 Johanne M. Pentier1
Johanne M. Pentier1
- 1Institute of Infection and Immunity, Cardiff University School of Medicine, Heath Park, Cardiff, Wales, UK
- 2Human Immunity Laboratory, Queensland Institute of Medical Research, Brisbane, QLD, Australia
- 3School of Medicine, The University of Queensland, Brisbane, QLD, Australia
Introduction
T-cells recognize small peptide fragments (p) cradled in multiple major histocompatibility complex (MHC) molecules, termed human leukocyte antigens (HLA) in humans. These membrane-integral pMHC molecules are present on surface of all nucleated cells and allow T-cells to detect aberrant intracellular activity, be this infection with microorganisms or abnormal host biochemistry such as neoplastic division. Scanning of pMHC molecules occurs via the αβ T-cell receptor (TCR), a clonotypic, heterodimeric, and membrane-integral molecule on the T-cell surface (Miles et al., 2011a). TCRs engage pMHC molecules via six highly flexible complementarity determining region (CDR) loops and, upon productive docking with a pMHC molecule, the TCR triggers a myriad of intracellular T-cell signaling cascades (Bridgeman et al., 2012). The binding strength (or affinity) between a TCR and a cognate pMHC is relatively weak across known biological systems with monomeric “dwell times” (or half-lives) typically measured in seconds or microseconds at physiological temperatures (Miles et al., 2010; Bridgeman et al., 2012; Smith et al., 2012). This is in contrast to numerous other biological interactions such as antibodies (van der Merwe and Davis, 2003), interleukins (Morton et al., 1994), lipoproteins (Misra et al., 2001), and structural membrane proteins (Matte et al., 2012) which have half-lives measured in hours-to-days. Overall, TCR/pMHC interactions are fleeting even by the dynamic standards of cell surface interactions (van der Merwe and Davis, 2003). The evolutionary rationale for this striking functional divide can only be speculated upon but likely pertains to the primary function of T-cells. T-cells must scan large numbers of pMHC on multiple cells in series in order to identify and eliminate threats quickly. Effective immunity requires that TCR scanning time must be minimal and antigen coverage maximal. Theoretical arguments dictate that maximal immune cover of possible foreign pMHC requires each TCR to recognize huge numbers of different peptides (Mason, 1998; Sewell, 2012). This theory is now supported up by direct experimental evidence that shows a single TCR can cross-recognize millions of pMHC molecules as well or better than the native antigen (Sewell, 2012; Wooldridge et al., 2012; Ekeruche-Makinde et al., 2013). Curiously, this extensive T-cell cross-reactivity is strictly compartmentalized based on peptide length (Ekeruche-Makinde et al., 2013).
An interesting consequence of the low antigen affinity and high antigen cross-reactivity characteristics of TCRs is that many, and perhaps all, cognate antigens could potentially be improved upon. Through rational structural modifications of native blueprint antigens we now know it is possible to engineer “optimal fit antigens” which exhibit logarithmic increases in affinity and immunogenicity. Compared with the native antigens, if these optimal antigens prove more effective at stimulating antigen-specific T-cell populations during experimental priming then the compounds may fundamentally redefine how we think about vaccine design.
Improving T-Cell Epitopes
T-cell epitopes can be optimized by: (i) improving antigen affinity for MHC; (ii) improving antigen affinity for TCR; and (iii) improving antigen pharmacology through synthetic biology. Enhancing the stability of the epitope within the MHC cleft is the simplest engineering strategy as MHC anchoring preferences have been determined from MS elution data (Rammensee et al., 1995) resulting in MHC-binding databases and MHC-binding prediction algorithms (Rammensee et al., 1999; Wang et al., 2011). In theory, the replacement of buried suboptimal anchor residues with optimal residues for MHC-binding will produce a more stable pMHC complex and improved recognition of peptide. Recent studies have shown that things are not straightforward and this simple strategy for antigen improvement requires careful evaluation. We now know that anchor residue-modified peptides can have minimal or no improvement on stability in the MHC cleft (Miles et al., 2011b). Additionally, previous work has shown that MHC-binding strength shows little correlation with immunogenicity (Assarsson et al., 2007). We have recently shown that anchor residue-modified peptides can substantially alter TCR binding in ways that are difficult to predict and thereby prime T-cells with altered TCR repertoires (Ekeruche-Makinde et al., 2012). These repertoire effects have clinical relevance as it was found that vaccination with anchor residue-modified peptides was less effective than vaccination with natural peptides at priming tumor-specific T-cells in patients (Speiser et al., 2008).
