 Giovanni Antonio Maria Povoleri1,2
Giovanni Antonio Maria Povoleri1,2 Cristiano Scottà1,2 Estefania Andrea Nova-Lamperti1,2
Cristiano Scottà1,2 Estefania Andrea Nova-Lamperti1,2 Susan John2,3
Susan John2,3 Giovanna Lombardi1,2
Giovanna Lombardi1,2 Behdad Afzali1,2*
Behdad Afzali1,2*
- 1Medical Research Council Centre for Transplantation, King’s College London, London, UK
- 2National Institute for Health Research Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London, London, UK
- 3Department of Immunobiology, King’s College London, London, UK
Physiological health must balance immunological responsiveness against foreign pathogens with tolerance toward self-components and commensals. Disruption of this balance causes autoimmune diseases/chronic inflammation, in case of excessive immune responses, and persistent infection/immunodeficiency if regulatory components are overactive. This homeostasis occurs at two different levels: at a resting state to prevent autoimmune disease, as autoreactive effector T-cells (Teffs) are only partially deleted in the thymus, and during inflammation to prevent excessive tissue injury, contract the immune response, and enable tissue repair. Adaptive immune cells with regulatory function (“regulatory T-cells”) are essential to control Teffs. Two sets of regulatory T cell are required to achieve the desired control: those emerging de novo from embryonic/neonatal thymus (“thymic” or tTregs), whose function is to control autoreactive Teffs to prevent autoimmune diseases, and those induced in the periphery (“peripheral” or pTregs) to acquire regulatory phenotype in response to pathogens/inflammation. The differentiation mechanisms of these cells determine their commitment to lineage and plasticity toward other phenotypes. tTregs, expressing high levels of IL-2 receptor alpha chain (CD25), and the transcription factor Foxp3, are the most important, since mutations or deletions in these genes cause fatal autoimmune diseases in both mice and men. In the periphery, instead, Foxp3+ pTregs can be induced from naïve precursors in response to environmental signals. Here, we discuss molecular signatures and induction processes, mechanisms and sites of action, lineage stability, and differentiating characteristics of both Foxp3+ and Foxp3− populations of regulatory T cells, derived from the thymus or induced peripherally. We relate these predicates to programs of cell-based therapy for the treatment of autoimmune diseases and induction of tolerance to transplants.
Introduction
Physiological health requires a balance between immunological responsiveness against foreign pathogens and tolerance toward self-components and commensals. The immune system must guarantee this homeostatic balance, since its disruption leads to autoimmune diseases (AID) and chronic inflammation in the event of excessive immune reactivity, on the one hand, and persistent infection(s) and immunodeficiency on the other (Figure 1).
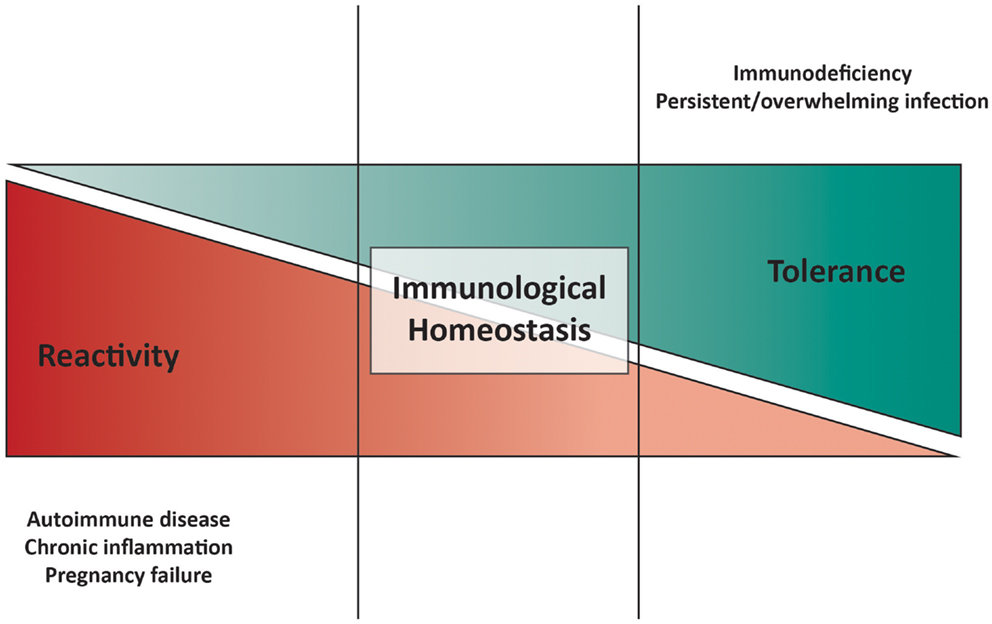
Figure 1. Model of immunological homeostasis. Disturbance of the balance between pro-inflammatory and anti-inflammatory mechanisms is shown at either end of the cartoon. On the one hand, excessive immune responsiveness and/or deficiency in tolerogenic mechanisms can lead to autoimmune diseases, chronic inflammation, and pregnancy failure. On the other hand, weak immune responsiveness and/or excessive tolerance-inducing machineries can result in in immunodeficiency, characterized by recurrent, and/or overwhelming infections.
Regulation of immune responses occurs concurrently at two different levels: in the “pathogen-free” environment (where “danger” is inherently internal), to maintain tolerance to self-components, and in the “pathogen-containing” environment (where “danger” is external), to prevent excessive tissue injury, contract the immune response and enable tissue repair.
Central selection of the T cell repertoire imparts intrinsic autoreactivity to adaptive immunity as only T cells capable of recognizing self-MHC are positively selected for survival. Thus, despite negative selection of strongly autoreactive thymocytes, the mature immune system can clearly be demonstrated to contain T cells with autospecificity (Muraro et al., 1997), necessitating active regulation of these cells in the periphery. That autoreactive cells exist in the neonatal circulation imparts an obligate requirement for the presence of regulation from birth, a function ascribed to non-redundant “thymically derived” regulatory T cells (tTregs).
Although tTregs have been the focus of the “immunoregulation” literature in recent years, the adult T cell pool also contains a series of other T cells with regulatory function, many of which are induced to develop suppressive phenotypes in the periphery in response to antigenic challenges and the local micro-environment. Such “induced” regulatory T cells include induced Foxp3+ (iTregs) and Foxp3− (Th3, Tr1, iTr35, and CD8+CD28−) populations of cells.
In this review, we describe the origins and functions of different T cells with regulatory function, detailing their properties. An important note that is worth highlighting at the outset is one of semantics. In this review, we refer to CD4+CD25hiFoxP3+ regulatory T cells as “Tregs.” As there have been recent calls for greater clarity in the nomenclature of FoxP3+ regulatory T cells (Abbas et al., 2013) (Table 1), we refer to thymically derived Tregs as tTregs and peripherally derived Tregs as pTregs. All in vitro induced FoxP3+ Tregs we will call iTregs. All other inducible regulatory T cell populations will be referred to by their current internationally accepted names, such as Tr1 cells.

Table 1. Recommendations for Treg cell nomenclature.
Foxp3+ Regulatory T Cells
The relative importance of centrally derived tolerance-inducing T cells was established by experiments between the late 1960s and early 1980s where it was observed that thymectomy of mice on the third day of life resulted in organ-specific autoimmune diseases [the exact target organ(s) depending on the mouse strain used]. However, this did not occur if neonatal mice were thymectomized on days 1 or 7 (Nishizuka and Sakakura, 1969; Kojima et al., 1976, 1980; Taguchi and Nishizuka, 1981) and day 3 thymectomized mice would not develop autoimmunity after infusion of thymocytes (Sakaguchi et al., 1982). These experiments suggested that autoreactive T cells exit the thymus in the first 3 days of life followed a few days later by a population of suppressor cells that control the autoreactive cohort. These experiments were followed by the first descriptions of Tregs by Sakaguchi et al. (1995, 1996) as a circulating subset of CD4+ T cells expressing high levels of CD25 (the IL-2 receptor α-chain), which could prevent the development of multi-organ autoimmune diseases (thyroiditis, gastritis, insulitis, sialoadenitis, adrenalitis, oophoritis, glomerulonephritis, and polyarthritis) and/or rodent graft-versus-host disease (GVHD)-like wasting disease in thymectomized mice, by adoptive transfer (Suri-Payer et al., 1998). This was an advance on previous observations that had identified the “rescuing” population as Thy1+(CD90+) Lyt1+(CD5+) Lyt2− (CD8a−) Lyt3−(CD8b−) (Sakaguchi et al., 1982) CD45RBlo (Morrissey et al., 1993). As CD25 correlates positively with CD5 and negatively with CD45RB, the identification of CD25 expression as a surface marker for Tregs was biologically plausible. The subsequent identification of humans and mice deficient in CD4+CD25hi cells (as a result of mutations in the FOXP3 and Foxp3 genes respectively – see below), which develop severe autoimmune diseases (Sakaguchi et al., 1995, 1996; Chatila et al., 2000; Wildin et al., 2001) strongly suggests that these cells have a critical and non-redundant regulatory role in the maintenance of self-tolerance.
Although CD25 expression was the original defining feature of Tregs, CD25 is also expressed by antigen-experienced and recently activated conventional T cells with non-regulatory properties (effector T cells, “Teff”). As a result, CD25 is of greatest sensitivity when used to identify Tregs from naïve T cell populations, such as human umbilical cord blood, or antigen-naïve animals. Thus, in antigen-experienced mammals, only the top 2–5% of CD25 expressing CD4+ cells (CD25,hi) contains genuine Tregs (Baecher-Allan et al., 2001). Since the descriptions of Tregs, therefore, a number of additional markers have been proposed as Treg-specifying, including cytotoxic T-lymphocyte antigen-4 (CTLA-4) (Wing et al., 2008; Sakaguchi et al., 2009), GITR (glucocorticoid-induced TNF receptor family related protein; TNFRSF18) (Shimizu et al., 2002), CD39 (Deaglio et al., 2007), HLA-DR (Baecher-Allan and Hafler, 2006), CD45RA (Miyara et al., 2009), and low expression of CD127 (the IL-7 receptor α-chain) (Liu et al., 2006). While these markers will not be the focus of this review, it is important to note that none can be used as unambiguous identifiers of human Tregs; however, they often identify subsets of Tregs with different (quantities or mechanisms of) suppressive functions, implying that there is considerable heterogeneity in human populations of Tregs. Such heterogeneity and the lack of specific markers for the Treg lineage remain the cornerstone of debates regarding whether Tregs are in fact a distinct T cell lineage and/or a possibility in the life cycle of many different T cells.
