

- Section of Rheumatology, Department of Pediatrics, Medical College of Wisconsin, Milwaukee, WI, USA
CD4+ CD25+ Foxp3+ regulatory T (Treg) cells are essential to the balance between pro- and anti-inflammatory responses. There are two major subsets of Treg cells, “natural” Treg (nTreg) cells that develop in the thymus, and “induced” Treg (iTreg) cells that arise in the periphery from CD4+ Foxp3− conventional T cells and can be generated in vitro. Previous work has established that both subsets are required for immunological tolerance. Additionally, in vitro-derived iTreg cells can reestablish tolerance in situations where Treg cells are decreased or defective. This review will focus on iTreg cells, drawing comparisons to nTreg cells when possible. We discuss the molecular mechanisms of iTreg cell induction, both in vivo and in vitro, review the Foxp3-dependent and -independent transcriptional landscape of iTreg cells, and examine the proposed suppressive mechanisms utilized by each Treg cell subset. We also compare the T cell receptor repertoire of the Treg cell subsets, discuss inflammatory conditions where iTreg cells are generated or have been used for treatment, and address the issue of iTreg cell stability.
Introduction
Early insights into the existence of a subset of T cells capable of exhibiting dominant tolerance, or suppression of other cells in a paracrine manner, came from work done in neonatal thymectomy models. Neonatal thymectomy of newborn mice between days 2 and 4 of life resulted in various organ-specific T cell-mediated autoimmune diseases that could be prevented by CD4+ CD25+ T cells (Nishizuka and Sakakura, 1969; Sakaguchi et al., 1982, 1995; Asano et al., 1996). The discovery of mutations in the X chromosome-encoded gene Foxp3 in human patients suffering from immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome and in the mutant scurfy mice led to recent advances in regulatory T (Treg) cell biology (Chatila et al., 2000; Bennett et al., 2001; Brunkow et al., 2001; Wildin et al., 2001). Expression of the forkhead/winged helix transcription factor Foxp3 ultimately identifies Treg cells and is essential for the acquisition of suppressive function (Lin et al., 2007; Zheng and Rudensky, 2007). Conditional deletion of Foxp3 via retroviral expression of Cre in mature Treg cells resulted in the loss of Treg cell suppressive function and the gain of effector T cell properties, suggesting that continuous expression of Foxp3 is required for maintenance of the Treg cell phenotype (Williams and Rudensky, 2007). Furthermore, in a system where Treg cells express the human diphtheria toxin receptor, chronic diphtheria toxin-mediated ablation of Treg cells resulted in death from lympho- and myeloproliferative disease, confirming the continued need for Treg cells throughout the lifespan of normal mice (Kim et al., 2007).
These CD4+ CD25+ Foxp3+ Treg cells, which account for ∼10% of peripheral CD4+ T cells, are essential to the balance between pro- and anti-inflammatory responses at mucosal surfaces. There are two subsets of Treg cells, “natural” Treg (nTreg) cells and “induced” Treg (iTreg) cells. While nTreg cells develop as a distinct lineage in the thymus, iTreg cells arise from peripheral naïve conventional T (Tconv) cells and can be generated in vitro (Curotto de Lafaille and Lafaille, 2009). The focus of this review is iTreg cells, their mechanisms of generation, transcriptional profiles, TCR repertoires, potential for immunotherapy, and their stability in vivo.
In Vivo and In Vitro Generation of iTreg Cells
CD4+ Tconv cells isolated from lymphoid organs and peripheral blood can be induced to express Foxp3 in vitro by T cell activation in the presence of TGF-β1 and IL-2 (Chen et al., 2003; Davidson et al., 2007). Following these important observations, several studies documented the development of functionally suppressive iTreg cells in vivo, either in a tolerogenic setting or arising during inflammation (Table 1). The emergence of iTreg cells has been observed in cases where antigens are encountered in the absence of optimal costimulation. This includes antigen delivery through intravenous injection (Thorstenson and Khoruts, 2001) and continuous infusion minipumps (osmotic pumps) (Apostolou and von Boehmer, 2004), or by the administration of non-depleting anti-CD4 antibodies (Cobbold et al., 2004). Oral administration of antigen leads to the development of iTreg cells that are functionally suppressive in a mouse model of asthma and are required to establish oral tolerance (Mucida et al., 2005; Curotto de Lafaille et al., 2008). Suboptimally activated dendritic cells support iTreg cell development. For example, dendritic cells targeted with low dose antigens by anti-DEC-205 (dendritic and epithelial cells, 205 kDa, multilectin endocytic receptor) antibodies (Kretschmer et al., 2005) and tolerogenic dendritic cells, residing in the small intestine lamina propria and mesenteric lymph node (Coombes et al., 2007; Sun et al., 2007), promote iTreg cell generation. In addition, several studies have demonstrated that the commensal microbiota contribute to iTreg cell development (Round and Mazmanian, 2010; Atarashi et al., 2011; Geuking et al., 2011). Alternatively, iTreg cells can be generated during states of chronic inflammation. Examples where chronic inflammation may support iTreg development include mouse models of asthma (Curotto de Lafaille et al., 2008; Weiss et al., 2012), colitis that occurs during T cell expansion in a lymphopenic environment (Haribhai et al., 2009), adoptive transfer immunotherapy for the treatment of Foxp3-deficiency (Haribhai et al., 2011), and infection with intestinal parasites (Grainger et al., 2010). The extent of iTreg cell development in locations other than mucosal tissues is not as well documented. However, recently iTreg cell development has been demonstrated to occur locally in immune privileged sites such as the spinal cords of mice with experimental autoimmune encephalomyelitis (Weiss et al., 2012) and in the eye (Zhou et al., 2012; McPherson et al., 2013). Tissue-specific Foxp3 induction has also been demonstrated in response to a neo-self antigen restricted to the pancreas (Thompson et al., 2011). These data generally support the biological relevance of the mechanisms that generate and sustain iTreg cells.
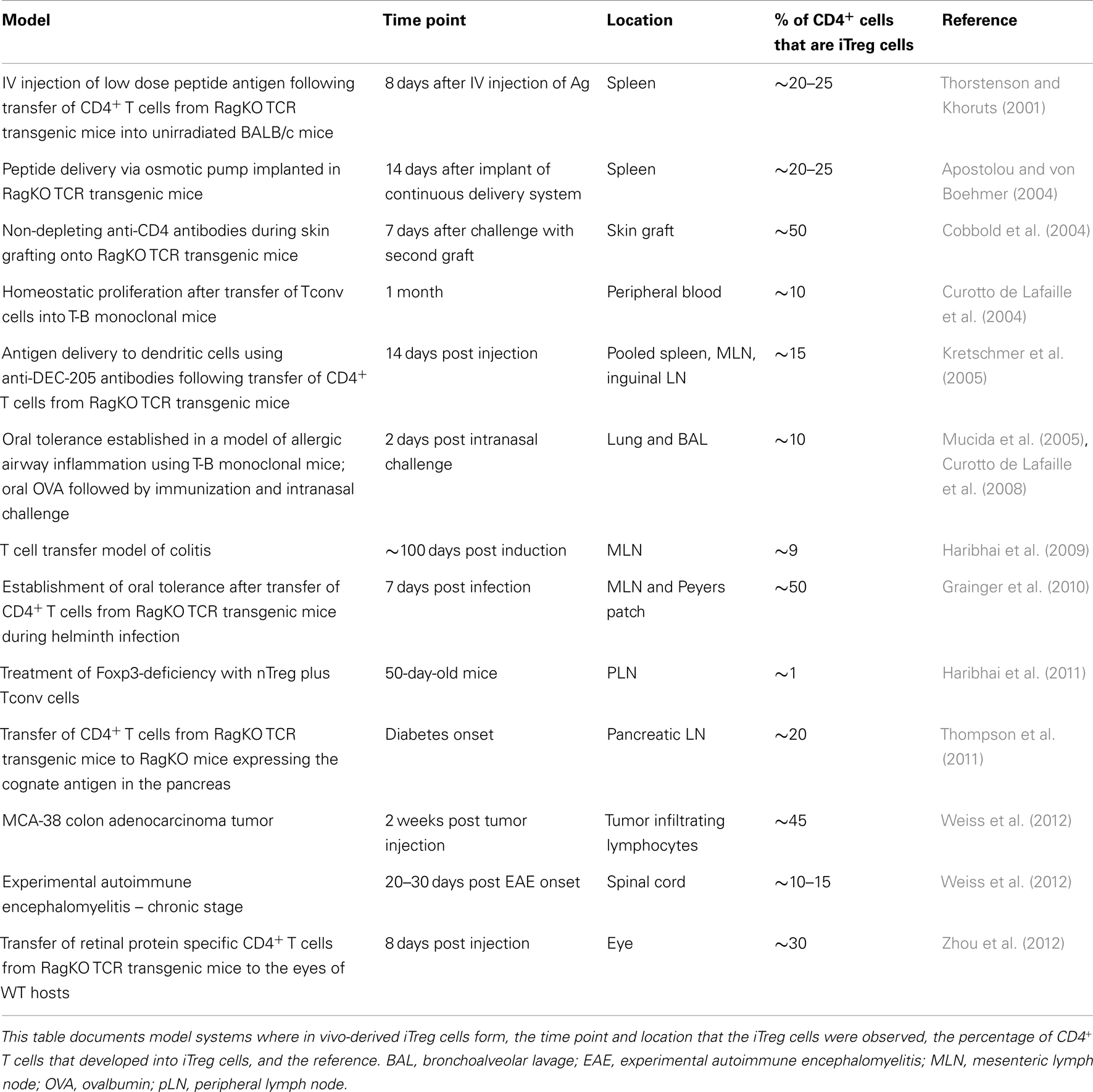
Table 1. Models generating in vivo-derived iTreg cells.
