 Anna Mari Lone1,2,3
Anna Mari Lone1,2,3 Kjetil Taskén1,2,3,4*
Kjetil Taskén1,2,3,4*
- 1Centre for Molecular Medicine Norway, Nordic EMBL Partnership, University of Oslo and Oslo University Hospital, Oslo, Norway
- 2Biotechnology Centre, University of Oslo, Oslo, Norway
- 3K.G. Jebsen Inflammation Research Centre, University of Oslo, Oslo, Norway
- 4Department of Infectious Diseases, Oslo University Hospital, Oslo, Norway
Eicosanoids are inflammatory mediators primarily generated by hydrolysis of membrane phospholipids by phospholipase A2 to ω-3 and ω-6 C20 fatty acids that next are converted to leukotrienes (LTs), prostaglandins (PGs), prostacyclins (PCs), and thromboxanes (TXAs). The rate-limiting and tightly regulated lipoxygenases control synthesis of LTs while the equally well-controlled cyclooxygenases 1 and 2 generate prostanoids, including PGs, PCs, and TXAs. While many of the classical signs of inflammation such as redness, swelling, pain, and heat are caused by eicosanoid species with vasoactive, pyretic, and pain-inducing effects locally, some eicosanoids also regulate T cell functions. Here, we will review eicosanoid production in T cell subsets and the inflammatory and immunoregulatory functions of LTs, PGs, PCs, and TXAs in T cells.
Introduction
The eicosanoids constitute a large and expanding family of lipid signaling molecules derived from ω-3 and ω-6 C20 fatty acids (Smith, 1989; Funk, 2001). This conversion of membrane phospholipids into potent signaling mediators provides an efficient way for cells to respond to various stimuli that require a cellular response. As part of a complex network of regulators controlling a number of important physiological properties including smooth muscle tone, vascular permeability, and platelet aggregation, eicosanoids have also been implicated in a wide array of pathophysiological processes and diseases, including inflammation, autoimmunity, allergy, HIV, and cancer (Harizi et al., 2008; Greene et al., 2011; Bertin et al., 2012). While eicosanoids, in particular prostaglandins, were originally thought of primarily as proinflammatory mediators given their high expression in inflamed tissues and ability to induce inflammatory symptoms, this picture has over time become more nuanced. It is now recognized that these lipids can have both pro- and anti-inflammatory roles by regulating the immune response (Tilley et al., 2001).
While some eicosanoids are produced from eicosapentaenoic acid (EPA, 20:5 ω-3) (Wada et al., 2007) or dihomo-γ-linolenic acid (DGLA, 20:3 ω-6), the majority arise from processing of arachidonic acid (AA, 20:4 ω-6) (Harizi et al., 2008). AA-derived eicosanoids comprise the P-450 epoxygenase-generated hydroxyeicosatetraenoic acids (HETEs) and epoxides, the lipoxygenase (LOX) – generated hydroperoxyeicosatetraenoic acids (HPETEs), lipoxins (LXs), and leukotrienes (LTs), and the cyclooxygenase (COX)-produced prostanoids (see Figure 1 for overview of biosynthetic pathways). The prostanoids are perhaps the most well-known class of eicosanoids and include the prostaglandins (PGs) PGD2, PGE2, and PGF2α as well as prostacyclin (PC/PGI2) and thromboxane (TXA2). Together with the leukotrienes, the AA-derived prostanoids will be the major focus of this article.
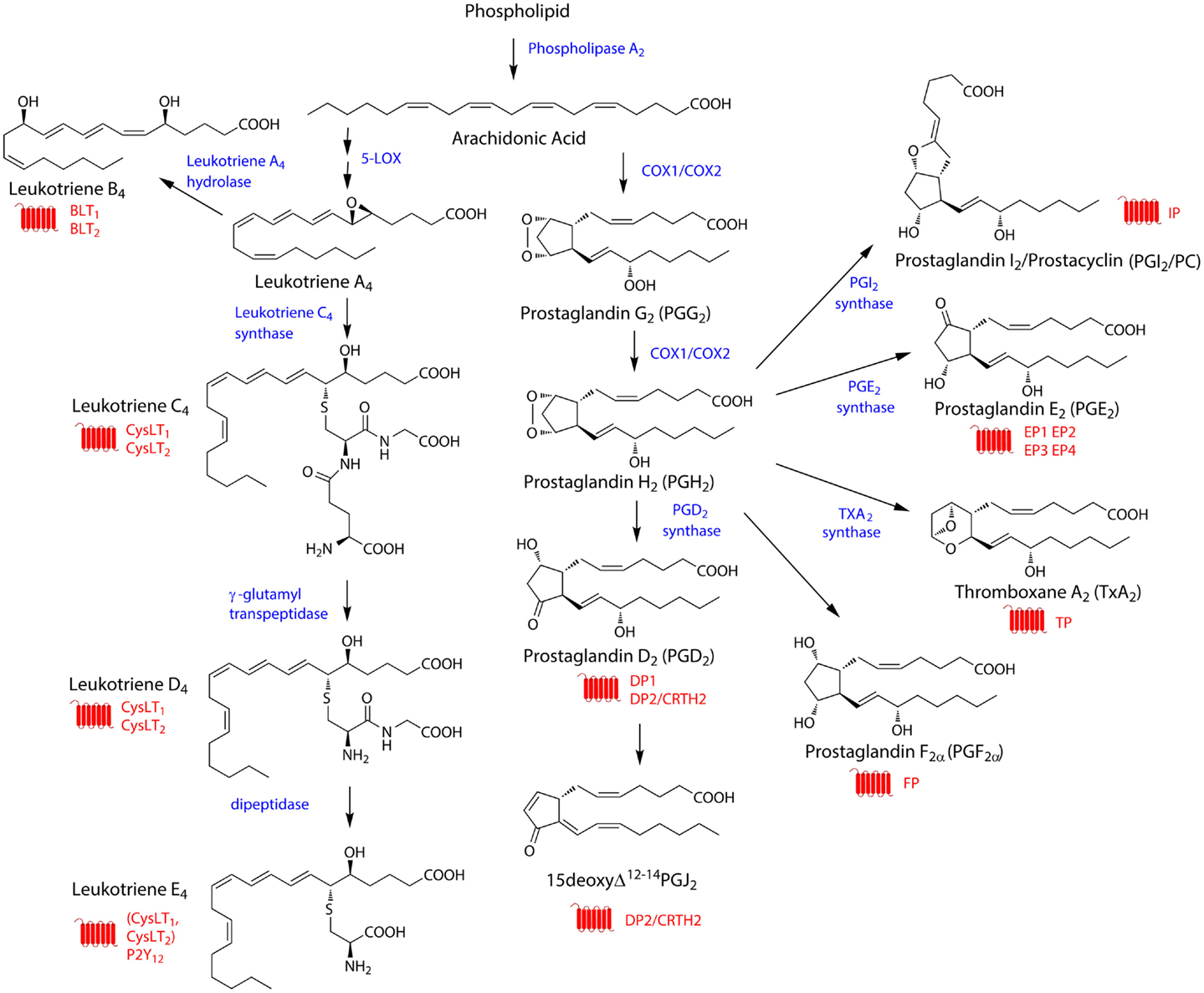
Figure 1. General overview of synthesis pathways for eicosanoids. The biosynthetic pathway for the arachidonic acid-derived eicosanoids described in this article. The figure shows the structures of the relevant eicosanoids (black), and indicates the enzymes involved in their biosynthesis (blue), as well as the GPCRs through which these eicosanoids signal (red). PGF2α can be synthesized through a number of different pathways.
Constitutive eicosanoid production is normally low, with the rate-limiting factor being the availability of free fatty acids, in particular AA. Free fatty acids are generated from membrane glycerophospholipids by phospholipase A2s (PLA2s) (Kudo and Murakami, 2002; Leslie, 2004; Burke and Dennis, 2009) in response to stimuli such as increased Ca2+ levels or phosphorylation (Kudo and Murakami, 2002). This elevation in intracellular free fatty acid levels, in particular that of arachidonic acid, then allows eicosanoid biosynthesis to proceed. In the case of prostanoid biosynthesis, AA is converted into PGG2 and then PGH2 through the actions of COX-1 and COX-2 (also known as PGH synthases 1 and 2). These enzymes act first as cyclooxygenases to create PGG2 and then as peroxidases to reduce the peroxide in PGG2 to an alcohol in PGH2 (Smith et al., 2000, 2011). Both PGG2 and PGH2 are thought to be transient intermediates and their production constitutes the committed step in prostanoid biosynthesis. PGH2 is then converted into one of four possible downstream signaling molecules (Figure 1). Prostacyclin synthase (PGIS) converts PGH2 to PGI2, hematopoietic (H-PGDS) or lipocalin-type (L-PGDS) PGD2 synthase convert PGH2 into PGD2, TXA2 synthase (TXAS) converts PGH2 into TXA2, and membrane-bound (mPGES-1 or -2) or cytosolic (cPGES) PGE2 synthases convert PGH2 into PGE2. PGF2α can be synthesized through a number of different pathways (Basu, 2010; Smith et al., 2011).
On the other hand, in leukotriene biosynthesis, AA is not processed by COX enzymes, but instead by 5-LOX, which with the help of 5-LOX -activating protein (FLAP) converts AA first into 5-HPETE and then into LTA4, an inactive intermediate and precursor for other leukotrienes. LTA4 can either be converted into LTB4 by LTA4 hydrolase or into LTC4 by LTC4 synthase, which conjugates a glutathione to LTA4 (Yokomizo, 2011). LTC4 can then be converted sequentially to LTD4 by gamma-glutamyl transpeptidase and LTE4 by dipeptidases (Brink et al., 2003).
By signaling through their receptors on the surface of T cells, eicosanoids have an important role in regulating many aspects of T lymphocyte function, usually through autocrine or paracrine signaling (Tilley et al., 2001; Sakata et al., 2010a). It has also recently become evident that T cells provide a source of these short-lived signaling mediators in compartments such as lymph nodes and spleen and in lymphoid infiltrates. In the present review, we will summarize the evidence for the production of and signaling by these molecules in T cells, especially in the context of the regulation of immunomodulatory or inflammatory functions.
Biosynthesis of and Signaling by Eicosanoids in T Lymphocytes
PGG2 and PGH2
Production of PGG2 and PGH2 proceeds through the actions of PLA2 and COX-1 or COX-2. While there has been some discussion about which PLA2 variant(s) are most relevant for eicosanoid biosynthesis, it is generally agreed that cytosolic PLA2α (cPLA2α) plays a major role in this process, with the Ca2+-independent PLA2 (iPLA2) more involved in membrane homeostasis and secreted PLA2 (sPLA2) regulating extracellular phospholipids (Murakami et al., 2011). Expression of cPLA2α in T cells has been controversial, with some groups finding no evidence for it in peripheral blood monocytes (Roshak et al., 2000) while others have observed mRNA but no effect of inhibiting this enzyme on AA production in Jurkat T cells (Tessier et al., 2002) and yet others observe this protein both in Jurkat T cells and peripheral blood lymphocytes (Burgermeister et al., 2003) (see Figure 1 for overview of biosynthetic enzymes and Table 1 for their expression in T cells).

Table 1. Eicosanoid synthesis in T cells.
Some groups have concluded that other PLA2 variants may also be active in T cells, with evidence for both iPLA2 (Roshak et al., 2000; Tessier et al., 2002) and sPLA2 (Tessier et al., 2002) being present and active in T lymphocytes. Interestingly, an sPLA2 isoform has also been shown to be expressed in and enhance the function of regulatory T cells (Treg), but this effect was found to be independent of the enzyme’s catalytic activity (von Allmen et al., 2009).
Alternatively, arachidonic acid can be released from membrane phospholipids by phospholipase D (PLD) (Liscovitch et al., 2000; Melendez and Allen, 2002), which has been shown to be inducible in human T cells (Bacon et al., 1995, 1998; Exton, 1999). Diacylglycerol (DAG) lipase has also been shown to play a role in the release of AA in lymphocytes (Cifone et al., 1995).
COX-1 and COX-2, which are capable of converting AA into PGH2, are both expressed in CD3+CD4+ cells and in Jurkat T cells. COX-1 is expressed constitutively in T cells and does not change in response to T cell activation (Pablos et al., 1999). In contrast, COX-2 is normally expressed at low levels but significantly upregulated in response to T cell activation (Iniguez et al., 1999; Pablos et al., 1999; Feldon et al., 2006). A study from this lab further demonstrated that during differentiation of adaptive Tregs, these cells also begin expressing COX-2 and producing PGE2 (Mahic et al., 2006).
In the context of a discussion of the cellular localizations of PLA2 and COX enzymes, it is worth noting that transcellular eicosanoid biosynthesis has recently been proposed as a mechanism whereby the entire biosynthetic pathway for a given eicosanoid need not be present in one particular cell. Instead, the synthesis may begin in one cell, followed by the transfer of a synthetic intermediate to a different cell where the final product is synthesized. PGH2, LTA4, and arachidonic acid have all been proposed as possible intermediates transported between cells, suggesting that in some cases, PLA2 (and COX enzymes) could be present in one cell and the remaining synthases required for prostanoid or leukotriene synthesis in another (Folco and Murphy, 2006; Sala et al., 2010). For PGH2, it has also been proposed that two distinct pathways for PGH2 synthesis exist: one for production of PGH2 to be converted into downstream prostanoids in the usual manner and one for production of untransformed PGH2 to be released for signaling functions.