Forkhead Box P3, the Key Transcription Factor of Tregs
The Scurfy mouse (sf), an X-linked mutant strain, described in 1949 [cit. loc (Russell et al., 1959)], exhibits a series of autoimmune features including skin scaliness, diarrhea, and death (between 2 and 4 weeks after birth) in association with CD4+ T cell hyper-proliferation, multi-organ CD4+ cell infiltration (Blair et al., 1994) and over-production of several inflammatory cytokines (Kanangat et al., 1996). This fatal autoimmune lymphoproliferative syndrome was found to map to a gene locus on the X chromosome called Foxp3, which was described as a new member of the forkhead/winged-helix family of transcription factors (TF) (Brunkow et al., 2001). The Foxp3 gene is highly conserved between species and a mutation in the human gene, FOXP3, was identified as the causative factor responsible for the human equivalent of Scurfy, the Immunodysregulation, Polyendocrinopathy, and Enteropathy, X-linked syndrome (IPEX), also known as X-linked autoimmunity and allergic dysregulation syndrome (XLAAD) (Chatila et al., 2000; Bennett et al., 2001; Hori et al., 2003). Both mouse and human diseases have deficient circulating Tregs, which suggests that Foxp3 and FOXP3 are essential for normal Treg development in the two species respectively. This position is strengthened by the failure of Foxp3 knockout mice to develop circulating Tregs; these animals develop a Scurfy-like syndrome from which they can be rescued by the adoptive transfer of Tregs from a Foxp3 replete animal (Fontenot et al., 2003). Furthermore, ectopic or over-expression of Foxp3 in CD4+CD25− mouse cells results in the development of a Treg phenotype (Fontenot et al., 2003; Hori et al., 2003; Khattri et al., 2003). In mice, Foxp3 expression is a good phenotypic marker of Tregs (Fontenot et al., 2005c; Wan and Flavell, 2005); in humans, however, FOXP3 does not allow the unambiguous identification of Tregs (Ziegler, 2006) as it is induced during TCR stimulation in conventional CD4+ T cells (Walker et al., 2003; Gavin et al., 2006; Wang et al., 2007) (in much the same manner as CD25) and there has been some debate as to whether the induced CD4+CD25+FOXP3+ population is suppressive or anergic (Walker et al., 2003; Gavin et al., 2006).
Although Foxp3 may function as a transcriptional inhibitor through associations with NFAT, NF-κB, and RORγt (Schubert et al., 2001; Bettelli et al., 2005; Zhou et al., 2008a), its biological function is still incompletely understood and will be discussed in an accompanying review in this series. However, it is worth mentioning that the concept of Foxp3 as a “lineage-specifying factor” of Tregs is an over-simplification, as suggested by three lines of evidence: (i) Foxp3 is not sufficient in itself to determine the full Treg transcriptional profile (Hill et al., 2007); (ii) Foxp3 is expressed by (human) Teffs following activation, without imparting the phenotype associated with Tregs; (iii) humans with IPEX syndrome have heterogeneous T cell abnormalities, including dysfunction in Teffs (Bacchetta et al., 2006).
Thymically Derived Tregs
Thymic education of T cells is a two step process involving, first, positive selection of thymocytes recognizing self-MHC and, second, negative selection of T cells with T cell receptors (TCRs) of high avidity for class I and class II MHC molecules presenting self-antigens. Thus, duration and avidity of the TCR interaction with self-peptide-MHC complexes on antigen-presenting cells (APC) determine thymocyte fate. Thymocytes that bind with high avidity undergo programed cell death in an attempt to limit autoreactivity in the periphery, while thymocytes with low avidity for self-MHC:peptide are selected as effector T cells (Teff).
A thymic origin for Tregs was suggested by the neonatal thymectomy-induced autoimmunity models described above (reviewed in Shevach, 2000). In addition, neonatal infection of BALB/c mice with superantigen-expressing murine mammary virus (MMV) results in increased numbers of Vβ6+ Tregs (Papiernik et al., 1998), which implies that thymocyte interaction with antigen preferentially favors Treg differentiation. Indeed, interactions between TCR and MHC class II peptides are essential for normal tTreg development (Sakaguchi et al., 2008; Josefowicz and Rudensky, 2009), an assertion which is consistent with the observation that Tregs express molecules associated with an activated state in Teffs (CD5, CD25, CTLA-4, and Foxp3) and the binding of TCR/CD28-coupled TFs (e.g., NFAT and AP1) to the Foxp3 promoter (Mantel et al., 2006). Thus, mice engineered for high antigen expression, e.g., influenza haemaglutinin (HA), and TCR specificity for that HA (i.e., I-Ed-restricted TCR specific for HA) develop large numbers of Tregs (Jordan et al., 2001), indicating that self-agonist ligands, contrary to inducing clonal deletion, or anergy, cause central development of Tregs. These observations are corroborated by a high degree of self-reactivity (against MHC/peptide complexes expressed on APCs) in Tregs compared to other CD4+ populations (Romagnoli et al., 2002). This demonstrates a biased thymically imprinted TCR repertoire based on recognition of self-MHC-peptide, suggesting that negative selection in the thymus is incomplete, with thymocytes having TCR-MHC:self-peptide interactions of intermediate strength escaping deletion and differentiating into cells with a regulatory phenotype (Tregs) (reviewed in Singer et al., 2008; Josefowicz et al., 2012). There is now significant evidence that tTreg development is self-antigen driven, with the tTreg population being largely autoreactive (Hsieh et al., 2004, 2006; Picca and Caton, 2005). The high similarity between the TCR repertoire of Tregs found within the thymus and Tregs isolated from the circulation (Hsieh et al., 2006; Wong et al., 2007) is, therefore, indicative of thymic Treg émigrés making a significant contribution to the peripheral Treg pool.
A number of additional cues are required for thymic induction of Tregs, notably those providing co-stimulation or IL-2R-γc cytokine family signaling. The importance of γc cytokines to tTreg development is highlighted by the absence of this population from the thymus and periphery of IL-2R-γc knockout animals (Fontenot et al., 2005b) and spontaneous development of autoimmune diseases in mice lacking IL-2Rβ (CD122), which can be prevented by infusion of donor Tregs (Suzuki et al., 1995; Malek et al., 2002). Although no single member of this cytokine family (IL-2, IL-7, or IL-15) is non-redundant in the thymic induction of tTregs, the most significant defect is observed in IL-2−/− or CD25−/− animals, in which Foxp3 expression is reduced by 50% in thymocytes and animals succumb to lethal autoimmune diseases (Sadlack et al., 1993; Willerford et al., 1995; Fontenot et al., 2005b). IL-2, the most important γc family member for tTreg induction (Fontenot et al., 2003; Hori et al., 2003) activates Stat5 through γc chain-associated Janus Kinase (JAK) 3; pY-Stat5 subsequently binds to the promoter region of Foxp3 to positively regulate the gene (Zorn et al., 2006; Burchill et al., 2007; Yao et al., 2007). As expected, Jak3−/− and Stat5−/− mice have few or no circulating Foxp3+ cells (Mayack and Berg, 2006; Yao et al., 2007). Of note, developing Treg-precursors in the thymus are highly attuned to IL-2 as they express CD25 and thus have a competitive advantage in the IL-2-poor environment of the thymus. Thus, even suboptimal IL-2Rβ signaling, for example through mutations of Y → F (tyrosine to phenylalanine) at key sites binding Shc or Stat5, is sufficient to support normal tTreg (but not iTreg) development (Yu et al., 2009; Cheng et al., 2013).
Co-stimulation through CD28 is particularly important for tTreg development as both CD80/CD86 and CD28 knockout animals (Salomon et al., 2000; Tai et al., 2005) have striking tTreg deficiency. Signals transduced through the TCR and CD28 that are clearly important in thymic Treg lineage commitment include both the NF-κB and Ras-Raf-MAPK pathways. This is demonstrated through inhibition of tTreg development by disruptions to components of either the NF-κB, e.g. Bcl10, PKCθ, CARMA1, IκB kinase 2, c-Rel, TRAF6 (Schmidt-Supprian et al., 2003, 2004; Barnes et al., 2009; Isomura et al., 2009; Long et al., 2009; Grigoriadis et al., 2011; Shimo et al., 2011; Schuster et al., 2012), or the Ras-Raf-MAPK pathways, such as RasGRP1 and Raf (Willoughby et al., 2007; Chen et al., 2008).
In contrast to previous reports suggesting that TGF-β is not required for the thymic induction of thymocytes (Marie et al., 2005; Li et al., 2006), conditional ablation of TGF-βRI in double-positive (CD4+CD8+; DP) thymocytes does result in a temporary reduction of Foxp3+ thymocytes in neonatal mice (Liu et al., 2008), suggesting that central Treg selection may be enhanced by TGF-β signaling. In contrast, Akt signaling in developing thymocytes suppresses Treg development through mTOR (Haxhinasto et al., 2008), in a manner akin to iTregs (see below).
These observations are consistent with a step-wise model (Burchill et al., 2008; Lio and Hsieh, 2008) in which Tregs are selected from late-stage, single-positive (CD4+) thymocytes (Fontenot et al., 2005a), whose TCRs engage high affinity ligands (Sakaguchi et al., 2008) presented by either medullary or cortical thymic epithelial cells (mTECs or cTECs) in the context of MHC class II (Aschenbrenner et al., 2007; Liston et al., 2008b) and in the presence of CD28 co-stimulation (Tai et al., 2005). Thus, TCR/CD28 engagement induces expression of CD25 by thymocytes, sensitizing them to IL-2, which instructs Foxp3 and CD25 expression in a Stat5-regulated manner (Burchill et al., 2008; Lio and Hsieh, 2008). However, there is also some evidence that Tregs may, in fact, be induced to differentiate at a much earlier, double-positive (CD4+CD8+), developmental stage before agonist selection (Pennington et al., 2006). This is consistent with demonstrations in K14-Aβb mice that, similar to other CD4+ T cells, positive selection on thymic cortical epithelium is sufficient for Treg differentiation from DP precursors (Bensinger et al., 2001).
Peripherally Induced Tregs
There is also significant evidence showing that, like other CD4+ lineages, Tregs can be generated from CD4+ naïve precursors in the periphery. Here, host detection of infection and tissue injury initiates events that result in recruitment and differentiation of CD4+ T helper (Th) lymphocytes to functions suited to removal/containment of the noxious stimulus. Specific signaling pathways essential for differentiation, expression of key TFs, specific cytokines, and surface molecules distinguish distinct CD4+ Th lineages from each other. Thus, pluripotent naïve CD4+ T cells (Thp) are induced to “commit” to particular lineages by mode of stimulation, antigen concentration, co-stimulation, and cytokine milieu (Constant and Bottomly, 1997) through distinct pathways, including, but not exclusively, Stat1/Stat4 (Th1), Stat6 (Th2), Stat5 (Treg), and Stat3 (Th17) (Zhu et al., 2010). Each lineage is then characterized by expression of its own cytokine profile: IFN-γ (Th1), IL-4 (Th2), and IL-17 (Th17); dominant TFs: T-bet (Th1), Gata-3 (Th2), Foxp3 (Treg), and Rorc (Th17) (Zheng and Flavell, 1997; Szabo et al., 2000, 2002; Fontenot et al., 2003; Wan and Flavell, 2005; Ivanov et al., 2006) and chemokine receptors: CCR5 and CXCR3 (Th1), CRTH2 and CCR4 (Th2), and CCR6 (Th17) (Figure 2). Individual lineages are specialized to promote specific biological functions, for example, immunity against intracellular microorganisms (Th1), humoral immunity to control helminthic and other extracellular pathogens (Th2), clearance of extracellular bacteria, and fungi at mucosal surfaces (Th17) and regulation of immune system activation (Tregs) (Zhou et al., 2009a; Zhu et al., 2010). Thus, detection of “danger” is a key event in the initiation of this cascade and recruitment and differentiation of the most appropriate Th lineage(s) is the key determinant of pathogen removal/persistence and tissue repair/healing during immune responses.