Multiple signaling pathways converge to influence the efficiency of iTreg cell generation. Specific TCR affinity and TCR-derived signals, costimulatory molecules, and cytokines promote optimal in vivo iTreg cell development. Low doses of high affinity ligands promote iTreg cell generation by creating a decreased aggregate TCR stimulation as compared to Tconv cells (Kretschmer et al., 2005; Gottschalk et al., 2010). Strong CD28 costimulation (Semple et al., 2011) and CTLA-4 blockade (Zheng et al., 2006) are detrimental to de novo induction of Foxp3 whereas activation of Tconv cells under conditions of suboptimal costimulation promotes the induction of Foxp3. Furthermore, signaling via the programed death (PD) 1-PD-ligand (PD-L) pathway promotes both the induction and maintenance of iTreg cells (Francisco et al., 2009). TCR-dependent activation of the PI3K-AKT-mTOR axis is an important negative regulator of peripheral Treg cell differentiation. AKT inhibits Foxo proteins, which normally facilitate Foxp3 induction (Kerdiles et al., 2010; Ouyang et al., 2010). Therefore, enhancing AKT signaling, either by overexpression (Haxhinasto et al., 2008) or by deletion of negative regulators of AKT, such as phosphatase and tensin homolog (PTEN) (Sauer et al., 2008) or the E3 ubiquitin ligase Cbl-b that degrades the regulatory subunit of PI3K (Wohlfert et al., 2006; Harada et al., 2010), adversely impacts iTreg cell development. Alternatively, inhibition of PI3K or mTOR enhances iTreg cell development (Battaglia et al., 2005; Sauer et al., 2008). Blockade of signals through the C3aR and C5aR complement receptors also decreases signaling through the PI3K-AKT-mTOR pathway thereby enhancing autoinductive signaling by TGF-β1 to generate iTreg cells (Strainic et al., 2013).
Both TGF-β1 and IL-2 are required for iTreg cell induction. TGF-β1 signaling promotes the binding of NFAT and Smad3 to the conserved non-coding sequence-1 (CNS1) enhancer and ultimately stimulates histone acetylation and Foxp3 induction (Tone et al., 2008). These data are further supported by the observation that CNS1 deletion impairs iTreg cell generation in gut-associated lymphoid tissues (Zheng et al., 2010). TGF-β1 also limits DNA methyltransferase I recruitment to the Foxp3 locus, a molecule that normally functions to prohibit promiscuous Foxp3 induction after TCR stimulation (Josefowicz et al., 2009). IL-2 is likewise required for iTreg generation in vitro (Davidson et al., 2007). In vivo, IL-2 has a role in Treg cell survival (D’Cruz and Klein, 2005), proliferation (Fontenot et al., 2005a), and stability (Chen et al., 2011) therefore a role for in vivo induction has been more difficult to parse out. Perhaps in support of a role for induction, cells in the periphery that are poised to develop into iTreg cells require only IL-2 for Foxp3 induction (Schallenberg et al., 2010). IL-2 also functions to limit the polarization of activated CD4+ T cells into the Th17 lineage (Laurence et al., 2007). Similar to IL-2, all-trans retinoic acid restricts reciprocal Th17 polarization (Xiao et al., 2008). CD103+ gut-derived tolerogenic dendritic cells, which play an important role in the generation of iTreg cells serve as a source of retinoic acid (Coombes et al., 2007). Activation of the aryl hydrocarbon receptor by the ligands 2,3,7,8-tetrachlorodibenzo-p-dioxin and 2-(1’H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester supports the generation of functional, stable iTreg cells by promoting both the generation of retinoic-acid producing tolerogenic dendritic cells and demethylation of the Foxp3 promoter (Quintana et al., 2008, 2010; Singh et al., 2011). In summary, antigenic TCR stimulation with low dose/high affinity ligands, suboptimal costimulation, TGF-β1, IL-2, and retinoic acid all facilitate the induction of Foxp3 expression in peripheral CD4+ Tconv cells in vivo.
Transcriptional Landscape and Function of iTreg Cells Versus nTreg Cells
The pivotal role of the X-linked gene Foxp3 in the identity of a Treg cell prompted investigation into the Foxp3-dependent and -independent programs of the Treg cell transcriptional signature. Mice possessing an altered Foxp3 locus, in which DNA encoding EGFP was inserted in frame into exon 11 at the C-terminal end of the Foxp3 locus (Foxp3ΔEGFP), express a non-functional ΔFoxp3-EGFP fusion protein that is devoid of the nuclear localization sequence and residues involved in DNA binding (Lin et al., 2007). In heterozygous Foxp3ΔEGFP± female mice, which have random inactivation of one of the two X chromosomes, the frequency of thymocytes expressing the non-functional ΔFoxp3-EGFP fusion protein was similar to thymocytes expressing normal Foxp3. The EGFP+ cells from these mice also expressed several Treg cell-associated molecules, such as CD25, CTLA-4, GITR, and CD44, but were not suppressive and produced Th1- and Th2-associated cytokines. Many transcripts commonly found in Treg cells were identified by gene array in the EGFP+ cells, these included Il2ra, Ctla4, and Itgae. The expression of additional genes suggestive of a cytotoxic effector program, such as Gzma, Gzmb, and Gzmk, and genes encoding chemokine receptors such as Cxcr6, were also observed (Lin et al., 2007).
In a separate set of studies, cells destined to be Treg cells were marked with an in frame insertion of GFP into a Foxp3 locus disrupted by a stop codon. This resulted in Foxp3 transcription, but not translation, and also allowed for the separation of Foxp3-dependent and independent factors (Gavin et al., 2007). As a result, several characteristic Treg cell markers, such as CD25, CD44, CTLA-4, GITR, and ICOS, were found to be Foxp3-independent. Although several hallmark Treg cell markers were found, suppressive activity was lost in the absence of Foxp3 protein. These studies confirmed that Foxp3 suppressive function and stability are dependent on a functional Foxp3 protein. Together, they suggest that some aspects of commitment to the Treg cell lineage begin independently of a functional Foxp3 protein.
Fundamental work from Hill et al. (2007) combined gene expression profiles of Treg cells obtained under many different conditions and identified a canonical Treg cell signature. This study confirmed previous work, in that it identified Treg cell-associated genes that were not correlated with Foxp3 expression, but they also organized the Treg signature into several co-regulated gene clusters influenced by a defined set of factors. This Treg cell transcriptional signature provides a framework for comparison of Treg cells derived by alternative methods or in varying anatomical locations. Treg cells found in different anatomical locations within the same individual have unique TCR repertoires, variations in their cell surface phenotypes, and distinct gene expression profiles (Lathrop et al., 2008; Feuerer et al., 2010). These findings are consistent with the idea that subsets of Treg cells exist, and that Treg cell suppressive activity may be finely tuned to the microenvironment. Currently there is no consistent, reliable marker to distinguish nTreg and iTreg cells in vivo, although in some systems Helios (Thornton et al., 2010) and Neuropilin 1 (Nrp1) (Weiss et al., 2012; Yadav et al., 2012) have been suggested to specifically identify nTreg cells. Others have determined that expression of Helios, an Ikaros family transcription factor, results from more general T cell activation and proliferation (Akimova et al., 2011). Nrp1 is a receptor for TGF-β1 and has been reported to activate the latent form of TGF-β1 and promote Treg cell activity (Glinka and Prud’homme, 2008). Under homeostatic conditions, this marker seems to reliably distinguish nTreg cells from iTreg cells; however, iTreg cells present in inflammatory conditions can express Nrp1 (Weiss et al., 2012). The lack of a suitable surface marker has hampered the ability to effectively distinguish the characteristics of the two subsets in a host without using a transfer model to mark the populations.
Many studies have compared the transcriptional signatures of nTreg and iTreg cells in an attempt to distinguish the two subsets. Given that a portion of the Treg cell signature is Foxp3-independent, it was not surprising that the transcriptional signature of iTreg cells derived in vitro did not fully recapitulate the observed nTreg cell genetic signature (Haribhai et al., 2009; Feuerer et al., 2010). On the other hand, iTreg cells that were allowed to develop in vivo were more similar to nTreg cells than their in vitro-derived counterparts (Feuerer et al., 2010; Haribhai et al., 2011). However, nTreg cells and in vitro-derived iTreg cells that are stably maintained in vivo for approximately 3 months share similar transcriptional profiles (Schmitt et al., 2012). This included the expression of many genes associated with Treg cell suppressive function such as Il2ra, Ctla4, Gzmb, and Il10. Thus, the transcriptional signature of in vitro-derived iTreg cells and nTreg cells, although much different immediately after generation in vitro, converge as the in vitro-derived iTreg cells are selected and maintained in vivo. While the collective gene expression data suggest that the two Treg subsets share similar suppressive mechanisms, the observed requirement for both subsets in maintaining tolerance hints that distinct suppressive mechanisms that play discrete roles, either in different anatomical locations or in different types of inflammation, may yet be identified. Indeed, a recent study uncovered four “Treg cell-representative regions” which included regions of Foxp3, Tnfrsf18, Ctla4, and Ikzf4 that display demethylation patterns in nTreg cells that are distinct from those observed in Tconv and iTreg cells. This nTreg cell-specific methylation pattern is instrumental in establishing Treg cell-type gene expression (Ohkura et al., 2012). Additionally, recent work demonstrated an important role for Foxo1 in establishing the Foxp3-independent Treg cell transcriptional program, in part by inhibiting IFN-γ expression in Treg cells (Ouyang et al., 2012).
The interaction of Foxp3 with several different molecules is important for Treg cell transcriptional activity. The Foxp3 gene has numerous structural domains including a transcriptional repression domain at the N-terminus, followed by a zinc finger domain, a leucine zipper domain, and a forkhead DNA binding domain. A series of serendipitous discoveries using a Foxp3GFP (Foxp3tm2Ayr) fusion protein to mark Treg cells, in which GFP is fused to the amino terminus of Foxp3 (Fontenot et al., 2005b), revealed altered autoimmune disease phenotypes. The Foxp3GFP fusion protein reduces or eliminates the interaction of the N-terminal domain of the Foxp3 gene with Eos, Tip60, HDAC7, and HIF-1α; however, distal interactions with NFAT, AML1/Runx-1, RORα, and IRF4 are maintained or enhanced. As a result, the transcriptional activity of Treg cells was altered leading to accelerated type 1 diabetes in disease prone NOD mice (Bettini et al., 2012) while protecting mice from autoimmune arthritis in the K/BxN model (Darce et al., 2012).