PGG2 is a transient intermediate in prostanoid biosynthesis, with a half-life of about 5 min in aqueous solution at 37 °C, pH 7.4 and significantly shorter – on the order of seconds – in plasma (Corey et al., 1975). Although there have been some suggestions that PGG2 may have a biological function (Kuehl et al., 1977; Seidel et al., 2001) it is primarily considered an ephemeral intermediate without independent signaling functions. There is no evidence for a signaling role of this species in T cells.
In the case of PGH2, this is also an unstable endoperoxide species with comparable half-life to that of PGG2 (Corey et al., 1975). Although no specific receptor has been identified for this species either, there is some evidence that it can interact with other prostanoid receptors, including the DP and CRTH2 receptors (Schuligoi et al., 2009) as well as the TP receptor (Saito et al., 2003). Because of the rapid conversion of both PGG2 and PGH2 to other prostanoid species, however, it has been challenging to unequivocally prove that there is a direct action of these intermediate species on any of the prostanoid receptors. Several of the receptors thought to be activated by PGH2, in particular CRTH2, are known to be expressed on T cells, but so far it has not been demonstrated that PGH2 has a biologically relevant role in activating these receptors when expressed on T cells in vivo (See Figure 1 for overview of receptors and Table 2 for overview of expression of eicosanoid receptors in T cells).
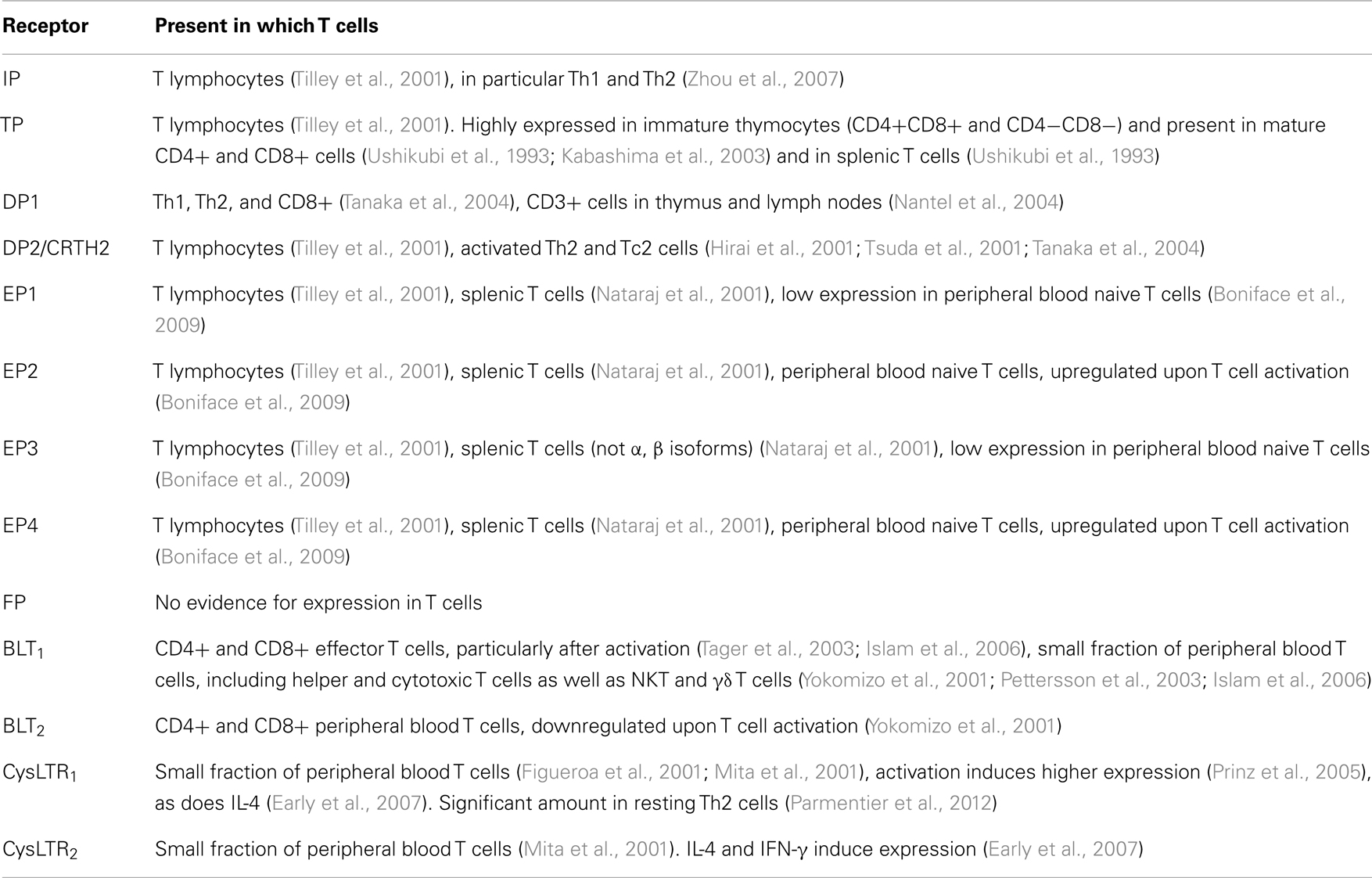
Table 2. Eicosanoid receptors in T cells.
PGI2/PC
Prostaglandin I2 was originally characterized as an inhibitor of platelet aggregation and a potent vasodilator (Boswell et al., 2011) and its analogs are used as treatments for pulmonary hypertension (Olschewski et al., 2004). Recently it has also been shown that this molecule has important roles in immune regulation (Boswell et al., 2011) and some studies suggest that treatment with PGI2 analogs may improve early graft viability in liver transplant patients, partly by reducing levels of inflammatory cytokines (Barthel et al., 2012).
While PGIS is expressed in some immune cells, in particular follicular dendritic cells (FDCs) (Lee et al., 2005; Boswell et al., 2011), there is no direct evidence for expression of this synthase in T cells. It has, however, been shown that lymphocytes are able to produce PGI2 through a transcellular mechanism when co-cultured with human vascular endothelial cells (HUVECs) (Merhi-Soussi et al., 2000) and that a similar mechanism appears to be operating between platelets and lymphocytes (Wu et al., 1987), although in neither of these cases were T cells specifically implicated.
The PGI2 receptor, IP, can be either Gs or Gq-coupled, leading to either increases in intracellular cyclic AMP (cAMP) levels through Gs-coupling, which can trigger cAMP-PKA signaling pathways or, through Gq-coupling, to the initiation of other signaling cascades (Woodward et al., 2011). IP is expressed on T cells, in particular cells of the Th1 and Th2 lineages (Zhou et al., 2007). Signaling through the IP receptor on these cells leads to inhibited cytokine secretion – in particular, IFNγ production in Th1 cells is abrogated and Th2 cells express less IL-4, IL-10, and IL-13 after IP stimulation. These results are mirrored by studies in IP knockout mice, where IL-4 and IFNγ production by splenocytes, which includes some T cells, was significantly higher in sensitized IP KO mice than in WT mice (Takahashi et al., 2002). With the exception of IL-10, where other studies have also shown upregulation in response to IP signaling (Jaffar et al., 2002), these downregulated cytokines are proinflammatory, and PGI2 is generally considered to be an anti-inflammatory and immune suppressive prostaglandin.
This inhibitory effect of IP signaling on cytokine production from Th1 and Th2 cells appears to be mediated by a cAMP-PKA pathway, since the PKA inhibitor Rp-8-Br-cAMPS significantly reduces the IP-stimulation induced effects on cytokine production. Further, it is accompanied by a reduction in nuclear-factor kappa-light-chain-enhancer of activated B cells (NF-κB), a transcription factor known to enhance expression of IFNγ and IL-4 (Zhou et al., 2007). While signaling through the IP receptor has a direct negative regulatory effect on Th1 and Th2 function, it appears to promote differentiation into the Th17 lineage (Boswell et al., 2011; Truchetet et al., 2012). This effect is partially due to a reduction in IL-12 expression and/or an increase in IL-23 from dendritic cells (DCs) or monocytes, thus perturbing the IL-23 to IL-12 ratio and favoring Th17 cell differentiation (Boswell et al., 2011; Truchetet et al., 2012; Zhou et al., 2012). It appears that this pathway is IP-specific and proceeds through a PKA pathway (Truchetet et al., 2012). In addition, the favoring of the Th17 lineage during T cell differentiation upon IP-stimulation appears also to be due to inhibited secretion of IL-4 (Zhou et al., 2012), a cytokine known to promote Th2 and antagonize Th17 development (Boswell et al., 2011).
In addition to its role in regulating T cell differentiation, PGI2 also has an important role in mediating FDC-T cell interactions in the germinal centers. FDC-produced PGI2 has been shown both to inhibit T cell proliferation and to protect T cells from TCR-mediated activation-induced death (AICD) (Lee et al., 2005, 2008), thus improving the current understanding of why T cells don’t proliferate or undergo AICD in germinal centers.
TXA2 and TXB2
Thromboxane A2 is a proinflammatory, short-lived (half-life ∼30 s (Remuzzi et al., 1994)) prostanoid primarily produced in platelets, but also in activated monocytes, macrophages, and DCs (Narumiya, 2003) through the actions of thromboxane A2 synthase. There is limited evidence for TXAS expression in T cells and this synthase was found to be absent in thymic lymphocytes (Ushikubi et al., 1993). However, TXA2’s stable and inactive downstream metabolite, TXB2, is produced by helper T cells in response to isoproterenol stimulation (Genaro et al., 1992) and another study found very low levels of TXB2 produced from CD4+ cells (Kabashima et al., 2002), suggesting that TXAS could be present at low levels in certain T cells.
By signaling through the TP receptor, which is coupled to Gq, TXA2 activates protein kinase C (PKC) and raises intracellular calcium levels (Narumiya, 2003; Woodward et al., 2011). TXA2 is best known for causing vasoconstriction and platelet aggregation and promotes fibrosis and scarring by regulating extracellular matrix protein levels (Thomas et al., 2003). The TP receptor is known to be highly expressed in immature thymocytes (CD4+CD8+ and CD4−CD8−) and to a lesser extent in CD4+CD8− and CD4−CD8+ thymocytes (Ushikubi et al., 1993; Kabashima et al., 2003). Splenic T cells also express lower amounts of the TP receptor. In line with this, immune regulatory functions for TXA2 have been proposed. Signaling through the TP receptor has been shown to cause apoptosis in immature thymocytes, in particular in CD4+CD8+ cells (Ushikubi et al., 1993), suggesting a potential role in T cell maturation. In other T cell populations, it has been suggested that signaling through the TP receptor could affect T cell proliferation (Kelly et al., 1979; Ceuppens et al., 1985), with a recent study showing that TXA2 signaling through the TP receptor inhibits anti-CD3 stimulated T cell proliferation (Thomas et al., 2003). Interestingly, proliferation in response to PMA and ionomycin, which produces a robust intracellular calcium response and bypasses the normal T cell activation mechanism, is not affected in TP-deficient cells (Thomas et al., 2003) or in cells treated with a TP agonist (Kabashima et al., 2003). These results indicate that TXA2 signaling through the TP receptor may play an important role in the initial activation of T cells by antigen-presenting cells (APCs), in particular DCs, but not in the later downstream intracellular signaling. TP signaling further attenuates DC-T cell interactions by promoting chemokinesis of naïve T cells and inhibiting DC-T cell adhesion, thus playing an important role in adaptive immunity (Kabashima et al., 2003).
Thromboxane A2 signaling has been implicated in anti-graft immune responses, with allografts eliciting higher levels of TXA2 than isografts (Gibbons et al., 1987). Mice deficient in the TP receptor have been shown to display weaker anti-allograft immune responses (Thomas et al., 2003) and blocking TXA2 synthesis pharmacologically has been shown to reduce alloreactive immune responses in vitro (Ruiz et al., 1992) and at least temporarily improve allograft survival and function in vivo by limiting cytotoxic T cell activity (Ruiz et al., 1989). On the other hand, in models of induced unresponsiveness to allografts by thymic injection of MHC allopeptides, TXA2 signaling abrogation through synthesis inhibition or receptor antagonists blocked the unresponsive state, suggesting that TXA2 signaling in the thymus is involved in mediating immune tolerance in this situation, possibly by leading to apoptosis of alloactivated T cells circulating through the thymus (Remuzzi et al., 1994). Together, these data suggests an important role for TXA2-TP signaling in T cells in the thymus, in particular in T cell maturation, activation by DCs and in anti-allograft immune responses.
