

- Department of Medicine, Radboud University Nijmegen Medical Center, Nijmegen Institute for Infection, Inflammation and Immunity (N4i), Nijmegen, Netherlands
The interleukin-1 (IL 1) family of ligands is associated with acute and chronic inflammation, and plays an essential role in the non-specific innate response to infection. The biological properties of IL 1 family ligands are typically pro-inflammatory. The IL 1 family has 11 family members and can be categorized into subfamilies according to the length of their precursor and the length of the propiece for each precursor (Figure 1). The IL 1 subfamily consists of IL 1α, IL 1β, and IL 33, with the longest propieces of the IL 1 family. IL 18 and IL 37 belong to the IL 18 subfamily and contain smaller propieces than IL 1 and IL-33. Since IL 37 binds to the IL 18Rα chain it is part of the IL 18 subfamily, however it remains to be elucidated how the propiece of IL 37 is removed. IL 36α, β, and γ as well as IL 36 Ra belong to the IL 36 subfamily. In addition, IL 38 likely belongs to this family since it has the ability to bind to the IL 36R. The IL 36 subfamily has the shortest propiece. The one member of the IL 1 family that cannot be categorized in these subfamilies is IL 1 receptor antagonist (IL 1Ra), which has a signal peptide and is readily secreted. In the present review we will describe the biological functions of the IL-1F members and new insights in their biology.
Introduction
The interleukin-1 (IL-1) family of ligands is associated with acute and chronic inflammation, and plays an essential role in the non-specific innate response to infection. The biological properties of IL-1 family ligands are typically pro-inflammatory. The IL-1 family has 11 family members and can be categorized into subfamilies according to the length of their precursor and the length of the propiece for each precursor (Figure 1). The IL-1 subfamily consists of IL-1α, IL-1β, and IL-33, with the longest propieces of the IL-1 family. IL-18 and IL-37 belong to the IL-18 subfamily and contain smaller propieces then IL-1 and IL-33. Since IL-37 binds to the IL-18Rα chain it is part of the IL-18 subfamily, however it remains to be elucidated how the propiece of IL-37 is removed. IL-36α, β, and γ as well as IL-36 Ra belong to the IL-36 subfamily. In addition, IL-38 likely belongs to this family since it has the ability to bind to the IL-36R. The IL-36 subfamily has the shortest propiece. The one member of the IL-1 family that cannot be categorized in these subfamilies is IL-1 receptor antagonist (IL-1Ra), which has a signal peptide and is readily secreted. In the present review we will describe the biological functions of the IL-1F members and new insights in their biology.
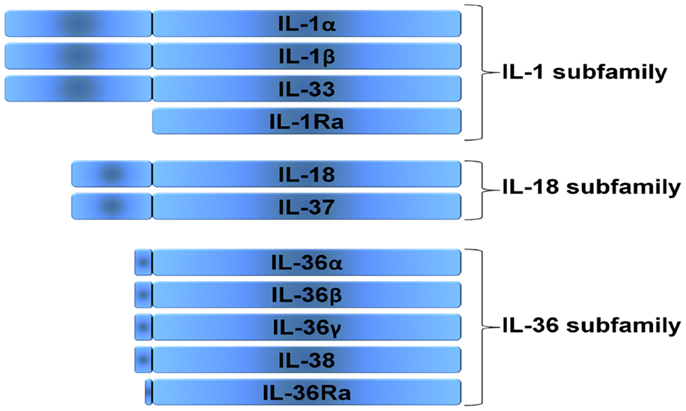
Figure 1. Subfamilies according to the length of their precursor. Three families can be distinguished in the IL-1 family, the IL-1 subfamily, the IL-18 subfamily, and the IL-36 subfamily. The IL-1 receptor antagonist (IL-1Ra) cannot be categorized in these subfamilies, since it has a signal peptide and is readily secreted.
The IL-1 Subfamily
Interleukin-1α
IL-1α does not have a signal peptide, binds to nuclear DNA, and is released from the cell upon death after which it can bind to the IL-1R1 receptor as either an unprocessed precursor or a processed protein. Primary cells such as keratinocytes, thymic epithelium, hepatocytes, endothelial cells, fibroblasts, and the epithelial cells of mucus membranes contain constitutive levels of intracellular IL-1α precursor (Hacham et al., 2002). Furthermore, precursor IL-1α can be found on the surface of several cells, particularly on monocytes and B-lymphocytes, referred to as membrane bound IL-1α (Kurt-Jones et al., 1985). Membrane bound IL-1α is biologically active (Kaplanski et al., 1994), and its biological activities are neutralized by antibodies specific to IL-1α. Endothelial cells undergoing stress-induced apoptosis release membrane apoptotic body-like particles containing full-length IL-1α precursor and the processed mature form (Berda-Haddad et al., 2011). When injected into mice, apoptotic body-like particles containing the IL-1α precursor induce neutrophilic infiltration that can be prevented by neutralization of IL-1α (Berda-Haddad et al., 2011). Although the IL-1α precursor is biologically active, the processed form is more active. The processing of the IL-1α precursor is accomplished by calpain II, a membrane-associated, calcium-dependent cysteine protease (Miller et al., 1994), and calcium influx induces IL-1α secretion of the processed form (Gross et al., 2012).
It has been proposed that IL-1α acts as an autocrine growth factor since the intracellular regulating normal cellular differentiation, particularly in epithelial and ectodermal cells. In support of this concept, neutralizing intracellular IL-1α reduces senescence in endothelial cells (Maier et al., 1990), and constitutive IL-1α precursor can bind to HAX-1 in fibroblasts that subsequently translocates as a complex to the nucleus (Kawaguchi et al., 2006). Although these data support the concept that IL-1α can act as an autocrine growth factor, it should be noted that mice deficient in IL-1α show no defects in growth and development, including skin, fur, epithelium, and gastrointestinal function (Horai et al., 1998). However, since mice deficient in IL-1α still retain the N-terminal propiece (Werman et al., 2004) and this N-terminal propiece of IL-1α has been shown to bind HAX-1 (Yin et al., 2001) it could still be that the propiece of IL-1α is responsible for the proposed autocrine growth factor function of IL-1α.
IL-1α plays an important role in sterile inflammation. Upon necrotic cell death the IL-1α precursor is released (Carmi et al., 2009; Cohen et al., 2010) and binds to the IL-1 receptor on nearby tissue macrophages and epithelial cells (Luheshi et al., 2011; Rider et al., 2011). This will trigger a pro-inflammatory response characterized by neutrophilic influx that is followed by influx of monocytes (Rider et al., 2011). This is underlined by the observation that extracts of tumor cells induce neutrophilic inflammation, which does not occur in mice deficient in IL-1RI and that can be prevented by neutralization of IL-1α (Chen et al., 2007). Thus, IL-1α, either the unprocessed precursor or the cleaved form can be seen as an alarmin (Chan et al., 2012). Furthermore, platelets also contain IL-1α (Hawrylowicz et al., 1989), and platelet-derived IL-1α has been described to be important in brain injury in stroke models (Thornton et al., 2010) and in atherosclerosis (Gawaz et al., 2000).
In mice fed a high-fat diet, serum amyloid A protein, a marker of inflammation in atherogenesis, was markedly lower in IL-1α-deficient mice compared to wild type or IL-1β-deficient mice (Kamari et al., 2007). IL-1α-deficient mice had significantly higher levels of non-high density lipoprotein cholesterol. The beneficial effect of IL-1α deficiency was due to hematopoietic cells transferred from the bone marrow of IL-1α-deficient mice resulting in a reduction in aortic lesion size twice that observed in mice transplanted with IL-1β-deficient bone marrow cells. Therefore, IL-1α appears to play an important role in the pathogenesis of lipid-mediated atherogenesis and this may be due to an effect of membrane IL-1α.
Interleukin-1β
IL-1β is a highly inflammatory cytokine as reviewed in Dinarello (2011a), and is primarily a product of monocytes, macrophages, and dendritic cells (DC) as well as B-lymphocytes and NK cells. Caspase-1, an intracellular cysteine protease, is responsible for the conversion of inactive IL-1β precursor into the active cytokine (Figure 2). Caspase-1 likewise needs to be processed in order to become active. This activation of caspase-1 is dependent on a complex of intracellular proteins termed the inflammasome (Agostini et al., 2004; Martinon et al., 2009). One critical component of the inflammasome is NLRP3, also termed cryopyrin since the gene was initially discovered in patients with “familial cold auto-inflammatory syndrome,” a genetic disease characterized by fevers and elevated acute phase proteins following exposure to cold (Hoffman et al., 2001). Human blood monocytes contain constitutively active caspase-1, which is dependent on the presence of the key components of the inflammasome, namely ASC and NLRP3 (Netea et al., 2009). By contrast, other cells, such as macrophages and DC, need an additional trigger to activate caspase-1 (Netea et al., 2009). Non-caspase-1 mechanisms also exist to generate active forms of IL-1β. Sterile inflammation induces fever and increased production of hepatic acute phase proteins, which are absent in mice deficient in IL-1β, but present in mice deficient in caspase-1 (Fantuzzi et al., 1997a; Joosten et al., 2009). This observation can be explained by the fact that proteinase 3 from neutrophils can also process the IL-1β precursor extracellularly into an active cytokine (Coeshott et al., 1999; Joosten et al., 2009), as well as other proteases including elastase, matrix metalloprotease 9, and granzyme A (Figure 2).