Much work has been done to uncover the molecular mechanisms of Treg cell suppressive activity delineated by the transcriptional data. However, there have been few attempts to discriminate between the two subsets. Consequently, with regard to the specific mechanisms utilized to control inflammation, the “division of labor” between nTreg cells and iTreg cells remains largely unresolved (Curotto de Lafaille and Lafaille, 2009). In general, Treg cell suppression has been demonstrated to modify effector cell activity at several different stages within the immune response (Suri-Payer et al., 1998). Suppression by Treg cells can operate at the early stages, by limiting cell activation and proliferation. Initial studies using in vitro proliferation assays demonstrated the ability of Treg cells to control effector cell proliferation in an IL-2 dependent manner (Thornton and Shevach, 1998). Gene expression profiling of the suppressed CD4+ T cells subsequently showed the induction of genes involved in growth arrest or the inhibition of proliferation (Sukiennicki and Fowell, 2006). In the later stages of the immune response, Treg cells have been shown to control effector cell differentiation and function in the target tissues (Oldenhove et al., 2003; Sarween et al., 2004; DiPaolo et al., 2005). The ability of Treg cells to effectively control diverse types of inflammation has been associated with Treg cell upregulation of specific transcription factors (Campbell and Koch, 2011). Treg cell expression of T-bet, IRF4, and STAT3 contribute to the ability of Treg cells to control the associated Th1 (Koch et al., 2009), Th2 (Zheng et al., 2009), and Th17 (Chaudhry et al., 2009) polarized inflammation, respectively. In addition, Treg cell expression of GATA-3 is important for their accumulation at the site of inflammation as a Treg cell-specific deletion of GATA-3 led to a failure of Treg cell accumulation in tissues and the acquisition of effector cytokine production (Wohlfert et al., 2011). Thus, it appears that Treg cells possess the ability to express transcription factors associated with the type of inflammation they are controlling, which in turn provides them with the ability to adapt their suppressive program to the surroundings.
Various molecular mechanisms of Treg cell-mediated suppression have been proposed. These suppressive mechanisms fall into three broad categories: suppression mediated by cell–cell contact, metabolic disruption, and the secretion of inhibitory cytokines (Figure 1). Cell–cell contact suppression operates via molecules such as CTLA-4 (Wing et al., 2008) and LAG-3 (Liang et al., 2008), which may function to modulate the immunostimulatory capacity of dendritic cells. In addition, Treg cells secrete cytotoxic molecules such as Granzyme B, which is presumed to require cell–cell contact (Grossman et al., 2004; Gondek et al., 2005). Metabolic disruption can occur via the delivery of cAMP to effector T cells through gap junctions (Bopp et al., 2007). The ectoenzymes CD39 and CD73 on Treg cells generate adenosine, which binds the adenosine receptor 2A on effector T cells and increases intracellular cAMP to suppress their function (Deaglio et al., 2007). Lastly, the increased constitutive expression of CD25 on Treg cells may allow them to out-compete effector cells for the growth factor IL-2, leading to cytokine deprivation-induced apoptosis of the effector T cells (de la Rosa et al., 2004; Pandiyan et al., 2007). Inhibitory cytokines such as TGF-β1 (Powrie et al., 1996), IL-35 (Collison et al., 2007), and IL-10 (Asseman et al., 1999) have been implicated in Treg cell suppressive function, and may serve to specifically dampen the activation of antigen presenting cells or inhibit effector T cell proliferation.
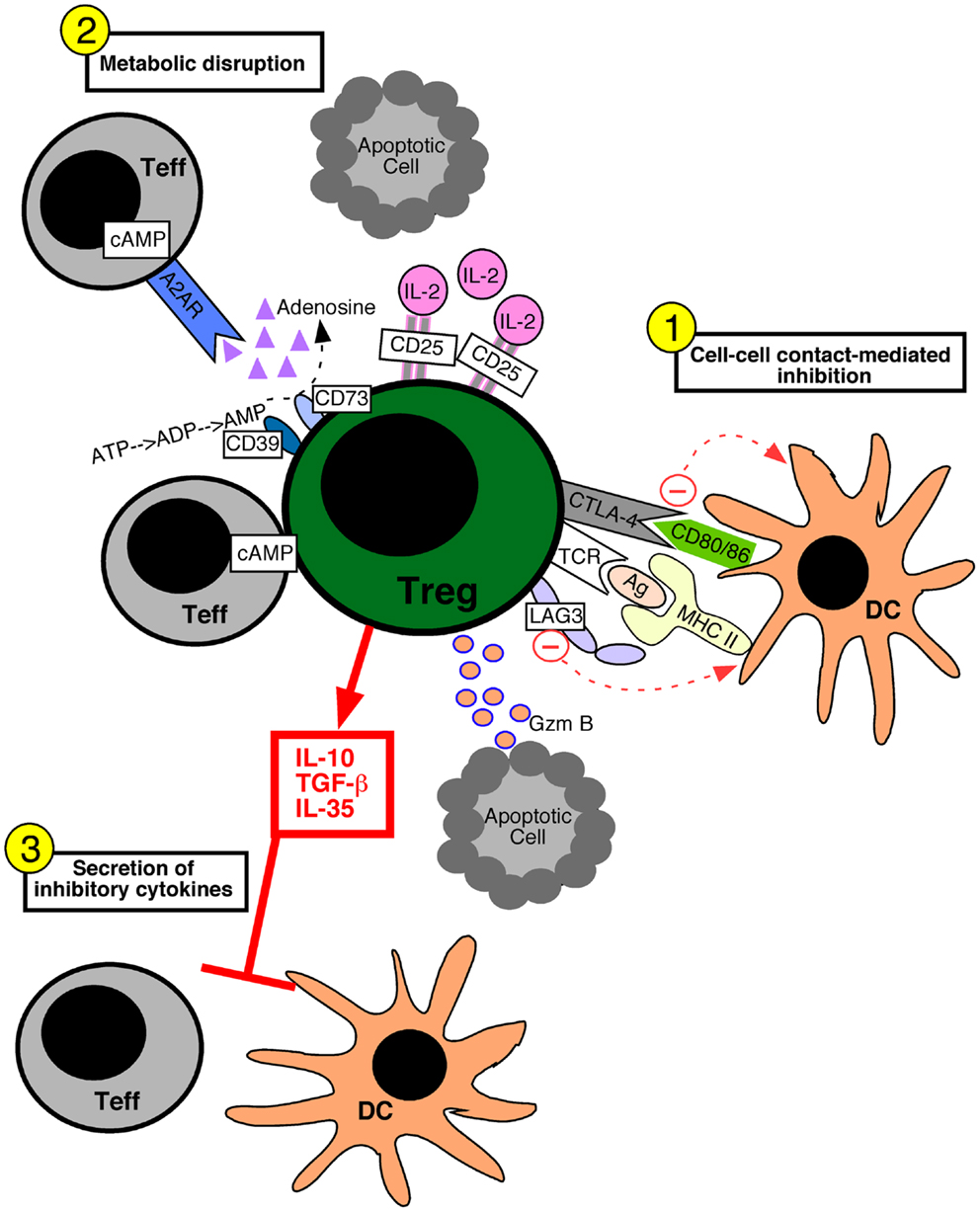
Figure 1. Mechanisms of regulatory T cell-mediated suppression. Regulatory T (Treg) cells can utilize several different suppressive mechanisms falling into three broad categories: (1) cell–cell contact-mediated suppression, (2) the metabolic disruption of effector T (Teff) cells, and (3) the secretion of inhibitory cytokines. (1) Contact-mediated suppression dampens the immunostimulatory properties of dendritic cells (DC) and occurs via the engagement of Treg cell inhibitory receptors such as CTLA-4 and LAG-3 with CD80/86 and MHC molecules on the DC, respectively. Delivery of granzyme B (Gzm B) to Teff cells leads to apoptosis. (2) Metabolic disruption of effector T cells is mediated by Treg cell delivery of cAMP to effector T cells via gap junctions, the generation of adenosine by the Treg cell ectoenzymes CD39 and CD73 which acts on Teff cell adenosine receptors (A2AR), and by Treg cell consumption of IL-2 thereby depriving Teff cells of growth factors. (3) Treg cells secrete inhibitory cytokines such as IL-10, IL-35, and TGF-β1, which inhibit both T cells and DCs.
The immunomodulatory cytokine IL-10 has been studied extensively in relation to Treg cell biology. IL-10 is particularly important for Treg cells at environmental interfaces, as a Treg cell-specific inactivation of IL-10 results in spontaneous colitis, heightened immune-mediated lung hyperreactivity, and increased skin sensitivity (Rubtsov et al., 2008). Treg cell-derived IL-10 controls Th17 cells and a unique population of T cells displaying features of both Th1 and Th17 cells (Th1 + Th17) in a transfer model of colitis (Huber et al., 2011). In a model where Foxp3-deficiency was treated with nTreg cells plus in vivo-derived iTreg cells, gene expression profiling revealed that both Treg cell types over-expressed Il10 as compared to naïve Tconv cells, suggesting a possible role for IL-10 as an iTreg cell mechanism of suppression (Haribhai et al., 2011). Recently, it was demonstrated that IL-10 produced by iTreg cells could replace nTreg cell-derived IL-10 in the cure of experimental colitis. Reversal of the experimental conditions was similarly effective, defining the novel principle of reciprocal compensation between Treg cell subsets, which was necessary to establish tolerance in this model (Schmitt et al., 2012). This work also demonstrated that iTreg cells limited the frequency of ex-iTreg cells adopting a Th1, Th17, or Th1 + Th17 cell fate, in concordance with previous data looking at the function of nTreg cells (Huber et al., 2011). Thus, it is possible that under certain circumstances, both Treg subsets must possess the ability to operate via the same mechanism. Further studies are needed to determine whether the principle of reciprocal compensation is model-specific or can be globally applied in situations where a known Treg cell defect exists.
