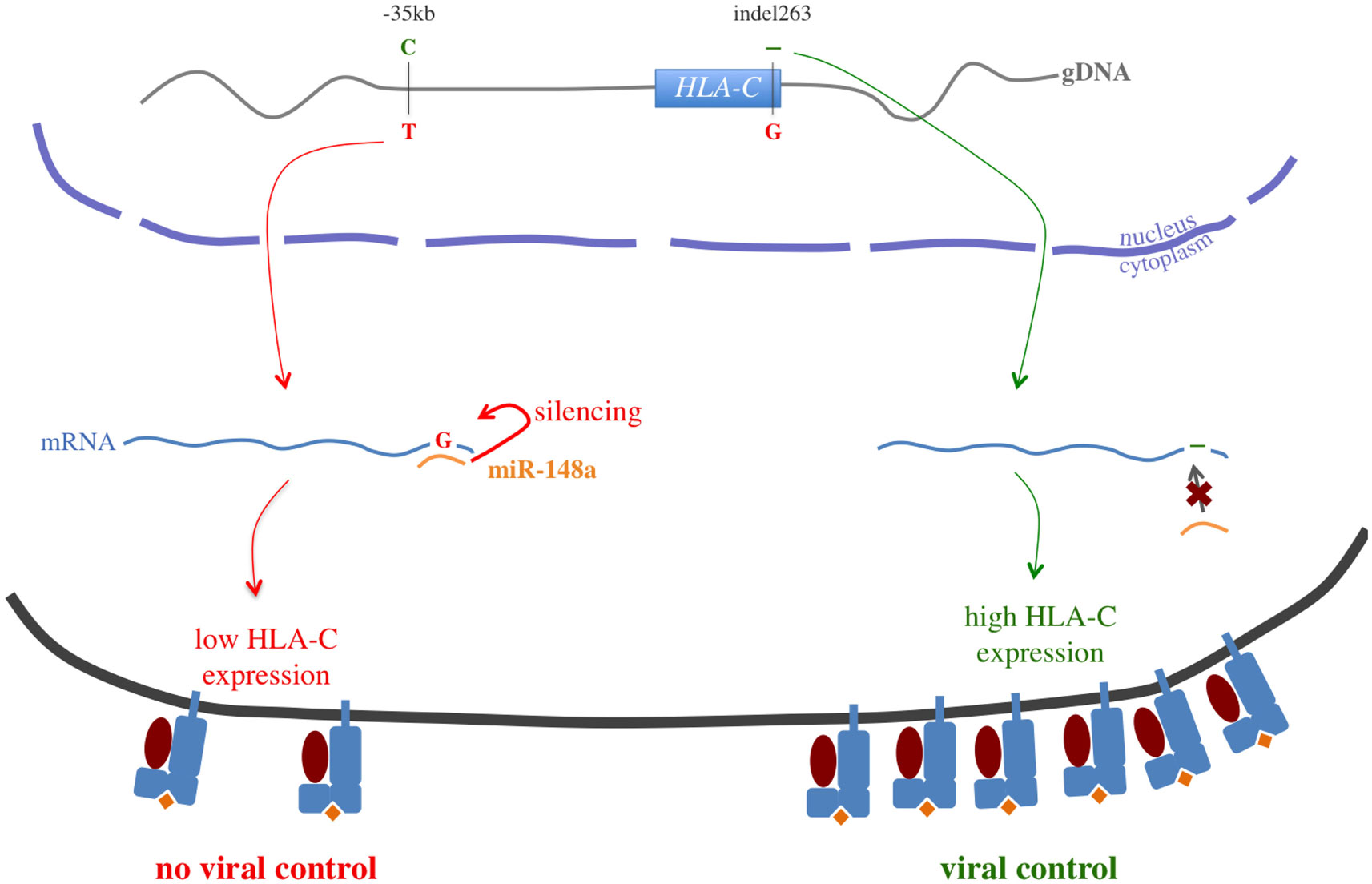
Figure 5. −35 kb HLA-C functional story. This picture illustrates the strong linkage disequilibrium between −35 kb HLA-C (rs9264942) and the 3′UTR indel263 (rs67384697): the −35 kb-C allele is strongly correlated with the deletion, when the −35 kb-T allele is strongly correlated with the insertion of a G nucleotide. If the mRNA carries the −35 kb-T/insertion haplotype, the human miR-148a can bind to the HLA-C 3′UTR and silence its expression, leading to a low HLA-C cell surface expression. However, if the mRNA carries the −35 kb-C/deletion haplotype, miR-148a cannot bind efficiently to the HLA-C 3′UTR anymore, and HLA-C cell surface expression is increased. This mechanism would mostly explain the association between the −35 kb HLA-C polymorphism and viral control.
Finally, a set of chromosome 6 SNPs in the ZNRD1/RNF39 locus was associated with AIDS progression independently from the HCP5 rs2395029 and −35 kb HLA-C signals. ZNRD1 and RNF39 encode for an RNA polymerase I subunit and for an unknown ring finger protein, respectively. ZNRD1 expression was significantly correlated to several SNPs associated with CD4+ T-cell depletion (Stranger et al., 2005). In addition, ZNRD1 was identified as 1 of the 273 host proteins required for HIV-1 infection and replication in a genome-wide siRNA screen (Brass et al., 2008). An independent study confirmed that the down-regulation of ZNRD1 through siRNA or shRNA impaired HIV-1 replication at the transcription level (Ballana et al., 2010), suggesting strongly that ZNRD1 is the causal factor for the ZNRD1/RNF39 associations.