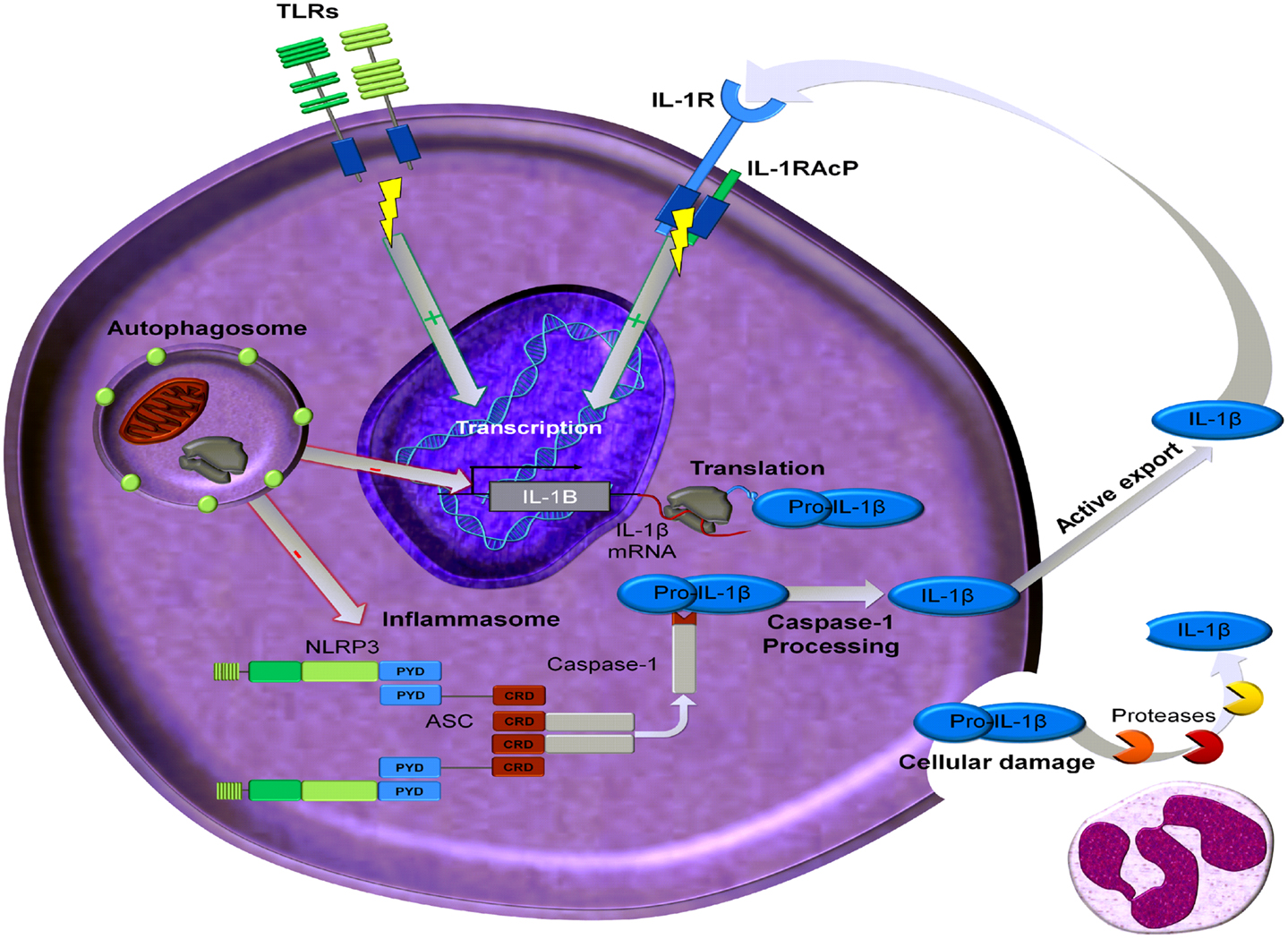
Figure 2. Processing and regulation of IL-1β. Transcription of IL-1β mRNA can be induced by ligands that activate Toll like receptors (TLR) or by IL-1 ligands (IL-1α and IL-1β). This transcription can be regulated by autophagy. The precursor of IL-1β, namely pro-IL-1β, can be processed by active caspase-1 which is part of the inflammsome. In addition proteases, predominantly derived from neutrophils, can cleave extracellular pro-IL-1β, which will be present extracellularly in the setting of damaged inflammatory cells. This processed mature and bioactive IL-1β can then induce its own transcription.
Recently, autophagy has been reported to regulate IL-1β production (Saitoh et al., 2008). Autophagy is an ancient process of recycling cellular components, such as cytosolic organelles and protein aggregates, through degradation mediated by lysosomes. Autophagy is activated in conditions of cell stress, hypoxia, starvation, or growth factor deprivation, and it promotes cell survival by generating free metabolites and energy through degradation of the endogenous cellular components (Klionsky, 2007). In addition to its role in the pathophysiology of aging, cancer, and neurodegenerative diseases, autophagy can also modulate inflammatory responses (Schmid and Munz, 2007). A role for autophagy in production of pro-inflammatory cytokines, particularly IL-1β, has emerged with the reported link between ATG16L1 (an autophagy gene) function and IL-1β production. Macrophages from ATG16L1-deficient mice produce higher levels of IL-1β and IL-18 after stimulation with lipopolysaccharide (LPS) (Saitoh et al., 2008). These data suggest that higher activation of caspase-1 in ATG16L1-deficient mice accounts for the higher production of these caspase-1 dependent cytokines. Indeed, additional studies in ATG16L1-deficient mice point toward a regulatory effect of autophagy on caspase-1 activation through modulating the NLRP3 inflammasome (Saitoh et al., 2008; Tschopp and Schroder, 2010; Nakahira et al., 2011; Zhou et al., 2011). Autophagy can thus regulate IL-1β production by influencing caspase-1 activity (Figure 2). Furthermore, autophagosomes present in mouse macrophages can specifically degradate IL-1β precursor (Harris et al., 2011). Moreover, inhibition of autophagy in human primary monocytes leads to increased production of IL-1β (Crisan et al., 2011) (Figure 2). However, in the same cells TNFα production was decreased by autophagy inhibition, suggesting that there are divergent effects of autophagy on the production of these two important pro-inflammatory cytokines. Interestingly, in human cells IL-1β mRNA transcription is elevated when autophagy is inhibited, whereas no effects can be observed on caspase-1 activation (Saitoh et al., 2008; Crisan et al., 2011; Harris et al., 2011). Despite these differences between mouse and human cells, autophagy modulates IL-1β production and inhibition of autophagy increases the production of IL-1β.
The potent inflammatory function of IL-1β is underlined by diseases that are specifically associated with IL-1β production. Although it has been described that IL-1 can play an important role in neurodegenerative diseases such as Alzheimer’s disease, developmental diseases such as schizophrenia (Meyer, 2011) or even stress (Goshen and Yirmiya, 2009) most disease states that have been clearly linked with IL-1β mediated inflammation fall into the category of auto-inflammatory diseases, which are to be distinguished from the classic autoimmune diseases. Although inflammation is common to both auto-inflammatory and autoimmune diseases, in the case of IL-1-mediated disease, there is no evidence for a role of the adaptive immune system in its induction. Persons with activating mutations in one of the key genes that control the activation of caspase-1, namely NLRP3 (cryopyrin), can develop life-threatening systemic inflammation, which can be reversed by blocking IL-1β. Interestingly, in patients with these mutations in NLRP3, there is a decrease in steady state levels of pro-caspase-1 mRNA with IL-1Ra treatment (Goldbach-Mansky et al., 2006), suggesting that IL-1β stimulates its own production and processing. Other studies supporting this concept of IL-1-induced IL-1 have been reported (Goldbach-Mansky et al., 2006; Gattorno et al., 2007; Greten et al., 2007; Boni-Schnetzler et al., 2008). This explains the auto-inflammatory nature of these IL-1 mediated diseases, namely an initial trigger induces the production of IL-1β, which thereafter can induce itself. Type 2 diabetes appears to be an example of an auto-inflammatory disease where glucose induces IL-1β production from the insulin-producing beta cell and IL-1β induces the beta cell to produce its own IL-1β (Maedler et al., 2002).
No spontaneous disease has been reported in mice deficient in IL-1β. However, upon challenge, IL-1β-deficient mice differ significantly in their responses from wild-type mice. The most dramatic is the response to local inflammation induced by a local irritant. IL-1β-deficient mice will not have an acute phase response, develop anorexia, and have no fever within the first 24 h (Zheng et al., 1995; Fantuzzi et al., 1997b). These effects have also been observed in the same model using anti-IL-1R type I antibodies in wild-type mice (Zheng et al., 1995; Fantuzzi et al., 1997b). Reduced inflammation has also been observed in IL-1β-deficient mice that are exposed to zymosan-induced peritonitis (Fantuzzi et al., 1997b). IL-1β-deficient mice injected with LPS have little or no expression of leptin mRNA or leptin protein (Faggioni et al., 1998), however they do have elevated febrile responses to LPS, IL-1β, or IL-1α when compared to wild-type mice (Fantuzzi et al., 1996).