T Cell Receptor Repertoire of the Treg Cell Subsets
In the thymus, developing Tconv cells and nTreg cell precursors have unique affinity requirements (Jordan et al., 2001; Apostolou et al., 2002). Induction of Foxp3 requires an agonist self-peptide, and the frequency of nTreg cells that develop is directly proportional to the strength of the signal (Relland et al., 2009). Furthermore, autoreactivity of the nTreg cell compartment has been demonstrated, despite a normal response to central tolerance mechanisms (Romagnoli et al., 2002; Hsieh et al., 2004). Given the observed bias of nTreg cells to self antigen, several studies have sought to compare the TCR repertoires of nTreg and Tconv cells. Studies that have reported differences between nTreg and Tconv repertoires have analyzed the TCRα complementarity determining region (CDR3) of mice with fixed transgenic TCRβ chains and a restricted Tcra locus. In these reports, the nTreg and Tconv TCR repertoires were found to be equally diverse, however the degree of observed overlap between the two populations varied (Hsieh et al., 2004, 2006; Pacholczyk et al., 2006, 2007; Wong et al., 2007b). In a separate system with limited diversity, the repertoire of the nTreg cells responding to a foreign antigen was found to be more limited and clonally distinct compared to Tconv cells also responding to the antigen (Relland et al., 2012). In contrast to the self-specificity seen in the nTreg cell population, iTreg cells are thought to be specific for foreign antigen, given that the iTreg cell population is derived from the Tconv cell pool. Therefore, it was not surprising that the iTreg cell TCR repertoire shared minimal overlap with that of nTreg cells (Haribhai et al., 2011). This limited overlap may in part contribute to the requirement for both nTreg and iTreg cells in the resolution of autoimmune diseases, as the combination provides a more diverse TCR repertoire. Evidence from a handful of TCR repertoire studies suggests that iTreg formation in non-mucosal tissues, such as in the central nervous system of mice with experimental autoimmune encephalomyelitis (Liu et al., 2009) and in the pancreas of diabetic mice (Wong et al., 2007a), may be limited. Minimal TCR repertoire overlap was observed between Tconv and Treg cells at these locations, supporting a role for Treg cell recruitment rather than induction. Furthermore, mice that lack iTreg cells due to a genetic ablation of the intronic Foxp3 enhancer CNS1 maintain tolerance to systemic and tissue-specific antigens but develop inflammation at the mucosal interfaces of the lung and gastrointestinal tract (Josefowicz et al., 2012). Interestingly, CNS1 deficient mice also display increased fetal resorption due to a lack of fetal alloantigen-specific iTreg cells (Samstein et al., 2012). These data support the notion that iTreg cell TCRs may function to expand tolerance to non-self antigens, particularly those present at mucosal interfaces.
To gain further insight into the TCR specificity of Treg cells, several groups have created TCR transgenic mice that harbor a TCR derived from a Treg cell (Bautista et al., 2009; Leung et al., 2009). Interestingly, nTreg cells were only efficiently generated when the transgenic cells were present at a low clonal frequency. These studies suggested that the development of nTreg cells is a saturable process that plateaus, most likely due to intraclonal competition for MHC/peptide complexes. However, recent work has demonstrated that a limited, fixed pool of in vitro-derived iTreg cells contains a large number of clones with TCRs that can be maintained within the iTreg cell niche, and mice receiving equivalent numbers of the same iTreg cells maintained distinct clones (Schmitt et al., 2012). This is in agreement with previous work demonstrating that high TCR diversity is important for the optimal function of Treg cells in a model of experimental acute GVHD (Fohse et al., 2011). In contrast to the nTreg cell niche, the iTreg cell niche is probably not constrained by the number of available antigens, given the proposed specificity for non-self and the complexity of the microbiome. This suggests that the size of the iTreg cell population may not be limited by TCR specificity, but may be determined by other factors such as the number of tolerogenic antigen presenting cells (Coombes et al., 2007), local concentrations of TGF-β1 (Marie et al., 2005; Li et al., 2006) and IL-2 (Chen et al., 2011), and signaling via the PD 1–PD-L pathway (Francisco et al., 2009). In addition, members of the Tumor Necrosis Factor Receptor superfamily expressed on Treg cells, including GITR (Ray et al., 2012) and OX40 (Piconese et al., 2010), have also been shown to be important for Treg cell proliferative fitness. Increased IL-2 signaling, via administration of IL-2 immune complexes or through constitutive STAT5b signaling, allows for Treg cell division in the absence of TCR signaling (Zou et al., 2012). During the treatment of autoimmune conditions, such as experimental colitis, high levels of IL-2 could allow for the maintenance of a diverse population of iTreg cells. Other cell types, such as IL-10-producing CXCR1+ macrophages in the lamina propria are important for Treg cell proliferation in the setting of oral tolerance, and may contribute to the size and composition of the iTreg cell population (Hadis et al., 2011). It is also likely that the nTreg cell subset dictates the size of the iTreg cell niche, because in the absence of nTreg cells, the iTreg cell compartment expands ∼fivefold (Haribhai et al., 2009). A recent study demonstrated that in vitro-derived iTreg cells cotransferred with naïve T cells into Rag−/− hosts were not effective in preventing colitis and many of the iTreg cells had decreased Foxp3, CTLA-4, and CD25 expression (Ohkura et al., 2012). Thus, cooperation between nTreg and iTreg cells, which is essential to establish tolerance, could therefore influence the composition of the iTreg cell niche. Manipulation of the factors implicated in shaping the iTreg cell niche may provide a mechanism to control the size, specificity, and/or function of the iTreg cell compartment.
Immunotherapy with iTreg Cells
Statistics published by the National Institutes of Health indicate that chronic autoimmune disease affects ∼5–8% of the U.S. population, an estimated 14.7–23.5 million individuals, and the prevalence is rising (NIAID, 2005). Existing therapeutic approaches are inadequate and current research efforts must focus on restoring the balance between pro- and anti-inflammatory responses. A decrease in Treg cell numbers and/or function has been associated with many human autoimmune diseases (Long and Buckner, 2011). Currently, ex vivo expanded nTreg cells are being used in umbilical cord blood transplantation clinical trials, where the benefit to risk ratio is high due to the risk of life-threatening GVHD (Brunstein et al., 2011). Although nTreg cells were functionally suppressive in vivo after several rounds of stimulation and expansion, the optimal ≥1:1 nTreg to peripheral blood mononuclear cell ratio could not be achieved (Hippen et al., 2011b). Therefore iTreg cells, which can be generated in large numbers ex vivo and have been shown to operate in a xenogeneic model of GVHD, may offer an alternative to nTreg cells (Hippen et al., 2011a). The ability of iTreg cells to be generated in large numbers makes them an attractive alternative for the treatment of human autoimmune disorders unresponsive to current approaches (Trzonkowski et al., 2009; Brunstein et al., 2011; Di Ianni et al., 2011; Hippen et al., 2011a). In vitro-derived iTreg cells are functionally suppressive in animal models of inflammatory bowel disease (Fantini et al., 2006), diabetes (Weber et al., 2006), autoimmune gastritis (DiPaolo et al., 2007), experimental autoimmune encephalitis (Selvaraj and Geiger, 2008), and Foxp3-deficiency (Huter et al., 2008). Notably, in vitro-derived iTreg cells contribute to tolerance in disease models where in vivo-derived iTreg cells are absent (Haribhai et al., 2009, 2011). Moreover, iTreg cells can be used to augment and restore regulatory networks in situations where nTreg cells are exhausted or defective (Schmitt et al., 2012). Yet, in many of these models the specific Treg cell suppressive mechanism that is important and functional at an individual site of inflammation remains poorly understood. It is likely that iTreg cells can operate via multiple means but that particular suppressive mechanisms may vary in importance in each autoimmune disease or in different stages of the same disease.
A recent phase 1/2a clinical study conducted in 20 patients with refractory Crohn’s disease demonstrated a clinically significant effect of a single infusion of Treg cells in 40% of patients 5 weeks post-infusion (Desreumaux et al., 2012). Patients’ cells were expanded in vitro in response to ovalbumin (OVA) and cloned by limiting dilution to generate IL-10–producing OVA-specific Treg cells. Whether these cells are functioning purely as IL-10–producing T regulatory (Tr1) cells or as Foxp3+ iTreg cells is unclear, as ∼60% of the OVA-Treg cells expressed Foxp3. For human cells, in vitro activation leads to Foxp3 expression within 48 h, peaking at 4–6 days, and diminishing by 10–14 days post-activation, leaving only a fraction of cells Foxp3+(Pillai et al., 2007). Bonafide human Foxp3+ Treg cells can be identified by characteristic epigenetic changes within the Foxp3 locus (Baron et al., 2007), however, tracking of the transferred cells was not feasible in this case (Desreumaux et al., 2012). Regardless of this caveat, both Foxp3+ and Foxp3− IL-10-producing regulatory cells can control pathogenic T helper cells in mouse models of intestinal inflammation (Huber et al., 2011). This initial clinical study provides the groundwork for additional research into adoptive transfer immunotherapy for autoimmune diseases refractory to current therapies.
Another issue with adoptive transfer immunotherapy is the in vivo stability of iTreg cells. In a model of experimental colitis, iTreg cells were recovered from successfully treated mice (Haribhai et al., 2009; Schmitt et al., 2012). Conversely, in a mouse model of GVHD these cells did not persist (Beres et al., 2011). Perhaps, the degree of ongoing inflammation will hamper the efficacy of these cells for therapy. If relevant clones could be pre-selected, enhancing the possibility that these cells will be expanded and/or maintained via interactions with their cognate ligand in vivo, this may increase the usefulness of iTreg cells. Further, excessive regulation may hamper normal immune responses to invading organisms, thus a fine balance between limiting disease progression and impeding natural responses to infectious agents needs to be established.
Stability of iTreg Cells
The self-specificity of nTreg cell TCRs creates the potential for autoimmunity that is averted by stable Foxp3 expression. Several recent studies have scrutinized the stability of nTreg cells, both long term and in pro-inflammatory conditions. Indeed, there is some disparity in the reports regarding Treg cell plasticity. On one hand, both nTreg and iTreg cells were shown to convert to a pro-inflammatory Th17 phenotype in the presence of IL-6, IL-1, and TGF-β1 in vitro (Yang et al., 2008). These “exFoxp3” cells were also tracked in a study using Foxp3-GFP-Cre BAC transgenic mice bred to mice that expresses YFP from the Rosa26 promoter after removal of a loxP-“stop” cassette (Rosa26-loxP-Stop-loxP-YFP). In this model, all cells that expressed Foxp3 at any time during their lifespan deleted the “stop” cassette and remained YFP+, thus marking “exFoxp3” cells with a YFP+ Foxp3− phenotype. These “exFoxp3” cells produced pro-inflammatory cytokines, were pathogenic, and the TCR repertoire analysis suggested that they were derived from both nTreg and iTreg cells (Zhou et al., 2009). In contrast to this report, a group that used an inducible labeling system found the nTreg cell population to be stable throughout the lifespan of the mouse and in the setting of Listeria infection, lymphopenia, and autoimmune inflammation (Rubtsov et al., 2010). In this model, mice with a Foxp3-eGFP-Cre-ERT2 (ERT2, mutated human estrogen receptor ligand-binding domain) fusion protein were bred to mice in which the Rosa26 locus contains a loxP site-flanked STOP cassette followed by YFP. The GFP-CreERT2 fusion protein is normally sequestered in the cytosol, but administration of tamoxifen allows nuclear localization and constitutive, heritable labeling of a cohort of Treg cells with YFP. The differences observed between these studies were attributed to the caveats with the BAC transgenic system, in which cells that transiently expressed Foxp3, prior to stabilization, would be labeled (Miyao et al., 2012). The latest labeling system revealed that mouse T cells can upregulate Foxp3 during activation (Miyao et al., 2012), as observed with human T cells (Pillai et al., 2007), and that this promiscuous Foxp3 expression accounts for the documented instability of the Treg cell lineage. Taken together, these results demonstrate that nTreg cells express Foxp3 in a stable, heritable fashion.