PGD2 and 15-Deoxy-Δ12,14-PGJ2
PGD2 is produced by activated mast cells in response to allergen exposure and is thought to play an important role in mediating allergic inflammation by acting as a vasodilator, recruiter of eosinophils, basophils, and Th2 cells, modulator of Th2 production, and bronchoconstrictor (Pettipher et al., 2007). It also has important roles in regulating sleep, platelet aggregation, smooth muscle contraction, and reproduction (Saito et al., 2002; Woodward et al., 2011). Beyond mast cells, a few other cell types also produce PGD2 from PGH2 through one of the two types of PGD synthase, L-PGDS and H-PGDS (Joo and Sadikot, 2012). The former is not known to be expressed in T cells, while the latter is expressed in certain T cells under specific conditions. In particular, activated COX-2-expressing T cells have been shown to express H-PGDS and thereby produce PGD2 and likely the downstream PGD2 processing product 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2) (Feldon et al., 2006). It appears that H-PGDS is particularly prevalent in activated Th2 and Tc2 cells but not Th1 cells (Tanaka et al., 2000; Herlong and Scott, 2006). As for the synthesis of 15d-PGJ2, no specific synthase has been described and few details are known about the dehydration steps leading to its formation from PGD2 (Scher and Pillinger, 2005).
PGD2 can signal through either the DP1 or DP2/CRTH2 receptor, while 15d-PGJ2 signals through the DP2 receptor (Harris et al., 2002a; Schuligoi et al., 2010). The DP1 receptor is Gs-coupled and its activation leads to increases in intracellular cAMP and PKA activation and can also lead to increased intracellular calcium levels (Woodward et al., 2011). This receptor was shown to be expressed in certain malignant T cell lines (Harris and Phipps, 2002), but was not detected in normal peripheral blood T cells in this study (Harris and Phipps, 2002). However, other groups have found DP1 to be constitutively expressed in both Th1, Th2 and CD8+ cells (Tanaka et al., 2004) and to be present in CD3+ cells in the thymus and lymph nodes (Nantel et al., 2004). CRTH2 has little sequence homology with other prostanoid receptors, being more closely related to the N-formyl peptide receptor subfamily of receptors (Hirai et al., 2001). This receptor is Gi-coupled, leading to increases in intracellular calcium and inhibition of cAMP formation in response to signaling (Hirai et al., 2001). It is thought to be mainly expressed in activated Th2 and Tc2 cells (Tsuda et al., 2001; Tanaka et al., 2004) and has also been detected in a subset of infiltrating T cells in patients suffering from polyposis, a severe form of rhinosinusitis (Nantel et al., 2004). Interestingly, when heterologously expressed, DP and CRTH2 can form heterodimers, where DP enhances the signaling by the CRTH2 receptor. In these heterodimers, when DP signaling is pharmacologically blocked, CRTH2 function is also inhibited, but not vice versa (Sedej et al., 2012). In addition to signaling through the cell surface receptors DP1 and CRTH2, 15d-PGJ2 and PGD2 can also bind the nuclear hormone receptor transcription factor peroxisome proliferator-activated receptor gamma (PPAR-γ) (Forman et al., 1995; Kliewer et al., 1995; Harris et al., 2002a; Feldon et al., 2006). By activating PPAR-γ, these prostanoids induce differentiation of fibroblasts into fat cells, and it has been shown that this can be pathophysiologically relevant. For instance, in the case of Graves’ disease, activated T cells infiltrate the eye orbit and by producing PGD2 and 15d-PGJ2, cause the differentiation of fibroblasts in the eye orbit to adipocytes, leading to disfiguration and sometimes blindness (Feldon et al., 2006).
Both PGD2 and 15d-PGJ2 affect cytokine production from T cells. In particular, 15-dPGJ2 is often thought of as an anti-inflammatory prostaglandin, in part due to its enhancement of PPARγ’s anti-inflammatory effects (Harris et al., 2002a; Scher and Pillinger, 2005). However, 15-dPGJ2 can also induce secretion of IL-8, a cytokine with chemotactic and angiogenic effects, from activated T cells, suggesting a proinflammatory role of this prostaglandin as well (Harris et al., 2002b). This effect is not PPAR-γ-dependent, but instead proceeds through a mitogen-activated protein kinase (MAPK) and NF-κB pathway, possibly by first binding an extracellular receptor such as CRTH2.
PGD2 has a well-established role in regulating cytokine secretion from Th2 cells. In particular, PGD2 produced in mast cells stimulates IL-4, IL-5, and IL-13 secretion from Th2 cells and this process is believed to be important in the pathophysiology of allergic inflammations (Xue et al., 2009a). It has been demonstrated that phosphoinositide 3-kinase (PI3K) and Ca2+/calcineurin/nuclear factor of activated T cells (NFAT) signaling pathways downstream of CRTH2 are both important in regulating PGD2-induced cytokine production (Xue et al., 2007) and that LTE4 enhances the PGD2-CRTH2-mediated secretion of cytokines from Th2 cells (Xue et al., 2012). Another study confirmed the effect of PGD2 receptor signaling on cytokine secretion and further noted that while signaling through CRTH2 increases secretion of IL-2, IL-4, IL-5, and IL-13 as well as the proinflammatory proteins CD11b and CD40L in Th2 cells, signaling through DP1 reduces the number of CD4+ and CD8+ cells expressing IFNγ and IL-2 (Tanaka et al., 2004). By thus promoting Th2 function and suppressing Th1 functions, PGD2 signaling may have an overall effect of promoting Th2 function, which could be relevant in allergic responses, where Th2 activity is elevated. PGD2 can have further inhibitory effects on cytokine secretion, for instance in invariant natural killer T (iNKT) cells, where PGD2 signals through DP1 and PKA to inhibit the production of IFNγ, but not IL-4, the other major cytokine produced in this cell type (Torres et al., 2008). Thus, PGD2 signaling also contributes to regulating the innate immune system.
While signaling through PGD2 receptors apparently has a role in driving Th2-type processes as described above, 15-dPGJ2 may have a role in resolving certain Th1-driven responses by inhibiting the proinflammatory NF-κB pathway (Trivedi et al., 2006). Also, 15-dPGJ2 is able to inhibit IL-2 production in T cells by promoting an interaction between PPARγ and NFAT, a crucial transcription factor for IL-2 production, which prevents NFAT from binding to the IL-2 promoter (Yang et al., 2000).
Aside from pro- and anti-inflammatory effects of PGD2 and 15-dPGJ2 mediated by cytokine secretion, these prostaglandins also affect T cell function by regulating proliferation and apoptosis. Both PGD2 and 15-dPGJ2 are capable of inducing apoptosis in T cells through a PPARγ-dependent mechanism (Harris and Phipps, 2001, 2002; Harris et al., 2002b). It has also been reported that 15-dPGJ2 and to a lesser extent PGD2 can induce apoptosis in Jurkat T cells through a non-PPARγ dependent mechanism involving activation of the mitochondrial apoptosis pathway (Nencioni et al., 2003). In other situations, PGD2 can also have anti-apoptotic effects. For instance, in the case of apoptosis induced by cytokine deprivation, PGD2 signaling through the CRTH2 receptor inhibits apoptosis in Th2 cells, suggesting that this pathway may hinder resolution of allergic inflammation (Xue et al., 2009b). In T lymphocytes, 15-dPGJ2 can also inhibit proliferation by acting as a PPARγ ligand (Clark et al., 2000; Yang et al., 2000; Harris and Phipps, 2001; Nencioni et al., 2003). However, only TCR-mediated and not IL-2 induced proliferation is affected by 15-dPGJ2 treatment (Clark et al., 2000).
Signaling through the PGD2 receptors also plays an important role in the chemotaxis of T cells. When PGD2 acts on the CRTH2 receptor on Th2 cells, this induces chemotactic migration of the Th2 cells (Hirai et al., 2001), probably through a PI3K pathway (Xue et al., 2007), providing a possible mechanism for recruitment of Th2 cells to sites of allergic inflammation, for instance in asthma (Luster and Tager, 2004). It has been demonstrated that blocking the CRTH2 receptor pharmacologically inhibits the trafficking of lymphocytes, including T cells, to the inflamed airways in a model of chronic obstructive pulmonary disease (COPD), presenting a possible new strategy for treating this disease (Stebbins et al., 2010). Further chemoattractive effects of PGD2 on T cells is the CRTH2-mediated recruitment of Th2 and Tc2 cells to the materno-fetal interface, where they are thought to increase in early pregnancy (Saito et al., 2002) and PGD2’s ability to promote transendothelial migration of memory T cells across blood vascular endothelial cells and lymphatic vascular endothelial cells (Ahmed et al., 2011).
PGE2
PGE2 is the most abundant prostanoid found in the body and has important roles in reproduction, gastro-intestinal function, the immune system, cardiovascular function, and the central nervous system (Sreeramkumar et al., 2012). It is present in large amounts in many cancers, in particular colorectal and lung cancers, where it stimulates tumor growth by inhibiting apoptosis, inducing Tregs and promoting metastasis, cell invasion, and angiogenesis (Bergmann et al., 2007; Wang et al., 2007; Greenhough et al., 2009; Mandapathil et al., 2010; Brudvik et al., 2011; Nakanishi et al., 2011). PGE2 is produced from PGH2 through one of three different PGE2 synthases – cytosolic (cPGES) or microsomal (mPGES-1 and mPGES-2). While cPGES and mPGES-2 are constitutively expressed, mPGES-1 is inducible in response to mitogenic or proinflammatory stimuli and is often upregulated in concert with COX-2 (Scher and Pillinger, 2005). In terms of expression in T cells, little is known except that it has been demonstrated that adaptive Tregs express COX-2 and produce PGE2 upon differentiation (Mahic et al., 2006), implying that they must also express a PGES. This production of PGE2 from adaptive Tregs has implications both in cancer and chronic infectious diseases.
PGE2 can signal through any of its four receptors – EP1, EP2, EP3, EP4 – often with opposing effects (Breyer et al., 2001; Harris et al., 2002a; Woodward et al., 2011; Sreeramkumar et al., 2012). EP2 and EP4 receptors are Gs-coupled and lead to increased intracellular cAMP levels and PKA signaling. The EP1 receptor is Gq-coupled and results in increased intracellular calcium levels. In the case of the EP3 receptor, three main isoforms of this receptor exist – EP3 α, β, and γ – and they can signal through different G proteins, but it appears that the major pathway is through Gi, which leads to decreased intracellular cAMP levels. Messenger RNA for all the different PGE2 receptors, with the exception of the EP3 α and β isoforms, is present in murine splenic T cells (Nataraj et al., 2001). In naive T cells isolated from peripheral blood, EP2 and EP4 receptors appear to be the most abundant and are upregulated in response to activation (Boniface et al., 2009).
Through its receptors, PGE2 controls T cell function in a variety of ways and a number of recent reviews have addressed this topic (Harris et al., 2002a; Brudvik and Tasken, 2012; Sreeramkumar et al., 2012). First, it appears to differentially regulate apoptosis in T cells depending on the subpopulation and condition of the cells. In particular, CD4+CD8+ thymocytes undergo apoptosis when stimulated by PGE2 in vivo (Mastino et al., 1992), but may also be protected against activation-induced cell death by this prostanoid (Goetzl et al., 1995). Similarly, while apoptosis is stimulated in resting mature T cells (Pica et al., 1996), activation-induced cell death is inhibited (Porter and Malek, 1999; Pace et al., 2007). PGE2 also has other known negative regulatory functions in T cells. It is known to influence the function of CD8+ cells through the inhibitory complex CD94/NKG2A (Zeddou et al., 2005) and the cytotoxicity of gamma delta T cells through a cAMP-PKA pathway (Martinet et al., 2010). An anti-proliferative effect is also well documented. Through the EP2 (Nataraj et al., 2001) and possibly the EP4 (Kabashima et al., 2002) receptor, PGE2 can inhibit T cell proliferation in CD4+ and CD8+ cells (Goodwin et al., 1977; Hendricks et al., 2000). It has also been shown that PGE2 inhibits the proliferation of double-negative Tregs (Lee et al., 2009). It appears that proliferation is inhibited in these cells by a negative regulatory effect of increased intracellular cAMP levels resulting from EP2 or EP4 stimulation on IL-2 synthesis and IL-2 receptor expression, resulting in diminished IL-2-stimulated proliferation responses (Farrar et al., 1987; Mary et al., 1987; Rincon et al., 1988; Anastassiou et al., 1992). Other possible mechanisms of inhibition of proliferation include downregulation of the transferrin receptor (Chouaib et al., 1985), inhibiting intracellular Ca2+ increase and inositol phosphate production in response to T cell activation (Chouaib et al., 1987; Lerner et al., 1988; Choudhry et al., 1999) and preventing K+ movements which would dampen signaling via G proteins (Bastin et al., 1990).