Interleukin-1β enhances T-cell activation and recognition of antigen. The specificity of this response was however not known initially. IL-1β, together with IL-6, and TGFβ have been reported to induce the development of Th17 cells, while IL-23 has been reported to be important for the maintenance of Th17 cells (Weaver et al., 2006; Dong, 2008; van de Veerdonk et al., 2009). The combination of IL-23 and IL-1β induces the development of human Th17 cells (Wilson et al., 2007). Interestingly, these cells also released IFNγ, displaying a phenotype common to both Th17 and Th1 cells (Wilson et al., 2007). The strong capacity of IL-1 to induce Th17 differentiation has also been linked to the induction and release of prostaglandins (Dinarello, 2011b). PGE2 are inducers of Th17 induction and inhibitors of cyclooxygenase decrease IL-17 production (Chizzolini et al., 2008; Smeekens et al., 2010). Furthermore, engagement of the aryl hydrocarbon receptor, a pathway demonstrated to be crucial for the generation of Th17 cells, has been shown to strongly induce IL-1β (Henley et al., 2004). IL-1β is also required for the production of IL-17 by NKT cells (Moreira et al., 2011) and of IL-22 from NK cells (Hughes et al., 2010).
Cytokines belonging to the IL-1 family have also been described to modulate neurons and functions of the central nervous system (CNS). For example IL-1β and its antagonist IL-1Ra have been extensively described for their ability to act within the CNS as modulators of hippocampal memory, as well as involvement in neuronal death (Yirmiya and Goshen, 2011; Hanamsagar et al., 2012). From these studies it is clear that in some context these cytokines that belong to the IL-1 family not only exert a pathological role but also play a role in homeostasis. These emerging observations underscore that the functions of inflammatory cytokines such a IL-1β are not only confined to the classical inflammatory response.
Interleukin-33
IL-33 belongs to the IL-1 subfamily, and was formerly termed IL-1F11. IL-33Rα is the ligand binding chain for IL-33 (Schmitz et al., 2005), and the co-receptor for IL-33 is the IL-1RAcP, which is also the co-receptor for IL-1α and IL-1β. The IL-33Rα chain is similar to IL-1R1, since it can bind the ligand but requires the IL-1RAcP to signal (Ali et al., 2007; Chackerian et al., 2007). The structure of IL-33 is closer to IL-18 than to IL-1β. Initially, IL-33 was considered closely related to IL-1β and IL-18 because the IL-33 precursor contains a caspase-1 site (Schmitz et al., 2005). However studies have revealed that caspase-1 cleaving of IL-33 actually results in loss of IL-33 activity and that the full-length IL-33 precursor can bind to IL-33Rα and is active (Cayrol and Girard, 2009). In addition, it has been reported that the caspase-1 cleavage site is similar to the consensus sequence for caspase-3 and that intracellular IL-33 precursor is a substrate for caspase-3 (Cayrol and Girard, 2009). Precursor IL-33 can also be processed by neutrophil proteinase 3 (PR3) into a biological active form of IL-33, however increasing the incubation time of PR3 will decrease the biological activity of IL-33 (Bae et al., 2012). Next to PR3 cleavage, neutrophil elastase and cathepsin G can cleave the IL-33 precursor, which results in the generation of IL-33 with different N-termini and varying levels of activity (Lefrancais et al., 2012). Thus, extracellular IL-33 is released as a precursor and can be processed by neutrophil enzymes which will generates active forms with varying levels of activity.
The dominant biological activity of IL-33 is the induction of Th2 cytokines, IL-4, IL-5, and IL-13 as well as other properties anticipated for a Th2 type cytokine. Therefore, the role of IL-33 in lung inflammation such as allergic type asthma has been studied extensively. Administration of IL-33 into the airways triggers an immediate allergic response in the lung of naïve mice and worsens the response in mice sensitized to antigen (Louten et al., 2011). When human IL-33 is administered to mice eosinophilic infiltration is a prominent finding in the lung and in allergic rhinitis as well as allergic conjunctivitis (Matsuba-Kitamura et al., 2010). Interestingly, it was recently described that interleukin-1α can control allergic sensitization to inhaled house dust mite (HDM) via the epithelial release of IL-33 (Willart et al., 2012). Mice deficient in IL-33Rα do not develop a Th2 response to Schistosoma egg antigen, and mice deficient in IL-33, are highly susceptible to Strongyloides venezuelensis (Yasuda et al., 2012). This infection induces a unique class of cells called natural helper cells or nuocytes, which produce IL-5 and IL-13 upon activation by IL-33, which results in eosinophilic infiltration into the lungs. This pulmonary eosinophilic inflammation causes damage that is IL-33 and IL-5 dependent (Yasuda et al., 2012).
Other impressive pathological findings such as changes in the arterial walls and intestinal tissues have also been observed when human IL-33 is injected in mice (Schmitz et al., 2005; Kim et al., 2012). In mice deficient in IL-33Rα, there is myocardial hypertrophy, ventricle dilation, and fibrosis of the heart suggesting that IL-33 plays a protective role in the heart (Sanada et al., 2007). Moreover, elevated levels of the extracellular domain of IL-33Rα predict outcomes in patients that have had a myocardial infarction (Sanada et al., 2007). Furthermore, administration of recombinant IL-33 inhibits the phosphorylation of IκB and reduces hypertrophy and fibrosis in a model of cardiomyocyte hypertrophy (Sanada et al., 2007). One of the more challenging aspects is the role of the IL-33 signaling pathway in the ApoE deficient mouse model of atherosclerosis. Arterial wall plaques of ApoE deficient mice on a high-fat diet contain IL-33 and IL-33Rα. Atherosclerotic plaques were markedly reduced when these mice were treated with IL-33, however when Insoluble IL-33Rα was administered to neutralize IL-33 signaling the disease worsened (Miller et al., 2008).
Clearly IL-33 has properties that go beyond its role of inducing Th2 responses. For example, IL-33 can induce potent CD8(+) T-cell (CTL) responses to replicating, prototypic RNA, and DNA viruses in mice (Bonilla et al., 2012). Moreover, IL-33 is identical to a nuclear factor which is dominantly expressed in high endothelial venules (HEV) called NF-HEV (Carriere et al., 2007). Constitutive nuclear localization of IL-33 has also been reported in several other cell types such as type II lung epithelial cells (Yasuda et al., 2012), epithelial cells (Moussion et al., 2008), and pancreatic stellate cells (Masamune et al., 2010). IL-33 binding to DNA and acting as a nuclear factor resembles closely the IL-1α binding to chromatin and IL-1α functioning as a nuclear factor (Stevenson et al., 1997; Werman et al., 2004; Cohen et al., 2010). IL-33 precursor can bind NF-κB p65 and IL-1β-induced TNFα is reduced in cells overexpressing the IL-33 precursor (Ali et al., 2011). These data suggest that next to the ability of IL-33 to induce T-cell responses, IL-33 possesses anti-inflammatory activity which appears to be dependent on nuclear sequestration (Cohen et al., 2010).
The IL-18 and IL-37 Subfamily
Interleukin-18
Interleukin-18 is extensively reviewed in this issue of Frontiers in Immunology by Dr. C. Dinarello.
Interleukin-37
IL-37, formerly termed IL-1F7, lacks a signal peptide and has a caspase-1 site. IL-37 can translocate to the nucleus following stimulation, similar to IL-1α and IL-33 (Sharma et al., 2008). Inhibition of caspase-1 markedly reduces nuclear entry of IL-37 (Sharma et al., 2008), suggesting that IL-37 translocates to the nucleus after caspase-1 processing, and acts as a transcriptional modulator reducing the production of LPS-stimulated pro-inflammatory cytokines. It must be noted that the secretion of IL-37 has not been documented with any certainty. It is likely that the IL-37 precursor exits the cell during cell death and that this precursor suppresses LPS-induced IL-1β, IL-6, and TNFα (Nold et al., 2010). It was from the first reports on IL-37 that recombinant IL-37 bound to the IL-18Rα (Pan et al., 2001; Kumar et al., 2002). In IL-37 transgenic mice this binding of IL-37 to IL-18Rα has also been observed (Nold et al., 2011), and it has been reported that IL-37 specifically binds to the third domain of the IL-18Rα (Bufler et al., 2002). However, IL-37 does not act as a classical receptor antagonist for IL-18, despite these studies showing binding of IL-37 to the IL-18Rα chain. High concentrations of IL-37 do not inhibit recombinant IL-18-induced IFNγ, and recombinant IL-37 modestly reduces IL-18-induced IFNγ in the presence of low concentrations of IL-18BP (Bufler et al., 2002).