Analysis of the Foxp3 locus revealed three intronic elements within the proximal CNS that influence the composition, stability, and size of the Treg cell compartment (Zheng et al., 2010). To determine the function of the CNS elements in vivo, individual deletions of each CNS element were created. These analyses revealed CNS1, which contains binding sites for NFAT, RAR/RXR, and Smad3, to be particularly important for the development of iTreg cells. In CNS1 knockout mice the efficiency of in vivo and in vitro generation of iTreg cells was reduced. CNS2 was shown to be important in the heritable maintenance of Foxp3. The CpG motifs in CNS2, also known as the Treg cell-specific demethylated region (TSDR), are demethylated in nTreg cells, but not in iTreg cells produced in vitro (Floess et al., 2007; Polansky et al., 2008) (Table 2). Interestingly, in vitro-derived iTreg cells that were stably maintained in vivo for ∼3 months could achieve at least partial demethylation of the TSDR (Schmitt et al., 2012). Treatment with inhibitors of DNA methyltransferases (Polansky et al., 2008; Lal et al., 2009) and histone deacetylases (Tao et al., 2007) can enhance the stability of Foxp3 expression. In a similar fashion, progesterone (Lee et al., 2012), rapamycin (Battaglia et al., 2005), and retinoic acid (Mucida et al., 2009) promote iTreg cell stability and/or generation, and could be incorporated into in vitro induction protocols to create stable iTreg cells for immunotherapy. After demethylation, a Foxp3-Runx-1-CBFb complex is recruited to CNS2 and may represent an important lineage specification event (Zheng et al., 2010). Since demethylation is required for the complex to bind, and iTreg cells generally fail to fully demethylate the TSDR, a lack of binding of this complex may account for their reduced stability. CNS2 is demethylated in GFP+Foxp3-null T cells (TFN) expressing a Foxp3 reporter “null” allele (Foxp3gfpko), suggesting that Foxp3 binding is not required for demethylation of the TSDR (Zheng et al., 2010). Rather, it appears that TCR stimulation is essential to establish the Treg cell-specific CpG hypomethylation patterns (Ohkura et al., 2012). The last CNS element observed, CNS3, is important for Foxp3 induction in the thymus and periphery. Formation of a c-Rel containing enhanceosome, in cooperation with NFAT, CREB, p65, and Smad3, may potentiate Foxp3 induction (Rudensky, 2011). In addition to the demethylation pattern observed in CNS2 of Foxp3, three other “Treg cell-representative regions” were identified and included regions of Tnfrsf18, Ctla4, and Ikzf4 that display distinct demethylation patterns in nTreg, Tconv, and iTreg cells and are essential to establish lineage stability (Ohkura et al., 2012). In addition to its roles in iTreg cell generation and proliferation, IL-2 signaling is important for iTreg cell stability in vivo (Chen et al., 2011). Also, expression of the suppressor of cytokine signaling-2 (SOCS2) protein plays a role in preventing IL-4-dependent iTreg instability (Knosp et al., 2013). In summary, a complex, regulated series of interactions with Foxp3 are required for the establishment of Treg cell stability.
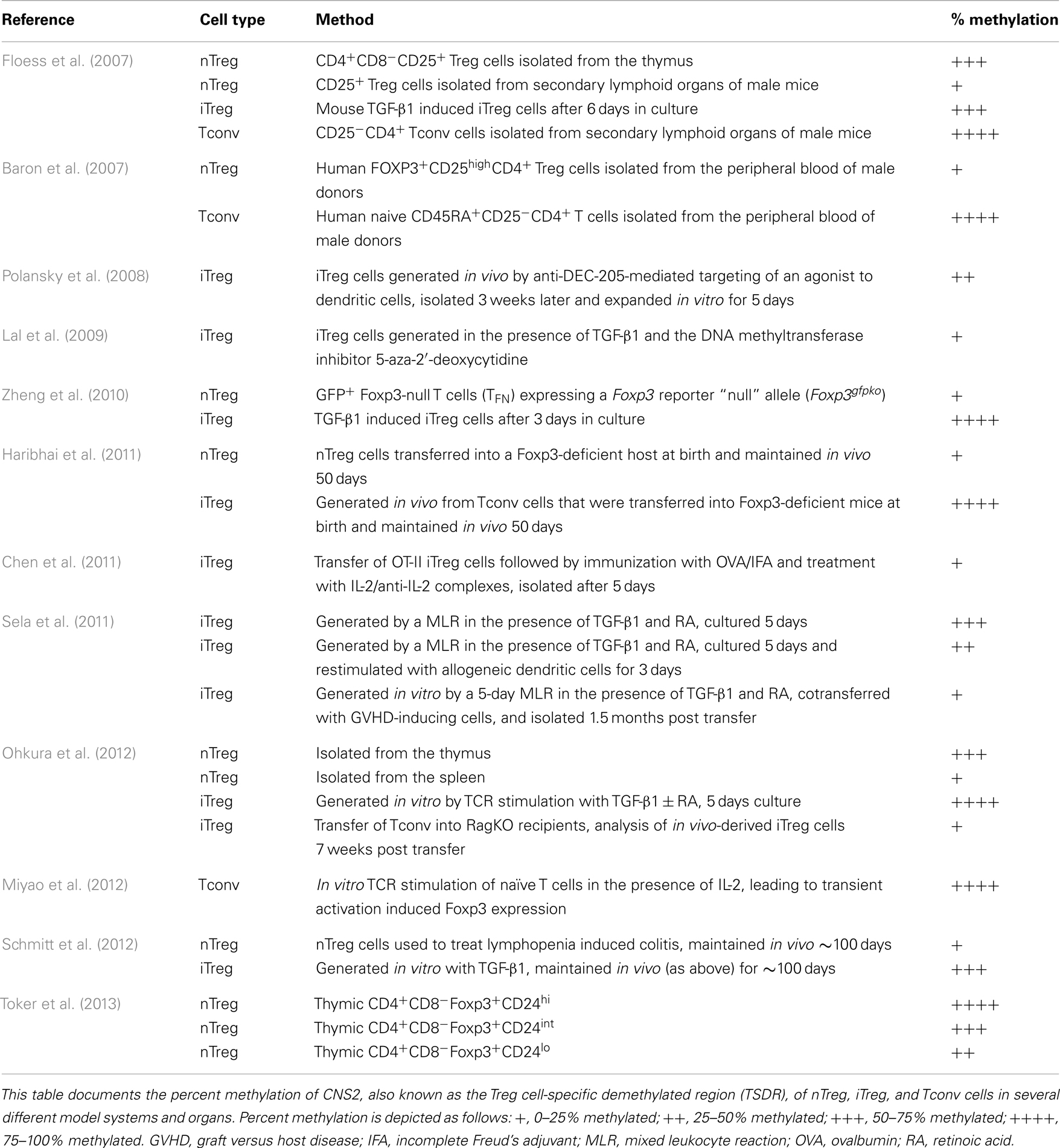
Table 2. Summary of CNS2 methylation status in CD4+ T cell populations.
Conclusion
In conclusion, recent work has established the importance of iTreg cells to the maintenance of immunological tolerance. As a population, iTreg cells share many characteristics with nTreg cells, but the observed differences in their respective TCR repertoires may lead to differential function and location, creating a need for both subsets. Future studies will look to establish additional surface markers to distinguish the subsets so that conclusive studies regarding the function and stability of the iTreg cell population can be conducted. An enhanced understanding of the origin and function of iTreg cells will promote future studies examining the translational potential of these cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are supported by the National Institutes of Health Grants RO1 AI073731 and RO1 AI085090 (Calvin B. Williams), a Senior Research Award #2858 from the Crohn’s and Colitis Foundation of America (Calvin B. Williams), the D.B. and Marjorie Reinhart Family Foundation (Calvin B. Williams), and the Children’s Hospital of Wisconsin (Calvin B. Williams). Erica G. Schmitt is a member of the MCW-MSTP which is partially supported by a T32 Grant from NIGMS, GM080202. The authors thank Dominic Co, Dipica Haribhai, and Christopher Mayne for critical review of the manuscript.
References
Akimova, T., Beier, U. H., Wang, L., Levine, M. H., and Hancock, W. W. (2011). Helios expression is a marker of T cell activation and proliferation. PLoS ONE 6:e24226. doi:10.1371/journal.pone.0024226
Apostolou, I., Sarukhan, A., Klein, L., and von Boehmer, H. (2002). Origin of regulatory T cells with known specificity for antigen. Nat. Immunol. 3, 756–763.