Recent studies have provided additional information about the intracellular signaling pathways initiated by PGE2 through which T cell function and proliferation is affected. In particular, a combined phosphoflow/phosphoproteomics approach allowed for the collection of detailed information about phosphorylation cascades initiated in response to different amounts of PGE2 stimulation in different T cell populations (Oberprieler et al., 2010). Furthermore, a pathway was described in effector T cells where signaling through EP2 or EP4, with its concomitant increase in cAMP levels, leads to PKA activation and, through an EBP50-Ezrin-PAG scaffolded process, phosphorylation of the C-terminal Src kinase (Csk). Phosphorylated Csk in turn inhibits Lck-mediated phosphorylation of the TCR complex, thus inhibiting TCR signaling and T cell proliferation and function (Vang et al., 2001; Ruppelt et al., 2007; Mosenden and Tasken, 2011) (see Figure 2 for schematic depiction of the PGE2-cAMP-PKA-Csk inhibitory pathway in T cells). This pathway is of particular relevance during inflammatory responses or cancer, where production of PGE2 is increased. It has been shown that disrupting this pathway in cells by molecular or genetic means prevents PGE2 – mediated inhibition of effector T cell function (Carlson et al., 2006; Ruppelt et al., 2007; Stokka et al., 2010). In mice, disrupting this pathway by overexpressing a PKA anchoring disruptor also leads to an increase in effector T cell function, as evidenced by increased signaling, enhanced IL-2 secretion, and reduced sensitivity to PGE2-mediated inhibition of T cell function. These mice also have improved resistance to murine AIDS, an immunodeficiency disease induced by the murine leukemia virus where the PKA-Csk pathway is hyperactivated (Mosenden et al., 2011). In mice with murine AIDS, this pathway can also be targeted with COX-2 inhibitors (Rahmouni et al., 2004). Interestingly, the PKA-Csk pathway is upregulated in several immunodeficiency diseases, as well as cancer, suggesting that targeting this pathway may be of therapeutic interest (Rahmouni et al., 2004; Brudvik and Tasken, 2012; Brudvik et al., 2012). In particular, targeting this pathway with COX-2 inhibitors in patients with HIV infection appears to give significant patient benefit in clinical intervention trials as evident from regulation of surrogate parameters such as CD38 and immune function parameters such as lymphoproliferation and T cell-dependent vaccine responses (Johansson et al., 2004; Kvale et al., 2006; Pettersen et al., 2011).
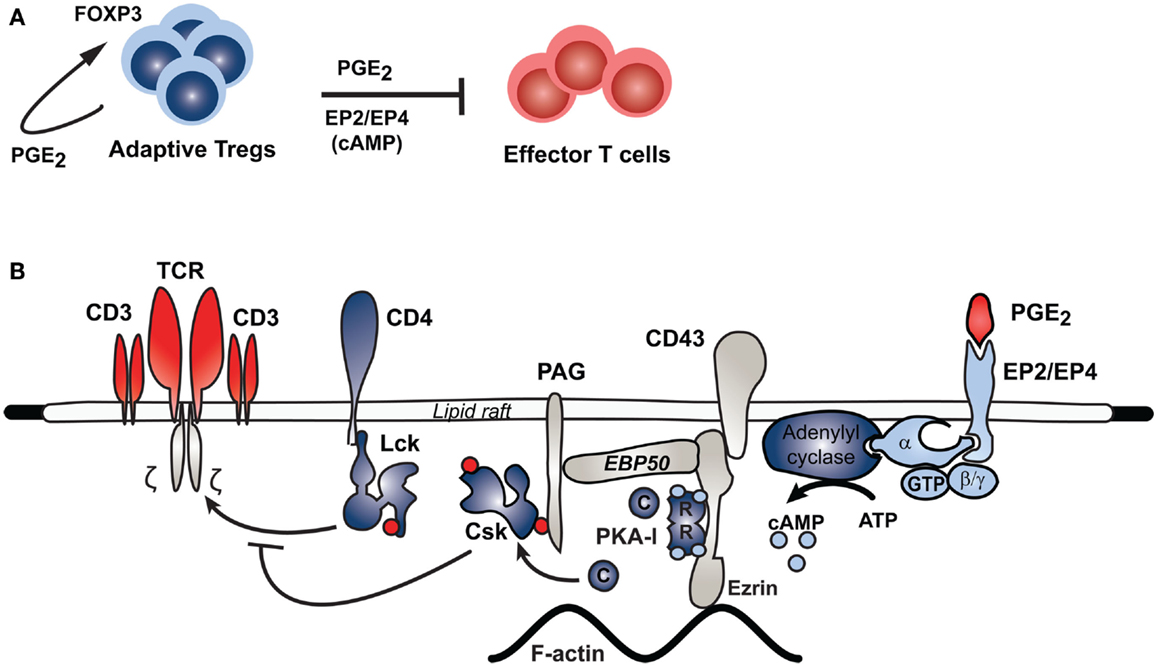
Figure 2. Inhibitory pathway of PGE2 in effector T cells. PGE2 mediates Treg inhibition of effector T cell function through a PKA-mediated pathway. (A) In response to continuous antigen exposure, for instance in cancer and HIV, adaptive regulatory T cells express COX-2 and produce PGE2, which stimulates FOXP3 expression in these cells. The Treg-derived PGE2 can signal through the EP2 and EP4 receptors on effector T cells to inhibit the function of these cells through the pathway shown in (B). Binding of PGE2 to its receptors on effector T cells stimulates adenylyl cyclase activity, which increases intracellular cAMP levels and thus activates PKA. Aided by an Ezrin-EBP50-PAG scaffold, PKA phosphorylates Csk, which in turn phosphorylates Lck to inhibit its activity. Lck normally acts to promote TCR signaling; thus Lck inhibition through this PGE2-initiated pathway inhibits TCR signaling in effector T cells.
As described above, PGE2 can influence the production and secretion of IL-2 from T cells, but it also influences the production of many other cytokines and contributes to T cell differentiation. In particular, it has been proposed that PGE2 signaling promotes a Th2 cell fate (Betz and Fox, 1991). In line with this, PGE2 has been shown to downregulate expression of IFNγ in T cells (Aandahl et al., 2002), indicating less differentiation to a Th1 cell type, with the caveat that recent studies have shown that in the presence of strengthened TCR stimulation, the Th1 cell fate can actually be promoted by PGE2 (Yao et al., 2009). In contrast, Th2-derived cytokines including IL-4, IL-5, IL-10, and IL-13 are unaffected or upregulated in response to PGE2 signaling (Betz and Fox, 1991; Snijdewint et al., 1993; Demeure et al., 1997). Furthermore, the IL-12 receptor is downregulated on T cells in response to PGE2, further promoting a Th2 cell fate (Wu et al., 1998). PGE2 has also been proposed to play a role in the differentiation of Th17 and Tregs. There is some debate about the role of PGE2 in the differentiation and expansion of Th17 cells (Sakata et al., 2010b), with some studies finding an inhibitory role in mouse Th17 differentiation (Chen et al., 2009) and others finding a promoting role in human Th17 differentiation (Boniface et al., 2009). There seems to be general agreement that Th17 IL-23-mediated expansion is enhanced by PGE2, however (Chizzolini et al., 2008; Boniface et al., 2009; Napolitani et al., 2009). In Treg differentiation the majority of reports seem to suggest an enhancing effect (Baratelli et al., 2005; Sharma et al., 2005; Mahic et al., 2006; Bryn et al., 2008), although some have found PGE2 to have an inhibitory effect on this process (Chen et al., 2009). Due to its role in promoting Treg differentiation and inhibiting effector T cell function and proliferation, PGE2 has traditionally been considered an immunosuppressant, but with recent studies showing a possible enhancing effect of this eicosanoid on Th17 and Th1 differentiation, some have argued that the picture is more nuanced (Sakata et al., 2010b; Sreeramkumar et al., 2012).
PGF2α
PGF2α has important functions in reproduction, inflammation, cardiovascular function, and other (patho)physiological processes (Simmons et al., 2004; Basu, 2007, 2010; Woodward et al., 2011). This prostaglandin can be synthesized through a number of different pathways (Basu, 2010), but there appears to be no evidence for any PGF2α synthesis in T cells. Evidence for a role of PGF2α signaling in T cells is also very limited, although a recent study demonstrated a role for this prostaglandin in promoting Th17 differentiation during allergic lung inflammation (Li et al., 2011). In this study, the authors propose that PGF2α together with PGI2 promotes differentiation of Th17 cells – proinflammatory cells and major contributors in allergic responses – from naïve CD4+ cells by signaling through their respective receptors in an autocrine fashion.
Leukotrienes LTA4, LTB4, LTC4, LTD4, LTE4
The first step in leukotriene biosynthesis, conversion of arachidonic acid to the unstable epoxide intermediate LTA4, is catalyzed by 5-LOX, an enzyme shown to occur in human T cell lines as well as in purified peripheral blood T cells (Cook-Moreau et al., 2007). 5-LOX expression is found across a wide range of T cells, including naive and memory helper and cytotoxic T cells as well as TCR–γδ cells (Cook-Moreau et al., 2007). However, some have noted that T lymphocytes require exogenous arachidonic acid in order to synthesize leukotrienes (Cook-Moreau et al., 2007). This is interesting in light of the proposed transcellular eicosanoid biosynthesis mechanism, and it has also been shown that LTA4 can act as the transferred intermediate metabolite in some systems (Folco and Murphy, 2006; Sala et al., 2010). LTB4 synthesis, which proceeds through LTA4 hydrolase, and LTC4 synthesis, which proceeds through LTC4 synthase, both occur in Jurkat T cells upon CD2, CD3, and CD28 crosslinking (Cook-Moreau et al., 2007). In primary T cells, synthesis of LTB4 and LTC4 was only found to occur if cells were stimulated by CD3 crosslinking and supplied with exogenous arachidonic acid (Cook-Moreau et al., 2007). Depending on the stimulation protocol, others have also detected the production of LTB4 or the cysteinyl leukotrienes (LTC4, LTD4, LTE4) in various T cell lines and primary cells (Cifone et al., 1995; Los et al., 1995). It should be noted that LTD4 and LTE4 are typically generated extracellularly after export of LTC4 from the producing cell.
LTB4 can signal through either of its two receptors, the high affinity BLT1 receptor and the low affinity BLT2 receptor (Yokomizo et al., 1997, 2000), which couple to Gi or Gq to exert their function (Back et al., 2011). BLT1 is expressed on CD4+ and CD8+ effector T cells, particularly shortly after activation (Tager et al., 2003; Islam et al., 2006). In peripheral blood T cells in healthy humans, BLT1 is found on a small fraction of the population, including both helper and cytotoxic T cells as well as NKT and γδ T cells (Yokomizo et al., 2001; Pettersson et al., 2003; Islam et al., 2006), and can expand in response to acute inflammation. The BLT2 receptor is more ubiquitously expressed across tissues, with very high expression levels in the spleen (Yokomizo et al., 2000). One study found no evidence for BLT2 expression in naive CD4+ cells or Th0, Th1, or Th2 cells 7 days after activation (Tager et al., 2003), while others have shown it to be present on both CD4+ and CD8+ peripheral blood T cells, but downregulated in response to T cell activation (Yokomizo et al., 2001).
In T cells, LTB4 is primarily known for its role in chemotaxis, but it has also been shown to have other functions, for instance in differentiation and proliferation. In chemotaxis, LTB4 signals through the BLT1 receptor on CD4+ or CD8+ cells to mediate cell movement, which is of particular relevance during T cell recruitment to airways and lungs in asthma (Tager et al., 2003; Luster and Tager, 2004; Gelfand and Dakhanna, 2006), after lung transplants (Medoff et al., 2005) and in various inflammatory settings (Goodarzi et al., 2003; Ott et al., 2003). In addition, signaling through BLT1 appears to enable adhesion of T cells to epithelial cells (Tager et al., 2003), which is important for migration into tissues. In T cell differentiation, LTB4 has been shown to promote Th17 and inhibit Treg generation, which may be of relevance in autoimmune diseases such as rheumatoid arthritis (Chen et al., 2009). However, it should be noted that early reports from 1985 had suggested that LTB4 may have an immunoregulatory role by inducing so-called suppressor T cells (Yamaoka and Kolb, 1993; Morita et al., 1999) but this has not been revisited since the definition of Treg. Proliferation and cytokine production in T cells can also be affected by LTB4. In particular, treatment with a BLT1 antagonist was shown to inhibit cytokine (IL-2, IFN-γ, IL-4) secretion and proliferation of T cells in response to activation (Rolapleszczynski, 1985), while LTB4 stimulation enhanced IL-5 production (Gualde et al., 1985), suggesting that LTB4 promotes T cell activation.
For the cysteinyl leukotrienes LTC4, LTD4, and LTE4, two receptors have been discovered, CysLT1 and CysLT2 (Brink et al., 2003; Kanaoka and Boyce, 2004; Singh et al., 2010; Back et al., 2011; Laidlaw and Boyce, 2012). These receptors can bind all the cysteinyl leukotrienes, albeit with significantly higher affinity for LTC4 and LTD4 than LTE4 (Laidlaw and Boyce, 2012). Recently, further receptors involved in cysteinyl leukotriene signaling have been identified, in particular GPR17, which is a ligand-independent negative regulator of CysLT1, as well as the LTE4-specific P2Y12 (Austen et al., 2009; Maekawa et al., 2009; Paruchuri et al., 2009; Laidlaw and Boyce, 2012). While it is unclear whether these latter two receptors are expressed in T cells, the CysLT1 and CysLT2 receptors have been shown to be expressed in a small fraction of peripheral blood T cells (Figueroa et al., 2001; Mita et al., 2001). Activation of the T cells induces higher expression of the CysLT1 receptor (Prinz et al., 2005), as does certain mutations in the linker for activation of T cells (LAT) (Prinz et al., 2005). It also appears that expression of this receptor is significantly higher in resting Th2 cells than in Th1 cells or activated Th2 cells (Parmentier et al., 2012). Interestingly, both receptors can also be upregulated in response to inflammatory stimuli. In particular, IL-4 induces expression of both receptors on T cells, while IFN-γ specifically upregulates expression of the CysLT2 receptor (Early et al., 2007). Presumably, this upregulation has the effect of making T cells more responsive to cysteinyl leukotriene signaling in inflammatory environments.