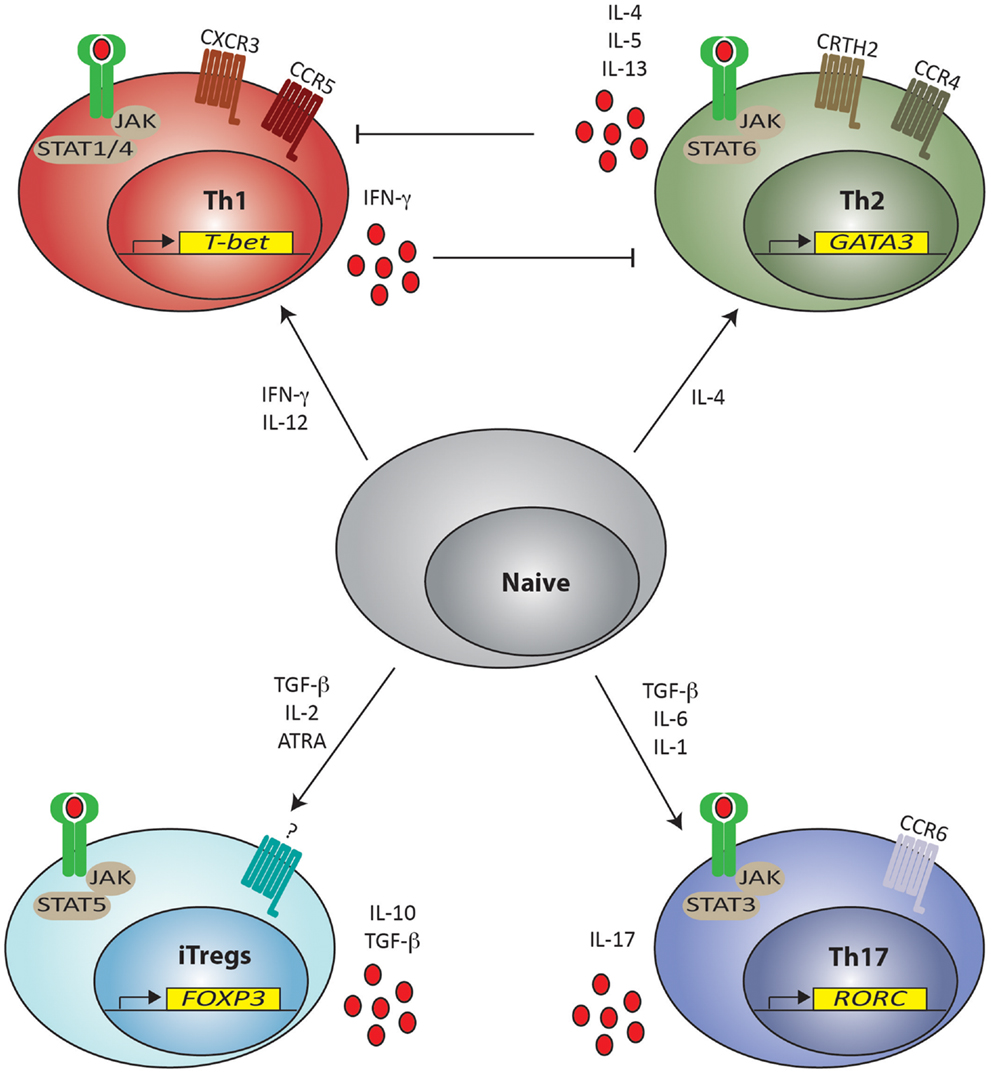
Figure 2. Naïve T helper cell differentiation pathways for lineage commitment in the periphery. T helper cells (Th) can be induced from naïve CD4+ cells to differentiate toward Th1, Th2, Th17 and Treg phenotypes depending on the cytokine milieu present in the environment. Presence of IFN-γ and IL-12 promote skewing toward Th1 commitment by signaling through STAT1 and STAT4, respectively. Th1 cells are characterized by expression of T-bet, chemokine receptors CCR5 and CXCR3 and produce IFN-γ, which inhibit Th2 differentiation. Th2 cell commitment is instead promoted by IL-4 via STAT6 signaling. Th2 committed cells express GATA-3, chemokine receptors CCR4 and CRTH2 and secrete IL-4, IL-5, and IL-13, which in turn inhibit Th1 differentiation. Development of both iTreg and Th17 phenotypes requires the presence of TGF-β, but the proinflammatory IL-6 and IL-1 preferentially skews the response toward a Th17 phenotype through STAT3 signaling leading to the expression of RORC. Th17 cells express the chemokine receptor CCR6 and secrete IL-17. iTregs are instead induced in the presence of TGF-β together with IL-2 (and ATRA) through STAT5 signaling, leading to the expression of Foxp3 and can secrete IL-10 and TGF-β. A definite chemokine receptor for iTregs has not yet been clearly described. JAK = Janus kinase; STAT = Signal Transducer and Activator of Transcription. ATRA = all-trans retinoic acid.
The conditions favoring peripheral induction of Tregs (pTregs) include suboptimal dendritic cell (DC) activation, sub-immunogenic doses of agonist peptide, mucosal administration of peptide and presence of appropriate cytokines, notably TGF-β and IL-2 (Chen et al., 2003; Apostolou and von Boehmer, 2004; Kretschmer et al., 2005; Selvaraj and Geiger, 2007; Siewert et al., 2008; Zheng et al., 2008). The greater the strength of the TCR/MHC:peptide interaction and co-stimulation, the greater the requirement for tolerance-inducing cytokines, specifically TGF-β and IL-2, to induce a regulatory, as opposed to, effector phenotype in Thp. Some of this effect is related to the ability of high concentrations of TGF-β to down-regulate receptors for other cytokines, including IL-6 (Zheng et al., 2008), and the ability of IL-2-activated Stat5 to inhibit loci of other lineages (Laurence et al., 2007), implying that efficient pTreg differentiation is at least partially contingent on inhibition of differentiation to alternate Th lineages. This assertion is supported by evidence that the presence of cytokines required for T cell skewing to alternate Th lineages, such as IL-12 (to Th1) and IL-6 (to Th17) (Figure 2) preferentially foster development of those lineages in contrast to iTreg through induction of lineage-specifying Stat proteins and TFs (Wei et al., 2007).
Of particular note, it appears that not all Thp can differentiate in the periphery to Tregs (Hsieh et al., 2004; Lathrop et al., 2008). Instead, recent evidence indicates that either the thymus may remain a site of Treg differentiation during immune responses (Zelenay et al., 2010) or that recent thymic émigrés are, in fact, the precursors of pTregs (Paiva et al., 2013). Speculatively, the implication is that either T cells with certain TCR specificities are more suited to differentiate into Tregs (presumably due to higher than average TCR avidity for self-MHC:peptide) or that signals received by pTreg-precursors in the thymus ensure that the Foxp3 locus is epigenetically in a state ready for gene transcription in the periphery. Nevertheless, pTregs suppress antigen driven CD4+ T-cell expansion and both Th1 and Th2 cytokine production in vitro in a manner akin to tTregs (Chen et al., 2003); the paucity of distinguishing phenotypic and functional characteristics between tTregs and pTregs is one argument for a high degree of similarity in the signals required for their induction.
The requirement for low level TCR signaling for induction of pTregs is highlighted by experiments in which in vivo Foxp3 induction in Thp inversely correlates with the dose of immunogen (Kretschmer et al., 2005) and in which augmenting TCR signaling by removing an inhibitory E3 ubiquitin ligase (Chiang et al., 2000) inhibits Foxp3 induction (Wohlfert et al., 2006). Consistent with this, in vitro iTreg induction is inhibited by increasing concentrations of activating anti-CD3 (Kim and Rudensky, 2006) whereas premature termination of TCR signaling soon after T cell activation or inhibition of the PI3 Kinase/Akt/mTOR pathway downstream of TCR signaling augments iTreg induction (Sauer et al., 2008). Similarly, while CD28 signaling is critical for central selection of Tregs (Salomon et al., 2000), peripheral pTreg induction is, in contrast, inhibited by strong CD28 ligation (Kim and Rudensky, 2006; Benson et al., 2007), which explains why mice deficient in CTLA-4, an inhibitor of T cell activation, have impaired pTreg induction (Zheng et al., 2006). Similarly, anaphylatoxin receptor signaling activates the mTOR pathway; thus C3ar1−/− or C5ar1−/− mice have impaired mTOR signaling and take on an iTreg phenotype in response to TGF-β more readily than wild-type T cells. Antagonism of C3aR and C5aR in human naïve CD4+ T cells induces functional iTregs (Strainic et al., 2012).
TGF-β directly regulates the Foxp3 gene through both TGF-β-inducible early gene 1 (TIEG1) and Mothers Against Decapentaplegic 3 (Smad3), which bind at promoter and enhancer regions in the Foxp3 gene to upregulate its expression (Tone et al., 2008; Venuprasad et al., 2008) (see below). Notch-pathway mediated signals synergize with TGF-β to enhance Foxp3 expression by recruiting Notch1, CSL, and Smad proteins to promoter regions of the Foxp3 gene (Samon et al., 2008). In vivo, DC populations producing local TGF-β are clearly sufficient to induce iTregs (Benson et al., 2007; Yamazaki et al., 2008).
The presence of all-trans retinoic acid (ATRA) in the Thp environment synergizes with TGF-β to promote iTreg development; this effect is sufficient to allow iTreg development even in the presence of high levels of co-stimulation (Benson et al., 2007). While receptor-ligand-mediated gene transcription is retinoic acid receptor (RAR)-α dependent (Elias et al., 2007; Hill et al., 2008), ATRA promotes iTreg differentiation both directly, through inhibition of differentiation to alternative lineages, notably Th17 (Elias et al., 2007; Mucida et al., 2007; Xiao et al., 2008), and indirectly, through the inhibition of environmental cytokines produced by CD44hi effector memory T cells, especially IL-4 and IFN-γ (Hill et al., 2008), which support the development of alternative Th lineages. ATRA, moreover, imprints a gut-homing phenotype on iTregs (α4β7+CCR9+) (Benson et al., 2007). This is noteworthy as CD103+CD11c+ DCs present in lamina propria of small and large bowel, mesenteric lymph nodes, and Peyer’s patches induce an identical gut-homing phenotype (Annacker et al., 2005; Johansson-Lindbom et al., 2005) and the development of iTregs through secretion of local TGF-β and ATRA (Coombes et al., 2007; Sun et al., 2007). Such local milieu for the induction of iTregs might reflect the need to control immune responses directed against antigens expressed by local microbiota and ingested food and may provide an evolutionary link between iTregs and commensal bacteria. This may explain why several studies have reported a reduction in lamina propria Tregs of mice housed in germ-free environments (Strauch et al., 2005; Östman et al., 2006; Ishikawa et al., 2008), which may be related to the specific organisms that are present or absent from the “germ-free” environment (Ivanov et al., 2008).