T-cell epitopes can also be improved by optimizing contact interface between the peptide and TCR. In its simplest form this can be achieved by scanning recognition of a monosubstituted analog library (MAL) (Burrows et al., 1992, 1995; Zaremba et al., 1997; Tangri et al., 2001; Kjer-Nielsen et al., 2003; Burrows, 2004; Bulek et al., 2012). MALs substitute one of all available proteogenic amino acids across each position of a peptide backbone. While this approach can rapidly identify optimal antigens it is expensive as it requires the manufacture a unique defined analog library for each epitope examined. Combinatorial peptide libraries (CPLs) provide more flexible approach for the identification of optimal ligands (Borras et al., 2002). These very large, mixture-based compound libraries are synthesized in positional scanning format so that just one position along the peptide backbone is a fixed amino acid and all other positions are degenerate, with degenerate positions containing any one of 19 proteogenic amino acids (cysteine is excluded to reduce disulfide bond formation within the compound mixtures). Scanning a CPL across a T-cell clone can quantitatively map which residue/s are preferred by the TCR along a peptide backbone even if true antigen specificity of the clone is unknown. Replacing native residues with preferred residues identified by CPL can significantly increase epitope affinity and immunogenicity (see table below). The advantage of CPL scanning is that the same compound library can be used for any T-cell from any system although it important to use a library of correct peptide length (Ekeruche-Makinde et al., 2012).
An alternative strategy for T-cell epitope optimization focuses on improving compound pharmacology by artificially engineering a compound to mimic a peptide (a peptidomimetic). The motive behind this strategy is that proteogenic amino acid-based polypeptides are susceptible to acid degradation and rapid proteolytic cleavage and have half-lives of less than 5 min in the presence of proteases (Guichard et al., 1994) or human serum (Stemmer et al., 1999). Replacing proteogenic amino acids with synthetic subunits generates resilience to proteases and has potential to vastly improve compound shelf life and in vivo bioavailability during prophylactic and therapeutic applications. Many synthetic subunits can be substituted for proteogenic amino acids to impede proteolysis. These include D-amino acids (Bartnes et al., 1997), β-amino acids (Webb et al., 2005), psi-bonded amino acids (Stemmer et al., 1999), and the shifting of the R group by one atom to create poly-N-substituted glycines (peptoids) where the side chains are appended to the nitrogen atom of the peptide backbone (Gocke et al., 2009).
Why Optimize T-Cell Epitopes?
We know that the number of antigen-specific T-cells within the host has fundamental relevance for disease control. For example the number of virus-specific T-cells generated during primary infection shows an inverse correlation with viral load (Ogg et al., 1998; Bharadwaj et al., 2001) and, in multiple cancer vaccine trials, the number of tumor-specific T-cells circulating within a patient and at tumor site/s correlates with clinical response (Lonchay et al., 2004; Rosenberg et al., 2004). While T-cell numbers alone do not determine disease outcome per se, global numbers are central in tipping control toward the host. With this in mind, engineering potent new compounds aimed at amplifying defined subsections of cellular immunity is the first step in manipulating the T-cell compartment for the purposes of rational vaccine design and therapeutic intervention. Such endeavors are particular important for cancer (self) antigens which generally exhibit very low immunogenicity (Chatten and Bathe, 2010).