Apostolou, I., and von Boehmer, H. (2004). In vivo instruction of suppressor commitment in naive T cells. J. Exp. Med. 199, 1401–1408. doi:10.1084/jem.20040249
Asano, M., Toda, M., Sakaguchi, N., and Sakaguchi, S. (1996). Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 184, 387–396. doi:10.1084/jem.184.2.387
Asseman, C., Mauze, S., Leach, M. W., Coffman, R. L., and Powrie, F. (1999). An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 190, 995–1004. doi:10.1084/jem.190.7.995
Atarashi, K., Tanoue, T., Shima, T., Imaoka, A., Kuwahara, T., Momose, Y., et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. doi:10.1126/science.1198469
Baron, U., Floess, S., Wieczorek, G., Baumann, K., Grutzkau, A., Dong, J., et al. (2007). DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur. J. Immunol. 37, 2378–2389. doi:10.1002/eji.200737594
Battaglia, M., Stabilini, A., and Roncarolo, M. G. (2005). Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 105, 4743–4748. doi:10.1182/blood-2004-10-3932
Bautista, J. L., Lio, C. W., Lathrop, S. K., Forbush, K., Liang, Y., Luo, J., et al. (2009). Intraclonal competition limits the fate determination of regulatory T cells in the thymus. Nat. Immunol. 10, 610–617. doi:10.1038/ni.1739
Bennett, C. L., Christie, J., Ramsdell, F., Brunkow, M. E., Ferguson, P. J., Whitesell, L., et al. (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21. doi:10.1038/83713
Beres, A., Komorowski, R., Mihara, M., and Drobyski, W. R. (2011). Instability of Foxp3 expression limits the ability of induced regulatory T cells to mitigate graft versus host disease. Clin. Cancer Res. 17, 3969–3983. doi:10.1158/1078-0432.CCR-10-3347
Bettini, M. L., Pan, F., Bettini, M., Finkelstein, D., Rehg, J. E., Floess, S., et al. (2012). Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity 36, 717–730. doi:10.1016/j.immuni.2012.03.020
Bopp, T., Becker, C., Klein, M., Klein-Hessling, S., Palmetshofer, A., Serfling, E., et al. (2007). Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J. Exp. Med. 204, 1303–1310. doi:10.1084/jem.20062129
Brunkow, M. E., Jeffery, E. W., Hjerrild, K. A., Paeper, B., Clark, L. B., Yasayko, S. A., et al. (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27, 68–73. doi:10.1038/83784
Brunstein, C. G., Miller, J. S., Cao, Q., McKenna, D. H., Hippen, K. L., Curtsinger, J., et al. (2011). Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 117, 1061–1070. doi:10.1182/blood-2010-07-293795
Campbell, D. J., and Koch, M. A. (2011). Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat. Rev. Immunol. 11, 119–130. doi:10.1038/nri2916
Chatila, T. A., Blaeser, F., Ho, N., Lederman, H. M., Voulgaropoulos, C., Helms, C., et al. (2000). JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J. Clin. Invest. 106, R75–R81. doi:10.1172/JCI11679
Chaudhry, A., Rudra, D., Treuting, P., Samstein, R. M., Liang, Y., Kas, A., et al. (2009). CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326, 986–991. doi:10.1126/science.1172702
Chen, Q., Kim, Y. C., Laurence, A., Punkosdy, G. A., and Shevach, E. M. (2011). IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J. Immunol. 186, 6329–6337. doi:10.4049/jimmunol.1100061
Chen, W., Jin, W., Hardegen, N., Lei, K. J., Li, L., Marinos, N., et al. (2003). Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886. doi:10.1084/jem.20030152
Cobbold, S. P., Castejon, R., Adams, E., Zelenika, D., Graca, L., Humm, S., et al. (2004). Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J. Immunol. 172, 6003–6010.
Collison, L. W., Workman, C. J., Kuo, T. T., Boyd, K., Wang, Y., Vignali, K. M., et al. (2007). The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450, 566–569. doi:10.1038/nature06306
Coombes, J. L., Siddiqui, K. R., Arancibia-Carcamo, C. V., Hall, J., Sun, C. M., Belkaid, Y., et al. (2007). A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764. doi:10.1084/jem.20070590
Darce, J., Rudra, D., Li, L., Nishio, J., Cipolletta, D., Rudensky, A. Y., et al. (2012). An N-terminal mutation of the Foxp3 transcription factor alleviates arthritis but exacerbates diabetes. Immunity 36, 731–741. doi:10.1016/j.immuni.2012.04.007
Davidson, T. S., Dipaolo, R. J., Andersson, J., and Shevach, E. M. (2007). Cutting edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J. Immunol. 178, 4022–4026.
D’Cruz, L. M., and Klein, L. (2005). Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat. Immunol. 6, 1152–1159. doi:10.1038/ni1264
de la Rosa, M., Rutz, S., Dorninger, H., and Scheffold, A. (2004). Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34, 2480–2488. doi:10.1002/eji.200425274
Curotto de Lafaille, M. A., Kutchukhidze, N., Shen, S., Ding, Y., Yee, H., and Lafaille, J. J. (2008). Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity 29, 114–126. doi:10.1016/j.immuni.2008.05.010
Curotto de Lafaille, M. A., and Lafaille, J. J. (2009). Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity 30, 626–635. doi:10.1016/j.immuni.2009.05.002
Curotto de Lafaille, M. A., Lino, A. C., Kutchukhidze, N., and Lafaille, J. J. (2004). CD25- T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J. Immunol. 173, 7259–7268.
Deaglio, S., Dwyer, K. M., Gao, W., Friedman, D., Usheva, A., Erat, A., et al. (2007). Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265. doi:10.1084/jem.20062512
Desreumaux, P., Foussat, A., Allez, M., Beaugerie, L., Hebuterne, X., Bouhnik, Y., et al. (2012). Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 143, 1207–1217 e1201–1202. doi:10.1053/j.gastro.2012.07.116
Di Ianni, M., Falzetti, F., Carotti, A., Terenzi, A., Castellino, F., Bonifacio, E., et al. (2011). Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 117, 3921–3928. doi:10.1182/blood-2010-10-311894
DiPaolo, R. J., Brinster, C., Davidson, T. S., Andersson, J., Glass, D., and Shevach, E. M. (2007). Autoantigen-specific TGF beta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J. Immunol. 179, 4685–4693.
DiPaolo, R. J., Glass, D. D., Bijwaard, K. E., and Shevach, E. M. (2005). CD4+CD25+ T cells prevent the development of organ-specific autoimmune disease by inhibiting the differentiation of autoreactive effector T cells. J. Immunol. 175, 7135–7142.
Fantini, M. C., Becker, C., Tubbe, I., Nikolaev, A., Lehr, H. A., Galle, P., et al. (2006). Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut 55, 671–680. doi:10.1136/gut.2005.072801
Feuerer, M., Hill, J. A., Kretschmer, K., von Boehmer, H., Mathis, D., and Benoist, C. (2010). Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc. Natl. Acad. Sci. U.S.A. 107, 5919–5924. doi:10.1073/pnas.1002006107
Floess, S., Freyer, J., Siewert, C., Baron, U., Olek, S., Polansky, J., et al. (2007). Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 5:e38. doi:10.1371/journal.pbio.0050038
Fohse, L., Suffner, J., Suhre, K., Wahl, B., Lindner, C., Lee, C. W., et al. (2011). High TCR diversity ensures optimal function and homeostasis of Foxp3+ regulatory T cells. Eur. J. Immunol. 41, 3101–3113. doi:10.1002/eji.201141986
Fontenot, J. D., Rasmussen, J. P., Gavin, M. A., and Rudensky, A. Y. (2005a). A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 6, 1142–1151. doi:10.1038/ni1263
Fontenot, J. D., Rasmussen, J. P., Williams, L. M., Dooley, J. L., Farr, A. G., and Rudensky, A. Y. (2005b). Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22, 329–341. doi:10.1016/j.immuni.2005.01.016
Francisco, L. M., Salinas, V. H., Brown, K. E., Vanguri, V. K., Freeman, G. J., Kuchroo, V. K., et al. (2009). PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029. doi:10.1084/jem.20090847
Gavin, M. A., Rasmussen, J. P., Fontenot, J. D., Vasta, V., Manganiello, V. C., Beavo, J. A., et al. (2007). Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445, 771–775. doi:10.1038/nature05543
Geuking, M. B., Cahenzli, J., Lawson, M. A., Ng, D. C., Slack, E., Hapfelmeier, S., et al. (2011). Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 34, 794–806. doi:10.1016/j.immuni.2011.03.021
Glinka, Y., and Prud’homme, G. J. (2008). Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J. Leukoc. Biol. 84, 302–310. doi:10.1189/jlb.0208090
Gondek, D. C., Lu, L. F., Quezada, S. A., Sakaguchi, S., and Noelle, R. J. (2005). Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol. 174, 1783–1786.
Gottschalk, R. A., Corse, E., and Allison, J. P. (2010). TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J. Exp. Med. 207, 1701–1711. doi:10.1084/jem.20091999
Grainger, J. R., Smith, K. A., Hewitson, J. P., McSorley, H. J., Harcus, Y., Filbey, K. J., et al. (2010). Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J. Exp. Med. 207, 2331–2341. doi:10.1084/jem.20101074
Grossman, W. J., Verbsky, J. W., Tollefsen, B. L., Kemper, C., Atkinson, J. P., and Ley, T. J. (2004). Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 104, 2840–2848. doi:10.1182/blood-2004-03-0859
Hadis, U., Wahl, B., Schulz, O., Hardtke-Wolenski, M., Schippers, A., Wagner, N., et al. (2011). Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246. doi:10.1016/j.immuni.2011.01.016
Harada, Y., Elly, C., Ying, G., Paik, J. H., Depinho, R. A., and Liu, Y. C. (2010). Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J. Exp. Med. 207, 1381–1391. doi:10.1084/jem.20100004
Haribhai, D., Lin, W., Edwards, B., Ziegelbauer, J., Salzman, N. H., Carlson, M. R., et al. (2009). A central role for induced regulatory T cells in tolerance induction in experimental colitis. J. Immunol. 182, 3461–3468. doi:10.4049/jimmunol.0802535
Haribhai, D., Williams, J. B., Jia, S., Nickerson, D., Schmitt, E. G., Edwards, B., et al. (2011). A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 35, 109–122. doi:10.1016/j.immuni.2011.03.029
Haxhinasto, S., Mathis, D., and Benoist, C. (2008). The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 205, 565–574. doi:10.1084/jem.20071477
Hill, J. A., Feuerer, M., Tash, K., Haxhinasto, S., Perez, J., Melamed, R., et al. (2007). Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800. doi:10.1016/j.immuni.2007.09.010
Hippen, K. L., Merkel, S. C., Schirm, D. K., Nelson, C., Tennis, N. C., Riley, J. L., et al. (2011a). Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am. J. Transplant. 11, 1148–1157. doi:10.1111/j.1600-6143.2011.03558.x
Hippen, K. L., Merkel, S. C., Schirm, D. K., Sieben, C. M., Sumstad, D., Kadidlo, D. M., et al. (2011b). Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci. Transl. Med. 3, 83ra41. doi:10.1126/scitranslmed.3001809
Hsieh, C. S., Liang, Y., Tyznik, A. J., Self, S. G., Liggitt, D., and Rudensky, A. Y. (2004). Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity 21, 267–277. doi:10.1016/j.immuni.2004.07.009
Hsieh, C. S., Zheng, Y., Liang, Y., Fontenot, J. D., and Rudensky, A. Y. (2006). An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat. Immunol. 7, 401–410. doi:10.1038/ni1318
Huber, S., Gagliani, N., Esplugues, E., O’Connor, W. Jr., Huber, F. J., Chaudhry, A., et al. (2011). Th17 cells express interleukin-10 receptor and are controlled by Foxp3 and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565. doi:10.1016/j.immuni.2011.01.020
Huter, E. N., Punkosdy, G. A., Glass, D. D., Cheng, L. I., Ward, J. M., and Shevach, E. M. (2008). TGF-beta-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur. J. Immunol. 38, 1814–1821. doi:10.1002/eji.200838346
Jordan, M. S., Boesteanu, A., Reed, A. J., Petrone, A. L., Holenbeck, A. E., Lerman, M. A., et al. (2001). Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2, 301–306. doi:10.1038/86302
Josefowicz, S. Z., Niec, R. E., Kim, H. Y., Treuting, P., Chinen, T., Zheng, Y., et al. (2012). Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399. doi:10.1038/nature10772
Josefowicz, S. Z., Wilson, C. B., and Rudensky, A. Y. (2009). Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J. Immunol. 182, 6648–6652. doi:10.4049/jimmunol.0803320
Kerdiles, Y. M., Stone, E. L., Beisner, D. R., McGargill, M. A., Ch’en, I. L., Stockmann, C., et al. (2010). Foxo transcription factors control regulatory T cell development and function. Immunity 33, 890–904. doi:10.1016/j.immuni.2010.12.002
Kim, J. M., Rasmussen, J. P., and Rudensky, A. Y. (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 8, 191–197. doi:10.1038/ni1428
Knosp, C. A., Schiering, C., Spence, S., Carroll, H. P., Nel, H. J., Osbourn, M., et al. (2013). Regulation of Foxp3+ inducible regulatory T cell stability by SOCS2. J. Immunol. 190, 3235–3245. doi:10.4049/jimmunol.1201396
Koch, M. A., Tucker-Heard, G., Perdue, N. R., Killebrew, J. R., Urdahl, K. B., and Campbell, D. J. (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602. doi:10.1038/ni.1731
Kretschmer, K., Apostolou, I., Hawiger, D., Khazaie, K., Nussenzweig, M. C., and von Boehmer, H. (2005). Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 6, 1219–1227. doi:10.1038/ni1265
Lal, G., Zhang, N., van der Touw, W., Ding, Y., Ju, W., Bottinger, E. P., et al. (2009). Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 182, 259–273.