There is some functional evidence for a role of cysteinyl leukotriene signaling in T cells. For one, these molecules appear to be important in Th2 cells where, as mentioned above, the CysLT1 receptor is present in significant amounts. It has been demonstrated that LTE4, through a montelukast-sensitive pathway, indicating CysLT1 involvement, enhances PGD2-mediated cytokine secretion in isolated Th2 cells (Xue et al., 2012). In line with this, another CysLT1 antagonist, pranlukast, inhibits production of Th2 cytokines, in particular IL-3, IL-4, GM-CSF and possibly IL-5, from peripheral blood mononuclear cells of patients with bronchial asthma (Tohda et al., 1999). Further roles in Th2 cells include a demonstrated effect of LTD4 on the induction of calcium signaling as well as chemotaxis in these cells, both processes being CysLT1-specific (Parmentier et al., 2012). Cysteinyl leukotriene signaling in Th2 cells may also be involved in disease. For instance, it has been suggested that cysteinyl leukotrienes may enhance GM-CSF stimulated Th2 functions in atopic asthmatic patients in vivo (Faith et al., 2008). There has also been a suggested role for the cysteinyl leukotrienes in T cell-mediated late airway responses to allergen challenge, since treatment with the CysLT1 antagonist pranlukast inhibits these responses (Hojo et al., 2000).
Conclusion
Eicosanoids are an important class of lipid signaling mediators and have long been studied for their proinflammatory functions. In recent years, however, it has become evident that these molecules not only promote inflammation, but can occasionally also act as anti-inflammatory agents and have more complex and nuanced roles in the regulation of immune and inflammatory responses. Here, we have summarized the evidence for the expression of and signaling by some important eicosanoids, the AA-derived prostanoids and the leukotrienes, in T lymphocytes. These lipid mediators regulate a number of functions in T cells, including proliferation, apoptosis, cytokine secretion, differentiation, chemotaxis, and more. Through these processes, eicosanoids regulate a wide array of physiological processes, ranging from inflammatory processes such as asthma and allergies, to immune regulation and involvement in graft rejection, as well as diseases such as cancer and AIDS. There is significant interest in targeting some of these pathways for therapeutic gain and it is therefore crucial to develop a complete understanding of all the different physiological functions of these important signaling mediators.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aandahl, E. M., Moretto, W. J., Haslett, P. A., Vang, T., Bryn, T., Tasken, K., et al. (2002). Inhibition of antigen-specific T cell proliferation and cytokine production by protein kinase a type I. J. Immunol. 169, 802–808.
Ahmed, S. R., McGettrick, H. M., Yates, C. M., Buckley, C. D., Ratcliffe, M. J., Nash, G. B., et al. (2011). Prostaglandin D-2 regulates CD4(+) memory T cell trafficking across blood vascular endothelium and primes these cells for clearance across lymphatic endothelium. J. Immunol. 187, 1432–1439. doi:10.4049/jimmunol.1100299
Anastassiou, E. D., Paliogianni, F., Balow, J. P., Yamada, H., and Boumpas, D. T. (1992). Prostaglandin-E2 and other cyclic AMP-elevating agents modulate IL-2 and IL-2R-alpha gene-expression at multiple levels. J. Immunol. 148, 2845–2852.
Austen, K. F., Maekawa, A., Kanaoka, Y., and Boyce, J. A. (2009). The leukotriene E-4 puzzle: finding the missing pieces and revealing the pathobiologic implications. J. Allergy Clin. Immunol. 124, 406–416. doi:10.1016/j.jaci.2009.05.046
Back, M., Dahlen, S. E., Drazen, J. M., Evans, J. F., Serhan, C. N., Shimizu, T., et al. (2011). International Union of Basic and Clinical Pharmacology. LXXXIV: leukotriene receptor nomenclature, distribution, and pathophysiological functions. Pharmacol. Rev. 63, 539–584. doi:10.1124/pr.110.004184
Bacon, K. B., Floresromo, L., Life, P. F., Taub, D. D., Premack, B. A., Arkinstall, S. J., et al. (1995). IL-8-induced signal transduction in T-lymphocytes involves receptor-mediated activation of phospholipase-C and phospholipase-D. J. Immunol. 154, 3654–3666.
Bacon, K. B., Schall, T. J., and Dairaghi, D. J. (1998). RANTES activation of phospholipase D in Jurkat T cells: requirement of GTP-binding proteins ARF and RhoA. J. Immunol. 160, 1894–1900.
Baratelli, F., Lin, Y., Zhu, L., Yang, S. C., Heuze-Vourch, N., Zeng, G., et al. (2005). Prostaglandin E-2 induces FOXP3 gene expression and T regulatory cell function in human CD4(+) T cells. J. Immunol. 175, 1483–1490.
Barthel, E., Rauchfuss, F., Hoyer, H., Habrecht, O., Jandt, K., Gotz, M., et al. (2012). Impact of stable PGI(2) analog iloprost on early graft viability after liver transplantation: a pilot study. Clin. Transplant. 26, E38–E47. doi:10.1111/j.1399-0012.2011.01516.x
Bastin, B., Payet, M. D., and Dupuis, G. (1990). Effects of modulators of adenylyl cyclase on interleukin-2 production, cytosolic Ca-2+ elevation, and K+ channel activity in Jurkat T-cells. Cell. Immunol. 128, 385–399. doi:10.1016/0008-8749(90)90035-P
Basu, S. (2007). Novel cyclooxygenase-catalyzed bioactive prostaglandin F-2 alpha from physiology to new principles in inflammation. Med. Res. Rev. 27, 435–468. doi:10.1002/med.20098
Basu, S. (2010). Bioactive eicosanoids: role of prostaglandin F-2 alpha and F-2-isoprostanes in inflammation and oxidative stress related pathology. Mol. Cells 30, 383–391. doi:10.1007/s10059-010-0157-1
Bergmann, C., Strauss, L., Zeidler, R., Lang, S., and Whiteside, T. L. (2007). Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res. 67, 8865–8873. doi:10.1158/0008-5472.CAN-07-0767
Bertin, J., Barat, C., Methot, S., and Tremblay, M. J. (2012). Interactions between prostaglandins, leukotrienes and HIV-1: possible implications for the central nervous system. Retrovirology 9, doi:10.1186/1742-4690-9-4
Betz, M., and Fox, B. S. (1991). Prostaglandin-E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 146, 108–113.
Boniface, K., Bak-Jensen, K. S., Li, Y., Blumenschein, W. M., McGeachy, M. J., McClanahan, T. K., et al. (2009). Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J. Exp. Med. 206, 535–548. doi:10.1084/jem.20082293
Boswell, M. G., Zhou, W. S., Newcomb, D. C., and Peebles, R. S. (2011). PGI(2) as a regulator of CD4+subset differentiation and function. Prostaglandins Other Lipid Mediat. 96, 21–26. doi:10.1016/j.prostaglandins.2011.08.003
Breyer, R. M., Bagdassarian, C. K., Myers, S. A., and Breyer, M. D. (2001). Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 41, 661–690. doi:10.1146/annurev.pharmtox.41.1.661
Brink, C., Dahlen, S. E., Drazen, J., Evans, J. F., Hay, D. W. P., Nicosia, S., et al. (2003). International Union of Pharmacology – XXXVII. Nomenclature for leukotriene and lipoxin receptors. Pharmacol. Rev. 55, 195–227. doi:10.1124/pr.55.1.8
Brudvik, K. W., Henjum, K., Aandahl, E. M., Bjornbeth, B. A., and Tasken, K. (2012). Regulatory T-cell-mediated inhibition of antitumor immune responses is associated with clinical outcome in patients with liver metastasis from colorectal cancer. Cancer Immunol. Immunother. 61, 1045–1053. doi:10.1007/s00262-011-1174-4
Brudvik, K. W., Paulsen, J. E., Aandahl, E. M., Roald, B., and Tasken, K. (2011). Protein kinase A antagonist inhibits beta-catenin nuclear translocation, c-Myc and COX-2 expression and tumor promotion in Apc(Min/+) mice. Mol. Cancer 10, doi:10.1186/1476-4598-10-149
Brudvik, K. W., and Tasken, K. (2012). Modulation of T cell immune functions by the prostaglandin E2-cAMP pathway in chronic inflammatory states. Br. J. Pharmacol. 166, 411–419. doi:10.1111/j.1476-5381.2011.01800.x
Bryn, T., Yaqub, S., Mahic, M., Henjum, K., Aandahl, E. M., and Tasken, K. (2008). LPS-activated monocytes suppress T-cell immune responses and induce FOXP3+T cells through a COX-2-PGE(2)-dependent mechanism. Int. Immunol. 20, 235–245. doi:10.1093/intimm/dxm134
Burgermeister, E., Endl, J., and Scheuer, W. V. (2003). Activation of cytosolic phospholipase A(2) in human T-lymphocytes involves inhibitor-kappa B and mitogen-activated protein kinases. Eur. J. Pharmacol. 466, 169–180. doi:10.1016/S0014-2999(03)01492-4
Burke, J. E., and Dennis, E. A. (2009). Phospholipase A(2) biochemistry. Cardiovasc. Drugs Ther. 23, 49–59. doi:10.1007/s10557-008-6132-9
Carlson, C. R., Lygren, B., Berge, T., Hoshi, N., Wong, W., Tasken, K., et al. (2006). Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. J. Biol. Chem. 281, 21535–21545.
Ceuppens, J. L., Vertessen, S., Deckmyn, H., and Vermylen, J. (1985). Effects of thromboxane-A2 on lymphocyte proliferation. Cell. Immunol. 90, 458–463. doi:10.1016/0008-8749(85)90210-2
Chen, H. Y., Qin, J., Wei, P., Zhang, J. G., Li, Q. X., Fu, L. H., et al. (2009). Effects of leukotriene B4 and prostaglandin E2 on the differentiation of murine Foxp3+T regulatory cells and Th17 cells. Prostaglandins Leukot. Essent. Fatty Acids 80, 195–200. doi:10.1016/j.plefa.2009.01.006
Chizzolini, C., Chicheportiche, R., Alvarez, M., de Rham, C., Roux-Lombard, P., Ferrari-Lacraz, S., et al. (2008). Prostaglandin E(2) synergistically with interleukin-23 favors human Th17 expansion. Blood 112, 3696–3703. doi:10.1182/blood-2008-05-155408
Chouaib, S., Robb, R. J., Welte, K., and Dupont, B. (1987). Analysis of prostaglandin-E2 effect on lymphocyte-T activation – abrogation of prostaglandin-E2 inhibitory effect by the tumor promotor 12.0 tetradecanoyl phorbol-13 acetate. J. Clin. Invest. 80, 333–340. doi:10.1172/JCI113077
Chouaib, S., Welte, K., Mertelsmann, R., and Dupont, B. (1985). Prostaglandin-E2 acts at 2 distinct pathways of lymphocyte-T activation – inhibition of interleukin-2 production and down-regulation of transferrin receptor expression. J. Immunol. 135, 1172–1179.
Choudhry, M. A., Hockberger, P. E., and Sayeed, M. M. (1999). PGE(2) suppresses mitogen-induced Ca2+ mobilization in T cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 277, R1741–R1748.
Cifone, M. G., Cironi, L., Santoni, A., and Testi, R. (1995). Diacylglycerol lipase activation and 5-lipoxygenase activation and translocation following TCR/CD3 triggering in T-cells. Eur. J. Immunol. 25, 1080–1086. doi:10.1002/eji.1830250433
Clark, R. B., Bishop-Bailey, D., Estrada-Hernandez, T., Hla, T., Puddington, L., and Padula, S. J. (2000). The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J. Immunol. 164, 1364–1371.
Cook-Moreau, J. M., Hojeij, Y. E. M., Barriere, G., Rabinovitch-Chable, H. C., Faucher, K. S., Sturtz, F. G., et al. (2007). Expression of 5-lipoxygenase (5-LOX) in T lymphocytes. Immunology 122, 157–166. doi:10.1111/j.1365-2567.2007.02621.x
Corey, E. J., Nicolaou, K. C., Machida, Y., Malmsten, C. L., and Samuelsson, B. (1975). Synthesis and biological properties of a 9,11-azo-prostanoid - highly active biochemical mimic of prostaglandin endoperoxides. Proc. Natl. Acad. Sci. U.S.A. 72, 3355–3358. doi:10.1073/pnas.72.9.3355
Demeure, C. E., Yang, L. P., Desjardins, C., Raynauld, P., and Delespesse, G. (1997). Prostaglandin E-2 primes naive T cells for the production of anti-inflammatory cytokines. Eur. J. Immunol. 27, 3526–3531. doi:10.1002/eji.1830271254
Early, S. B., Barekzi, E., Negri, J., Hise, K., Borish, L., and Steinke, J. W. (2007). Concordant modulation of cysteinyl leukotriene receptor expression by IL-4 and IFN-gamma on peripheral immune cells. Am. J. Respir. Cell Mol. Biol. 36, 715–720. doi:10.1165/rcmb.2006-0252OC
Exton, J. H. (1999). Regulation of phospholipase D. Biochim. Biophys. Acta 1439, 121–133. doi:10.1016/S1388-1981(99)00089-X
Faith, A., Fernandez, M. H., Caulfield, J., Loke, T. K., Corrigan, C., O’Connor, B., et al. (2008). Role of cysteinyl leukotrienes in human allergen-specific Th2 responses induced by granulocyte macrophage-colony stimulating factor. Allergy 63, 168–175. doi:10.1111/j.1398-9995.2007.01531.x
Farrar, W. L., Evans, S. W., Rapp, U. R., and Cleveland, J. L. (1987). Effects of antiproliferative cyclic-AMP on interleukin-2-stimulated gene-expression. J. Immunol. 139, 2075–2080.