Examples of T-Cell Epitope Optimization
Table 1 presents examples of MHC-anchor based (Parkhurst et al., 1996; Bernatchez et al., 2011), MAL-based (Burrows et al., 1992; Zaremba et al., 1997; Tangri et al., 2001; Terasawa et al., 2002), CPL-based (La Rosa et al., 2001; Blondelle et al., 2008; Ekeruche-Makinde et al., 2012; Wooldridge et al., 2012), and synthetic ligand-based (Mézière et al., 1997; Stemmer et al., 1999; Webb et al., 2005) T-cell epitope optimization across mice and humans. Collectively, this work demonstrates that T-cell epitopes can be readily optimized across both foreign and self peptide “blueprints” to increase sensitivity to T-cells and/or increase compound stability. Both MAL-based and CPL-based platforms appear equally effective at generating optimized ligands and have been used to increase antigen sensitivity between 100- and 10,000-fold relative to the native epitope blueprint. It is of particular interest to note that it is not just self self-derived epitopes that can have their immunogenicities amplified logarithmically. One CPL study (La Rosa et al., 2001) generated optimal ligands based on the HLA-A*-0201-binding epitope NLVPMVATV (A2-NLV) blueprint from human cytomegalovirus (HCMV) which were 10,000-fold more active. This achievement is striking given that the native A2-NLV epitope is one of the most immunogenic across the HCMV proteome (Khan et al., 2004). Thus, even very good T-cell antigens can be improved upon by modification in amino acid sequence. There is now accumulating evidence demonstrating that T-cells recognize a variety of molecular shapes presented by self MHC molecules (Bridgeman et al., 2012). These molecular shapes need not be made from L-amino acids as demonstrated by recognition of peptidomimetics. To date, such compounds have been made by rational design and have failed to exceed the potency of optimal peptide ligands. It is possible that the screening of combinatorial chemistries so that T-cells can select their recognition preference may overcome this limitation. While these synthetic compounds are unable, at this time, to exceed the sensitivity of the model epitope during completive titrations, they are considerably more resistant to digestion from proteases and serum and demonstrate remarkable biostability.
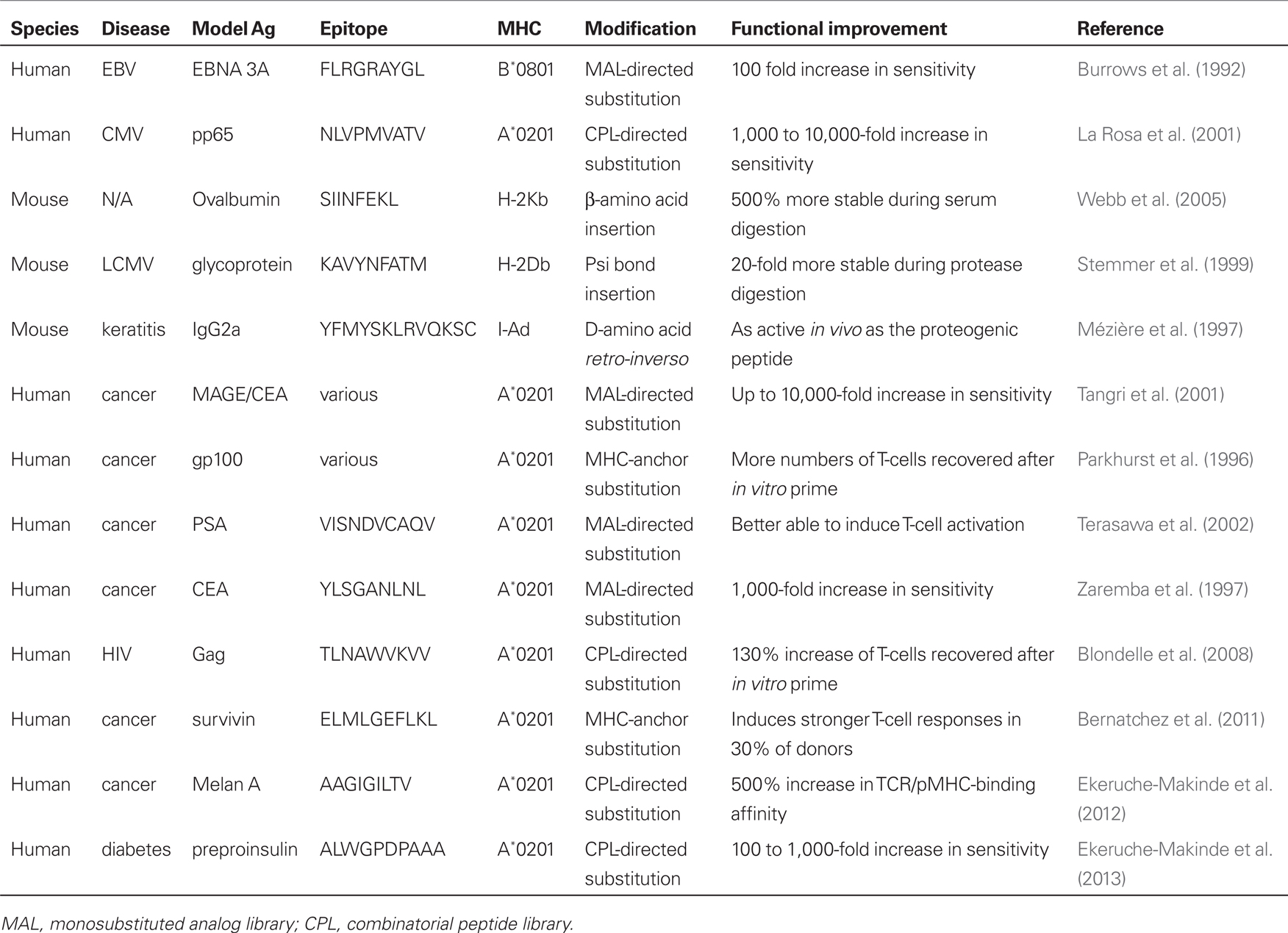
Table 1. Examples of T-cell epitope optimization.
Where Next?