Regulation of Foxp3 Gene Expression
Epigenetic mechanisms, such as DNA methylation, histone modification, nucleosome positioning, as well as microRNAs (miRNAs), are essential for control of gene expression (Baltimore et al., 2008; Wilson et al., 2009; Cedar and Bergman, 2011). Chromatin remodeling has a role in determining the accessibility of genes by transcriptional activators or repressors. In particular, methylated DNA sequences are “silenced”, while opening of the locus for transcription is linked to demethylation. For comprehensive reviews, the reader is referred to (Wilson et al., 2009; Cedar and Bergman, 2011). Foxp3 gene expression is controlled by four elements, containing conserved non-coding sequences (CNS). The first is in the promoter region, two are in the first intron (CNS1 and CNS2, at 2 and 4.5 kb downstream of the transcriptional start site (TSS) of murine Foxp3, respectively) and the fourth (CNS3, at 7 kb downstream of the TSS of murine Foxp3) is in the second intron. These sites are regulated by epigenetic modifications that determine chromatin structure and DNA methylation, altering the accessibility of the gene locus to TFs. Known TF binding and epigenetic modifications at these sites are shown in Figure 3.
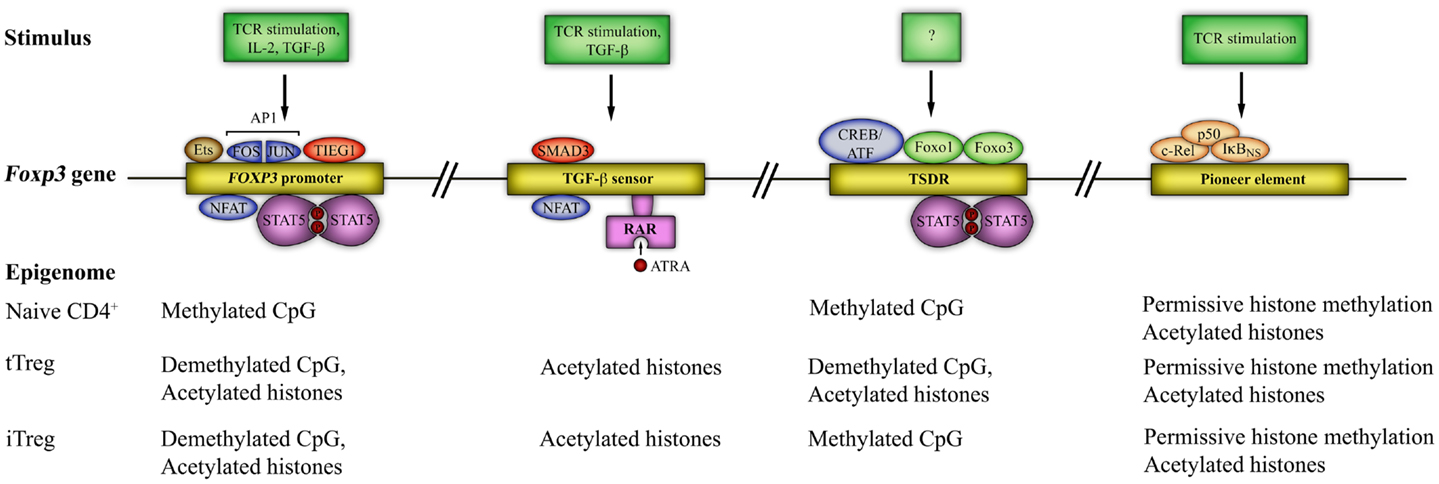
Figure 3. The Foxp3 epigenome and transcription factor binding sites. Four distinct regions of the Foxp3 gene are susceptible to epigenetic modification. These are the Foxp3 promoter and three other conserved non-coding sequences (CNS): CNS1 – the TGF-β sensor/enhancer, CNS2 – Treg-cell-specific demethylation region (TSDR), and CNS3 – a Foxp3 pioneer element. Epigenetic modifications include histone acetylation/deacetylation and CpG methylation/demethylation and are shown at each locus. Known transcription factor binding sites and the signals required for access to each region are also shown. STAT5 = signal transducer and activator of transcription 5, SMAD3 = small body size mothers against decapentaplegic 3, CREB = cAMP-response-element binding protein, ATF = activating transcription factor, NFAT = nuclear factor of activated T cells, AP1 = activator protein 1 (a dimer of FOS and JUN), TIEG1 = TGF-β-inducible early gene 1; Ets = E-twenty six; RAR = retinoic acid receptor; ATRA = all-trans retinoic acid; Foxo = forkhead box o.
Three important caveats should be noted here. The first is that emerging evidence suggests a role for Foxp3 binding within enhancer elements in the Foxp3 gene, exploiting enhancers “established” by Foxp3 predecessors, such as Foxo1 (Samstein et al., 2012). These data are not included in Figure 3. The second is that Foxp3 expression alone is insufficient for establishment of the Treg lineage; rather, the development of a Treg-specific genome-wide methylation pattern (“nTreg-Me”) in addition to Foxp3 expression is critical (Ohkura et al., 2012). Thus, nTreg-Me is independent of Foxp3 expression, but necessary for Foxp3+ cells to acquire the genome-wide transcriptional profile, stability, and functional characteristics of the Treg lineage (suppressive capability) (Ohkura et al., 2012). Interestingly, in vitro induced iTregs lack the nTreg-Me pattern, whilst in vivo generated iTreg gradually develop it after TCR stimulation (Ohkura et al., 2012). This difference in stability of Foxp3 expression between tTregs and iTregs could then be attributed to epigenetic differences at the Foxp3 locus, as detailed below. The third, as has been elegantly described recently in the mouse, is that differentiation of both tTregs and iTregs is critically dependent on transcriptional repression of alternate lineages through the expression of the Bach2 TF (Roychoudhuri et al., 2013). Thus, animals deficient in this TF are unable to generate Tregs and succumb to spontaneous autoimmune disease (Roychoudhuri et al., 2013). Interestingly, this TF is also linked to multiple autoimmune diseases in man.
Foxp3 promoter
CpG motifs in the Foxp3 promoter are basally demethylated in resting Tregs, but partially methylated in conventional naïve CD4+ T cells (Kim and Leonard, 2007; Janson et al., 2008). In addition, histones in this region are more highly acetylated in Tregs than in naïve T cells (Mantel et al., 2006; Kim and Leonard, 2007). As a result, the Foxp3 promoter is more accessible for the binding of TFs, such as NFAT, AP1, STAT5, TIEG1, and Ets1 and 2 [recently described to bind the Foxp3 promoter (Fayyad-Kazan et al., 2010)] in Tregs than in conventional T cells. In mice, the Foxp3 promoter in conventional T cells remains methylated following TCR activation, albeit at a lower level than at baseline (Janson et al., 2008), and demethylation requires activation in the presence of TGF-β (Kim and Leonard, 2007; Janson et al., 2008); these structural effects limit and promote access for induction of Foxp3 transcription respectively.
CNS1 (TGF-β-sensitive enhancer element)
CNS1, an “enhancer” region in the Foxp3 locus, contains binding sites for NFAT and Smad3 and is in an accessible, histone-acetylated, state in both tTregs and iTregs (Tone et al., 2008) but not in naïve, resting T cells. This area has no CpG motifs, therefore the sole epigenetic modification at this locus is through histone acetylation. RAR and RXR (retinoid X receptor) bind within CNS1 and are responsible for increased histone acetylation, permitting greater Smad3 binding (Xu et al., 2010), thus, explaining some of the direct effects of ATRA in iTreg induction. CNS1 knockout animals demonstrate normal tTreg development, but have impaired iTreg induction (Zheng et al., 2010); therefore, CNS1 is redundant in thymic Treg selection, but is essential for peripheral induction of Tregs, consistent with the role of TGF-β in pTreg generation.
CNS2 (“Treg-specific demethylated region”, TSDR)
A third, highly conserved, CpG dinucleotide-rich region in both mouse and human Th cells, termed the “Treg-specific demethylated region” (TSDR), is completely demethylated in nTregs, but methylated in conventional T cells (Baron et al., 2007; Floess et al., 2007). In tTregs, this area also contains acetylated histones (H3Ac and H4Ac) (Floess et al., 2007) and TF binding sites, which in the demethylated state bind Stat5, CREB/ATF (Yao et al., 2007; Nagar et al., 2008), Foxo1, and Foxo3 (Ouyang et al., 2010), which also bind the Foxp3 promoter (Harada et al., 2010; Ouyang et al., 2010). Interestingly, Foxp3 induction by TGF-β is associated with only partial demethylation of the TSDR, an unstable state that reverses upon restimulation (Floess et al., 2007). Thus, iTregs contain methylated CpGs. The TSDR was initially described as having enhancer activity (Kim and Leonard, 2007). However, given quantitatively similar Foxp3 expression in iTregs and tTregs despite large differences in methylation state at the TSDR, it is unlikely that it acts as an enhancer element; instead, TSDR demethylation appears critical for stable Foxp3 expression (Floess et al., 2007; Nagar et al., 2008; Polansky et al., 2008). Indeed, pharmacological inhibition of DNA methyltransferase-1 (Dnmt-1) in conventional T cells, using the covalent inhibitor 5-azacytidine (5AzadC), followed by activation through the TCR, results in stable expression of Foxp3, in contrast to the transient Foxp3 expression seen with TCR activation alone (Kim and Leonard, 2007; Nagar et al., 2008; Polansky et al., 2008; Josefowicz et al., 2009). Similarly, CNS2-deficient animals have reduced Treg numbers only after 6 months of age (Zheng et al., 2010), suggesting that CNS2 is redundant for expression of Foxp3 but critical for its stable maintenance. Of note, demethylated CNS2 acts as a binding site for Foxp3 in a Runx1- and Cbf-β-dependent manner (Zheng et al., 2010), which may serve as a mechanism for stable Foxp3 expression in mature Tregs. The signals controlling methylation/demethylation at the TSDR are currently unknown, but given the difference between iTregs and tTregs in Foxp3 stability, it is likely that demethylation at this locus is thymically initiated.