Feldon, S. E., O’Loughlin, C. W., Ray, D. M., Landskroner-Eiger, S., Seweryniak, K. E., and Phipps, R. P. (2006). Activated human T lymphocytes express cyclooxygenase-2 and produce proadipogenic prostaglandins that drive human orbital fibroblast differentiation to adipocytes. Am. J. Pathol. 169, 1183–1193. doi:10.2353/ajpath.2006.060434
Figueroa, D. J., Breyer, R. M., Defoe, S. K., Kargman, S., Daugherty, B. L., Waldburger, K., et al. (2001). Expression of the cysteinyl leukotriene 1 receptor in normal human lung and peripheral blood leukocytes. Am. J. Respir. Crit. Care Med. 163, 226–233. doi:10.1164/ajrccm.163.1.2003101
Folco, G., and Murphy, R. C. (2006). Eicosanoid transcellular biosynthesis: from cell-cell interactions to in vivo tissue responses. Pharmacol. Rev. 58, 375–388. doi:10.1124/pr.58.3.8
Forman, B. M., Tontonoz, P., Chen, J., Brun, R. P., Spiegelman, B. M., and Evans, R. M. (1995). 15-deoxy-delta(12,14)-prostaglandin J(2) is a ligand for the adipocyte determination factor PPAR-gamma. Cell 83, 803–812.
Funk, C. D. (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875. doi:10.1126/science.294.5548.1871
Gelfand, E. W., and Dakhanna, A. (2006). CD8(+) T lymphocytes and leukotriene B-4: novel interactions in the persistence and progression of asthma. J. Allergy Clin. Immunol. 117, 577–582. doi:10.1016/j.jaci.2005.12.1340
Genaro, A. M., Sterinborda, L., Gorelik, G., and Borda, E. (1992). Prostanoid synthesis in lymphocyte subpopulations by adrenergic and cholinergic receptor stimulation. Int. J. Immunopharmacol. 14, 1145–1151. doi:10.1016/0192-0561(92)90049-Q
Gibbons, C. P., Wiley, K. N., Lindsey, N. J., Fox, M., Beck, S., Slater, D. N., et al. (1987). Cortical and vascular prostaglandin synthesis during renal-allograft rejection in the rat. Transplantation 43, 472–478. doi:10.1097/00007890-198704000-00003
Goetzl, E. J., An, S. Z., and Zeng, L. (1995). Specific suppression by prostaglandin-E(2) of activation-induced apoptosis of human CD4(+)CD8(+) T-lymphoblasts. J. Immunol. 154, 1041–1047.
Goodarzi, K., Goodarzi, M., Tager, A. M., Luster, A. D., and von Andrian, U. H. (2003). Leukotriene B-4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat. Immunol. 4, 965–973. doi:10.1038/ni972
Goodwin, J. S., Bankhurst, A. D., and Messner, R. P. (1977). Suppression of human T-cell mitogenesis by prostaglandin – existence of a prostaglandin-producing suppressor cell. J. Exp. Med. 146, 1719–1734. doi:10.1084/jem.146.6.1719
Greene, E. R., Huang, S., Serhan, C. N., and Panigrahy, D. (2011). Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 96, 27–36. doi:10.1016/j.prostaglandins.2011.08.004
Greenhough, A., Smartt, H. J. M., Moore, A. E., Roberts, H. R., Williams, A. C., Paraskeva, C., et al. (2009). The COX-2/PGE(2) pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 30, 377–386. doi:10.1093/carcin/bgp014
Gualde, N., Atluru, D., and Goodwin, J. S. (1985). Effects of lipoxygenase metabolites of arachidonic-acid on proliferation of human T-cells and T-cell subsets. J. Immunol. 134, 1125–1129.
Harizi, H., Corcuff, J. B., and Gualde, N. (2008). Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol. Med. 14, 461–469. doi:10.1016/j.molmed.2008.08.005
Harris, S. G., Padilla, J., Koumas, L., Ray, D., and Phipps, R. P. (2002a). Prostaglandins as modulators of immunity. Trends Immunol. 23, 144–150. doi:10.1016/S1471-4906(01)02154-8
Harris, S. G., Smith, R. S., and Phipps, R. P. (2002b). 15-deoxy-Delta(12,14)-PGJ(2) induces IL-8 production in human T cells by a mitogen-activated protein kinase pathway. J. Immunol. 168, 1372–1379.
Harris, S. G., and Phipps, R. P. (2001). The nuclear receptor PPAR gamma is expressed by mouse T lymphocytes and PPAR gamma agonists induce apoptosis. Eur. J. Immunol. 31, 1098–1105. doi:10.1002/1521-4141(200104)31:4<1098::AID-IMMU1098>3.0.CO;2-I
Harris, S. G., and Phipps, R. P. (2002). Prostaglandin D-2, its metabolite 15-d-PGJ(2), and peroxisome proliferator activated receptor-gamma agonists induce apoptosis in transformed, but not normal, human T lineage cells. Immunology 105, 23–34. doi:10.1046/j.0019-2805.2001.01340.x
Hendricks, A., Leibold, W., Kaever, V., and Schuberth, H. J. (2000). Prostaglandin E-2 is variably induced by bacterial super-antigens in bovine mononuclear cells and has a regulatory role for the T cell proliferative response. Immunobiology 201, 493–505. doi:10.1016/S0171-2985(00)80069-8
Herlong, J. L., and Scott, T. R. (2006). Positioning prostanoids of the D and J series in the immunopathogenic scheme. Immunol. Lett. 102, 121–131. doi:10.1016/j.imlet.2005.10.004
Hirai, H., Tanaka, K., Yoshie, O., Ogawa, K., Kenmotsu, K., Takamori, Y., et al. (2001). Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J. Exp. Med. 193, 255–261. doi:10.1084/jem.193.2.255
Hojo, M., Suzuki, M., Maghni, K., Hamid, Q., Powell, W. S., and Martin, J. G. (2000). Role of cysteinyl leukotrienes in CD4+ T cell-driven late allergic airway responses. J. Pharmacol. Exp. Ther. 293, 410–416.
Iniguez, M. A., Punzon, C., and Fresno, M. (1999). Induction of cyclooxygenase-2 on activated T lymphocytes: regulation of T cell activation by cyclooxygenase-2 inhibitors. J. Immunol. 163, 111–119.
Islam, S. A., Thomas, S. Y., Hess, C., Medoff, B. D., Means, T. K., Brander, C., et al. (2006). The leukotriene B-4 lipid chemoattractant receptor BLT1 defines antigen-primed T cells in humans. Blood 107, 444–453. doi:10.1182/blood-2005-06-2362
Jaffar, Z., Wan, K. S., and Roberts, K. (2002). A key role for prostaglandin I-2 in limiting lung mucosal Th2, but not Th1, responses to inhaled allergen. J. Immunol. 169, 5997–6004.
Johansson, C. C., Bryn, T., Aandahl, E. M., Areklett, M. A., Aukrust, P., Tasken, K., et al. (2004). Treatment with type-2 selective and non-selective cyclooxygenase inhibitors improves T-cell proliferation in HIV-infected patients on highly active antiretroviral therapy. AIDS 18, 951–952. doi:10.1097/00002030-200404090-00015
Joo, M., and Sadikot, R. T. (2012). PGD synthase and PGD(2) in immune response. Mediators Inflamm. 2012:503128 doi:10.1155/2012/503128
Kabashima, K., Murata, T., Tanaka, H., Matsuoka, T., Sakata, D., Yoshida, N., et al. (2003). Thromboxane A(2) modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat. Immunol. 4, 694–701. doi:10.1038/ni943
Kabashima, K., Saji, T., Murata, T., Nagamachi, M., Matsuoka, T., Segi, E., et al. (2002). The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J. Clin. Invest. 109, 883–893. doi:10.1172/JCI14459
Kanaoka, Y., and Boyce, A. A. (2004). Cysteinyl leukotrienes and their receptors: cellular distribution and function in immune and inflammatory responses. J. Immunol. 173, 1503–1510.
Kelly, J. P., Johnson, M. C., and Parker, C. W. (1979). Effect of inhibitors of arachidonic-acid metabolism on mitogenesis in human lymphocytes – possible role of thromboxanes and products of the lipoxygenase pathway. J. Immunol. 122, 1563–1571.
Kliewer, S. A., Lenhard, J. M., Willson, T. M., Patel, I., Morris, D. C., and Lehmann, J. M. (1995). A prostaglandin J(2) metabolite binds peroxisome proliferator-activated receptor-gamma and promotes adipocyte differentiation. Cell 83, 813–819. doi:10.1016/0092-8674(95)90194-9
Kudo, I., and Murakami, M. (2002). Phospholipase A(2) enzymes. Prostaglandins Other Lipid Mediat. 68–69, 3–58. doi:10.1016/S0090-6980(02)00020-5
Kuehl, F. A., Humes, J. L., Egan, R. W., Ham, E. A., Beveridge, G. C., and Vanarman, C. G. (1977). Role of prostaglandin endoperoxide PGG2 in inflammatory processes. Nature 265, 170–173. doi:10.1038/265170a0
Kvale, D., Ormaasen, V., Kran, A. M. B., Johansson, C. C., Aukrust, P., Aandahl, E. M., et al. (2006). Immune modulatory effects of cyclooxygenase type 2 inhibitors in HIV patients on combination antiretroviral treatment. AIDS 20, 813–820. doi:10.1097/01.aids.0000218544.54586.f1
Laidlaw, T. M., and Boyce, J. A. (2012). Cysteinyl leukotriene receptors, old and new; implications for asthma. Clin. Exp. Allergy 42, 1313–1320. doi:10.1111/j.1365-2222.2012.03982.x
Lee, B. P. L., Juvet, S. C., and Zhang, L. (2009). Prostaglandin E2 signaling through E prostanoid receptor 2 impairs proliferative response of double negative regulatory T cells. Int. Immunopharmacol. 9, 534–539. doi:10.1016/j.intimp.2009.01.023
Lee, I. Y., Cho, W., Kim, J., Park, C. S., and Choe, J. (2008). Human follicular dendritic cells interact with T cells via expression and regulation of cyclooxygenases and prostaglandin E and I Synthases. J. Immunol. 180, 1390–1397.
Lee, I. Y., Ko, E. M., Kim, S. H., Jeoung, D. I., and Choe, J. (2005). Human follicular dendritic cells express prostacyclin synthase: a novel mechanism to control T cell numbers in the germinal center. J. Immunol. 175, 1658–1664.
Lerner, A., Jacobson, B., and Miller, R. A. (1988). Cyclic-AMP concentrations modulate both calcium flux and hydrolysis of phosphatidylinositol phosphates in mouse lymphocytes-T. J. Immunol. 140, 936–940.
Leslie, C. C. (2004). Regulation of arachidonic acid availability for eicosanoid production. Biochem. Cell Biol. 82, 1–17. doi:10.1139/o03-080
Li, H., Bradbury, J. A., Dackor, R. T., Edin, M. L., Graves, J. P., DeGraff, L. M., et al. (2011). Cyclooxygenase-2 regulates Th17 cell differentiation during allergic lung inflammation. Am. J. Respir. Crit. Care Med. 184, 37–49. doi:10.1164/rccm.201010-1637OC
Liscovitch, M., Czarny, M., Fiucci, G., and Tang, X. Q. (2000). Phospholipase D: molecular and cell biology of a novel gene family. Biochem. J. 345, 401–415. doi:10.1042/0264-6021:3450401
Los, M., Schenk, H., Hexel, K., Baeuerle, P. A., Droge, W., and Schulzeosthoff, K. (1995). IL-2 gene-expression and NF-kappa-B activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J. 14, 3731–3740.
Luster, A. D., and Tager, A. M. (2004). T-cell trafficking in asthma: lipid mediators grease the way. Nat. Rev. Immunol. 4, 711–724. doi:10.1038/nri1438
Maekawa, A., Balestrieri, B., Austen, K. F., and Kanaoka, Y. (2009). GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D-4. Proc. Natl. Acad. Sci. U.S.A. 106, 11685–11690. doi:10.1073/pnas.0905364106
Mahic, M., Yaqub, S., Johansson, C. C., Tasken, K., and Aandahl, E. M. (2006). FOXP3(+)CD4(+)CD25(+) adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E-2-dependent mechanism. J. Immunol. 177, 246–254.
Mandapathil, M., Szczepanski, M. J., Szajnik, M., Ren, J., Jackson, E. K., Johnson, J. T., et al. (2010). Adenosine and prostaglandin E-2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J. Biol. Chem. 285, 27571–27580. doi:10.1074/jbc.M110.127100
Martinet, L., Jean, C., Dietrich, G., Fournie, J. J., and Poupot, R. (2010). PGE(2) inhibits natural killer and gamma delta T cell cytotoxicity triggered by NKR and TCR through a cAMP-mediated PKA type I-dependent signaling. Biochem. Pharmacol. 80, 838–845. doi:10.1016/j.bcp.2010.05.002
Mary, D., Aussel, C., Ferrua, B., and Fehlmann, M. (1987). Regulation of interleukin-2 synthesis by cAMP in human T-cells. J. Immunol. 139, 1179–1184.