A mouse homolog for human IL-37 has not been identified, therefore a strain of transgenic mice has been generated to study the in vivo biological function of IL-37 (Nold et al., 2010). No obvious phenotype in homozygous IL-37 transgenic mice (IL-37 tg) mice has been observed and breed normally. Importantly, these mice do not constitutively express mRNA levels of IL-37, which is most likely due to a functional instability sequence found in IL-37 that limits the half-life of IL-37 mRNA (Bufler et al., 2004). Upon stimulation with IL-1β or LPS, expression of IL-37 increases after 4–24 h and the IL-37 precursor can be found in peripheral blood cells isolated from the transgenic mice (Nold et al., 2003). Compared to wild-type mice, IL-37 transgenic mice are protected against LPS challenge (Nold et al., 2010). They display significantly less hypothermia, acidosis, hyperkalemia, hepatitis, and dehydration during LPS challenge. In addition, IL-6 and TNFα production is significantly less in whole blood cultures from IL-37 transgenic mice when stimulated by IL-1β or the combination of IL-12 plus IL-18. This anti-inflammatory activity of IL-37 is not limited to a reduction of the cytokines and chemokines, also DC isolated from the spleen of IL-37 transgenic mice have a marked reduction in their expression of CD86 and MHC II after LPS challenge (Nold et al., 2010). IL-37 transgenic mice subjected to dextran sulfate sodium (DSS)-induced colitis have significantly less clinical disease compared to wild-type mice (McNamee et al., 2011). A decreased leukocyte recruitment into the colonic lamina propria was observed in IL-37Tg mice which was associated with decreased proinflammatory cytokine production. Wild-type mice reconstituted with bone marrow from IL-37 transgenic mice were protected from colitis, suggesting that IL-37 originating from hematopoietic cells is sufficient to exert protective anti-inflammatory effects.
The IL-36 Subfamily
The IL-1 family members IL-1F5, IL-1F6, IL-1F8, IL-1F9, and IL-1F10 are now termed IL-36Ra, IL-36α, IL-36β, IL-36γ, and IL-38 respectively (Dinarello et al., 2010). Each member of the IL-36 subfamily binds to the IL-1Rpr2, now termed IL-36R (Towne et al., 2004). The IL-36 subfamily is closely related to the IL-1 subfamily because similar to the IL-1α and IL-1β and IL-33, the IL-36R forms a signaling complex with the IL-1RAcP (Towne et al., 2004; Ali et al., 2007).
Interleukin-36α, β, γ (IL-36)
IL-36α, IL-36β, and IL-36γ all have agonistic characteristics and signal through the IL-36R (Towne et al., 2004; Magne et al., 2006; Chustz et al., 2011). These IL-36 cytokines are mainly expressed in keratinocytes, bronchial epithelium, brain tissue, and monocytes/macrophages (Smith et al., 2000; Barksby et al., 2009). LPS derived from E. coli or P. gingivalis specifically induces expression of IL-36γ, but not IL-36α or IL-36β in THP-1 cells (Barksby et al., 2009). Peripheral blood lymphocytes are able to express IL-36γ in response to α-particles, which can be used for targeted cancer therapy (Turtoi et al., 2010), and T-lymphocytes have been reported to express IL-36α and IL-36β (Smith et al., 2000; Li et al., 2010; Vigne et al., 2011). Interestingly, it has recently been shown that γδ T-cells can express IL-36β under specific conditions (Yang et al., 2010). IL-36 cytokines like IL-1 and IL-18 also need to be processed in order to gain full bioactivity, although the enzyme responsible still remains to be determined (Towne et al., 2011).
IL-36β can induce expression of itself, and thus an autocrine/paracrine loop similar to IL-1 also seems to be present in the IL-36 subfamily of cytokines (Dinarello et al., 1987; Carrier et al., 2011). IL-36α, IL-36β, and IL-36γ can induce IL-17 and TNF expression in keratinocytes, which can be synergized by the cytokine IL-22 (Carrier et al., 2011). Furthermore, several reports indicate that epidermal growth factor signaling regulates the expression of IL-36α and IL-36β in the skin (Yang et al., 2010; Franzke et al., 2012) suggesting an important role of the agonists IL-36α and IL-36β in skin homeostasis. In line with this is the observation that transgenic mice which overexpress the IL-36α gene in basal keratinocytes display acanthosis and hyperkeratosis of the skin, which are characteristics of psoriatic skin lesions (Blumberg et al., 2007). IL-36 cytokine expression in bronchial epithelial cells can be induced by several pro-inflammatory stimuli (Vos et al., 2005; Chustz et al., 2011). In human lung fibroblasts, IL-36γ induces the chemokine IL-8 and the Th17 chemokine CCL20 (Chustz et al., 2011), suggesting that IL-36 cytokines can contribute to pro-inflammatory responses and in particular neutrophilic airway inflammation.
Furthermore, the IL-36R is highly expressed on microglial cells and astrocytes (Lovenberg et al., 1996; Berglöf et al., 2003; Wang et al., 2005). Murine IL-36β is expressed in neuron cells and in glial cells, but cannot be upregulated by LPS or IL-1β stimulation (Berglöf et al., 2003; Wang et al., 2005). Intraventricular injection of recombinant non-processed mouse IL-36β does not induce any of the classical IL-1 like responses such as fever or modification of food intake and body weight in mice (Berglöf et al., 2003). However, it must be noted that these studies have been performed with non-processed IL-36 agonists, which is shown to have significantly less bioactivity compared to its processed form (Towne et al., 2011).
IL-36 Receptor Antagonist
IL-36Ra shares homology with IL-1Ra but is unable to bind to the IL-1R1 since it significantly differs in loop conformations from IL-1Ra (Dunn et al., 2003). IL-36Ra inhibits IL-36γ-induced NFκB activation (Debets et al., 2001; Towne et al., 2004) in a way similar to IL-1Ra (Towne et al., 2011). However unlike IL-1Ra, IL-36Ra needs to be processed in order to gain antagonistic properties (Towne et al., 2011). Interestingly, IL-36Ra itself can induce mRNA of IL-4 and protein expression in glia cells, which can be attenuated by anti-SIGIRR antibodies. Moreover, the anti-inflammatory action IL-36Ra in vivo in the brain is dependent on IL-4 and SIGIRR (Costelloe et al., 2008). IL-36Ra reduces fungal-induced Th17 responses, however not in a classical dose-dependent manner (van de Veerdonk et al., 2012; Gresnigt et al., 2013). These reports suggest that IL-36Ra might be able to recruit the anti-inflammatory IL-1 orphan receptor SIGIRR and activate an anti-inflammatory signaling pathway, and thus does not act as a classical receptor antagonist such as IL-1Ra.
The importance of the biological activity of IL-36Ra in regulating skin inflammation has been demonstrated by several reports. IL-36Ra deficiency exacerbates skin lesions in IL-36α transgenic mice (Blumberg et al., 2007). Phorbol ester treatment of mouse skin overexpressing IL-36α results in an inflammatory condition with macroscopic and histological similarities to human psoriasis (Blumberg et al., 2010), and characteristic inflammation of human psoriatic skin transplanted into immunodeficient mice is dependent on the IL-36R (Blumberg et al., 2010). In patients with psoriasis anti-TNF treatment results in decreased expression of the IL-36 agonists and IL-36Ra, which was associated with improved clinical outcome (Johnston et al., 2011). This increased expression of IL-36 agonists correlates with Th17 cytokines in human psoriatic skin lesions, although the expression of IL-36Ra by IL-17-stimulated keratinocytes derived from patients with psoriasis does not differ from healthy controls (Carrier et al., 2011; Muhr et al., 2011). Moreover it has recently been shown that mutations in IL-36RN can cause a rare life-threatening disease called general pustular psoriasis (GPP) (Marrakchi et al., 2011; Onoufriadis et al., 2011; Sugiura et al., 2012; Farooq et al., 2013). The currently found mutations in IL-36RN lead to introduction of a premature stop-codon, frameshift mutation, or an amino acid substitution which were found to result in a misfolded IL-36Ra protein that is less stable and poorly expressed (Marrakchi et al., 2011; Sugiura et al., 2012; Farooq et al., 2013). The misfolded IL-36Ra has less affinity with the IL-36R compared to the wild-type IL-36Ra protein, and therefore is not able to dampen IL-36R-mediated inflammation (Marrakchi et al., 2011; Sugiura et al., 2012; Farooq et al., 2013). These data indicate that IL-36Ra is a receptor antagonist, and that IL-36 signaling plays a significant role in regulating skin inflammation.
IL-36 cytokines might play a significant role in joint disease. Remarkably, only IL-36β is expressed joints of mice and humans (Magne et al., 2006). Interestingly, IL-36β can be measured in the serum of healthy human volunteers, but when serum IL-36β concentrations of healthy volunteers are compared to serum concentrations in rheumatoid arthritis there were no significant differences observed (Magne et al., 2006). However, a recent study showed that the IL-36R was not involved in the inflammatory response in a mouse model of collagen induced arthritis (Lamacchia et al., 2013). In a Caucasian cohort polymorphisms in IL-36β have been associated with spondylitis ankylopoetica, but not this association was not observed in Asian cohorts (Wu and Gu, 2007; Kim et al., 2008).
IL-36γ expression in the lung is significantly increased compared to non-challenged mice in a murine model of HDM-induced allergic inflammation. When recombinant IL-36γ is given intratracheally, this will result in neutrophil influx, but not eosinophilic influx in the lungs of mice, suggesting that IL-36γ is more involved in the regulation of neutrophilic airway inflammation (Chustz et al., 2011; Ramadas et al., 2011, 2012). Bronchial epithelial cells from patients with asthma that were infected with rhinovirus show a higher expression of IL-36γ compared to infected cells from healthy controls (Bochkov et al., 2010). These data support the concept that IL-36 cytokines might also play a significant role in regulating airway inflammation.