It is clear that optimal T-cell epitopes can be generated with relative ease but the big question of whether they really work as therapeutic and prophylactic vaccines remains unanswered. Future studies should determine whether optimal T-cell epitopes can outperform native ligands during in vitro priming from healthy and patient samples. We also need to determine what effects priming with optimal T-cell epitopes has on the responding T-cell repertoire, both in terms of polyfunctionality and TCR usage. Studies that directly test whether optimal T-cell epitopes can outperform native ligands during in vivo priming of naïve animals are required. Importantly, these responses must be tested in relevant in vivo models of infection. It is hoped that optimal epitopes can be found that amplify greater numbers of T-cells than the native ligand alone and that T-cells maintain the desired functional profiles required for effective immunity. The development of effective synthetic T-cell epitopes will be particularly exciting as these epitopes have many attractive characteristics. First, they can be engineered to show absolute resistance to temperature fluctuations and environmental degradation, effectively nullifying the need for cold chain storage and transport. Indeed, the cold chain transport of vaccines costs WHO–UNICEF around USD 11.5 billion every year (Wolfson et al., 2008). Second, the acid- and protease-resistant properties of synthetic compounds raise the possibility of orally active administration. Here, the needle-free nature of compound release would considerably decrease logistical problems with vaccine service delivery, significantly decrease systems costs and is likely to increase vaccine acceptance rates. T-cell sensitivity toward the first generation of “designed” synthetic epitopes has been slightly weaker when compared with proteogenic epitopes. However, we believe that that there is ample scope to improve upon proteogenic epitopes by screening the right sorts of combinatorial chemistry. Our recent preliminary data with such a synthetic epitope shows that it can produce strong responses when given orally and that the T-cells induced against this compound protect humanized mice against a lethal challenge with influenza. Further studies are required in order to see whether such an approach can be extended to other T-cell epitopes.
In summary, optimization of T-cell epitopes can be achieved using a number of different techniques. Whether the resultant compounds can be used as effective prophylactic or therapeutic vaccines in humans remains to be determined. However, such approaches may allow the precise targeting of the most effective T-cell clonotypes (Ekeruche-Makinde et al., 2012) in vivo and could have the potential to change the way we build vaccines and immunotherapies in the future.
References
Assarsson, E., Sidney, J., Oseroff, C., Pasquetto, V., Bui, H. H., Frahm, N., et al. (2007). A quantitative analysis of the variables affecting the repertoire of T cell specificities recognized after vaccinia virus infection. J. Immunol. 178, 7890–7901.