In addition to these three loci, all securely replicated associations revealed by GWAS (red in Figure 2) require further functional studies in order to fully understand their causal relationship with HIV control or disease pathogenesis. Potential associations (light blue in Figure 2) require independent replication to support further functional studies.
Alternative Explorations
Beyond the main GWAS results described above, we believe that other strategies and HIV phenotypes deserve to be mentioned.
Low-frequency SNPs (<5%) are predicted to have larger effect size and contribute to phenotype, but are often overlooked in GWAS because of power. An interesting study re-analyzed data from three European GWAS focusing on these neglected low-frequency SNPs (Le Clerc et al., 2011). The comparison of 365 LTNP with 1,394 HIV-negative controls on 8,584 low-frequency SNPs revealed two independent associations: the HCP5 rs2395029 signal (P = 8.54 × 10−15), and the RICH2 rs2072255 SNP (P = 3.30 × 10−6). RICH2 is a factor contributing to the externalization of BST-2/Tetherin (Rollason et al., 2009), which prevents HIV-1 virion budding and release (Tokarev et al., 2009). The RICH2 rs2072255 SNP is in full LD with a coding polymorphism predicted to impact splicing, which could thus alter RICH2 expression and its subsequent effect on HIV production.
Beyond HIV/AIDS primary incomes, GWAS investigating metabolic, and age-related complications are also emerging: atherosclerosis (Shrestha et al., 2010), chronic liver disease (Rauch et al., 2010), kidney disease (Kopp et al., 2008), neurocognitive disorder (Levine et al., 2012). Understanding the genetic factors contributing to these conditions is essential since they tend to occur more frequently and at an earlier age in HIV-infected subjects compared to the general population.
Finally, a number of alternative strategies have been deployed outside genetics in order to discover some of the host factors involved in viral control, AIDS progression or HIV replication. We can mention transcriptomic (mRNA expression) studies (Giri et al., 2006; Rotger et al., 2010; Salgado et al., 2011; Vigneault et al., 2011), proteomic studies (Coiras et al., 2006; Chan et al., 2007; Molina et al., 2007; Ringrose et al., 2008), or siRNA and shRNA screens (Brass et al., 2008; Konig et al., 2008; Zhou et al., 2008; Yeung et al., 2009). Some of these studies will be further discussed in Santa-Marta et al. (2013).
Discussion and Future Perspectives
The GWAS focusing on viral control identified HLA as the primary region involved in HIV control in both European and African derived populations. Even if the genetic variants highlighted in these two populations were different, it might suggest a similar mechanism of control, engaging notably the antigen presentation by the HLA-B molecule. A role for HLA-C was also unraveled in Europeans through a mechanism involving variation of expression level on the cell surface.
In addition to viral control (and not surprisingly), HLA is also the major region involved in long-term non-progression. However, the role for the HCP5/HLA-B*57 and −35 kb HLA-C SNPs on disease progression was mainly driven by their influence on early viral control. On the other hand, the independent ZNRD1 signal was associated with disease progression without impacting directly viral replication.
Even if the design of each cohort varies, the contribution of HLA variants was consistently replicated in the different studies focusing on viral control and/or disease progression. The differences observed between the studies were mostly due to different genotyping platforms. Indeed, Affymetrix and Illumina genotyping arrays have different human genome coverage: Affymetrix SNPs were selected for their genotyping accuracy and their regular physical spacing across the genome, while Illumina selected SNPs based on the LD (tagSNPs) from the HapMap populations, focused on gene-centric variations, and enriched the coverage in the highly polymorphic HLA region. In particular, the difference of coverage for the HLA region explains why the cohorts that were genotyped with the Affymetrix arrays (MACS and other natural history USA cohorts (Herbeck et al., 2010; Troyer et al., 2011) did not identify the HLA and ZNRD1 signals. Similarly, the early Illumina 300K chips did not cover the −35 kb HLA-C variant, when more dense Illumina chips (≥500 K) do. This explains why this variant was not identify by the ACS, GRIV, and PRIMO studies using Illumina 300K. However, when this variant is genotyped independently, the −35 kb HLA-C signal is replicated (van Manen et al., 2009).
Of course, the use of different genotyping arrays is not the only source of variation between the studies. As illustrated by Figures 1, 3, and 4, the cohort size varies across GWAS and this affects statistical power. This power concern might explain why the HLA region was not significant in the Hispanic population from the HIV controllers study (International HIV Controllers Study et al., 2010), although we cannot exclude that HLA is not involved in viral control in this population.
The exploration of different HIV phenotype is also a great source of variation in the design of all these GWAS. As an example, the examination of a progression phenotype defined according to time to AIDS or to AIDS-related death might explain why HLA was not identified as the primary regulator of progression in the ACS cohort (van Manen et al., 2011), although we cannot exclude that a lack of power, a difference of transmission route, or another unknown factor might account for the failure to confirm HLA.