Lathrop, S. K., Santacruz, N. A., Pham, D., Luo, J., and Hsieh, C. S. (2008). Antigen-specific peripheral shaping of the natural regulatory T cell population. J. Exp. Med. 205, 3105–3117. doi:10.1084/jem.20081359
Laurence, A., Tato, C. M., Davidson, T. S., Kanno, Y., Chen, Z., Yao, Z., et al. (2007). Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26, 371–381. doi:10.1016/j.immuni.2007.02.009
Lee, J. H., Lydon, J. P., and Kim, C. H. (2012). Progesterone suppresses the mTOR pathway and promotes generation of induced regulatory T cells with increased stability. Eur. J. Immunol. 42, 2683–2696. doi:10.1002/eji.201142317
Leung, M. W., Shen, S., and Lafaille, J. J. (2009). TCR-dependent differentiation of thymic Foxp3+ cells is limited to small clonal sizes. J. Exp. Med. 206, 2121–2130. doi:10.1084/jem.20091033
Li, M. O., Sanjabi, S., and Flavell, R. A. (2006). Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 25, 455–471. doi:10.1016/j.immuni.2006.07.011
Liang, B., Workman, C., Lee, J., Chew, C., Dale, B. M., Colonna, L., et al. (2008). Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J. Immunol. 180, 5916–5926.
Lin, W., Haribhai, D., Relland, L. M., Truong, N., Carlson, M. R., Williams, C. B., et al. (2007). Regulatory T cell development in the absence of functional Foxp3. Nat. Immunol. 8, 359–368. doi:10.1038/ni1445
Liu, X., Nguyen, P., Liu, W., Cheng, C., Steeves, M., Obenauer, J. C., et al. (2009). T cell receptor CDR3 sequence but not recognition characteristics distinguish autoreactive effector and Foxp3(+) regulatory T cells. Immunity 31, 909–920. doi:10.1016/j.immuni.2009.09.023
Long, S. A., and Buckner, J. H. (2011). CD4+FOXP3+ T regulatory cells in human autoimmunity: more than a numbers game. J. Immunol. 187, 2061–2066. doi:10.4049/jimmunol.1003224
Marie, J. C., Letterio, J. J., Gavin, M., and Rudensky, A. Y. (2005). TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med. 201, 1061–1067. doi:10.1084/jem.20042276
McPherson, S. W., Heuss, N. D., and Gregerson, D. S. (2013). Local “on-demand” generation and function of antigen-specific foxp3+ regulatory T cells. J. Immunol. 190, 4971–4981. doi:10.4049/jimmunol.1202625
Miyao, T., Floess, S., Setoguchi, R., Luche, H., Fehling, H. J., Waldmann, H., et al. (2012). Plasticity of foxp3(+) T cells reflects promiscuous foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 36, 262–275. doi:10.1016/j.immuni.2011.12.012
Mucida, D., Kutchukhidze, N., Erazo, A., Russo, M., Lafaille, J. J., and Curotto de Lafaille, M. A. (2005). Oral tolerance in the absence of naturally occurring Tregs. J. Clin. Invest. 115, 1923–1933. doi:10.1172/JCI24487
Mucida, D., Pino-Lagos, K., Kim, G., Nowak, E., Benson, M. J., Kronenberg, M., et al. (2009). Retinoic acid can directly promote TGF-beta-mediated Foxp3(+) Treg cell conversion of naive T cells. Immunity 30, 471–472. doi:10.1016/j.immuni.2009.03.008 [author reply 472–473].
NIAID. (2005). Progress in Autoimmune Diseases Research. Bethesda, MD: National Institutes of Heath Autoimmune Disease Coordinating Committee.
Nishizuka, Y., and Sakakura, T. (1969). Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science 166, 753–755. doi:10.1126/science.166.3906.753
Ohkura, N., Hamaguchi, M., Morikawa, H., Sugimura, K., Tanaka, A., Ito, Y., et al. (2012). T cell receptor stimulation-induced epigenetic changes and foxp3 expression are independent and complementary events required for Treg cell development. Immunity 37, 785–799. doi:10.1016/j.immuni.2012.09.010
Oldenhove, G., de Heusch, M., Urbain-Vansanten, G., Urbain, J., Maliszewski, C., Leo, O., et al. (2003). CD4+ CD25+ regulatory T cells control T helper cell type 1 responses to foreign antigens induced by mature dendritic cells in vivo. J. Exp. Med. 198, 259–266. doi:10.1084/jem.20030654
Ouyang, W., Beckett, O., Ma, Q., Paik, J. H., Depinho, R. A., and Li, M. O. (2010). Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat. Immunol. 11, 618–627. doi:10.1038/ni.1884
Ouyang, W., Liao, W., Luo, C. T., Yin, N., Huse, M., Kim, M. V., et al. (2012). Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 491, 554–559. doi:10.1038/nature11581
Pacholczyk, R., Ignatowicz, H., Kraj, P., and Ignatowicz, L. (2006). Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity 25, 249–259. doi:10.1016/j.immuni.2006.05.016
Pacholczyk, R., Kern, J., Singh, N., Iwashima, M., Kraj, P., and Ignatowicz, L. (2007). Nonself-antigens are the cognate specificities of Foxp3+ regulatory T cells. Immunity 27, 493–504. doi:10.1016/j.immuni.2007.07.019
Pandiyan, P., Zheng, L., Ishihara, S., Reed, J., and Lenardo, M. J. (2007). CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 8, 1353–1362. doi:10.1038/ni1536
Piconese, S., Pittoni, P., Burocchi, A., Gorzanelli, A., Care, A., Tripodo, C., et al. (2010). A non-redundant role for OX40 in the competitive fitness of Treg in response to IL-2. Eur. J. Immunol. 40, 2902–2913. doi:10.1002/eji.201040505
Pillai, V., Ortega, S. B., Wang, C. K., and Karandikar, N. J. (2007). Transient regulatory T-cells: a state attained by all activated human T-cells. Clin. Immunol. 123, 18–29. doi:10.1016/j.clim.2006.10.014
Polansky, J. K., Kretschmer, K., Freyer, J., Floess, S., Garbe, A., Baron, U., et al. (2008). DNA methylation controls Foxp3 gene expression. Eur. J. Immunol. 38, 1654–1663. doi:10.1002/eji.200838105
Powrie, F., Carlino, J., Leach, M. W., Mauze, S., and Coffman, R. L. (1996). A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med. 183, 2669–2674. doi:10.1084/jem.183.6.2669
Quintana, F. J., Basso, A. S., Iglesias, A. H., Korn, T., Farez, M. F., Bettelli, E., et al. (2008). Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71. doi:10.1038/nature06880
Quintana, F. J., Murugaiyan, G., Farez, M. F., Mitsdoerffer, M., Tukpah, A. M., Burns, E. J., et al. (2010). An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 107, 20768–20773. doi:10.1073/pnas.1009201107
Ray, A., Basu, S., Williams, C. B., Salzman, N. H., and Dittel, B. N. (2012). A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J. Immunol. 188, 3188–3198. doi:10.4049/jimmunol.1103354
Relland, L. M., Mishra, M. K., Haribhai, D., Edwards, B., Ziegelbauer, J., and Williams, C. B. (2009). Affinity-based selection of regulatory T cells occurs independent of agonist-mediated induction of Foxp3 expression. J. Immunol. 182, 1341–1350.
Relland, L. M., Williams, J. B., Relland, G. N., Haribhai, D., Ziegelbauer, J., Yassai, M., et al. (2012). The TCR repertoires of regulatory and conventional T cells specific for the same foreign antigen are distinct. J. Immunol. 189, 3566–3574. doi:10.4049/jimmunol.1102646
Romagnoli, P., Hudrisier, D., and van Meerwijk, J. P. (2002). Preferential recognition of self antigens despite normal thymic deletion of CD4(+)CD25(+) regulatory T cells. J. Immunol. 168, 1644–1648.