Bartnes, K., Hannestad, K., Guichard, G., and Briand, J. P. (1997). A retro-inverso analog mimics the cognate peptide epitope of a CD4+ T cell clone. Eur. J. Immunol. 27, 1387–1391. doi:10.1002/eji.1830270512
Bernatchez, C., Zhu, K., Li, Y., Andersson, H., Ionnides, C., Fernandez-Vina, M., et al. (2011). Altered decamer and nonamer from an HLA-A0201-restricted epitope of Surviving differentially stimulate T-cell responses in different individuals. Vaccine 29, 3021–3030. doi:10.1016/j.vaccine.2011.01.115
Bharadwaj, M., Burrows, S. R., Burrows, J. M., Moss, D. J., Catalina, M., and Khanna, R. (2001). Longitudinal dynamics of antigen-specific CD8+ cytotoxic T lymphocytes following primary Epstein-Barr virus infection. Blood 98, 2588–2589. doi:10.1182/blood.V98.8.2588
Blondelle, S. E., Moya-Castro, R., Osawa, K., Schroder, K., and Wilson, D. B. (2008). Immunogenically optimized peptides derived from natural mutants of HIV CTL epitopes and peptide combinatorial libraries. Biopolymers 90, 683–694. doi:10.1002/bip.21020
Borras, E., Martin, R., Judkowski, V., Shukaliak, J., Zhao, Y., Rubio-Godoy, V., et al. (2002). Findings on T cell specificity revealed by synthetic combinatorial libraries. J. Immunol. Methods 267, 79–97. doi:10.1016/S0022-1759(02)00142-4
Bridgeman, J. S., Sewell, A. K., Miles, J. J., Price, D. A., and Cole, D. K. (2012). Structural and biophysical determinants of alphabeta T-cell antigen recognition. Immunology 135, 9–18. doi:10.1111/j.1365-2567.2011.03515.x
Bulek, A. M., Cole, D. K., Skowera, A., Dolton, G., Gras, S., Madura, F., et al. (2012). Structural basis for the killing of human beta cells by CD8(+) T cells in type 1 diabetes. Nat. Immunol. 13, 283–289. doi: 10.1038/ni.2206
Burrows, S. R. (2004). Cross-reactive recognition of viral and self-peptides by a “public” T cell receptor expressed by cytotoxic T lymphocytes expanded in multiple unrelated individuals. Immunol. Lett. 93, 7–9. doi:10.1016/j.imlet.2004.03.001
Burrows, S. R., Rodda, S. J., Suhrbier, A., Geysen, H. M., and Moss, D. J. (1992). The specificity of recognition of a cytotoxic T lymphocyte epitope. Eur. J. Immunol. 22, 191–195. doi:10.1002/eji.1830220128
Burrows, S. R., Silins, S. L., Moss, D. J., Khanna, R., Misko, I. S., and Argaet, V. P. (1995). T cell receptor repertoire for a viral epitope in humans is diversified by tolerance to a background major histocompatibility complex antigen. J. Exp. Med. 182, 1703–1715. doi:10.1084/jem.182.6.1703
Chatten, C., and Bathe, O. F. (2010). Immunoregulatory cells of the tumor microenvironment. Front. Biosci. 15:291–308. doi:10.2741/3621
Ekeruche-Makinde, J., Clement, M., Cole, D. K., Edwards, E. S., Ladell, K., Miles, J. J., et al. (2012). T-cell receptor-optimized peptide skewing of the T-cell repertoire can enhance antigen targeting. J. Biol. Chem. 287, 37269–37281. doi:10.1074/jbc.M112.386409
Ekeruche-Makinde, J., Miles, J. J., van den, B., erg, H. A., Skowera, A., Cole, D. K., Dolton, G., et al. (2013). Peptide length determines the outcome of TCR/peptide-MHCI engagement. Blood 121, 1112–1123. doi:10.1182/blood-2012-06-437202
Gocke, A. R., Udugamasooriya, D. G., Archer, C. T., Lee, J., and Kodadek, T. (2009). Isolation of antagonists of antigen-specific autoimmune T cell proliferation. Chem. Biol. 16, 1133–1139. doi:10.1016/j.chembiol.2009.10.011
Guichard, G., Benkirane, N., Zeder-Lutz, G., van, R., egenmortel, M. H., Briand, J. P., and Muller, S. (1994). Antigenic mimicry of natural L-peptides with retro-inverso-peptidomimetics. Proc. Natl. Acad. Sci. U.S.A. 91, 9765–9769. doi:10.1073/pnas.91.21.9765
Khan, N., Hislop, A., Gudgeon, N., Cobbold, M., Khanna, R., Nayak, L., et al. (2004). Herpesvirus-specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a coresident EBV infection. J. Immunol. 173, 7481–7489.