CNS3 (Pioneer element)
CNS3 contains a DNase I hypersensitive site and is bound by c-Rel, IκBNS, and p50, members of the NF-κB family (Zheng et al., 2010; Schuster et al., 2012). Chromatin modifications at this site show permissive marks (H3K9/14Ac, H3K4me2, and H3K4me1) in Tregs, but also mono (H3K4me1) and di-(H3K4me2) methylation in Treg-precursors (CD4+CD8+ and CD4+CD8- thymocytes) (Zheng et al., 2010). As permissive marks are absent at CNS1 and 2 in Treg-precursors, this argues that CNS3 can bind TFs before both CNS1 and 2 during Treg induction and opens the Foxp3 locus to other TFs, thus acting as a pioneer element (Zheng et al., 2010). Indeed, CNS3−/− mice have significantly reduced Treg numbers, but normal per cell levels of Foxp3 in the remaining Tregs, supporting the assertion that CNS3 acts as a pioneer element. The importance of c-Rel binding is highlighted by the profound loss of Tregs seen in mice that are c-Rel deficient (Ruan et al., 2009; Zheng et al., 2010). It is possible that the binding of c-Rel acts as a chromatin opener or, that c-Rel, in co-operation with other TFs, e.g., p65, NFAT, CREB, and Smad3, enhances formation of an enhanceosome at the Foxp3 promoter (Ruan et al., 2009).
Distinguishing tTregs from iTregs
To date, no single marker has been identified to differentiate tTregs from iTregs and no definitive test to distinguish their in vivo functions. The Treg transcriptional profile is dominated by genes induced by cell activation alone (Hill et al., 2007) and has so far not yielded definitive markers to distinguish iTregs from tTregs despite early promise (Gavin et al., 2007; Zheng et al., 2007). Reports that tTregs exclusively express Helios, an Ikaros transcription factory family member (Thornton et al., 2010), have been challenged by the demonstration that Helios is induced during T cell activation and proliferation and then down-regulated (Akimova et al., 2011). Thus, expression of Helios cannot reliably differentiate iTregs from tTregs. Likewise, although TSDR demethylation could in theory distinguish Tregs that have received thymic induction from those induced in the periphery, in vivo generated iTregs can also efficiently demethylate the TSDR if given sufficient time (in this case, 6 weeks) (Polansky et al., 2008). Very recently, neuropilin-1 (Nrp-1) has been described by two groups as differentially expressed in murine tTregs and pTregs, being poorly expressed in the latter (Weiss et al., 2012; Yadav et al., 2012). Nrp-1 is a cell surface molecule mediating prolonged interactions between Tregs and DCs (Sarris et al., 2008), a receptor for TGF-β (Glinka and Prud’Homme, 2008) and vascular endothelial growth factor (Ferrara et al., 2003), which has previously been proposed as a Treg marker (Bruder et al., 2004; Hansen et al., 2012). Of interest, the lowest Nrp-1 expression was seen in in vivo generated pTregs compared to in vitro generated iTregs, presumably reflecting positive regulation of Nrp-1 by high dose TGF-β in vitro (Weiss et al., 2012). These observations have not yet been replicated in human Tregs, although Nrp-1+ Tregs have been identified in inflamed synovial fluid of patients with rheumatoid arthritis (E et al., 2012).
tTreg and iTreg Function
Tregs suppress target cells through a number of inhibitory mechanisms, including cell–cell contact-dependent inhibition (Takahashi et al., 1998; Thornton and Shevach, 1998; Ng et al., 2001), secretion of inhibitory cytokines (Powrie et al., 1996; Asseman et al., 1999; Belkaid et al., 2002; Maloy et al., 2003; Collison et al., 2007), cytolysis of target cells (Gondek et al., 2005; Cao et al., 2007), metabolic disruption (Deaglio et al., 2007), modulation of APC function (DiPaolo et al., 2007; Puccetti and Grohmann, 2007), and competition for environmental IL-2 (Pandiyan et al., 2007). Such redundancy suggests that the mode of suppression may be context dependent and directed by the degree and mode of inflammation. While details of these mechanisms falls outside the scope of this review, it is noteworthy that they are not mutually exclusive, and while not necessarily limited to a single “delivery system”, are mostly compatible with a cell-to-cell contact deployment package. For example, Tregs can deliver suppressive factors like cyclic adenosine monophosphate (cAMP) into conventional T-cells via gap junctions (Bopp et al., 2007), they can modulate APC function through membrane-bound suppressive TGF-β (Nakamura et al., 2001), through negative signaling by cytotoxic T-lymphocyte antigen-4 (CTLA-4) (Read et al., 2006) or lymphocyte-activation gene 3 (LAG3) (Huang et al., 2004).
So far, no distinct functional differences have been conclusively demonstrated between tTregs and iTregs, suggesting that the mechanistic repertoire of Treg function is specified by lineage and not mode of induction. Indeed, iTregs are as potent as tTregs in protecting from autoimmune diseases by preventing the antigen-presenting capacity of DCs to autoreactive Teffs (DiPaolo et al., 2007). As only a small proportion of the transcriptional profile of Tregs can be explained by expression of Foxp3, and the majority by T cell activation and survival signals (Hill et al., 2007), it is not surprising perhaps that function is also lineage and not induction-specific. As argued above, it is generally accepted that tTregs function to prevent the development of autoimmune diseases and that iTregs limit inflammation to neo-antigens, such as bowel commensal. iTregs can clearly be generated and are essential and sufficient to mediate oral tolerance in response to dietary antigens in animals devoid of tTregs (Mucida et al., 2005; Curotto de Lafaille et al., 2008). Although these experiments have been conducted under highly non-physiological conditions, the same mechanisms of iTreg induction in the periphery may explain the persistence of alternative neo-antigens, such as pathogenic organisms (Robertson and Hasenkrug, 2006; Wohlfert and Belkaid, 2008) or neoplastic cells (Zou, 2006).
Treg Plasticity
Emerging concepts of mammalian Th cell polarization have recently challenged traditional models of terminal differentiation, suggesting that Th lineage commitment is not as irreversible as previously thought and that lineage reprograming to alternate lineages can be achieved through the expression of key TFs and appropriate epigenetic modifications in lineage-specifying genes. For in-depth reviews, please see (O’Shea and Paul, 2010; Hirahara et al., 2011; Nakayamada et al., 2012). Several reports in the literature suggest that Tregs retain significant plasticity, with the capacity to express TFs and signature cytokines, particularly, of Th1 (Koch et al., 2009) and Th17 (Koenen et al., 2008; Beriou et al., 2009; Afzali et al., 2010) cells.
The ability of Tregs to express key TFs of alternate lineages may license them to efficiently regulate inflammation generated by those Th lineages. For example, T-bet expression by Tregs induces CXCR3 expression, licensing Treg trafficking to sites of Th1-mediated inflammation to control Th1 cells (Koch et al., 2009). Likewise, expression of IFN-γ by Tregs may be a surrogate marker for T-bet expression and licensing for suppression of Th1 inflammation (Feng et al., 2011). Expression of interferon regulatory factor-4 (IRF4), a TF essential for Th2 (Rengarajan et al., 2002) and Th17 (Brüstle et al., 2007) cell differentiation directs Tregs to selectively regulate Th2 responses (Zheng et al., 2009). Selective ablation of Stat3, critically required for Th17 differentiation (Figure 2), in Tregs results in uncontrolled Th17-dependent responses (Chaudhry et al., 2009).
On the other hand, plasticity in Tregs may indicate a potential to assume an effector phenotype and to contribute to inflammation (Zhou et al., 2009b). In particular, as Treg and Th17 differentiation from naïve Thp are reciprocally linked (Bettelli et al., 2006; Mangan et al., 2006; Veldhoen et al., 2006; De Jong et al., 2010) (see Figure 2) and the two lineages have opposing functions (Afzali et al., 2007), lineage reprograming from one to the other could have significant implications for the development of autoimmune diseases and for programs of Treg-based cell therapy in humans. It is certainly possible that there may be a threshold of expression and/or activation of Th-specific TFs in Foxp3+ Tregs allowing them to act either as lineage-specific regulators or contributors to effector responses. There remains still considerable controversy regarding Treg plasticity and lineage reprograming as even complex, and elegant, fate-mapping murine models (Zhou et al., 2009b; Hori, 2010, 2011; Rubtsov et al., 2010) have produced divergent results.
Mechanistically, the epigenome of many “terminally differentiated” Th cells shows considerable flexibility in accessibility of genes of alternate lineages to TFs. This is elegantly described in the study of Wei et al. (2009) showing a rather flexible signature of genome-wide H3K4me3 and H3K27me3 maps of naïve, Th1, Th2, Th17, iTreg, and tTreg cells. In this study, the methylation of loci for signature cytokines conformed broadly to that expected from lineage commitment; however, the epigenome of “master” TFs showed significant flexibility, presenting both permissive and repressive modifications in the various Th subsets, including bivalent epigenetic states. This suggests that the overall balance of epigenetic state determines cell differentiation and that bivalent modifications might allow specific lineage regulator gene loci to be activated under different polarizing conditions, thus reprograming Th cells into other lineages. For example, tTregs and iTregs both have repressive H3K27me3 marks at the Il17a locus. This is in contrast to permissive H3K4me3 at the Rorc locus in iTregs and bivalent chromatin at this locus in tTregs (Wei et al., 2009), potentially permitting co-expression of Foxp3 and RORγt after culture under Th17 polarizing conditions (Xu et al., 2007; Yang et al., 2008). Signals from the micro-environment are then clearly key to lineage stability. While much focus has been on the local cytokine cytokine milieu, recent data has also highlighted the role of local complement components, notably the anaphylatoxins C3a and C5a, which can signal through cognate receptors on Tregs to down-regulate Foxp3 expression by activating Akt (Kwan et al., 2013).
Given the difficulty in distinguishing pTregs from tTreg in a healthy host, no definitive experiment has yet conclusively shown a difference in Th17 plasticity between tTregs and iTregs in vivo (Zhou et al., 2009b). Human data is also inconclusive; while it appears that Th17 plasticity is restricted to a population of suppressive memory Tregs expressing the lectin receptor CD161 (Afzali et al., 2013; Pesenacker et al., 2013), divergent reports suggest that these cells are thymically derived (predominantly demethylated TSDR, Pesenacker et al., 2013) and peripherally induced [low Helios expression, virtual absence from umbilical cord blood and CD45RA- phenotype (Ayyoub et al., 2009; Afzali et al., 2013)].
miRNA and Tregs
Gene transcription events are also heavily influenced by microRNAs (miRNAs or miRs) and recent evidence supports the role of this class of molecules in Treg biology. miRNAs are an evolutionarily conserved class of pleiotropically acting small endogenous RNAs, about 23 nucleotides long, that play important gene-regulatory roles by pairing to the mRNAs of protein-coding genes to direct their post-transcriptional repression. miRNAs are predominantly transcribed by RNA polymerase II, which produces a primary transcript containing the mature miRNA sequence and a varying amount of flanking region (Lee et al., 2004). Two nucleases then process the miRNA: the first one, Drosha, cleaves the primary miRNA into a precursor miRNA (Han et al., 2006) that is exported from the nucleus by exportin 5 (Yi et al., 2003); after reaching the cytoplasm, the precursor miRNA is further processed by the other nuclease, Dicer, and is loaded into the RNA-induced silencing complex (RISC) (Chendrimada et al., 2007). Finally, a specific single strand of the miRNA duplex is selected as a guide to direct sequence-specific targeting of mRNA 3’ untranslated regions (UTRs) by RISC (Bartel, 2009). In mammalian cells, miRNAs silence genes mainly through binding of target mRNA leading to degradation of the mRNA; however, another mechanism of repression at a translational level has been reported, showing that miRNAs can inhibit either the initiation or the elongation stages of protein translation (reviewed in Pillai et al., 2007; Lodish et al., 2008). Interestingly, given the short sequence and non-stringent binding to target sequence, abiding to a Watson–Crick match, an individual miRNA is capable of regulating dozens of distinct mRNAs (Bartel, 2009). For a general review on miRNAs, please see (Chen and Rajewsky, 2007).