Mastino, A., Piacentini, M., Grelli, S., Favalli, C., Autuori, F., Tentori, L., et al. (1992). Induction of apoptosis in thymocytes by prostaglandin E2 in vivo. Dev. Immunol. 2, 263–271. doi:10.1155/1992/80863
Medoff, B. D., Seung, E., Wain, J. C., Means, T. K., Campanella, G. S. V., Islam, S. A., et al. (2005). BLT1-mediated T cell trafficking is critical for rejection and obliterative bronchiolitis after lung transplantation. J. Exp. Med. 202, 97–110. doi:10.1084/jem.20042481
Melendez, A. J., and Allen, J. M. (2002). Phospholipase D and immune receptor signalling. Semin. Immunol. 14, 49–55. doi:10.1006/smim.2001.0341
Merhi-Soussi, F., Dominguez, Z., Macovschi, O., Dubois, M., Savany, A., Lagarde, M., et al. (2000). Human lymphocytes stimulate prostacyclin synthesis in human umbilical vein endothelial cells. Involvement of endothelial cPLA(2). J. Leukoc. Biol. 68, 881–889.
Mita, H., Hasegawa, M., Saito, H., and Akiyama, K. (2001). Levels of cysteinyl leukotriene receptor mRNA in human peripheral leucocytes: significantly higher expression of cysteinyl leukotriene receptor 2 mRNA in eosinophils. Clin. Exp. Allergy 31, 1714–1723. doi:10.1046/j.1365-2222.2001.01184.x
Morita, H., Takeda, K., Yagita, H., and Okumura, K. (1999). Immunosuppressive effect of leukotriene B-4 receptor antagonist in vitro. Biochem. Biophys. Res. Commun. 264, 321–326. doi:10.1006/bbrc.1999.1523
Mosenden, R., Singh, P., Cornez, I., Heglind, M., Ruppelt, A., Moutschen, M., et al. (2011). Mice with disrupted type I protein kinase A anchoring in T cells resist retrovirus-induced immunodeficiency. J. Immunol. 186, 5119–5130. doi:10.4049/jimmunol.1100003
Mosenden, R., and Tasken, K. (2011). Cyclic AMP-mediated immune regulation – overview of mechanisms of action in T cells. Cell. Signal. 23, 1009–1016. doi:10.1016/j.cellsig.2010.11.018
Murakami, M., Taketomi, Y., Miki, Y., Sato, H., Hirabayashi, T., and Yamamoto, K. (2011). Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog. Lipid Res. 50, 152–192. doi:10.1016/j.plipres.2010.12.001
Nakanishi, M., Menoret, A., Tanaka, T., Miyamoto, S., Montrose, D. C., Vella, A. T., et al. (2011). Selective PGE(2) suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev. Res. (Phila.) 4, 1198–1208. doi:10.1158/1940-6207.CAPR-11-0188
Nantel, F., Fong, C., Lamontagne, S., Wright, D. H., Giaid, A., Desrosiers, M., et al. (2004). Expression of prostaglandin D synthase and the prostaglandin D-2 receptors DP and CRTH2 in human nasal mucosa. Prostaglandins Other Lipid Mediat. 73, 87–101. doi:10.1016/j.prostaglandins.2003.12.002
Napolitani, G., Acosta-Rodriguez, E. V., Lanzavecchia, A., and Sallusto, F. (2009). Prostaglandin E2 enhances Th17 responses via modulation of IL-17 and IFN-gamma production by memory CD4(+) T cells. Eur. J. Immunol. 39, 1301–1312. doi:10.1002/eji.200838969
Narumiya, S. (2003). Prostanoids in immunity: roles revealed by mice deficient in their receptors. Life Sci. 74, 391–395. doi:10.1016/j.lfs.2003.09.025
Nataraj, C., Thomas, D. W., Tilley, S. L., Nguyen, M., Mannon, R., Koller, B. H., et al. (2001). Receptors for prostaglandin E-2 that regulate cellular immune responses in the mouse. J. Clin. Invest. 108, 1229–1235. doi:10.1172/JCI200113640
Nencioni, A., Lauber, K., Grunebach, F., Van Parijs, L., Denzlinger, C., Wesselborg, S., et al. (2003). Cyclopentenone prostaglandins induce lymphocyte apoptosis by activating the mitochondrial apoptosis pathway independent of external death receptor signaling. J. Immunol. 171, 5148–5156.
Oberprieler, N. G., Lemeer, S., Kalland, M. E., Torgersen, K. M., Heck, A. J. R., and Tasken, K. (2010). High-resolution mapping of prostaglandin E-2-dependent signaling networks identifies a constitutively active PKA signaling node in CD8(+)CD45RO(+) T cells. Blood 116, 2253–2265. doi:10.1182/blood-2010-01-266650
Olschewski, H., Rose, F., Schermuly, R., Ghofrani, H. A., Enke, B., Olschewski, A., et al. (2004). Prostacyclin and its analogues in the treatment of pulmonary hypertension. Pharmacol. Ther. 102, 139–153. doi:10.1016/j.pharmthera.2004.01.003
Ott, V. L., Cambier, J. C., Kappler, J., Marrack, P., and Swanson, B. J. (2003). Mast cell-dependent migration of effector CD8(+) T cells through production of leukotriene B-4. Nat. Immunol. 4, 974–981. doi:10.1038/ni971
Pablos, J. L., Santiago, B., Carreira, P. E., Galindo, M., and Gomez-Reino, J. J. (1999). Cyclooxygenase-1 and -2 are expressed by human T cells. Clin. Exp. Immunol. 115, 86–90. doi:10.1046/j.1365-2249.1999.00780.x
Pace, E., Bruno, T. F., Berenger, B., Mody, C. H., Melis, M., Ferraro, M., et al. (2007). Elevated expression of prostaglandin receptor and increased release of prostaglandin E-2 maintain the survival of CD45RO(+) T cells in the inflamed human pleural space. Immunology 121, 427–436. doi:10.1111/j.1365-2567.2007.02593.x
Parmentier, C. N., Fuerst, E., McDonald, J., Bowen, H., Lee, T. H., Pease, J. E., et al. (2012). Human T(H)2 cells respond to cysteinyl leukotrienes through selective expression of cysteinyl leukotriene receptor 1. J. Allergy Clin. Immunol. 129, 1136–1142. doi:10.1016/j.jaci.2012.01.057
Paruchuri, S., Tashimo, H., Feng, C. L., Maekawa, A., Xing, W., Jiang, Y. F., et al. (2009). Leukotriene E-4-induced pulmonary inflammation is mediated by the P2Y(12) receptor. J. Exp. Med. 206, 2543–2555. doi:10.1084/jem.20091240
Pettersen, F. O., Torheim, E. A., Dahm, A. E. A., Aaberge, I. S., Lind, A., Holm, M., et al. (2011). An exploratory trial of cyclooxygenase type 2 inhibitor in HIV-1 infection: downregulated immune activation and improved t cell-dependent vaccine responses. J. Virol. 85, 6557–6566. doi:10.1128/JVI.00073-11
Pettersson, A., Richter, J., and Owman, C. (2003). Flow cytometric mapping of the leukotriene B-4 receptor, BLT1, in human bone marrow and peripheral blood using specific monoclonal antibodies. Int. Immunopharmacol. 3, 1467–1475. doi:10.1016/S1567-5769(03)00145-0
Pettipher, R., Hansel, T. T., and Armer, R. (2007). Antagonism of the prostaglandin D-2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat. Rev. Drug Discov. 6, 313–325. doi:10.1038/nrd2266
Pica, F., Franzese, O., Donofrio, C., Bonmassar, E., Favalli, C., and Garaci, E. (1996). Prostaglandin E(2) induces apoptosis in resting immature and mature human lymphocytes: a c-Myc-dependent and Bcl-2-independent associated pathway. J. Pharmacol. Exp. Ther. 277, 1793–1800.
Porter, B. O., and Malek, T. R. (1999). Prostaglandin E-2 inhibits T cell activation-induced apoptosis and Fas-mediated cellular cytotoxicity by blockade of Fas-ligand induction. Eur. J. Immunol. 29, 2360–2365. doi:10.1002/(SICI)1521-4141(199907)29:07<2360::AID-IMMU2360>3.0.CO;2-A
Prinz, I., Gregoire, C., Mollenkopft, H., Aguado, E., Wang, Y., Malissen, M., et al. (2005). The type 1 cysteinyl leukotriene receptor triggers calcium influx and chemotaxis in mouse alpha beta and gamma delta effector T cells. J. Immunol. 175, 713–719.
Rahmouni, S., Aandahl, E. M., Nayjib, B., Zeddou, M., Giannini, S., Verlaet, M., et al. (2004). Cyclo-oxygenase type 2-dependent prostaglandin E-2 secretion is involved in retrovirus-induced T-cell dysfunction in mice. Biochem. J. 384, 469–476. doi:10.1042/BJ20031859
Remuzzi, G., Noris, M., Benigni, A., Imberti, O., Sayegh, M. H., and Perico, N. (1994). Thromboxane A(2) receptor blocking abrogates donor-specific unresponsiveness to renal-allografts induced by thymic recognition of major histocompatibility allopeptides. J. Exp. Med. 180, 1967–1972. doi:10.1084/jem.180.5.1967
Rincon, M., Tugores, A., Lopezrivas, A., Silva, A., Alonso, M., Delandazuri, M. O., et al. (1988). Prostaglandin-E2 and the increase of intracellular cAMP inhibit the expression of interleukin-2 receptors in human T-cells. Eur. J. Immunol. 18, 1791–1796. doi:10.1002/eji.1830181121
Rolapleszczynski, M. (1985). Differential-effects of leukotriene-B-4 on T4+ and T8+ lymphocyte phenotype and immunoregulatory functions. J. Immunol. 135, 1357–1360.
Roshak, A. K., Capper, E. A., Stevenson, C., Eichman, C., and Marshall, L. A. (2000). Human calcium-independent phospholipase A(2) mediates lymphocyte proliferation. J. Biol. Chem. 275, 35692–35698. doi:10.1074/jbc.M002273200
Ruiz, P., Coffman, T. M., Klotman, P. E., and Sanfilippo, F. (1989). Association of chromic thromboxane inhibition with reduced insitu cytotoxic T-cell activity in rejecting rat renal-allografts. Transplantation 48, 660–666.
Ruiz, P., Rey, L., Spurney, R., Coffman, T., and Viciana, A. (1992). Thromboxane augmentation of alloreactive T cell function. Transplantation 54, 498–505. doi:10.1097/00007890-199209000-00021
Ruppelt, A., Mosenden, R., Groenholm, M., Aandahl, E. M., Tobin, D., Carlson, C. R., et al. (2007). Inhibition of T cell activation by cyclic adenosine 5’-monophosphate requires lipid raft targeting of protein kinase a type I by the A-kinase anchoring protein Ezrin. J. Immunol. 179, 5159–5168.
Saito, M., Tanabe, Y., Kudo, I., and Nakayama, K. (2003). Endothelium-derived prostaglandin H-2 evokes the stretch-induced contraction of rabbit pulmonary artery. Eur. J. Pharmacol. 467, 151–161. doi:10.1016/S0014-2999(03)01569-3
Saito, S., Tsuda, H., and Michimata, T. (2002). Prostaglandin D-2 and reproduction. Am. J. Reprod. Immunol. 47, 295–302. doi:10.1034/j.1600-0897.2002.01113.x
Sakata, D., Yao, C. C., and Narumiya, S. (2010a). Emerging roles of prostanoids in T cell-mediated immunity. IUBMB Life 62, 591–596. doi:10.1002/iub.356
Sakata, D., Yao, C. C., and Narumiya, S. (2010b). Prostaglandin E-2, an immunoactivator. J. Pharmacol. Sci. 112, 1–5. doi:10.1254/jphs.09R03CP
Sala, A., Folco, G., and Murphy, R. C. (2010). Transcellular biosynthesis of eicosanoids. Pharmacol. Rep. 62, 503–510.