Kjer-Nielsen, L., Clements, C. S., Purcell, A. W., Brooks, A. G., Whisstock, J. C., Burrows, S. R., et al. (2003). A structural basis for the selection of dominant alphabeta T cell receptors in antiviral immunity. Immunity 18, 53–64. doi:10.1016/S1074-7613(02)00513-7
La Rosa, C., Krishnan, R., Markel, S., Schneck, J. P., Houghten, R., Pinilla, C., et al. (2001). Enhanced immune activity of cytotoxic T-lymphocyte epitope analogs derived from positional scanning synthetic combinatorial libraries. Blood 97, 1776–1786. doi:10.1182/blood.V97.6.1776
Lonchay, C., van, derB. ruggenP., Connerotte, T., Hanagiri, T., Coulie, P., Colau, D., et al. (2004). Correlation between tumor regression and T cell responses in melanoma patients vaccinated with a MAGE antigen. Proc. Natl. Acad. Sci. U.S.A. 101(Suppl. 2), 14631–14638. doi:10.1073/pnas.0405743101
Mason, D. (1998). A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 19, 395–404. doi:10.1016/S0167-5699(98)01299-7
Matte, A., Bertoldi, M., Mohandas, N., An, X., Bugatti, A., Brunati, A. M., et al. (2012). Membrane association of peroxiredoxin-2 in red cells is mediated by the N-terminal cytoplasmic domain of band 3. Free Radic. Biol. Med. 55, 27–35. doi:10.1016/j.freeradbiomed.2012.10.543
Mézière, C., Viguier, M., Dumortier, H., Lo-Man, R., Leclerc, C., Guillet, J. G., et al. (1997). In vivo T helper cell response to retro-inverso peptidomimetics. J. Immunol. 159, 3230–3237.
Miles, J. J., Bulek, A. M., Cole, D. K., Gostick, E., Schauenburg, A. J., Dolton, G., et al. (2010). Genetic and structural basis for selection of a ubiquitous T cell receptor deployed in Epstein-Barr virus infection. PLoS Pathog. 6:e1001198. doi:10.1371/journal.ppat.1001198
Miles, J. J., Douek, D. C., and Price, D. A. (2011a). Bias in the alphabeta T-cell repertoire: implications for disease pathogenesis and vaccination. Immunol. Cell Biol. 89, 375–387. doi:10.1038/icb.2010.139
Miles, K. M., Miles, J. J., Madura, F., Sewell, A. K., and Cole, D. K. (2011b). Real time detection of peptide-MHC dissociation reveals that improvement of primary MHC-binding residues can have a minimal, or no, effect on stability. Mol. Immunol. 48, 728–732. doi:10.1016/j.molimm.2010.11.004
Misra, U. K., Adlakha, C. L., Gawdi, G., McMillian, M. K., Pizzo, S. V., and Laskowitz, D. T. (2001). Apolipoprotein E and mimetic peptide initiate a calcium-dependent signaling response in macrophages. J. Leukoc. Biol. 70, 677–683.
Morton, T. A., Bennett, D. B., Appelbaum, E. R., Cusimano, D. M., Johanson, K. O., Matico, R. E., et al. (1994). Analysis of the interaction between human interleukin-5 and the soluble domain of its receptor using a surface plasmon resonance biosensor. J. Mol. Recognit. 7, 47–55. doi:10.1002/jmr.300070107
Ogg, G. S., Jin, X., Bonhoeffer, S., Dunbar, P. R., Nowak, M. A., Monard, S., et al. (1998). Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science 279, 2103–2106. doi:10.1126/science.279.5359.2103
Parkhurst, M. R., Salgaller, M. L., Southwood, S., Robbins, P. F., Sette, A., Rosenberg, S. A., et al. (1996). Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J. Immunol. 157, 2539–2548.