Even if it makes it more difficult to compare results among studies, the diversity of phenotype might be advantageous when considering the discovery of the full spectrum of host factors involved in HIV/AIDS pathogenesis. Therefore, the alternative analysis of the GRIV LTNP after excluding individuals controlling viral load at very low levels revealed the importance of CXCR6 in long-term non-progression (Limou et al., 2010). This association being largely independent of viral control, it was logically not identified by the cohorts designed to specifically investigate viral control (Euro-CHAVI, HIV controllers, etc.). Another striking example of the asset of exploring different endpoints is the GRIV rapid progression GWAS. This underpowered analysis revealed associations with strong effect size (OR from 2.2 to 4.2) and biologically relevant candidate genes (Le Clerc et al., 2009). Rapid progressors are often depleted from cohorts (e.g., enrollment in Euro-CHAVI requires a stable viral load), and thus, the absence of a powerful replication cohort is a severe limitation for further investigation of this unique phenotype. Consequently, it might be very useful for the community to develop new cohorts exploring rapid progressors and other innovative phenotypes in the hope of drawing a complete picture of AIDS pathogenesis.
Genome-wide association studies exploring the HIV acquisition were mainly disappointing since no strong signal outside the CCR5-Δ32 homozygosity were identified. The lack of significant signals associated with HIV-1 acquisition indicates that common genetic variants with strong effect size are unlikely to play a role in HIV-1 susceptibility in these populations. Three and two GWAS were performed in African and European ancestry populations, respectively. Only the GRIV/ACS European GWAS identified an association with chromosome 8 CYP7B1 (Limou et al., 2012). The absence of confirmation in the European CHAVI014 Hemophilia cohort might be due to a lack of power (see Figure 4), to a difference of phenotype (GRIV/ACS used normal population controls when CHAVI014 used highly exposed HIV-seronegatives), to a difference of transmission route (GRIV/ACS were mostly infected through sexual contact when CHAVI014 were infected through blood-transfusion), to the identification of a false positive in the GRIV/ACS study, or to an unidentified factor.
Altogether, the HLA and chemokine receptors (CCR2/CCR5 and CXCR6) associations explain around 20% of the variance in viral control and/or disease progression, depending on the cohorts. In contrast, most GWAS for common diseases or traits (e.g., type 2 diabetes, height) have identified common variants with small to modest effect sizes explaining a small portion of the phenotypic variance (Lango Allen et al., 2010). With ever larger cohorts, variants with smaller effect sizes might be discovered, but new strategies are also needed to supplement standard genetic analyses.
First, common variants were widely tested in the context of HIV-infected populations of European ancestry. However, we cannot exclude that common variants with small to medium effect size were missed and buried below the very strict statistical threshold (type 2 errors or false negatives) – this is especially relevant in the context of the relatively modest sample size of the HIV/AIDS cohorts. Increasing study power by recruiting additional individuals can contribute to the discovery of new variants associated with HIV. The examination of a bigger sample size for the Euro-CHAVI cohort was indeed successful since it revealed additional HLA variants and confirmed the ZNRD1 signal (Fellay et al., 2009). An alternative solution is to focus on individuals with extreme phenotypes (such as the LTNP, rapid progressors, the highly exposed HIV-seronegatives), who represent a powerful contrasting tool enriched for genetic variants associated with the phenotype. Indeed, the GRIV cohort illustrates the power of a modest size cohort designed with very careful extreme phenotypes to identify new candidate genes (Le Clerc et al., 2009; Limou et al., 2009, 2010). Finally, combining several studies into large meta-analyses also increases power and can thus contribute to the discovery of new host factors. A collaborative effort was initiated in 2009 to create the international consortium on HIV genomics (ICHG) in order to combine the previously published GWAS and explore further the different phenotypes. The first phase of meta-analysis focusing on HIV-1 acquisition by comparing 10,000 HIV-infected subjects with 15,000 HIV-uninfected has been completed and did not identify any new signal beyond the resistance conferred by CCR5-Δ32 homozygotes, suggesting again that common host genetic variation has little influence on HIV susceptibility (McLaren et al., 2012b). The second phase of ICHG meta-analysis is on-going and will focus on phenotypes associated with viral control and disease progression.
African populations have also been explored during this first wave of GWAS, but not for the analysis of disease progression phenotypes. This oversight stems from funding and infrastructure limitations to support large prospective cohorts. In addition, African populations exhibit a higher genetic diversity and a lower LD level than populations outside of Africa, and therefore require arrays with a higher density of tagSNPs to ensure a genome-wide capture. The new generation of genotyping arrays now offers the simultaneous genotyping of up to 5 M tagSNPs, and incorporates the knowledge from the 1000 Genomes Project (new public catalog of human genetic variants with a frequency down to 1%) (1000 Genomes Project Consortium et al., 2012). Even if the coverage is still less effective for African populations than for European and Asian populations, the use of these new genotyping arrays might participate in the discovery of common and less common genetic variants associated with HIV-related outcomes in Africans.
Other HIV-infected populations with different ancestries have been widely disregarded in the host genetics field (e.g., Asian populations, admixed-populations such as Latinos and Hispanics). These populations may harbor different variants from Europeans that also influence HIV infection, replication, and pathogenesis.