MicroRNAs have been implicated as fundamental regulators of post-transcriptional programs and play a role in T-lymphocyte development, differentiation, and effector functions since they are differentially expressed, both spatially and temporally, in many types of immune cells (Lykken and Li, 2010). MicroRNA appear critical for the Treg phenotype, as conditional knockout of Dicer in CD4 cells (CD4CreDicer Δ/Δ animals) results in substantial depletion of tTregs and inhibits induction of Foxp3 in naïve CD4 T-cells by TGF-β (Cobb et al., 2006). These mice develop spontaneous autoimmune disease from about 3–4 months of age, in contrast to conditional knockout of Dicer in Foxp3+ cells (Foxp3CreDicer fl/fl), which results in spontaneous autoimmune disease that is fatal by 4 weeks of age (Liston et al., 2008a; Zhou et al., 2008b). In the latter model, Foxp3 expression is unstable and Tregs revert to an effector phenotype producing IL-4 and IFN-γ as part of the disease (Zhou et al., 2008b). Likewise, conditional disruption of Drosha in CD4 cells produces a very similar phenotype (Chong et al., 2008). Dicer and Drosha knockout, however, results in ablation of not only canonical miRNAs, but also that of other small cellular RNA species (e.g., siRNAs and shRNAs). That the phenotype of mice with ablated Dgcr8, an RNA-binding protein required in the processing of canonical miRNAs (Babiarz et al., 2008), resembles that of the Dicer deficient mice (Jeker et al., 2013) establishes that miRNAs are critical for normal Treg development in the thymus and the periphery and they are essential for normal Treg function. Conversely, Foxp3 also contributes to the miRNA signature of Tregs (Cobb et al., 2006; Rouas et al., 2009).
Of the miRNAs that are important for Treg function, only a few are known and the exact function(s) of these are still largely unknown. As a single miRNA can regulate potentially thousands of genes, small differences in miRNA profiles can have profound effects on T cell function. The miRNA machinery and miRNAs that are differentially expressed in Tregs, including those known to be direct Foxp3 targets are shown in Figure 4.
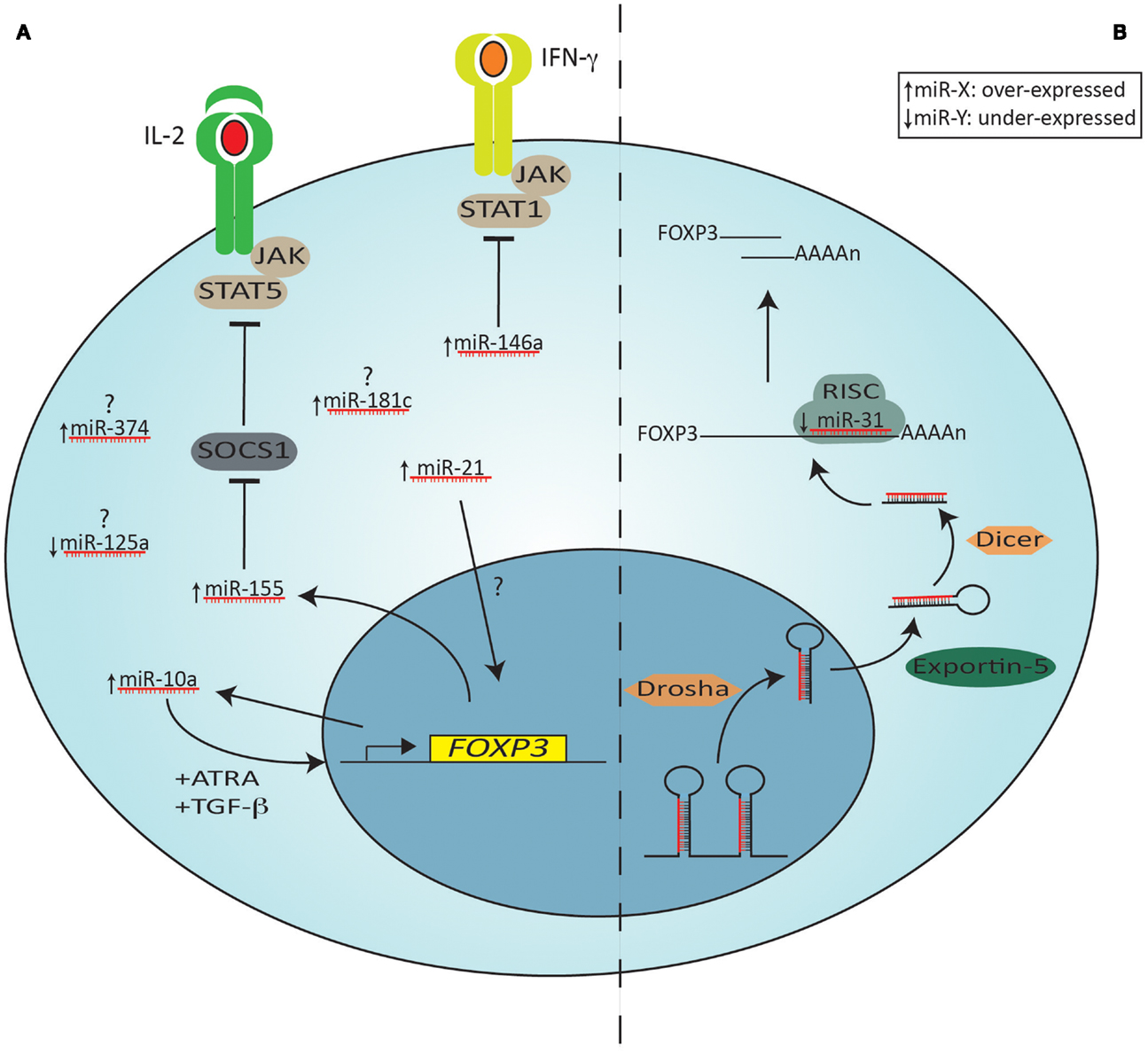
Figure 4. The miRNA machinery and miRNAs differentially expressed in Tregs compared to other CD4+ T cells. Shown on the left (A) are specific miRNAs that are over (arrow pointing up) or under (arrow pointing down)-expressed in Tregs compared to other Th cells. miRNAs that are over-expressed (miR-374, miR-181c) or under-expressed (miR-125a) without a known target or function in Tregs are indicated with a question mark. Some miRNAs are induced by Foxp3, leading to either down-regulation of a specific target (e.g., miR-155 repressing SOCS1) or inducing positive feedback on Foxp3 expression (miR-10a, in combination with ATRA and TGF-β). miR-21 is also an indirect positive regulator of Foxp3, but its mechanism of action is still unknown. Shown on the right (B) is the miRNA processing and targeting machinery, depicting miR-31, an under-expressed microRNA in Tregs, which targets the 3′ UTR of Foxp3 mRNA. The primary miRNA transcript is first processed in the nucleus by Drosha. The precursor is then exported from the nucleus by Exportin 5 and in the cytoplasm a second nuclease, Dicer, generates a double stranded miRNA. The functional strand is subsequently selectively loaded onto RISC. Binding of the mature miRNA to the 3′ UTRs of the target mRNA leads to its degradation. JAK = Janus kinase; STAT = Signal Transducer and Activator of Transcription; SOCS = suppressor of cytokine signaling proteins; RISC = RNA-induced silencing complex (RISC); UTR = untranslated region.
Amongst these, miR-31 is under-expressed in human Tregs while miR-21 is over-expressed in both human and mouse Tregs. Using lentiviral transduction studies, it can readily be seen that miR-31 and miR-21 have opposing effects on FOXP3/Foxp3 expression. Whilst miR-31 negatively regulates FOXP3 (it has a direct binding site in the 3′UTR of FOXP3 mRNA), mir-21 positively regulates FOXP3/Foxp3 in an indirect, but still not fully elucidated, manner (Rouas et al., 2009). Of interest, histone deacetylase inhibition using valproate reduces miR-31 and increases mir-21 as well as FOXP3 expression in human Teffs to levels seen in Tregs (Fayyad-Kazan et al., 2010). This change in miRNA profile is independent of the change in FOXP3 expression (Fayyad-Kazan et al., 2010).
MicroRNA-155, which has previously been studied in T and B cell biology (Baltimore et al., 2008), is a direct Foxp3 target (Marson et al., 2007; Zheng et al., 2007; Liston et al., 2008a), and highly expressed in Tregs. Mir-155 targets suppressor of cytokine signaling 1 (Socs1), enhancing Stat5 signaling. As a result, deletion of miR-155 results in limited Stat5 signaling, attenuating IL-2 signaling, manifesting as reduced thymic and peripheral Treg numbers (Lu et al., 2009). It may also target Foxo3a, albeit in a Treg cell line (Yamamoto et al., 2011).
Mir-146a is another microRNA prevalently expressed in Tregs that targets Stat1; deletion of mir-146a in Tregs causes a severe autoimmune phenotype akin to Dicer knockout animals, characterized by increased numbers of poorly functional Foxp3+ Tregs in the periphery (Lu et al., 2010). As thymic Treg numbers are unaltered, it is likely that the biological role of mir-146a is preferentially to regulate Treg gene expression in the periphery. Indeed, not only do miR-146a−/− Tregs fail to control Teffs in the periphery, but they also gain Th1-like properties, such as secretion of IFN-γ (Lu et al., 2010), as a result of failure to regulate Stat1 signaling (Tang et al., 2009; Lu et al., 2010).
None of miR-21, miR-31, miR-155 nor miR-146a have been shown to regulate gene expression preferentially in tTregs compared to iTregs or vice versa. Mir-10a, on the other hand, is preferentially expressed in tTregs, but poorly expressed in iTregs induced with TGF-β without ATRA (Jeker et al., 2012; Takahashi et al., 2012). Of note, expression of miR-10a is lowest in Tregs from animals prone to autoimmune disease, such as non-obese diabetic (NOD) mice, and in Tregs with unstable Foxp3 expression (Jeker et al., 2012). miR-10a expression in Tregs that lose Foxp3 expression is the same as in Teffs (Jeker et al., 2012). miR-10a is functionally linked to stabilization of Foxp3 expression (Jeker et al., 2012) and targets the transcriptional repressor Bcl-6 and corepressor Ncor2 to limit conversion of iTregs to Tfh (Takahashi et al., 2012). It also fine-tunes Thp fate decisions between iTreg and Th17 (Takahashi et al., 2012).