Rammensee, H., Bachmann, J., Emmerich, N. P., Bachor, O. A., and Stevanović, S. (1999). SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50, 213–219. doi:10.1007/s002510050595
Rammensee, H. G., Friede, T., and Stevanoviíc, S. (1995). MHC ligands and peptide motifs: first listing. Immunogenetics 41, 178–228. doi:10.1007/BF00172063
Rosenberg, S. A., Yang, J. C., and Restifo, N. P. (2004). Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 10, 909–915. doi:10.1038/nm1204-1279b
Sewell, A. K. (2012). Why must T cells be cross-reactive? Nat Rev. Immunol. 12, 669–677. doi:10.1038/nri3279
Smith, C., Miles, J. J., and Khanna, R. (2012). Advances in direct T-cell alloreactivity: function, avidity, biophysics and structure. Am. J. Transplant. 12, 15–26. doi:10.1111/j.1600-6143.2011.03863.x
Speiser, D. E., Baumgaertner, P., Voelter, V., Devevre, E., Barbey, C., Rufer, N., et al. (2008). Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proc. Natl. Acad. Sci. U.S.A. 105, 3849–3854. doi:10.1073/pnas.0800080105
Stemmer, C., Quesnel, A., Prévost-Blondel, A., Zimmermann, C., Muller, S., Briand, J. P., et al. (1999). Protection against lymphocytic choriomeningitis virus infection induced by a reduced peptide bond analogue of the H-2Db-restricted CD8(+) T cell epitope GP33. J. Biol. Chem. 274, 5550–5556. doi:10.1074/jbc.274.9.5550
Tangri, S., Ishioka, G. Y., Huang, X., Sidney, J., Southwood, S., Fikes, J., et al. (2001). Structural features of peptide analogs of human histocompatibility leukocyte antigen class I epitopes that are more potent and immunogenic than wild-type peptide. J. Exp. Med. 194, 833–846. doi:10.1084/jem.194.6.833
Terasawa, H., Tsang, K. Y., Gulley, J., Arlen, P., and Schlom, J. (2002). Identification and characterization of a human agonist cytotoxic T-lymphocyte epitope of human prostate-specific antigen. Clin. Cancer Res. 8, 41–53.
van der Merwe, P. A., and Davis, S. J. (2003). Molecular interactions mediating T cell antigen recognition. Annu. Rev. Immunol. 21, 659–684. doi:10.1146/annurev.immunol.21.120601.141036
Wang, P., Sidney, J., Sette, A., and Peters, B. (2011). A computational pipeline to generate MHC binding motifs. Immunome Res. 7, 3.
Webb, A. I., Dunstone, M. A., Williamson, N. A., Price, J. D., de, K. auweA., Chen, W., et al. (2005). T cell determinants incorporating beta-amino acid residues are protease resistant and remain immunogenic in vivo. J. Immunol. 175, 3810–3818.
Wolfson, L. J., Gasse, F., Lee-Martin, S. P., Lydon, P., Magan, A., Tibouti, A., et al. (2008). Estimating the costs of achieving the WHO-UNICEF global immunization vision and strategy, 2006-2015. Bull. World Health Organ. 86, 27–39. doi:10.2471/BLT.07.045096
Wooldridge, L., Ekeruche-Makinde, J., van, denB. ergH. A., Skowera, A., Miles, J. J., Tan, M. P., et al. (2012). A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem. 287, 1168–1177. doi:10.1074/jbc.M111.289488
Citations: Pentier JM, Sewell AK and Miles JJ (2013) Advances in T-cell epitope engineering. Front. Immunol. 4:133. doi: 10.3389/fimmu.2013.00133
Received: 10 May 2013; Accepted: 21 May 2013;
Published online: 05 June 2013.
Edited by:
Nilabh Shastri, University of California Berkeley, USACopyright: © 2013 Pentier, Sewell and Miles. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence:am9obi5taWxlc0BxaW1yLmVkdS5hdQ==