Beyond the common polymorphisms (with a frequency >5%) targeted during the first wave of GWAS, rare SNPs, indels, and structural variants might also contribute to HIV infection and AIDS progression. The new generation of genotyping arrays encompasses information for lower frequency SNPs (1–5%) from the 1000 Genomes Project. The use of this newer high density arrays combined with the next-generation sequencing (exome or genome) should capture rare SNPs, indels, and structural variants, and highlight new host factors associated with HIV infection course. Since next-generation sequencing is an expensive technology, focusing on extreme phenotypes to emphasize the genetic differences as in the vanguard GRIV cohort (Hendel et al., 1996) should prove a promising strategy (McLaren et al., 2011; Pelak et al., 2011).
The next challenges are to assess the computationally challenging gene × gene and gene × environment interactions, as well as the role of epigenetics in HIV disease. The incorporation of biological knowledge might aide in assessing these complex interplays under reasonable assumptions. Systems biology integrates information from multiple “omics” sources (genomics, transcriptomics, proteomics, etc.), and could thus disclose specific host pathways or networks contributing to the HIV-linked phenotypes.
Finally, we cannot forget that HIV course is also impacted by viral and environmental factors that may play a larger role than initially suspected (Wang, 2013).
Conclusion
The motivation for identifying genes associated with HIV/AIDS has gained momentum as effective vaccines and curative treatments have failed to materialize. The technologic breakthrough of genotyping arrays has opened up a prolific era in human genetics. Relatively to the discovery of hundreds of genes involved in dozen of complex diseases, the re-discovery of HLA and chemokine region in the HIV/AIDS field could seem quite disappointing. However, it is important to note that GWAS revealed that HLA is the primary region involved in viral control. In addition, two signals were discovered (CXCR6 and PARD3B), and candidate regions highlighted. New strategies to exploit the large amount of genomic data generated (e.g., meta-analyses, systems biology, etc.) and the generation of new data (e.g., new phenotypes, new populations, next-generation sequencing, etc.) will hopefully lead to the discovery of new host genetic associations that could lead, following the example of CCR5-Δ32, to the development of new antiretroviral therapy or vaccine.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are very grateful to Dr. Cheryl A. Winkler and Dr. Nicolas Vince for their advice and careful reading of the manuscript. This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Abbreviations
Genes: CCR2, C-C chemokine receptor 2; CCR5, C-C chemokine receptor 5; CXCR6, C-X-C chemokine receptor 6; CYP7B1, cytochrome P450, family 7, subfamily B, polypeptide 1; HCP5, HLA complex P5; HLA, human leukocyte antigen; KLF3, Kruppel-like factor 3; PARD3B, PAR-3 partitioning defective 3 homolog B; PRMT6, protein arginine methyltransferase 6; PROX1, prospero homeobox 1; RNF39, ring finger protein 39; RXRG, retinoid X receptor gamma; SOX5, SRY-box 5; TBC1D1, TBC1 domain family member 1; TGFBRAP1, transforming growth factor beta receptor associated protein 1; ZNRD1, zinc ribbon domain containing 1.
Cohorts: ACS, Amsterdam cohort study; ALIVE, AIDS link to intravenous experience; CHAVI, center for HIV/AIDS vaccine immunology; COS, Couples Observational Study; DCG, DC gay; DoD HIV NHS, Department of Defense HIV Natural History Study; GISHEAL, genetic and immunological study on HIV+ European and African LTNP; GRIV, genomics of resistance to immunodeficiency virus; HGDS, Hemophilia Growth and Development Study; LSOCA, longitudinal studies of the ocular complications of AIDS; MACS, multicenter AIDS cohort study; MHCS, multicenter Hemophilia cohort study; PPHHTS, Partners in Prevention HSV/HIV Transmission Study; PRIMO, primo infection; SFCC, San Francisco city clinic.
References
1000 Genomes Project Consortium, G. P., Abecasis, G. R., Auton, A., Brooks, L. D., DePristo, M. A., Durbin, R. M., et al. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65.
Allcock, R. J., Williams, J. H., and Price, P. (2001). The central MHC gene, BAT1, may encode a protein that down-regulates cytokine production. Genes Cells 6, 487–494.
An, P., and Winkler, C. A. (2010). Host genes associated with HIV/AIDS: advances in gene discovery. Trends Genet. 26, 119–131.
Ballana, E., Senserrich, J., Pauls, E., Faner, R., Mercader, J. M., Uyttebroeck, F., et al. (2010). ZNRD1 (zinc ribbon domain-containing 1) is a host cellular factor that influences HIV-1 replication and disease progression. Clin. Infect. Dis. 50, 1022–1032.
Bauman, D. R., Bitmansour, A. D., McDonald, J. G., Thompson, B. M., Liang, G., and Russell, D. W. (2009). 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. U.S.A. 106, 16764–16769.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. Ser. B 57, 289–300.
Brass, A. L., Dykxhoorn, D. M., Benita, Y., Yan, N., Engelman, A., Xavier, R. J., et al. (2008). Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926.
Carrington, M., Nelson, G. W., Martin, M. P., Kissner, T., Vlahov, D., Goedert, J. J., et al. (1999). HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283, 1748–1752.
Chan, E. Y., Qian, W. J., Diamond, D. L., Liu, T., Gritsenko, M. A., Monroe, M. E., et al. (2007). Quantitative analysis of human immunodeficiency virus type 1-infected CD4+ cell proteome: dysregulated cell cycle progression and nuclear transport coincide with robust virus production. J. Virol. 81, 7571–7583.