Thus, these studies show a defined requirement of miRNAs for the differentiation and suppressive function of Treg cells as well as their lineage stability. Differential expression of miRNAs in tTregs and iTregs could reflect divergent pathways of differentiation, functional properties or lineage stability.
Other, Foxp3−, T Cells with Regulatory Function
In addition to Tregs, a number of other inducible T cells have been described with regulatory properties. These include members of the CD4+ (Th3, Tr1, and iTr35) and CD8+ (CD8+CD28−) families. Amongst these, one of the most controversial is the T helper 3 (Th3) subset. This subset was described as an unusual Th2-like regulatory subset, which secretes TGF-β, derived from orally tolerized animals induced by mucosal stimulation with antigen (Chen et al., 1994). Thus, Th3 cells could be induced through cognate stimulation of CD4+ Thp by APC together with CD86 co-stimulation, particularly in the presence of TGF-β and IL-4 (Inobe et al., 1998; Seder et al., 1998; Weiner, 2001). Further growth and division of Th3 cells was dependent on IL-4 and TGF-β rather than IL-2, and some Th3 clones produced IL-4 and/or IL-10 together with TGF-β. Th3-mediated suppression, for the maintenance of oral tolerance, was described as mediated by TGF-β, secreted in response to CTLA-4 ligation (Chen et al., 1998). There is, thus, a degree of similarity between Th3 cells and iTregs given their peripheral (TGF-β-enhanced) induction, mucosal location and TGF-β-dependent function. The lack of iTreg- or Th3-specific markers effectively ensures that the two populations cannot at present be distinguished as disparate.
Type 1 regulatory T cells (Tr1) cells are Foxp3- regulatory T cells that are induced in the periphery in a TCR-dependent and antigen-specific manner through either repeated stimulation with antigen or encounter of antigen in the context of immature DCs (Jonuleit et al., 2000; Dhodapkar et al., 2001) or IL-10 (Groux et al., 1997), with or without IFN-α (Levings et al., 2001). Thus, potent Tr1 induction can be achieved through stimulation of human T cells with a subset of IL-10 producing tolerogenic DCs (DC-10) (Gregori et al., 2010). IL-10 produced by DC-10 stimulates HLA-G expression on target Thp; HLA-G subsequently binds ILT4 (immunoglobulin-like transcript 4) on the DC-10 to augment Tr1 induction (Gregori et al., 2010). Intriguingly, engagement of complement receptor CD46 induces IL-10 producing T-cells phenotypically similar to Tr1 cells (Kemper et al., 2003). Since such complement engagement occurs in vivo (Le Friec et al., 2012) and in vitro (Cardone et al., 2010) to induce Th1 cells before switch to a Tr1 phenotype, an interesting possibility remains that a “Tr1 phenotype” could also represent a final common pathway of activated T cells that have gone through an inflammatory phase and have entered a self-regulatory, IL-10 producing, phase required for wound healing and tissue repair. Indeed, this would comply with the fact that Th1 cells, Th2 cells, and Th17 can all produce IL-10, as is further discussed in an accompanying article in this series. Interestingly, ATRA inhibits IL-10 production, in contrast to augmentation of Foxp3 (Maynard et al., 2009); thus it is possible that induction of Tr1 cells and iTreg in an ATRA-containing environment are to an extent mutually exclusive.
Tr1 cells are anergic, proliferate poorly to antigen, produce little IL-2 or IL-4, but suppress through production of IL-10 and TGF-β (Groux et al., 1997). In addition, they secrete IFN-γ and IL-5; thus their cytokine profile is distinct from Th1, Th2 and classical Tregs (Groux et al., 1997). Although they do not constitutively express Foxp3 (Vieira et al., 2004), and can be generated in FOXP3 mutant patients with IPEX syndrome (Passerini et al., 2011), they are able to mediate their suppressive function through multiple mechanisms, such as engagement of CTLA-4 and Programed cell death 1 (PD1) (Akdis et al., 2004; Meiler et al., 2008), metabolic disruption through CD39 and CD73 (Mandapathil et al., 2010), and cytolysis of APCs through release of granzyme B and perforin (Magnani et al., 2011). Thus, they share functions in common with Foxp3+ Tregs. Until now, no reliable markers could successfully distinguish Tr1 cells from other IL-10 producing T cells. However, the recent description of co-expression of integrin α-subunit CD49b and lymphocyte-activation gene (LAG)-3 as identifiers of human and mouse Tr1 cells (Gagliani et al., 2013), will allow further specific characterization of Tr1 cell genesis and function as well as its relation to other IL-10 producing T cells.
IL-35 is a member of the IL-12 cytokine family (Figure 5) with inhibitory functions. It was originally described in murine cells as a heterodimeric suppressive cytokine secreted from Foxp3+ Tregs [the Ebi gene is a downstream target of Foxp3 (Collison et al., 2007)], without which the suppressive function of Tregs was significantly reduced, rendering Tregs incapable of controlling experimental inflammatory bowel disease (Collison et al., 2007). Secretion of IL-35 by Tregs is increased by co-culture with Teffs, subsequently enabling them to suppress Teffs separated by a semi-permeable membrane (Collison et al., 2009). In both man and mouse, IL-35 can induce the development of T cells that secrete IL-35, but not TGF-β or IL-10, and can then mediate suppression in an IL-35-dependent manner. These induced regulatory T cells have been termed iTr35 (Collison et al., 2010; Chaturvedi et al., 2011). iTr35 cells are hyporesponsive to restimulation and, like Tr1 cells (see below), don’t express the TF Foxp3. Moreover, they can be induced from Foxp3−/− murine Thp (Collison et al., 2010), showing that Foxp3 is neither required for their induction nor for their function. Of note, however, iTr35 cells have a gene transcriptional profile that is very similar to non-suppressive Teffs activated without IL-35 (though very different to Tregs) (Collison et al., 2010), suggesting that the induction of iTr35 cells, as with iTregs, is dominated by signals that are generic to T cell activation/survival and requires only modest transcriptional changes induced by IL-35. Although the exact role of IL-35 and iTr35 cells in immune physiology is not known, ectopic expression of IL-35 on pancreatic β-cells can protect against experimental autoimmune diabetes (Bettini et al., 2012) and can be expressed by other immune cells, such as CD8+CTLA-4+ T cells that can suppress tumor (prostate)-specific Teff responses (Olson et al., 2012). The induction of iTr35 cells by neighboring cells producing IL-35, such as Foxp3+ Tregs, may be important in providing at least partial explanations for the phenomenon of infectious tolerance (Waldmann et al., 2006), which hypothetically could be a key component in the success or failure of Treg-based programs of cell therapy.
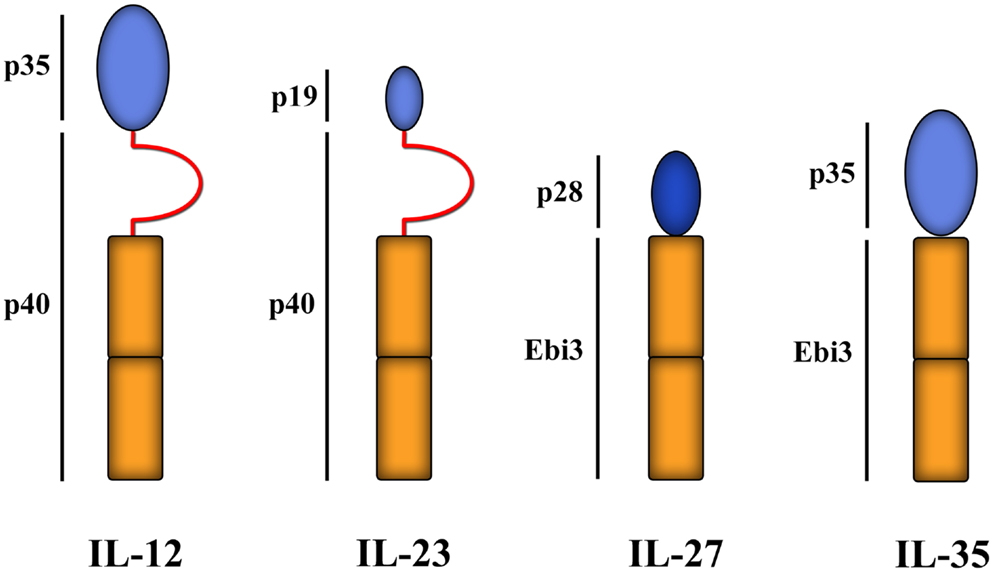
Figure 5. IL-12 family of cytokines. Members of the structurally related IL-12 family of cytokines all comprise of a helical subunit (depicted as blue ovals) and a cytokine receptor homology domain (depicted as orange rectangles) with or without an intervening immunoglobulin-like domain (red semi-circles). Thus far, four family members have been identified: IL-12, IL-23, IL-27, and IL-35. Ebi3 = Epstein–Barr-Virus-induced molecule 1.
Briefly, T-lymphocytes with regulatory functions are not only limited to the CD4+ population, but include some CD8+ populations as well. Gilliet and Liu, for instance, demonstrated that stimulation of naïve CD8+ T-cells with CD40 ligand-activated plasmacytoid DCs induced, in an IL-10-dependent manner, poorly proliferative CD8+ T-cells. These cells produced significant amounts of IL-10, low IFN-γ, no IL-4, IL-5, nor TGF-β, and suppressed CD8+ alloresponses through IL-10 (Gilliet and Liu, 2002). Likewise, repeated stimulation of CD8+ T cells with antigen can generate suppressive CD8+CD28− T cells (Jiang et al., 1998) that show significant overlap in molecular signature with Tregs (Foxp3, GITR, CTLA-4, CD25 for example) (Scotto et al., 2004). The loss of CD28 on CD8 cells, through repeated stimulation, is a well recognized phenomenon and occurs physiologically during aging (Weng et al., 2009) and correlates with poorer responses to vaccination (Saurwein-Teissl et al., 2002). These cells may have a variety of suppressive mechanisms that include inhibition of co-stimulatory molecules on T cells (Ciubotariu et al., 1998) or DCs (Li et al., 1999).
Cell-Based Therapy Using Tregs
The critical role played by Tregs in maintaining peripheral tolerance to self-antigens, thereby controlling autoimmune diseases, reveals the clinical potential of these cells, which can find extensive application to induce transplant tolerance (Wood and Sakaguchi, 2003; Hippen et al., 2011; Issa et al., 2011; Lombardi et al., 2011; Blazar et al., 2012; Tang et al., 2012). In this context, emerging data from animal models reveals that adoptive transfer of Tregs could ameliorate autoimmune diseases, graft-versus-host disease (GvHD) (Blazar et al., 2012) and also induce tolerance to solid organ transplants (Issa et al., 2011). These findings suggest that clinical therapy with human Tregs represents a promising strategy for treatment of autoimmune diseases or for induction of transplantation tolerance.