Interleukin-38
IL-1F10 has recently been renamed IL-38 (Dinarello et al., 2010). IL-38 shares 43% homology with IL-36Ra and 41% homology with IL-1Ra (Bensen et al., 2001). The IL-38 precursor lacks a signal peptide and is 152 amino acids in length, and the natural N-terminus is unknown (Bensen et al., 2001). There is no caspase-1 consensus cleavage site present in the precursor of IL-38. IL-38 is predominantly expressed in the skin and in proliferating B-cells of the tonsil (Lin et al., 2001). The allele combinations that include IL-38 polymorphisms are associated with psoriatic arthritis and ankylosing spondylitis (Chou et al., 2006; Rahman et al., 2006; Guo et al., 2010), suggesting that IL-38 plays a role in the pathogenesis of these inflammatory diseases. Moreover, and suggesting an important role for this cytokine in human cardiovascular disease, polymorphisms in IL-38 were associated with CRP concentrations in humans in addition to polymorphisms in CRP, IL-6 receptor, and NLRP3 that were also associated with CRP concentrations (Dehghan et al., 2011).
Although it has been reported earlier that IL-38 binds to the IL-1 receptor type I this binding affinity of recombinant IL-38 was low (Lin et al., 2001), and more recently it was demonstrated that IL-38 can bind to the IL-36R similar to IL-36Ra (van de Veerdonk et al., 2012). The only biological activity reported so far is that IL-38 can reduce Candida-induced T helper 17 responses (van de Veerdonk et al., 2012). Notably, the dose-response suppression of IL-38 as well as that of IL-36Ra of Candida-induced IL-22 and IL-17 was not similar to the classic dose-response of IL-1 receptor antagonist, because low concentrations were optimal for inhibiting IL-22 production (van de Veerdonk et al., 2012). A non-classical dose-response has now been observed for IL-36Ra, IL-37, and IL-38 activity and it remains to be determined what the underlying mechanism and biological significance is of these findings.
Interleukin-1 Receptor Antagonist
The IL-1 receptor is expressed in nearly all tissues and its antagonism prevents receptor binding of either IL-1α or IL-1β, therefore its biological function is as diverse as the roles of IL-1α and IL-1β apart and combined. IL-1Ra can inhibit these responses by binding to the IL-1R1 and preventing the recruitment of IL-1RAcP, which will block IL-1 signaling (Dinarello, 1996). The potent inhibitory effect of IL-1Ra and its importance as a regulating protein in IL-1-mediated inflammation is underlined by a disease called deficiency in interleukin-1 receptor antagonist (DIRA) (Aksentijevich et al., 2009). This disease is characterized by severe sterile multifocal osteomyelitis, periostitis, and pustulosis (Aksentijevich et al., 2009). The life-threatening overwhelming inflammation of skin and bones in these patients can be resolved by treatment with recombinant IL-1Ra. Next to treating this rare disease it should be highlighted that IL-1Ra as a recombinant molecule is successful and on the rise as a new therapeutic agent for many diseases. The use of blocking IL-1 is extensively reviewed in Dinarello et al. (2012), and treating auto-inflammatory diseases with IL-1Ra such as Muckle-wells or gout is highly effective, and a growing list of diseases in which blocking IL-1 signaling with IL-1Ra is growing (Dinarello et al., 2012).
Conclusion
It is becoming clear that most members of the IL-1 family primarily promote inflammation and enhance specific acquired immune responses, while some members can provide a brake on inflammation, such as IL-1Ra and IL-36Ra. We are just beginning to understand the biological function of the new IL-1 family members, IL-37 and the cytokines belonging to the IL-36 subfamily, and we are increasingly appreciating the potency of blocking IL-1 in disease. This underscores that long after the initial discovery of IL-1, the cytokine biology of the IL-1 family is still contributing to understanding pathology of disease and remains an exciting field to study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Charles Dinarello for his helpful comments. We thank Mark Gresnigt for drawing the figures. Frank L. van de Veerdonk was supported by a Veni grant of the Netherlands Organization for Scientific Research. Mihai G. Netea was supported by a Vici grant of the Netherlands Organization for Scientific Research.
References
Agostini, L., Martinon, F., Burns, K., McDermott, M. F., Hawkins, P. N., and Tschopp, J. (2004). NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325. doi:10.1016/S1074-7613(04)00046-9
Aksentijevich, I., Masters, S. L., Ferguson, P. J., Dancey, P., Frenkel, J., van Royen-Kerkhoff, A., et al. (2009). An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 360, 2426–2437. doi:10.1056/NEJMoa0807865
Ali, S., Huber, M., Kollewe, C., Bischoff, S. C., Falk, W., and Martin, M. U. (2007). IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc. Natl. Acad. Sci. U.S.A. 104, 18660–18665. doi:10.1073/pnas.0705939104
Ali, S., Mohs, A., Thomas, M., Klare, J., Ross, R., Schmitz, M. L., et al. (2011). The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J. Immunol. 187, 1609–1616. doi:10.4049/jimmunol.1003080
Bae, S., Kang, T., Hong, J., Lee, S., Choi, J., Jhun, H., et al. (2012). Contradictory functions (activation/termination) of neutrophil proteinase 3 enzyme (PR3) in interleukin-33 biological activity. J. Biol. Chem. 287, 8205–8213. doi:10.1074/jbc.M111.295055
Barksby, H. E., Nile, C. J., Jaedicke, K. M., Taylor, J. J., and Preshaw, P. M. (2009). Differential expression of immunoregulatory genes in monocytes in response to Porphyromonas gingivalis and Escherichia coli lipopolysaccharide. Clin. Exp. Immunol. 156, 479–487. doi:10.1111/j.1365-2249.2009.03920.x
Bensen, J. T., Dawson, P. A., Mychaleckyj, J. C., and Bowden, D. W. (2001). Identification of a novel human cytokine gene in the interleukin gene cluster on chromosome 2q12-14. J. Interferon Cytokine Res. 21, 899–904. doi:10.1089/107999001753289505
Berda-Haddad, Y., Robert, S., Salers, P., Zekraoui, L., Farnarier, C., Dinarello, C. A., et al. (2011). Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1alpha. Proc. Natl. Acad. Sci. U.S.A. 108, 20684–20689. doi:10.1073/pnas.1116848108
Berglöf, E., Andre, R., Renshaw, B. R., Allan, S. M., Lawrence, C. B., Rothwell, N. J., et al. (2003). IL-1Rrp2 expression and IL-1F9 (IL-1H1) actions in brain cells. J. Neuroimmunol. 139, 36–43. doi:10.1016/S0165-5728(03)00130-9
Blumberg, H., Dinh, H., Dean, C., Trueblood, E. S., Bailey, K., Shows, D., et al. (2010). IL-1RL2 and its ligands contribute to the cytokine network in psoriasis. J. Immunol. 185, 4354–4362. doi:10.4049/jimmunol.1000313
Blumberg, H., Dinh, H., Trueblood, E. S., Pretorius, J., Kugler, D., Weng, N., et al. (2007). Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J. Exp. Med. 204, 2603–2614. doi:10.1084/jem.20070157
Bochkov, Y., Hanson, K. M., Keles, S., Brockman-Schneider, R., Jarjour, N. N., and Gern, J. E. (2010). Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 3, 69–80. doi:10.1038/mi.2009.109
Bonilla, W. V., Frohlich, A., Senn, K., Kallert, S., Fernandez, M., Johnson, S., et al. (2012). The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science 335, 984–989. doi:10.1126/science.1215418
Boni-Schnetzler, M., Thorne, J., Parnaud, G., Marselli, L., Ehses, J. A., Kerr-Conte, J., et al. (2008). Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta-cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 93, 4065–4074. doi:10.1210/jc.2008-0396
Bufler, P., Azam, T., Gamboni-Robertson, F., Reznikov, L. L., Kumar, S., Dinarello, C. A., et al. (2002). A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc. Natl. Acad. Sci. U.S.A. 99, 13723–13728. doi:10.1073/pnas.212519099
Bufler, P., Gamboni-Robertson, F., Azam, T., Kim, S. H., and Dinarello, C. A. (2004). Interleukin-1 homologues IL-1F7b and IL-18 contain functional mRNA instability elements within the coding region responsive to lipopolysaccharide. Biochem. J. 381, 503–510. doi:10.1042/BJ20040217
Carmi, Y., Voronov, E., Dotan, S., Lahat, N., Rahat, M. A., Fogel, M., et al. (2009). The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J. Immunol. 183, 4705–4714. doi:10.4049/jimmunol.0901511
Carrier, Y., Ma, H. L., Ramon, H. E., Napierata, L., Small, C., O’Toole, M., et al. (2011). Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J. Invest. Dermatol. 131, 2428–2437. doi:10.1038/jid.2011.234
Carriere, V., Roussel, L., Ortega, N., Lacorre, D. A., Americh, L., Aguilar, L., et al. (2007). IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 282–287. doi:10.1073/pnas.0606854104
Cayrol, C., and Girard, J. P. (2009). The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. U.S.A. 106, 9021–9026. doi:10.1073/pnas.0812690106
Chackerian, A. A., Oldham, E. R., Murphy, E. E., Schmitz, J., Pflanz, S., and Kastelein, R. A. (2007). IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J. Immunol. 179, 2551–2555.