Chen, H., Ndhlovu, Z. M., Liu, D., Porter, L. C., Fang, J. W., Darko, S., et al. (2012). TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat. Immunol. 13, 691–700.
Coiras, M., Camafeita, E., Urena, T., Lopez, J. A., Caballero, F., Fernandez, B., et al. (2006). Modifications in the human T cell proteome induced by intracellular HIV-1 Tat protein expression. Proteomics 6(Suppl. 1), S63–S73.
Corrah, T. W., Goonetilleke, N., Kopycinski, J., Deeks, S. G., Cohen, M. S., Borrow, P., et al. (2011). Reappraisal of the relationship between the HIV-1-protective single-nucleotide polymorphism 35 kilobases upstream of the HLA-C gene and surface HLA-C expression. J. Virol. 85, 3367–3374.
Dalmasso, C., Carpentier, W., Meyer, L., Rouzioux, C., Goujard, C., Chaix, M. L., et al. (2008). Distinct genetic loci control plasma HIV-RNA and cellular HIV-DNA levels in HIV-1 infection: the ANRS Genome Wide Association 01 study. PLoS ONE 3:e3907. doi:10.1371/journal.pone.0003907
Dean, M., Carrington, M., Winkler, C., Huttley, G. A., Smith, M. W., Allikmets, R., et al. (1996). Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 273, 1856–1862.
Deng, H. K., Unutmaz, D., KewalRamani, V. N., and Littman, D. R. (1997). Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 388, 296–300.
Dulos, J., van der Vleuten, M. A., Kavelaars, A., Heijnen, C. J., and Boots, A. M. (2005). CYP7B expression and activity in fibroblast-like synoviocytes from patients with rheumatoid arthritis: regulation by proinflammatory cytokines. Arthritis Rheum. 52, 770–778.
Elahi, S., Dinges, W. L., Lejarcegui, N., Laing, K. J., Collier, A. C., Koelle, D. M., et al. (2011). Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat. Med. 17, 989–995.
Fellay, J. (2009). Host genetics influences on HIV type-1 disease. Antivir. Ther. (Lond.) 14, 731–738.
Fellay, J., Ge, D., Shianna, K. V., Colombo, S., Ledergerber, B., Cirulli, E. T., et al. (2009). Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 5:e1000791. doi:10.1371/journal.pgen.1000791
Fellay, J., Shianna, K. V., Ge, D., Colombo, S., Ledergerber, B., Weale, M., et al. (2007). A whole-genome association study of major determinants for host control of HIV-1. Science 317, 944–947.
Fowke, K. R., Nagelkerke, N. J., Kimani, J., Simonsen, J. N., Anzala, A. O., Bwayo, J. J., et al. (1996). Resistance to HIV-1 infection among persistently seronegative prostitutes in Nairobi, Kenya. Lancet 348, 1347–1351.
Germanov, E., Veinotte, L., Cullen, R., Chamberlain, E., Butcher, E. C., and Johnston, B. (2008). Critical role for the chemokine receptor CXCR6 in homeostasis and activation of CD1d-restricted NKT cells. J. Immunol. 181, 81–91.
Gillespie, G. M., Kaul, R., Dong, T., Yang, H. B., Rostron, T., Bwayo, J. J., et al. (2002). Cross-reactive cytotoxic T lymphocytes against a HIV-1 p24 epitope in slow progressors with B*57. AIDS 16, 961–972.
Gillespie, G. M., Stewart-Jones, G., Rengasamy, J., Beattie, T., Bwayo, J. J., Plummer, F. A., et al. (2006). Strong TCR conservation and altered T cell cross-reactivity characterize a B*57-restricted immune response in HIV-1 infection. J. Immunol. 177, 3893–3902.
Giri, M. S., Nebozhyn, M., Showe, L., and Montaner, L. J. (2006). Microarray data on gene modulation by HIV-1 in immune cells: 2000-2006. J. Leukoc. Biol. 80, 1031–1043.
Guergnon, J., Dalmasso, C., Broet, P., Meyer, L., Westrop, S. J., Imami, N., et al. (2012). Single-nucleotide polymorphism-defined class I and class III major histocompatibility complex genetic subregions contribute to natural long-term nonprogression in HIV infection. J. Infect. Dis. 205, 718–724.
Gunderson, K. L., Steemers, F. J., Lee, G., Mendoza, L. G., and Chee, M. S. (2005). A genome-wide scalable SNP genotyping assay using microarray technology. Nat. Genet. 37, 549–554.
Hendel, H., Caillat-Zucman, S., Lebuanec, H., Carrington, M., O’Brien, S., Andrieu, J. M., et al. (1999). New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J. Immunol. 162, 6942–6946.
Hendel, H., Cho, Y. Y., Gauthier, N., Rappaport, J., Schachter, F., and Zagury, J. F. (1996). Contribution of cohort studies in understanding HIV pathogenesis: introduction of the GRIV cohort and preliminary results. Biomed. Pharmacother. 50, 480–487.
Henrich, T. J., and Kuritzkes, D. R. (2013). HIV-1 entry inhibitors: recent development and clinical use. Curr. Opin. Virol. 3, 51–57.