Round, J. L., and Mazmanian, S. K. (2010). Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. U.S.A. 107, 12204–12209. doi:10.1073/pnas.0909122107
Rubtsov, Y. P., Niec, R. E., Josefowicz, S., Li, L., Darce, J., Mathis, D., et al. (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671. doi:10.1126/science.1191996
Rubtsov, Y. P., Rasmussen, J. P., Chi, E. Y., Fontenot, J., Castelli, L., Ye, X., et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558. doi:10.1016/j.immuni.2008.02.017
Rudensky, A. Y. (2011). Regulatory T cells and Foxp3. Immunol. Rev. 241, 260–268. doi:10.1111/j.1600-065X.2011.01018.x
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M., and Toda, M. (1995). Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164.
Sakaguchi, S., Takahashi, T., and Nishizuka, Y. (1982). Study on cellular events in post thymectomy autoimmune oophoritis in mice. I. Requirement of Lyt-1 effector cells for oocytes damage after adoptive transfer. J. Exp. Med. 156, 1565–1576. doi:10.1084/jem.156.6.1565
Samstein, R. M., Josefowicz, S. Z., Arvey, A., Treuting, P. M., and Rudensky, A. Y. (2012). Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell 150, 29–38. doi:10.1016/j.cell.2012.05.031
Sarween, N., Chodos, A., Raykundalia, C., Khan, M., Abbas, A. K., and Walker, L. S. (2004). CD4+CD25+ cells controlling a pathogenic CD4 response inhibit cytokine differentiation, CXCR-3 expression, and tissue invasion. J. Immunol. 173, 2942–2951.
Sauer, S., Bruno, L., Hertweck, A., Finlay, D., Leleu, M., Spivakov, M., et al. (2008). T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. U.S.A. 105, 7797–7802. doi:10.1073/pnas.0800928105
Schallenberg, S., Tsai, P. Y., Riewaldt, J., and Kretschmer, K. (2010). Identification of an immediate Foxp3(-) precursor to Foxp3(+) regulatory T cells in peripheral lymphoid organs of nonmanipulated mice. J. Exp. Med. 207, 1393–1407. doi:10.1084/jem.20100045
Schmitt, E. G., Haribhai, D., Williams, J. B., Aggarwal, P., Jia, S., Charbonnier, L. M., et al. (2012). IL-10 produced by induced regulatory T cells (iTregs) controls colitis and pathogenic ex-iTregs during immunotherapy. J. Immunol. 189, 5638–5648. doi:10.4049/jimmunol.1200936
Sela, U., Olds, P., Park, A., Schlesinger, S. J., and Steinman, R. M. (2011). Dendritic cells induce antigen-specific regulatory T cells that prevent graft versus host disease and persist in mice. J. Exp. Med. 208, 2489–2496. doi:10.1084/jem.20110466
Selvaraj, R. K., and Geiger, T. L. (2008). Mitigation of experimental allergic encephalomyelitis by TGF-beta induced Foxp3(+) regulatory T lymphocytes through the induction of anergy and infectious tolerance. J. Immunol. 180, 2830–2838.
Semple, K., Nguyen, A., Yu, Y., Wang, H., Anasetti, C., and Yu, X. Z. (2011). Strong CD28 costimulation suppresses induction of regulatory T cells from naive precursors through Lck signaling. Blood 117, 3096–3103. doi:10.1182/blood-2010-08-301275
Singh, N. P., Singh, U. P., Singh, B., Price, R. L., Nagarkatti, M., and Nagarkatti, P. S. (2011). Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE 6:e23522. doi:10.1371/journal.pone.0023522
Strainic, M. G., Shevach, E. M., An, F., Lin, F., and Medof, M. E. (2013). Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nat. Immunol. 14, 162–171. doi:10.1038/ni.2499
Sukiennicki, T. L., and Fowell, D. J. (2006). Distinct molecular program imposed on CD4+ T cell targets by CD4+CD25+ regulatory T cells. J. Immunol. 177, 6952–6961.
Sun, C. M., Hall, J. A., Blank, R. B., Bouladoux, N., Oukka, M., Mora, J. R., et al. (2007). Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204, 1775–1785. doi:10.1084/jem.20070602
Suri-Payer, E., Amar, A. Z., Thornton, A. M., and Shevach, E. M. (1998). CD4+CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J. Immunol. 160, 1212–1218.
Tao, R., De Zoeten, E. F., Ozkaynak, E., Chen, C., Wang, L., Porrett, P. M., et al. (2007). Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 13, 1299–1307. doi:10.1038/nm1652
Thompson, L. J., Valladao, A. C., and Ziegler, S. F. (2011). Cutting edge: de novo induction of functional Foxp3+ regulatory CD4 T cells in response to tissue-restricted self antigen. J. Immunol. 186, 4551–4555. doi:10.4049/jimmunol.1003573
Thornton, A. M., Korty, P. E., Tran, D. Q., Wohlfert, E. A., Murray, P. E., Belkaid, Y., et al. (2010). Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 184, 3433–3441. doi:10.4049/jimmunol.0904028
Thornton, A. M., and Shevach, E. M. (1998). CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188, 287–296. doi:10.1084/jem.188.2.287
Thorstenson, K. M., and Khoruts, A. (2001). Generation of anergic and potentially immunoregulatory CD25+CD4 T cells in vivo after induction of peripheral tolerance with intravenous or oral antigen. J. Immunol. 167, 188–195.
Toker, A., Engelbert, D., Garg, G., Polansky, J. K., Floess, S., Miyao, T., et al. (2013). Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. J. Immunol. 190, 3180–3188. doi:10.4049/jimmunol.1203473
Tone, Y., Furuuchi, K., Kojima, Y., Tykocinski, M. L., Greene, M. I., and Tone, M. (2008). Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 9, 194–202. doi:10.1038/ni1549
Trzonkowski, P., Bieniaszewska, M., Juscinska, J., Dobyszuk, A., Krzystyniak, A., Marek, N., et al. (2009). First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin. Immunol. 133, 22–26. doi:10.1016/j.clim
Weber, S. E., Harbertson, J., Godebu, E., Mros, G. A., Padrick, R. C., Carson, B. D., et al. (2006). Adaptive islet-specific regulatory CD4 T cells control autoimmune diabetes and mediate the disappearance of pathogenic Th1 cells in vivo. J. Immunol. 176, 4730–4739.
Weiss, J. M., Bilate, A. M., Gobert, M., Ding, Y., Curotto de Lafaille, M. A., Parkhurst, C. N., et al. (2012). Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J. Exp. Med. 209, 1723–1742. doi:10.1084/jem.20120914
Wildin, R. S., Ramsdell, F., Peake, J., Faravelli, F., Casanova, J. L., Buist, N., et al. (2001). X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 27, 18–20. doi:10.1038/83707
Williams, L. M., and Rudensky, A. Y. (2007). Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 8, 277–284. doi:10.1038/ni1437
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., et al. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275. doi:10.1126/science.1160062
Wohlfert, E. A., Gorelik, L., Mittler, R., Flavell, R. A., and Clark, R. B. (2006). Cutting edge: deficiency in the E3 ubiquitin ligase Cbl-b results in a multifunctional defect in T cell TGF-beta sensitivity in vitro and in vivo. J. Immunol. 176, 1316–1320.
Wohlfert, E. A., Grainger, J. R., Bouladoux, N., Konkel, J. E., Oldenhove, G., Ribeiro, C. H., et al. (2011). GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J. Clin. Invest. 121, 4503–4515. doi:10.1172/JCI57456
Wong, J., Mathis, D., and Benoist, C. (2007a). TCR-based lineage tracing: no evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J. Exp. Med. 204, 2039–2045. doi:10.1084/jem.20070822
Wong, J., Obst, R., Correia-Neves, M., Losyev, G., Mathis, D., and Benoist, C. (2007b). Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J. Immunol. 178, 7032–7041.
Xiao, S., Jin, H., Korn, T., Liu, S. M., Oukka, M., Lim, B., et al. (2008). Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J. Immunol. 181, 2277–2284.
Yadav, M., Louvet, C., Davini, D., Gardner, J. M., Martinez-Llordella, M., Bailey-Bucktrout, S., et al. (2012). Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med. 209, 1713–1722. doi:10.1084/jem.20120822
Yang, X. O., Nurieva, R., Martinez, G. J., Kang, H. S., Chung, Y., Pappu, B. P., et al. (2008). Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56. doi:10.1016/j.immuni.2008.05.007
Zheng, S. G., Wang, J. H., Stohl, W., Kim, K. S., Gray, J. D., and Horwitz, D. A. (2006). TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J. Immunol. 176, 3321–3329.
Zheng, Y., Chaudhry, A., Kas, A., Deroos, P., Kim, J. M., Chu, T. T., et al. (2009). Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356. doi:10.1038/nature07674
Zheng, Y., Josefowicz, S., Chaudhry, A., Peng, X. P., Forbush, K., and Rudensky, A. Y. (2010). Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812. doi:10.1038/nature08750
Zheng, Y., and Rudensky, A. Y. (2007). Foxp3 in control of the regulatory T cell lineage. Nat. Immunol. 8, 457–462. doi:10.1038/ni1455
Zhou, R., Horai, R., Silver, P. B., Mattapallil, M. J., Zarate-Blades, C. R., Chong, W. P., et al. (2012). The living eye “disarms” uncommitted autoreactive T cells by converting them to Foxp3(+) regulatory cells following local antigen recognition. J. Immunol. 188, 1742–1750. doi:10.4049/jimmunol.1102415
Zhou, X., Bailey-Bucktrout, S. L., Jeker, L. T., Penaranda, C., Martinez-Llordella, M., Ashby, M., et al. (2009). Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10, 1000–1007. doi:10.1038/ni.1774
Keywords: Treg cells, Treg stability, immunotherapy, Treg function, gene expression profiling, TCR repertoire
Citation: Schmitt EG and Williams CB (2013) Generation and function of induced regulatory T cells. Front. Immunol. 4:152. doi: 10.3389/fimmu.2013.00152
Received: 30 April 2013; Paper pending published: 14 May 2013;
Accepted: 04 June 2013; Published online: 19 June 2013.
Edited by:
Eyad Elkord, United Arab Emirates University, UAE; University of Salford and University of Manchester, UKReviewed by:
Joan Stein-Streilein, Schepens Eye Research Institute, USAJohan Verhagen, University of Bristol, UK
Copyright: © 2013 Schmitt and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Calvin B. Williams, Medical College of Wisconsin, 8701 Watertown Plank Road, MFRC Room 5052, Milwaukee, WI 53226, USA e-mail:Y3dpbGxpYW1AbWN3LmVkdQ==