Chan, J. K., Roth, J., Oppenheim, J. J., Tracey, K. J., Vogl, T., Feldmann, M., et al. (2012). Alarmins: awaiting a clinical response. J. Clin. Invest. 122, 2711–2719. doi:10.1172/JCI62423
Chen, C. J., Kono, H., Golenbock, D., Reed, G., Akira, S., and Rock, K. L. (2007). Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 13, 851–856. doi:10.1038/nm1603
Chizzolini, C., Chicheportiche, R., Alvarez, M., de Rham, C., Roux-Lombard, P., Ferrari-Lacraz, S., et al. (2008). Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood 112, 3696–3703. doi:10.1182/blood-2008-05-155408
Chou, C. T., Timms, A. E., Wei, J. C., Tsai, W. C., Wordsworth, B. P., and Brown, M. A. (2006). Replication of association of IL1 gene complex members with ankylosing spondylitis in Taiwanese Chinese. Ann. Rheum. Dis. 65, 1106–1109. doi:10.1136/ard.2005.046847
Chustz, R. T., Nagarkar, D. R., Poposki, J. A., Favoreto, S. Jr., Avila, P. C., Schleimer, R. P., et al. (2011). Regulation and function of the IL-1 family cytokine IL-1F9 in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 45, 145–153. doi:10.1165/rcmb.2010-0075OC
Coeshott, C., Ohnemus, C., Pilyavskaya, A., Ross, S., Wieczorek, M., Kroona, H., et al. (1999). Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc. Natl. Acad. Sci. U.S.A. 96, 6261–6266. doi:10.1073/pnas.96.11.6261
Cohen, I., Rider, P., Carmi, Y., Braiman, A., Dotan, S., White, M. R., et al. (2010). Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc. Natl. Acad. Sci. U.S.A. 107, 2574–2579. doi:10.1073/pnas.0915018107
Costelloe, C., Watson, M., Murphy, A., McQuillan, K., Loscher, C., Armstrong, M. E., et al. (2008). IL-1F5 mediates anti-inflammatory activity in the brain through induction of IL-4 following interaction with SIGIRR/TIR8. J. Neurochem. 105, 1960–1969. doi:10.1111/j.1471-4159.2008.05304.x
Crisan, T. O., Plantinga, T. S., van de Veerdonk, F. L., Farcas, M. F., Stoffels, M., Kullberg, B. J., et al. (2011). Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS ONE 6:e18666. doi:10.1371/journal.pone.0018666
Debets, R., Timans, J. C., Homey, B., Zurawski, S., Sana, T. R., Lo, S., et al. (2001). Two novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as an antagonist and agonist of NF-kappa B activation through the orphan IL-1 receptor-related protein 2. J. Immunol. 167, 1440–1446.
Dehghan, A., Dupuis, J., Barbalic, M., Bis, J. C., Eiriksdottir, G., Lu, C., et al. (2011). Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation 123, 731–738. doi:10.1161/CIRCULATIONAHA.110.948570
Dinarello, C., Arend, W., Sims, J., Smith, D., Blumberg, H., O’Neill, L., et al. (2010). IL-1 family nomenclature. Nat. Immunol. 11, 973. doi:10.1038/ni1110-973
Dinarello, C. A. (2011a). A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur. J. Immunol. 41, 1203–1217. doi:10.1002/eji.201141550
Dinarello, C. A. (2011b). Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732. doi:10.1182/blood-2010-07-273417
Dinarello, C. A., Ikejima, T., Warner, S. J., Orencole, S. F., Lonnemann, G., Cannon, J. G., et al. (1987). Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J. Immunol. 139, 1902–1910.
Dinarello, C. A., Simon, A., and van der Meer, J. W. (2012). Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652. doi:10.1038/nrd3800
Dong, C. (2008). TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat. Rev. Immunol. 8, 337–348. doi:10.1038/nri2295
Dunn, E. F., Gay, N. J., Bristow, A. F., Gearing, D. P., O’Neill, L. J., and Pei, X. Y. (2003). High-resolution structure of murine interleukin 1 homologue IL-1F5 reveals unique loop conformations for receptor binding specificity. Biochemistry 42, 10938–10944. doi:10.1021/bi0341197
Faggioni, R., Fantuzzi, G., Fuller, J., Dinarello, C. A., Feingold, K. R., and Grunfeld, C. (1998). IL-1 beta mediates leptin induction during inflammation. Am. J. Physiol. 274, R204–R208.
Fantuzzi, G., Ku, G., Harding, M. W., Livingston, D. J., Sipe, J. D., Kuida, K., et al. (1997a). Response to local inflammation of IL-1 beta-converting enzyme-deficient mice. J. Immunol. 158, 1818–1824.
Fantuzzi, G., Sacco, S., Ghezzi, P., and Dinarello, C. A. (1997b). Physiological and cytokine responses in interleukin-1β-deficient mice after zymosan-induced inflammation. Am. J. Physiol. 273, R400–R406.
Fantuzzi, G., Zheng, H., Faggioni, R., Benigni, F., Ghezzi, P., Sipe, J. D., et al. (1996). Effect of endotoxin in IL-1β-deficient mice. J. Immunol. 157, 291–296.
Farooq, M., Nakai, H., Fujimoto, A., Fujikawa, H., Matsuyama, A., Kariya, N., et al. (2013). Mutation analysis of the IL36RN gene in 14 Japanese patients with generalized pustular psoriasis. Hum. Mutat. 34, 176–183. doi:10.1002/humu.22203
Franzke, C.-W., Cobzaru, C., Triantafyllopoulou, A., Löffek, S., Horiuchi, K., Threadgill, D. W., et al. (2012). Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Exp. Med. 209, 1105–1119. doi:10.1084/jem.2011225820913c
Gattorno, M., Tassi, S., Carta, S., Delfino, L., Ferlito, F., Pelagatti, M. A., et al. (2007). Pattern of interleukin-1beta secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum. 56, 3138–3148. doi:10.1002/art.22842
Gawaz, M., Brand, K., Dickfeld, T., Pogatsa-Murray, G., Page, S., Bogner, C., et al. (2000). Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin-1-dependent mechanism. Implications for atherogenesis. Atherosclerosis 148, 75–85. doi:10.1016/S0021-9150(99)00241-5
Goldbach-Mansky, R., Dailey, N. J., Canna, S. W., Gelabert, A., Jones, J., Rubin, B. I., et al. (2006). Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 355, 581–592. doi:10.1056/NEJMoa055137
Goshen, I., and Yirmiya, R. (2009). Interleukin-1 (IL-1): a central regulator of stress responses. Front. Neuroendocrinol. 30:30–45. doi:10.1016/j.yfrne.2008.10.001
Gresnigt, M. S., Rosler, B., Jacobs, C. W., Becker, K. L., Joosten, L. A., van der Meer, J. W., et al. (2013). The IL-36 receptor pathway regulates Aspergillus fumigatus-induced Th1 and Th17 responses. Eur. J. Immunol. 43, 416–426. doi:10.1002/eji.201242711
Greten, F. R., Arkan, M. C., Bollrath, J., Hsu, L. C., Goode, J., Miething, C., et al. (2007). NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130, 918–931. doi:10.1016/j.cell.2007.07.009
Gross, O., Yazdi, A. S., Thomas, C. J., Masin, M., Heinz, L. X., Guarda, G., et al. (2012). Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36, 388–400. doi:10.1016/j.immuni.2012.01.018
Guo, Z. S., Li, C., Lin, Z. M., Huang, J. X., Wei, Q. J., Wang, X. W., et al. (2010). Association of IL-1 gene complex members with ankylosing spondylitis in Chinese Han population. Int. J. Immunogenet. 37, 33–37. doi:10.1111/j.1744-313X.2009.00889.x
Hacham, M., Argov, S., White, R. M., Segal, S., and Apte, R. N. (2002). Different patterns of interleukin-1alpha and interleukin-1beta expression in organs of normal young and old mice. Eur. Cytokine Netw. 13, 55–65.
Hanamsagar, R., Hanke, M. L., and Kielian, T. (2012). Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. 33, 333–342. doi:10.1016/j.it.2012.03.001
Harris, J., Hartman, M., Roche, C., Zeng, S. G., O’Shea, A., Sharp, F. A., et al. (2011). Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 286, 9587–9597. doi:10.1074/jbc.M110.202911
Hawrylowicz, C. M., Santoro, S. A., Platt, F. M., and Unanue, E. R. (1989). Activated platelets express IL-1 activity. J. Immunol. 143, 4015–4018.
Henley, D. V., Bellone, C. J., Williams, D. A., Ruh, T. S., and Ruh, M. F. (2004). Aryl hydrocarbon receptor-mediated posttranscriptional regulation of IL-1beta. Arch. Biochem. Biophys. 422, 42–51. doi:10.1016/j.abb.2003.11.022
Hoffman, H. M., Mueller, J. L., Broide, D. H., Wanderer, A. A., and Kolodner, R. D. (2001). Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 29, 301–305. doi:10.1038/ng756
Horai, R., Asano, M., Sudo, K., Kanuka, H., Suzuki, M., Nishihara, M., et al. (1998). Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J. Exp. Med. 187, 1463–1475. doi:10.1084/jem.187.9.1463
Hughes, T., Becknell, B., Freud, A. G., McClory, S., Briercheck, E., Yu, J., et al. (2010). Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity 32, 803–814. doi:10.1016/j.immuni.2010.06.007
Johnston, A., Xing, X., Guzman, A. M., Riblett, M., Loyd, C. M., Ward, N. L., et al. (2011). IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. 186, 2613–2622. doi:10.4049/jimmunol.1003162
Joosten, L. A., Netea, M. G., Fantuzzi, G., Koenders, M. I., Helsen, M. M., Sparrer, H., et al. (2009). Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 60, 3651–3662. doi:10.1002/art.25006
Kamari, Y., Werman-Venkert, R., Shaish, A., Werman, A., Harari, A., Gonen, A., et al. (2007). Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis 195, 31–38. doi:10.1016/j.atherosclerosis.2006.11.026
Kaplanski, G., Farnarier, C., Kaplanski, S., Porat, R., Shapiro, L., Bongrand, P., et al. (1994). Interleukin-1 induces interleukin-8 secretion from endothelial cells by a juxtacrine mechanism. Blood 84, 4242–4248.