Herbeck, J. T., Gottlieb, G. S., Winkler, C. A., Nelson, G. W., An, P., Maust, B. S., et al. (2010). Multistage genomewide association study identifies a locus at 1q41 associated with rate of HIV-1 disease progression to clinical AIDS. J. Infect. Dis. 201, 618–626.
Hetherington, S., Hughes, A. R., Mosteller, M., Shortino, D., Baker, K. L., Spreen, W., et al. (2002). Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet 359, 1121–1122.
International HIV Controllers Study, Pereyra, F., Jia, X., McLaren, P. J., Telenti, A., de Bakker, P. I., et al. (2010). The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330, 1551–1557.
Joubert, B. R., Lange, E. M., Franceschini, N., Mwapasa, V., North, K. E., and Meshnick, S. R. (2010). A whole genome association study of mother-to-child transmission of HIV in Malawi. Genome Med. 2, 17.
Kaslow, R. A., Carrington, M., Apple, R., Park, L., Munoz, A., Saah, A. J., et al. (1996). Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2, 405–411.
Kedzierska, K., and Crowe, S. M. (2001). Cytokines and HIV-1: interactions and clinical implications. Antivir. Chem. Chemother. 12, 133–150.
Kim, C. H., Kunkel, E. J., Boisvert, J., Johnston, B., Campbell, J. J., Genovese, M. C., et al. (2001). Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J. Clin. Invest. 107, 595–601.
Klein, M. R., van der Burg, S. H., Hovenkamp, E., Holwerda, A. M., Drijfhout, J. W., Melief, C. J., et al. (1998). Characterization of HLA-B57-restricted human immunodeficiency virus type 1 Gag- and RT-specific cytotoxic T lymphocyte responses. J. Gen. Virol. 79(Pt 9), 2191–2201.
Kloverpris, H. N., Harndahl, M., Leslie, A. J., Carlson, J. M., Ismail, N., van der Stok, M., et al. (2012). HIV control through a single nucleotide on the HLA-B locus. J. Virol. 86, 11493–11500.
Konig, R., Zhou, Y., Elleder, D., Diamond, T. L., Bonamy, G. M., Irelan, J. T., et al. (2008). Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135, 49–60.
Kopp, J. B., Smith, M. W., Nelson, G. W., Johnson, R. C., Freedman, B. I., Bowden, D. W., et al. (2008). MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat. Genet. 40, 1175–1184.
Kulkarni, S., Savan, R., Qi, Y., Gao, X., Yuki, Y., Bass, S. E., et al. (2011). Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature 472, 495–498.
Kulski, J. K., and Dawkins, R. L. (1999). The P5 multicopy gene family in the MHC is related in sequence to human endogenous retroviruses HERV-L and HERV-16. Immunogenetics 49, 404–412.
Landro, L., Damas, J. K., Halvorsen, B., Fevang, B., Ueland, T., Otterdal, K., et al. (2009). CXCL16 in HIV infection – a link between inflammation and viral replication. Eur. J. Clin. Invest. 39, 1017–1024.
Lane, J., McLaren, P. J., Dorrell, L., Shianna, K. V., Stemke, A., Pelak, K., et al. (2013). A genome-wide association study of resistance to HIV infection in highly exposed uninfected individuals with hemophilia A. Hum. Mol. Genet. 22, 1903–1910.
Langlade-Demoyen, P., Ngo-Giang-Huong, N., Ferchal, F., and Oksenhendler, E. (1994). Human immunodeficiency virus (HIV) nef-specific cytotoxic T lymphocytes in noninfected heterosexual contact of HIV-infected patients. J. Clin. Invest. 93, 1293–1297.
Lango Allen, H., Estrada, K., Lettre, G., Berndt, S. I., Weedon, M. N., Rivadeneira, F., et al. (2010). Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467, 832–838.
Le Clerc, S., Coulonges, C., Delaneau, O., Van Manen, D., Herbeck, J. T., Limou, S., et al. (2011). Screening low-frequency SNPS from genome-wide association study reveals a new risk allele for progression to AIDS. J. Acquir. Immune Defic. Syndr. 56, 279–284.
Le Clerc, S., Limou, S., Coulonges, C., Carpentier, W., Dina, C., Taing, L., et al. (2009). Genomewide association study of a rapid progression cohort identifies new susceptibility alleles for AIDS (ANRS Genomewide Association Study 03). J. Infect. Dis. 200, 1194–1201.
Levine, A. J., Service, S., Miller, E. N., Reynolds, S. M., Singer, E. J., Shapshak, P., et al. (2012). Genome-wide association study of neurocognitive impairment and dementia in HIV-infected adults. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 669–683.
Limou, S., Coulonges, C., Herbeck, J. T., van Manen, D., An, P., Le Clerc, S., et al. (2010). Multiple-cohort genetic association study reveals CXCR6 as a new chemokine receptor involved in long-term nonprogression to AIDS. J. Infect. Dis. 202, 908–915.
Limou, S., Delaneau, O., van Manen, D., An, P., Sezgin, E., Le Clerc, S., et al. (2012). Multicohort genomewide association study reveals a new signal of protection against HIV-1 acquisition. J. Infect. Dis. 205, 1155–1162.
Limou, S., Le Clerc, S., Coulonges, C., Carpentier, W., Dina, C., Delaneau, O., et al. (2009). Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02). J. Infect. Dis. 199, 419–426.