Scher, J. U., and Pillinger, M. H. (2005). 15d-PGJ(2): the anti-inflammatory prostaglandin? Clin. Immunol. 114, 100–109. doi:10.1016/j.clim.2004.09.008
Schuligoi, R., Sedej, M., Waldhoer, M., Vukoja, A., Sturm, E. M., Lippe, I. T., et al. (2009). Prostaglandin H-2 induces the migration of human eosinophils through the chemoattractant receptor homologous molecule of Th2 cells, CRTH2. J. Leukoc. Biol. 85, 136–145. doi:10.1189/jlb.0608387
Schuligoi, R., Sturm, E., Luschnig, P., Konya, V., Philipose, S., Sedej, M., et al. (2010). CRTH2 and D-type prostanoid receptor antagonists as novel therapeutic agents for inflammatory diseases. Pharmacology 85, 372–382. doi:10.1159/000313836
Sedej, M., Schroder, R., Bell, K., Platzer, W., Vukoja, A., Kostenis, E., et al. (2012). D-type prostanoid receptor enhances the signaling of chemoattractant receptor-homologous molecule expressed on T(H)2 cells. J. Allergy Clin. Immunol. 129, 492–500. doi:10.1016/j.jaci.2011.08.015
Seidel, S. D., Winters, G. M., Rogers, W. J., Ziccardi, M. H., Li, V., Keser, B., et al. (2001). Activation of the Ah receptor signaling pathway by prostaglandins. J. Biochem. Mol. Toxicol. 15, 187–196. doi:10.1002/jbt.16
Sharma, S., Yang, S. C., Zhu, L., Reckamp, K., Gardner, B., Baratelli, F., et al. (2005). Tumor cyclooxygenase-2/prostaglandin E-2-dependent promotion of FOXP3 expression and CD4(+)CD25(+) T regulatory cell activities in lung cancer. Cancer Res. 65, 5211–5220. doi:10.1158/0008-5472.CAN-05-0141
Simmons, D. L., Botting, R. M., and Hla, T. (2004). Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 56, 387–437. doi:10.1124/pr.56.3.3
Singh, R. K., Gupta, S., Dastidar, S., and Ray, A. (2010). Cysteinyl leukotrienes and their receptors: molecular and functional characteristics. Pharmacology 85, 336–349. doi:10.1159/000312669
Smith, W. L. (1989). The eicosanoids and their biochemical mechanisms of action. Biochem. J. 259, 315–324.
Smith, W. L., DeWitt, D. L., and Garavito, R. M. (2000). Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182. doi:10.1146/annurev.biochem.69.1.145
Smith, W. L., Urade, Y., and Jakobsson, P. J. (2011). Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 111, 5821–5865. doi:10.1021/cr2002992
Snijdewint, F. G. M., Kalinski, P., Wierenga, E. A., Bos, J. D., and Kapsenberg, M. L. (1993). Prostaglandin-E(2) differentially modulates cytokine secretion profiles of human T-helper lymphocytes. J. Immunol. 150, 5321–5329.
Sreeramkumar, V., Fresno, M., and Cuesta, N. (2012). Prostaglandin E-2 and T cells: friends or foes? Immunol. Cell Biol. 90, 579–586. doi:10.1038/icb.2011.75
Stebbins, K. J., Broadhead, A. R., Baccei, C. S., Scott, J. M., Truong, Y. P., Coate, H., et al. (2010). Pharmacological blockade of the DP2 receptor inhibits cigarette smoke-induced inflammation, mucus cell metaplasia, and epithelial hyperplasia in the mouse lung. J. Pharmacol. Exp. Ther. 332, 764–775. doi:10.1124/jpet.109.161919
Stokka, A. J., Mosenden, R., Ruppelt, A., Lygren, B., and Tasken, K. (2010). The adaptor protein EBP50 is important for localization of the protein kinase A-Ezrin complex in T-cells and the immunomodulating effect of cAMP. Biochem. J. 425, 381–388. doi:10.1042/BJ20091136
Tager, A. M., Bromley, S. K., Medoff, B. D., Islam, S. A., Bercury, S. D., Friedrich, E. B., et al. (2003). Leukotriene B-4 receptor BLT1 mediates early effector T cell recruitment. Nat. Immunol. 4, 982–990. doi:10.1038/ni970
Takahashi, Y., Tokuoka, S., Masuda, T., Hirano, Y., Nagao, M., Tanaka, H., et al. (2002). Augmentation of allergic inflammation in prostanoid IP receptor deficient mice. Br. J. Pharmacol. 137, 315–322. doi:10.1038/sj.bjp.0704872
Tanaka, K., Hirai, H., Takano, S., Nakamura, M., and Nagata, K. (2004). Effects of prostaglandin D-2 on helper T cell functions. Biochem. Biophys. Res. Commun. 316, 1009–1014. doi:10.1016/j.bbrc.2004.02.151
Tanaka, K., Ogawa, K., Sugamura, K., Nakamura, M., Takano, S., and Nagata, K. (2000). Cutting edge: differential production of prostaglandin D-2 by human helper T cell subsets. J. Immunol. 164, 2277–2280.
Tessier, C., Hichami, A., and Khan, N. A. (2002). Implication of three isoforms of PLA(2) in human T-cell proliferation. FEBS Lett. 520, 111–116. doi:10.1016/S0014-5793(02)02779-5
Thomas, D. W., Rocha, P. N., Nataraj, C., Robinson, L. A., Spurney, R. F., Koller, B. H., et al. (2003). Proinflammatory actions of thromboxane receptors to enhance cellular immune responses. J. Immunol. 171, 6389–6395.
Tilley, S. L., Coffman, T. M., and Koller, B. H. (2001). Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J. Clin. Invest. 108, 15–23. doi:10.1172/JCI13416
Tohda, Y., Nakahara, H., Kubo, H., Haraguchi, R., Fukuoka, M., and Nakajima, S. (1999). Effects of ONO-1078 (pranlukast) on cytokine production in peripheral blood mononuclear cells of patients with bronchial asthma. Clin. Exp. Allergy 29, 1532–1536. doi:10.1046/j.1365-2222.1999.00710.x
Torres, D., Paget, C., Fontaine, J., Mallevaey, T., Matsuoka, T., Maruyama, T., et al. (2008). Prostaglandin D(2) inhibits the production of IFN-gamma by invariant NK T cells: consequences in the control of b16 melanoma. J. Immunol. 180, 783–792.
Trivedi, S. G., Newson, J., Rajakariar, R., Jacques, T. S., Hannon, R., Kanaoka, Y., et al. (2006). Essential role for hematopoietic prostaglandin D2 synthase in the control of delayed type hypersensitivity. Proc. Natl. Acad. Sci. U.S.A. 103, 5179–5184. doi:10.1073/pnas.0507175103
Truchetet, M. E., Allanore, Y., Montanari, E., Chizzolini, C., and Brembilla, N. C. (2012). Prostaglandin I-2 analogues enhance already exuberant Th17 cell responses in systemic sclerosis. Ann. Rheum. Dis. 71, 2044–2050. doi:10.1136/annrheumdis-2012-201400
Tsuda, H., Michimata, T., Sakai, M., Nagata, K., Nakamura, M., and Saito, S. (2001). A novel surface molecule of Th2- and Tc2-type cells, CRTH2 expression on human peripheral and decidual CD4(+) and CD8(+) T cells during the early stage of pregnancy. Clin. Exp. Immunol. 123, 105–111. doi:10.1046/j.1365-2249.2001.01422.x
Ushikubi, F., Aiba, Y. I., Nakamura, K. I., Namba, T., Hirata, M., Mazda, O., et al. (1993). Thromboxane-A2 receptor is highly expressed in mouse immature thymocytes and mediates DNA fragmentation and apoptosis. J. Exp. Med. 178, 1825–1830. doi:10.1084/jem.178.5.1825
Vang, T., Torgersen, K. M., Sundvold, V., Saxena, M., Levy, F. O., Skalhegg, B. S., et al. (2001). Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J. Exp. Med. 193, 497–507. doi:10.1084/jem.193.4.497
von Allmen, C. E., Schmitz, N., Bauer, M., Hinton, H. J., Kurrer, M. O., Buser, R. B., et al. (2009). Secretory phospholipase A2-IID is an effector molecule of CD4(+)CD25(+) regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 106, 11673–11678. doi:10.1073/pnas.0812569106
Wada, M., DeLong, C. J., Hong, Y. H., Rieke, C. J., Song, I., Sidhu, R. S., et al. (2007). Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J. Biol. Chem. 282, 22254–22266. doi:10.1074/jbc.M703169200
Wang, M. T., Honn, K. V., and Nie, D. (2007). Cyclooxygenases, prostanoids, and tumor progression. Cancer Metastasis Rev. 26, 525–534. doi:10.1007/s10555-007-9096-5
Woodward, D. F., Jones, R. L., and Narumiya, S. (2011). International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol. Rev. 63, 471–538. doi:10.1124/pr.110.003517
Wu, C. Y., Wang, K. N., McDyer, J. F., and Seder, R. A. (1998). Prostaglandin E-2 and dexamethasone inhibit IL-12 receptor expression and IL-12 responsiveness. J. Immunol. 161, 2723–2730.
Wu, K. K., Papp, A. C., Manner, C. E., and Hall, E. R. (1987). Interaction between lymphocytes and platelets in the synthesis of prostacyclin. J. Clin. Invest. 79, 1601–1606. doi:10.1172/JCI112995
Xue, L., Barrow, A., and Pettipher, R. (2009a). Interaction between prostaglandin D-2 and chemoattractant receptor-homologous molecule expressed on Th2 cells mediates cytokine production by Th2 lymphocytes in response to activated mast cells. Clin. Exp. Immunol. 156, 126–133. doi:10.1111/j.1365-2249.2008.03871.x
Xue, L. Z., Barrow, A., and Pettipher, R. (2009b). Novel function of CRTH2 in preventing apoptosis of human Th2 cells through activation of the phosphatidylinositol 3-kinase pathway. J. Immunol. 182, 7580–7586. doi:10.4049/jimmunol.0804090
Xue, L. Z., Barrow, A., Fleming, V. M., Hunter, M. G., Ogg, G., Klenerman, P., et al. (2012). Leukotriene E-4 activates human Th2 cells for exaggerated proinflammatory cytokine production in response to prostaglandin D-2. J. Immunol. 188, 694–702. doi:10.4049/jimmunol.1102474
Xue, L. Z., Gyles, S. L., Barrow, A., and Pettipher, R. (2007). Inhibition of PI3K and calcineurin suppresses chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2)-dependent responses of Th2 lymphocytes to prostaglandin D-2. Biochem. Pharmacol. 73, 843–853. doi:10.1016/j.bcp.2006.11.021
Yamaoka, K. A., and Kolb, J. P. (1993). Leukotriene-B(4) induces interleukin-5 generation from human T-lymphocytes. Eur. J. Immunol. 23, 2392–2398. doi:10.1002/eji.1830231003
Yang, X. Y., Wang, L. H., Chen, T. S., Hodge, D. R., Resau, J. H., DaSilva, L., et al. (2000). Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPAR gamma) agonists – PPAR gamma co-association with transcription factor NFAT. J. Biol. Chem. 275, 4541–4544. doi:10.1074/jbc.275.7.4541
Yao, C. C., Sakata, D., Esaki, Y., Li, Y. X., Matsuoka, T., Kuroiwa, K., et al. (2009). Prostaglandin E-2-EP4 signaling promotes immune inflammation through T(H)1 cell differentiation and T(H)17 cell expansion. Nat. Med. 15, 633–640. doi:10.1038/nm.1968
Yokomizo, T. (2011). “Leukotriene B-4 receptors: novel roles in immunological regulations,” in Advances in Enzyme Regulation, Vol. 51, eds L. Cocco, G. Weber, and C. E. F. Weber (Amsterdam: Elsevier Science), 59–64.
Yokomizo, T., Izumi, T., Chang, K., Takuwa, Y., and Shimizu, T. (1997). A G-protein-coupled receptor for leukotriene B-4 that mediates chemotaxis. Nature 387, 620–624. doi:10.1038/42506
Yokomizo, T., Izumi, T., and Shimizu, T. (2001). Co-expression of two LTB4 receptors in human mononuclear cells. Life Sci. 68, 2207–2212. doi:10.1016/S0024-3205(01)01007-4
Yokomizo, T., Kato, K., Terawaki, K., Izumi, T., and Shimizu, T. (2000). A second leukotriene B-4 receptor, BLT2: a new therapeutic target in inflammation and immunological disorders. J. Exp. Med. 192, 421–431. doi:10.1084/jem.192.3.421
Zeddou, M., Greimers, R., de Valensart, N., Nayjib, B., Tasken, K., Boniver, J., et al. (2005). Prostaglandin E-2 induces the expression of functional inhibitory CD94/NKG2A receptors in human CD8(+) T lymphocytes by a cAMP-dependent protein kinase A type I pathway. Biochem. Pharmacol. 70, 714–724. doi:10.1016/j.bcp.2005.05.015
Zhou, W. S., Blackwell, T. S., Goleniewska, K., O’Neal, J. F., FitzGerald, G. A., Lucitt, M., et al. (2007). Prostaglandin I-2 analogs inhibit Th1 and Th2 effector cytokine production by CD4 T cells. J. Leukoc. Biol. 81, 809–817. doi:10.1189/jlb.0606375
Keywords: prostaglandins, leukotrienes, cyclooxygenase 2, regulatory T cells, cAMP, immunoregulation effect, inflammation mediators, inflammation
Citation: Lone AM and Taskén K (2013) Proinflammatory and immunoregulatory roles of eicosanoids in T cells. Front. Immunol. 4:130. doi: 10.3389/fimmu.2013.00130
Received: 15 April 2013; Paper pending published: 09 May 2013;
Accepted: 17 May 2013; Published online: 04 June 2013.
Edited by:
Karsten Sauer, The Scripps Research Institute, USAReviewed by:
António Gil Castro, University of Minho, PortugalAmnon Altman, La Jolla Institute for Allergy and Immunology, USA
Copyright: © 2013 Lone and Taskén. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Kjetil Taskén, Centre for Molecular Medicine Norway, Nordic EMBL Partnership, Oslo University Hospital, University of Oslo, POB 1137, Blindern, N-0318 Oslo, Norway e-mail:a2pldGlsLnRhc2tlbkBuY21tLnVpby5ubw==