Kawaguchi, Y., Nishimagi, E., Tochimoto, A., Kawamoto, M., Katsumata, Y., Soejima, M., et al. (2006). Intracellular IL-1alpha-binding proteins contribute to biological functions of endogenous IL-1alpha in systemic sclerosis fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 103, 14501–14506. doi:10.1073/pnas.0603545103
Kim, T.-J., Kim, T.-H., Lee, H.-J., Peddle, L., Rahman, P., Hu, P., et al. (2008). Interleukin 1 polymorphisms in patients with ankylosing spondylitis in Korea. J. Rheumatol. 35, 1603–1608.
Kim, Y. H., Yang, T. Y., Park, C. S., Ahn, S. H., Son, B. K., Kim, J. H., et al. (2012). Anti-IL-33 antibody has a therapeutic effect in a murine model of allergic rhinitis. Allergy 67, 183–190. doi:10.1111/j.1398-9995.2011.02735.x
Klionsky, D. J. (2007). Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931–937. doi:10.1038/nrm2245
Kumar, S., Hanning, C. R., Brigham-Burke, M. R., Rieman, D. J., Lehr, R., Khandekar, S., et al. (2002). Interleukin-1F7B (IL-1H4/IL-1F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine 18, 61–71. doi:10.1006/cyto.2002.0873
Kurt-Jones, E. A., Beller, D. I., Mizel, S. B., and Unanue, E. R. (1985). Identification of a membrane-associated interleukin-1 in macrophages. Proc. Natl. Acad. Sci. U.S.A. 82, 1204–1208. doi:10.1073/pnas.82.4.1204
Lamacchia, C., Palmer, G., Rodriguez, E., Martin, P., Vigne, S., Seemayer, C. A., et al. (2013). The severity of experimental arthritis is independent of IL-36 receptor signaling. Arthritis Res. Ther. 15, R38. doi:10.1186/ar4192
Lefrancais, E., Roga, S., Gautier, V., Gonzalez-de-Peredo, A., Monsarrat, B., Girard, J. P., et al. (2012). IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. U.S.A. 109, 1673–1678. doi:10.1073/pnas.1115884109
Li, Y., Messina, C., Bendaoud, M., Fine, D. H., Schreiner, H., and Tsiagbe, V. K. (2010). Adaptive immune response in osteoclastic bone resorption induced by orally administered Aggregatibacter actinomycetemcomitans in a rat model of periodontal disease. Mol. Oral Microbiol. 25, 275–292. doi:10.1111/j.2041-1014.2010.00576.x
Lin, H., Ho, A. S., Haley-Vicente, D., Zhang, J., Bernal-Fussell, J., Pace, A. M., et al. (2001). Cloning and characterization of IL-1HY2, a novel interleukin-1 family member. J. Biol. Chem. 276, 20597–20602. doi:10.1074/jbc.M010095200
Louten, J., Rankin, A. L., Li, Y., Murphy, E. E., Beaumont, M., Moon, C., et al. (2011). Endogenous IL-33 enhances Th2 cytokine production and T-cell responses during allergic airway inflammation. Int. Immunol. 23, 307–315. doi:10.1093/intimm/dxr006
Lovenberg, T. W., Crowe, P. D., Liu, C., Chalmers, D. T., Liu, X. J., Liaw, C., et al. (1996). Cloning of a cDNA encoding a novel interleukin-1 receptor related protein (IL 1R-rp2). J. Neuroimmunol. 70, 113–122. doi:10.1016/S0165-5728(96)00047-1
Luheshi, N. M., Kovacs, K. J., Lopez-Castejon, G., Brough, D., and Denes, A. (2011). Interleukin-1alpha expression precedes IL-1beta after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J. Neuroinflammation 8, 186. doi:10.1186/1742-2094-8-186
Maedler, K., Sergeev, P., Ris, F., Oberholzer, J., Joller-Jemelka, H. I., Spinas, G. A., et al. (2002). Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 110, 851–860. doi:10.1172/JCI15318
Magne, D., Palmer, G., Barton, J. L., Mézin, F., Talabot-Ayer, D., Bas, S., et al. (2006). The new IL-1 family member IL-1F8 stimulates production of inflammatory mediators by synovial fibroblasts and articular chondrocytes. Arthritis Res. Ther. 8, R80. doi:10.1186/ar1946
Maier, J. A. M., Voulalas, P., Roeder, D., and Maciag, T. (1990). Extension of the life span of human endothelial cells by an interleukin-1α antisense oligomer. Science 249, 1570–1574. doi:10.1126/science.2218499
Marrakchi, S., Guigue, P., Renshaw, B. R., Puel, A., Pei, X. Y., Fraitag, S., et al. (2011). Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 365, 620–628. doi:10.1056/NEJMoa1013068
Martinon, F., Mayor, A., and Tschopp, J. (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265. doi:10.1146/annurev.immunol.021908.132715
Masamune, A., Watanabe, T., Kikuta, K., Satoh, K., Kanno, A., and Shimosegawa, T. (2010). Nuclear expression of interleukin-33 in pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G821–G832. doi:10.1152/ajpgi.00178.2010
Matsuba-Kitamura, S., Yoshimoto, T., Yasuda, K., Futatsugi-Yumikura, S., Taki, Y., Muto, T., et al. (2010). Contribution of IL-33 to induction and augmentation of experimental allergic conjunctivitis. Int. Immunol. 22, 479–489. doi:10.1093/intimm/dxq035
McNamee, E. N., Masterson, J. C., Jedlicka, P., McManus, M., Grenz, A., Collins, C. B., et al. (2011). Interleukin 37 expression protects mice from colitis. Proc. Natl. Acad. Sci. U.S.A. 108, 16711–16716. doi:10.1073/pnas.1111982108
Meyer, U. (2011). Anti-inflammatory signaling in schizophrenia. Brain Behav. Immun. 25, 1507–1518. doi:10.1016/j.bbi.2011.05.014
Miller, A. C., Schattenberg, D. G., Malkinson, A. M., and Ross, D. (1994). Decreased content of the IL1 alpha processing enzyme calpain in murine bone marrow-derived macrophages after treatment with the benzene metabolite hydroquinone. Toxicol. Lett. 74, 177–184. doi:10.1016/0378-4274(94)90096-5
Miller, A. M., Xu, D., Asquith, D. L., Denby, L., Li, Y., Sattar, N., et al. (2008). IL-33 reduces the development of atherosclerosis. J. Exp. Med. 205, 339–346. doi:10.1084/jem.20071868
Moreira, A. P., Cavassani, K. A., Ismailoglu, U. B., Hullinger, R., Dunleavy, M. P., Knight, D. A., et al. (2011). The protective role of TLR6 in a mouse model of asthma is mediated by IL-23 and IL-17A. J. Clin. Invest. 121, 4420–4432. doi:10.1172/JCI44999
Moussion, C., Ortega, N., and Girard, J. P. (2008). The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS ONE 3:e3331. doi:10.1371/journal.pone.0003331
Muhr, P., Zeitvogel, J., Heitland, I., Werfel, T., and Wittmann, M. (2011). Expression of interleukin (IL)-1 family members upon stimulation with IL-17 differs in keratinocytes derived from patients with psoriasis and healthy donors. Br. J. Dermatol. 165, 189–193. doi:10.1111/j.1365-2133.2011.10302.x
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi:10.1038/ni.1980
Netea, M. G., Nold-Petry, C. A., Nold, M. F., Joosten, L. A., Opitz, B., van der Meer, J. H., et al. (2009). Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113, 2324–2335. doi:10.1182/blood-2008-03-146720
Nold, M., Hauser, I. A., Hofler, S., Goede, A., Eberhardt, W., Ditting, T., et al. (2003). IL-18BPa:Fc cooperates with immunosuppressive drugs in human whole blood. Biochem. Pharmacol. 66, 505–510. doi:10.1016/S0006-2952(03)00294-6
Nold, M. F., Nold-Petry, C. A., Zepp, J. A., Lo, C., Garlanda, C., Mantovani, A., et al. (2011). Interleukin 37 exerts its anti-inflammatory functions by associating with IL-18R alpha and SIGIRR. Cytokine 56, 12. doi:10.1016/j.cyto.2011.07.333 (ABSTRACT).