Lingappa, J. R., Petrovski, S., Kahle, E., Fellay, J., Shianna, K., McElrath, M. J., et al. (2011). Genomewide association study for determinants of HIV-1 acquisition and viral set point in HIV-1 serodiscordant couples with quantified virus exposure. PLoS ONE 6:e28632. doi:10.1371/journal.pone.0028632
Liu, R., Paxton, W. A., Choe, S., Ceradini, D., Martin, S. R., Horuk, R., et al. (1996). Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86, 367–377.
Ludlam, C. A., Tucker, J., Steel, C. M., Tedder, R. S., Cheingsong-Popov, R., Weiss, R. A., et al. (1985). Human T-lymphotropic virus type III (HTLV-III) infection in seronegative haemophiliacs after transfusion of factor VIII. Lancet 2, 233–236.
Mallal, S., Nolan, D., Witt, C., Masel, G., Martin, A. M., Moore, C., et al. (2002). Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 359, 727–732.
McLaren, P. J., Ripke, S., Pelak, K., Weintrob, A. C., Patsopoulos, N. A., Jia, X., et al. (2012a). Fine-mapping classical HLA variation associated with durable host control of HIV-1 infection in African Americans. Hum. Mol. Genet. 21, 4334–4347.
McLaren, P. J., Zagury, J. F., Fellay, J., and Consortium TIHA. (2012b). “Joining forces in HIV host genomics: the International HIV Acquisition Consortium (Poster 295),” in Conference on Retroviruses and Opportunistic Infections (CROI), Seattle, WA.
McLaren, P. J., Stahl, E. A., Rivas, M., Pereyra, F., Gupta, N., Walker, B., et al. (2011). “Deep sequencing and pooled association testing of 259 candidate genes reveals a role for rare variants associated with durable control of HIV-1 replication (Poster 393F),” in The American Society of Human Genetics (ASHG), Montreal.
Migueles, S. A., Sabbaghian, M. S., Shupert, W. L., Bettinotti, M. P., Marincola, F. M., Martino, L., et al. (2000). HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. U.S.A. 97, 2709–2714.
Miura, T., Brockman, M. A., Schneidewind, A., Lobritz, M., Pereyra, F., Rathod, A., et al. (2009). HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte [corrected] recognition. J. Virol. 83, 2743–2755.
Molina, L., Grimaldi, M., Robert-Hebmann, V., Espert, L., Varbanov, M., Devaux, C., et al. (2007). Proteomic analysis of the cellular responses induced in uninfected immune cells by cell-expressed X4 HIV-1 envelope. Proteomics 7, 3116–3130.
Pantaleo, G., and Fauci, A. S. (1996). Immunopathogenesis of HIV infection. Annu. Rev. Microbiol. 50, 825–854.
Paust, S., Gill, H. S., Wang, B. Z., Flynn, M. P., Moseman, E. A., Senman, B., et al. (2010). Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 11, 1127–1135.
Pelak, K., Goldstein, D. B., Walley, N. M., Fellay, J., Ge, D., Shianna, K. V., et al. (2010). Host determinants of HIV-1 control in African Americans. J. Infect. Dis. 201, 1141–1149.
Pelak, K., Shianna, K., Ge, D., and Goldstein, D. (2011). “Whole-genome sequencing to identify the genetic basis for resistance to HIV infection (Poster 410T),” in The American Society of Human Genetics (ASHG), Montreal.
Petrovski, S., Fellay, J., Shianna, K. V., Carpenetti, N., Kumwenda, J., Kamanga, G., et al. (2011). Common human genetic variants and HIV-1 susceptibility: a genome-wide survey in a homogeneous African population. AIDS 25, 513–518.
Ptak, R. G., Fu, W., Sanders-Beer, B. E., Dickerson, J. E., Pinney, J. W., Robertson, D. L., et al. (2008). Cataloguing the HIV type 1 human protein interaction network. AIDS Res. Hum. Retroviruses 24, 1497–1502.
Rauch, A., Kutalik, Z., Descombes, P., Cai, T., Di Iulio, J., Mueller, T., et al. (2010). Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology 138, 1338–45, 1345.e1–e7.
Reed, R., and Hurt, E. (2002). A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell 108, 523–531.
Rennert, P. D., Browning, J. L., Mebius, R., Mackay, F., and Hochman, P. S. (1996). Surface lymphotoxin alpha/beta complex is required for the development of peripheral lymphoid organs. J. Exp. Med. 184, 1999–2006.
Ringrose, J. H., Jeeninga, R. E., Berkhout, B., and Speijer, D. (2008). Proteomic studies reveal coordinated changes in T-cell expression patterns upon infection with human immunodeficiency virus type 1. J. Virol. 82, 4320–4330.
Rollason, R., Korolchuk, V., Hamilton, C., Jepson, M., and Banting, G. (2009). A CD317/tetherin-RICH2 complex plays a critical role in the organization of the subapical actin cytoskeleton in polarized epithelial cells. J. Cell Biol. 184, 721–736.