Nold, M. F., Nold-Petry, C. A., Zepp, J. A., Palmer, B. E., Bufler, P., and Dinarello, C. A. (2010). IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 11, 1014–1022. doi:10.1038/ni.1944
Onoufriadis, A., Simpson, M. A., Pink, A. E., Di Meglio, P., Smith, C. H., Pullabhatla, V., et al. (2011). Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 89, 432–437. doi:10.1016/j.ajhg.2011.07.022
Pan, G., Risser, P., Mao, W., Baldwin, D. T., Zhong, A. W., Filvaroff, E., et al. (2001). IL-1H, an interleukin 1-related protein that binds IL-18 receptor/IL-1Rrp. Cytokine 13, 1–7. doi:10.1006/cyto.2000.0799
Rahman, P., Sun, S., Peddle, L., Snelgrove, T., Melay, W., Greenwood, C., et al. (2006). Association between the interleukin-1 family gene cluster and psoriatic arthritis. Arthritis Rheum. 54, 2321–2325. doi:10.1002/art.21928
Ramadas, R. A., Ewart, S. L., Iwakura, Y., Medoff, B. D., and LeVine, A. M. (2012). IL-36alpha exerts pro-inflammatory effects in the lungs of mice. PLoS ONE 7:e45784. doi:10.1371/journal.pone.0045784
Ramadas, R. A., Ewart, S. L., Medoff, B. D., and LeVine, A. M. (2011). Interleukin-1 family member 9 stimulates chemokine production and neutrophil influx in mouse lungs. Am. J. Respir. Cell Mol. Biol. 44, 134–145. doi:10.1165/rcmb.2009-0315OC
Rider, P., Carmi, Y., Guttman, O., Braiman, A., Cohen, I., Voronov, E., et al. (2011). IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 187, 4835–4843. doi:10.4049/jimmunol.1102048
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B. G., Satoh, T., et al. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268. doi:10.1038/nature07383
Sanada, S., Hakuno, D., Higgins, L. J., Schreiter, E. R., McKenzie, A. N., and Lee, R. T. (2007). IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J. Clin. Invest. 117, 1538–1549. doi:10.1172/JCI30634
Schmid, D., and Munz, C. (2007). Innate and adaptive immunity through autophagy. Immunity 27, 11–21. doi:10.1016/j.immuni.2007.07.004
Schmitz, J., Owyang, A., Oldham, E., Song, Y., Murphy, E., McClanahan, T. K., et al. (2005). IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490. doi:10.1016/j.immuni.2005.09.015
Sharma, S., Kulk, N., Nold, M. F., Graf, R., Kim, S. H., Reinhardt, D., et al. (2008). The IL-1 family member 7b translocates to the nucleus and down-regulates proinflammatory cytokines. J. Immunol. 180, 5477–5482.
Smeekens, S. P., van de Veerdonk, F. L., van der Meer, J. W., Kullberg, B. J., Joosten, L. A., and Netea, M. G. (2010). The Candida Th17 response is dependent on mannan- and beta-glucan-induced prostaglandin E2. Int. Immunol. 22, 889–895. doi:10.1093/intimm/dxq442
Smith, D. E., Renshaw, B. R., Ketchem, R. R., Kubin, M., Garka, K. E., and Sims, J. E. (2000). Four new members expand the interleukin-1 superfamily. J. Biol. Chem. 275, 1169–1175. doi:10.1074/jbc.275.2.1169
Stevenson, F. T., Turck, J., Locksley, R. M., and Lovett, D. H. (1997). The N-terminal propiece of interleukin 1 alpha is a transforming nuclear oncoprotein. Proc. Natl. Acad. Sci. U.S.A. 94, 508–513. doi:10.1073/pnas.94.2.508
Sugiura, K., Takeichi, T., Kono, M., Ogawa, Y., Shimoyama, Y., Muro, Y., et al. (2012). A novel IL36RN/IL1F5 homozygous nonsense mutation, p.Arg10X, in a Japanese patient with adult-onset generalized pustular psoriasis. Br. J. Dermatol. 167, 699–701. doi:10.1111/j.1365-2133.2012.10953.x
Thornton, P., McColl, B. W., Greenhalgh, A., Denes, A., Allan, S. M., and Rothwell, N. J. (2010). Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood 115, 3632–3639. doi:10.1182/blood-2009-11-252643
Towne, J. E., Garka, K. E., Renshaw, B. R., Virca, G. D., and Sims, J. E. (2004). Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 279, 13677–13688. doi:10.1074/jbc.M400117200
Towne, J. E., Renshaw, B. R., Douangpanya, J., Lipsky, B. P., Shen, M., Gabel, C. A., et al. (2011). Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J. Biol. Chem. 286, 42594–42602. doi:10.1074/jbc.M111.267922
Tschopp, J., and Schroder, K. (2010). NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10, 210–215. doi:10.1038/nri2725
Turtoi, A., Brown, I., Schläger, M., and Schneeweiss, F. H. (2010). Gene expression profile of human lymphocytes exposed to (211) At alpha particles. Radiat. Res. 174, 125–136. doi:10.1667/RR1659.1
van de Veerdonk, F. L., Gresnigt, M. S., Kullberg, B. J., van der Meer, J. W., Joosten, L. A., and Netea, M. G. (2009). Th17 responses and host defense against microorganisms: an overview. BMB Rep. 42, 776–787. doi:10.5483/BMBRep.2009.42.12.776
van de Veerdonk, F. L., Stoeckman, A. K., Wu, G., Boeckermann, A. N., Azam, T., Netea, M. G., et al. (2012). IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 109, 3001–3005. doi:10.1073/pnas.1121534109
Vigne, S., Palmer, G., Lamacchia, C., Martin, P., Talabot-Ayer, D., Rodriguez, E., et al. (2011). IL-36R ligands are potent regulators of dendritic and T cells. Blood 118, 5813–5823. doi:10.1182/blood-2011-05-356873
Vos, J. B., van Sterkenburg, M., Rabe, K. F., Schalkwijk, J., Hiemstra, P. S., and Datson, N. (2005). Transcriptional response of bronchial epithelial cells to Pseudomonas aeruginosa: identification of early mediators of host defense. Physiol. Genomics 21, 324–336. doi:10.1152/physiolgenomics.00289.2004
Wang, P., Meinhardt, B., Andre, R., Renshaw, B. R., Kimber, I., Rothwell, N. J., et al. (2005). The interleukin-1-related cytokine IL-1F8 is expressed in glial cells, but fails to induce IL-1beta signalling responses. Cytokine 29, 245–250.
Weaver, C. T., Harrington, L. E., Mangan, P. R., Gavrieli, M., and Murphy, K. M. (2006). Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity 24, 677–688. doi:10.1016/j.immuni.2006.06.002
Werman, A., Werman-Venkert, R., White, R., Lee, J. K., Werman, B., Krelin, Y., et al. (2004). The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. U.S.A. 101, 2434–2439. doi:10.1073/pnas.0308705101
Willart, M. A., Deswarte, K., Pouliot, P., Braun, H., Beyaert, R., Lambrecht, B. N., et al. (2012). Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J. Exp. Med. 209, 1505–1517. doi:10.1084/jem.20112691
Wilson, N. J., Boniface, K., Chan, J. R., McKenzie, B. S., Blumenschein, W. M., Mattson, J. D., et al. (2007). Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 8, 950–957. doi:10.1038/ni1497
Wu, Z., and Gu, J. (2007). A meta-analysis on interleukin-1 gene cluster polymorphism and genetic susceptbility for ankylosing spondylitis. Zhonghua Yi Xue Za Zhi 87, 433–437.
Yang, J., Meyer, M., Müller, A.-K., Böhm, F., Grose, R., Dauwalder, T., et al. (2010). Fibroblast growth factor receptors 1 and 2 in keratinocytes control the epidermal barrier and cutaneous homeostasis. J. Cell Biol. 188, 935–952. doi:10.1083/jcb.200910126
Yasuda, K., Muto, T., Kawagoe, T., Matsumoto, M., Sasaki, Y., Matsushita, K., et al. (2012). Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc. Natl. Acad. Sci. U.S.A. 109, 3451–3456. doi:10.1073/pnas.1201042109
Yin, H., Morioka, H., Towle, C. A., Vidal, M., Watanabe, T., and Weissbach, L. (2001). Evidence that HAX-1 is an interleukin-1 alpha N-terminal binding protein. Cytokine 15, 122–137. doi:10.1006/cyto.2001.0891
Yirmiya, R., and Goshen, I. (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 25, 181–213. doi:10.1016/j.bbi.2010.10.015
Zheng, H., Fletcher, D., Kozak, W., Jiang, M., Hofmann, K., Conn, C. C., et al. (1995). Resistance to fever induction and impaired acute-phase response in interleukin-1β deficient mice. Immunity 3, 9–19. doi:10.1016/1074-7613(95)90154-X
Keywords: interleukin-1, cytokines, ligands, review, biology
Citation: van de Veerdonk FL and Netea MG (2013) New insights in the immunobiology of IL-1 family members. Front. Immunol. 4:167. doi: 10.3389/fimmu.2013.00167
Received: 29 March 2013; Accepted: 12 June 2013;
Published online: 08 July 2013.
Edited by:
Cecilia Garlanda, Istituto Clinico Humanitas, ItalyReviewed by:
Massimo Gadina, National Institute of Arthritis Musculoskeletal and Skin diseases-National Institutes of Health, USABarbara Viviani, University of Milan, Italy
Copyright: © 2013 van de Veerdonk and Netea. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Frank L. van de Veerdonk, Department of Medicine, Radboud University Nijmegen Medical Center, P.O. Box 9101, 6500HB Nijmegen, Netherlands e-mail:Zi52ZWVyZG9ua0BhaWcudW1jbi5ubA==