Rotger, M., Dang, K. K., Fellay, J., Heinzen, E. L., Feng, S., Descombes, P., et al. (2010). Genome-wide mRNA expression correlates of viral control in CD4+ T-cells from HIV-1-infected individuals. PLoS Pathog. 6:e1000781. doi:10.1371/journal.ppat.1000781
Rusinol, A. E., Thewke, D., Liu, J., Freeman, N., Panini, S. R., and Sinensky, M. S. (2004). AKT/protein kinase B regulation of BCL family members during oxysterol-induced apoptosis. J. Biol. Chem. 279, 1392–1399.
Saksena, N. K., Rodes, B., Wang, B., and Soriano, V. (2007). Elite HIV controllers: myth or reality? AIDS Rev. 9, 195–207.
Salgado, M., Lopez-Romero, P., Callejas, S., Lopez, M., Labarga, P., Dopazo, A., et al. (2011). Characterization of host genetic expression patterns in HIV-infected individuals with divergent disease progression. Virology 411, 103–112.
Samson, M., Libert, F., Doranz, B. J., Rucker, J., Liesnard, C., Farber, C. M., et al. (1996). Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382, 722–725.
Santa-Marta, M., Brito, P., Godinho-Santos, A., and Goncalves, J. (2013). Host proteins implicated in non-progression. Front. Immunol. (in press).
Shrestha, S., Irvin, M. R., Taylor, K. D., Wiener, H. W., Pajewski, N. M., Haritunians, T., et al. (2010). A genome-wide association study of carotid atherosclerosis in HIV-infected men. AIDS 24, 583–592.
Stiles, A. R., McDonald, J. G., Bauman, D. R., and Russell, D. W. (2009). CYP7B1: one cytochrome P450, two human genetic diseases, and multiple physiological functions. J. Biol. Chem. 284, 28485–28489.
Stranger, B. E., Forrest, M. S., Clark, A. G., Minichiello, M. J., Deutsch, S., Lyle, R., et al. (2005). Genome-wide associations of gene expression variation in humans. PLoS Genet. 1:e78. doi:10.1371/journal.pgen.0010078
Thomas, R., Apps, R., Qi, Y., Gao, X., Male, V., O’hUigin, C., et al. (2009). HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat. Genet. 41, 1290–1294.
Tokarev, A., Skasko, M., Fitzpatrick, K., and Guatelli, J. (2009). Antiviral activity of the interferon-induced cellular protein BST-2/tetherin. AIDS Res. Hum. Retroviruses 25, 1197–1210.
Troyer, J. L., Nelson, G. W., Lautenberger, J. A., Chinn, L., McIntosh, C., Johnson, R. C., et al. (2011). Genome-wide association study implicates PARD3B-based AIDS restriction. J. Infect. Dis. 203, 1491–1502.
van Manen, D., Delaneau, O., Kootstra, N. A., Boeser-Nunnink, B. D., Limou, S., Bol, S. M., et al. (2011). Genome-wide association scan in HIV-1-infected individuals identifying variants influencing disease course. PLoS ONE 6:e22208. doi:10.1371/journal.pone.0022208
van Manen, D., Kootstra, N. A., Boeser-Nunnink, B., Handulle, M. A., van’t Wout, A. B., and Schuitemaker, H. (2009). Association of HLA-C and HCP5 gene regions with the clinical course of HIV-1 infection. AIDS 23, 19–28.
Vigneault, F., Woods, M., Buzon, M. J., Li, C., Pereyra, F., Crosby, S. D., et al. (2011). Transcriptional profiling of CD4 T cells identifies distinct subgroups of HIV-1 elite controllers. J. Virol. 85, 3015–3019.
Wang, L., Zhu, J., Shan, S., Qin, Y., Kong, Y., Liu, J., et al. (2008). Repression of interferon-gamma expression in T cells by Prospero-related homeobox protein. Cell Res. 18, 911–920.
Yeung, M. L., Houzet, L., Yedavalli, V. S., and Jeang, K. T. (2009). A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 284, 19463–19473.
Yoon, W., Ma, B. J., Fellay, J., Huang, W., Xia, S. M., Zhang, R., et al. (2010). A polymorphism in the HCP5 gene associated with HLA-B*5701 does not restrict HIV-1 in vitro. AIDS 24, 155–157.
Keywords: genome-wide association study, SNP, HIV-1, viral control, long-term non-progression, chemokine receptors region, HLA
Citation: Limou S and Zagury J-F (2013) Immunogenetics: genome-wide association of non-progressive HIV and viral load control: HLA genes and beyond. Front. Immunol. 4:118. doi: 10.3389/fimmu.2013.00118
Received: 25 March 2013; Accepted: 04 May 2013;
Published online: 27 May 2013.
Edited by:
Nitin K. Saksena, Westmead Hospital, Australia; Westmead Millennium Institute, AustraliaReviewed by:
Aurelio Cafaro, Istituto Superiore di Sanità, ItalyNicolas Chomont, Vaccine and Gene Therapy Institute Florida, USA
Copyright: © 2013 Limou and Zagury. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Sophie Limou, Frederick National Laboratory, PO Box B, Building 560, Room 11-84, Frederick, MD 21702, USA. e-mail:c29waGllLmxpbW91QG5paC5nb3Y=;
Jean-François Zagury, Laboratoire Génomique, Bioinformatique et Applications, Equipe d’Accueil EA 4627, Conservatoire National des Arts et Métiers, 292 rue Saint Martin, 75003 Paris, France. e-mail:emFndXJ5QGNuYW0uZnI=