In solid organ transplant recipients, allo-reactive Teffs in the immune repertoire outnumber Tregs, causing inflammation and leading to graft rejection. So far, most, if not all, therapies aimed at preventing transplant rejection have targeted Teffs. However, another approach, artificially increasing Treg number to regulate Teffs (Figure 1), also has the potential to promote tolerance and facilitate graft survival (Safinia et al., 2013). This is supported by evidence showing that prolonged organ engraftment is essential to induce and expand allo-antigen-specific Tregs, favoring long-term acceptance (Hamano et al., 1996) and data that show better transplant outcomes when organs are infiltrated with greater numbers of Tregs.
There are effectively three strategies for using Tregs as therapeutic agents in humans. The first is introduction of freshly isolated donor Tregs into lymphopaenic hosts, an approach most attractive in the prevention of GvHD post-bone marrow transplantation (Di Ianni et al., 2011). The lymphopaenic environment supports expansion of infused Tregs in vivo and does not cause over-immunosuppression. Indeed, similar experiments in mice have shown that the animals are immunologically intact and able to respond to vaccination (Gaidot et al., 2011) and to control influenza virus infections (Bushell et al., 2005) using this approach.
The second approach involves the in vitro expansion of Tregs prior to infusion, a pre-requisite for infusion of large numbers of Tregs, since their numbers in the peripheral circulation are low. Using polyclonal activation and high doses of IL-2 to expand Tregs could provide the necessary number for therapeutic efficacy. However, intensive expansion protocols could compromise purity of Tregs at the end of the culture protocol. These limitations may be in part due to the presence of “contaminating” Teffs within bead-separated Treg preparations; however the capacity for conversion of human Tregs into IL-17-producing cells has also been well demonstrated (see above). To this aim, the application of tolerogenic approaches to both enhance Treg expansion in vitro and stabilize their suppressive phenotype has been investigated in recent years. Rapamycin, an mTOR kinase inhibitor, for example, selectively promotes expansion of suppressive human Tregs (Battaglia et al., 2006; Scotta et al., 2012). Likewise, culture of Tregs in vitro in the presence of ATRA also supports expansion of functional FOXP3+ human Tregs (Scotta et al., 2012). In contrast, only Treg cultures propagated in the absence of Rapamycin contain CD161+ Tregs, the precursor population of IL-17-producing Tregs (Tresoldi et al., 2011; Scotta et al., 2012). Thus, culture of Tregs with a combination of Rapamycin and clinically acceptable retinoic acid-related molecules may be a viable option to generate large numbers of suppressive and stable Tregs with limited IL-17 potential (Golovina et al., 2011; Scotta et al., 2012). However, among the first Treg-based cell therapy trials in humans (for the treatment of GvHD and type 1 diabetes mellitus respectively), two have used no drug supplementation (except for IL-2) during ex vivo expansion of Tregs and neither has reported unexpected side effects nor paradoxical exacerbation of disease in patients (Trzonkowski et al., 2009; Marek-Trzonkowska et al., 2012).
Neither of the first two approaches, however, makes a distinction between tTregs and iTregs as the starting population. Indeed, culture of contaminating Teffs in the presence of Rapamycin or ATRA during Treg expansion would support the development of iTregs, as discussed above. Thus, the third approach for Treg-based therapy is the induction of iTregs in vivo. As previously discussed, induction of Tregs in the periphery, whether Foxp3+ or Foxp3- can be achieved through a variety of means. Therapeutic options can therefore include administration of tolerogenic DCs that support the in vivo development of both iTregs and Tr1 cells (Naranjo-Gómez et al., 2011; Boks et al., 2012), injection of in vitro expanded Tr1 cells (Brun et al., 2009; Desreumaux et al., 2012) or the introduction of regulatory macrophages (Mregs – not discussed here).
Although these data provide only the earliest evidence for the clinical application of Tregs in cell therapy, a strategy to use these approaches in solid organ transplantation is under way. The ONE Study, for instance, is a multi-center phase I/II study, funded by the European Union FP7 program, investigating the safety of infusing ex vivo generated/expanded Tregs, Tr1 cells, Mregs and tolerogenic DC into kidney transplanted recipients. Altogether, about 200 patients will be enrolled in this clinical trial and, importantly, every center will use the same immunosuppressive protocol for both cell therapy as well as control arms. All patients will be extensively monitored, to obtain data regarding safety, pharmacodynamics, and efficacy of cell therapy, providing an extensive data set for future clinical trials.
Conclusion
Immunological homeostasis is a delicate balance in which both excessive and suboptimal responses can lead to pathological states. CD4+ T cells can differentiate to different Th subsets and promote either an inflammatory response (Th1, Th2, and Th17) or a regulatory one (Tregs). Are then Tregs always beneficial for the optimal resolution of homeostatic challenges? As always, when considering immunological homeostasis, the situation is similar to “Goldilocks and the three bears.” While Tregs are essential to prevent autoimmune disease (Asano et al., 1996) and minimize inflammatory immune responses against dietary antigens and commensal flora (Izcue et al., 2006), excessive Treg responses may facilitate tumor growth and chronic infections by limiting anti-tumor (Shimizu et al., 1999) or anti-pathogen responses (Sakaguchi, 2005). Thus, Tregs function must be tightly regulated to ensure responses are appropriate for each pathological scenario (reviewed in Belkaid, 2007).
Regulatory T cells are both centrally derived and peripherally induced and include both Foxp3+ and Foxp3− populations of cells. An understanding of the mechanisms of Treg induction, suppressive function and lineage stability is key to unraveling the causes underlying development of autoimmune diseases and the design of studies employing Tregs as therapeutic tools in the clinic. Important questions include which regulatory population(s) we should use, whether/how they should be expanded in vitro or induced in vivo, what role infectious tolerance will play, whether Treg plasticity will pose a significant problem and whether the epigenetic/miRNA profile should/could be exploited. On the other hand, lineage plasticity could in theory allow the conversion of effector Th1 and Th17 cells into functioning Tregs in a therapeutic manner. Increasing numbers of clinical trials are focusing on the use of Tregs in a clinical setting, suggesting that Treg-based therapy is considered as both a feasible and acceptable approach to treat inflammatory diseases, offering an alternative to standard pharmacological care. Answers to the questions posed here should, therefore, be forthcoming.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by grants from the National Institute for Health Research (Behdad Afzali, Giovanna Lombardi), Medical Research Council (Behdad Afzali, Susan John, and Giovanna Lombardi), the Academy of Medical Sciences (Behdad Afzali) Wellcome Trust (Behdad Afzali), the ONE Study (Cristiano Scottà and Giovanna Lombardi), the British Heart Foundation (Giovanna Lombardi), Kidney Patients’ Association and Guy’s and St Thomas’ Charity (Giovanna Lombardi). The research was also funded by the National Institute for Health Research (NIHR) Biomedical Research Center based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. The authors acknowledge the support of the MRC Center for Transplantation.
References
Abbas, A. K., Benoist, C., Bluestone, J. A., Campbell, D. J., Ghosh, S., Hori, S., et al. (2013). Regulatory T cells: recommendations to simplify the nomenclature. Nat. Immunol. 14, 307–308. doi:10.1038/ni.2554
Afzali, B., Lombardi, G., Lechler, R. I., and Lord, G. M. (2007). The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin. Exp. Immunol. 148, 32–46. doi:10.1111/j.1365-2249.2007.03356.x
Afzali, B., Mitchell, P., Lechler, R. I., John, S., and Lombardi, G. (2010). Translational mini-review series on Th17 cells: induction of interleukin-17 production by regulatory T cells. Clin. Exp. Immunol. 159, 120–130. doi:10.1111/j.1365-2249.2009.04038.x
Afzali, B., Mitchell, P. J., Edozie, F. C., Povoleri, G., Dowson, S., Demandt, L., et al. (2013). CD161 expression characterizes a sub-population of human regulatory T cells that produces IL-17 in a STAT3 dependent manner. Eur. J. Immunol. Available at: http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)1521-4141/accepted [Accessed May 16, 2013] doi:10.1002/eji.201243296
Akdis, M., Verhagen, J., Taylor, A., Karamloo, F., Karagiannidis, C., Crameri, R., et al. (2004). Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J. Exp. Med. 199, 1567–1575. doi:10.1084/jem.20032058
Akimova, T., Beier, U. H., Wang, L., Levine, M. H., and Hancock, W. W. (2011). Helios expression is a marker of T cell activation and proliferation. PLoS ONE 6:e24226. doi:10.1371/journal.pone.0024226
Annacker, O., Coombes, J. L., Malmstrom, V., Uhlig, H. H., Bourne, T., Johansson-Lindbom, B., et al. (2005). Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J. Exp. Med. 202, 1051–1061. doi:10.1084/jem.20040662
Apostolou, I., and von Boehmer, H. (2004). In vivo instruction of suppressor commitment in naive T cells. J. Exp. Med. 199, 1401–1408. doi:10.1084/jem.20040249
Asano, M., Toda, M., Sakaguchi, N., and Sakaguchi, S. (1996). Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 184, 387–396. doi:10.1084/jem.184.2.387
Aschenbrenner, K., D’Cruz, L. M., Vollmann, E. H., Hinterberger, M., Emmerich, J., Swee, L. K., et al. (2007). Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat. Immunol. 8, 351–358. doi:10.1038/ni1444
Asseman, C., Mauze, S., Leach, M. W., Coffman, R. L., and Powrie, F. (1999). An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 190, 995–1004. doi:10.1084/jem.190.7.995
Ayyoub, M., Deknuydt, F., Raimbaud, I., Dousset, C., Leveque, L., Bioley, G., et al. (2009). Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc. Natl. Acad. Sci. U.S.A. 106, 8635–8640. doi:10.1073/pnas.0900621106
Babiarz, J. E., Ruby, J. G., Wang, Y., Bartel, D. P., and Blelloch, R. (2008). Mouse ES cells express endogenous shRNAs, siRNAs, and other microprocessor-independent, dicer-dependent small RNAs. Genes Dev. 22, 2773–2785. doi:10.1101/gad.1705308
Bacchetta, R., Passerini, L., Gambineri, E., Dai, M., Allan, S. E., Perroni, L., et al. (2006). Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J. Clin. Invest. 116, 1713–1722. doi:10.1172/JCI25112
Baecher-Allan, C., Brown, J. A., Freeman, G. J., and Hafler, D. A. (2001). CD4+CD25 high regulatory cells in human peripheral blood. J. Immunol. 167, 1245–1253.
Baecher-Allan, C., and Hafler, D. A. (2006). Human regulatory T cells and their role in autoimmune disease. Immunol. Rev. 212, 203–216. doi:10.1111/j.0105-2896.2006.00417.x
Baltimore, D., Boldin, M. P., O’Connell, R. M., Rao, D. S., and Taganov, K. D. (2008). MicroRNAs: new regulators of immune cell development and function. Nat. Immunol. 9, 839–845. doi:10.1038/ni.f.209
Barnes, M. J., Krebs, P., Harris, N., Eidenschenk, C., Gonzalez-Quintial, R., Arnold, C. N., et al. (2009). Commitment to the regulatory T cell lineage requires CARMA1 in the thymus but not in the periphery. PLoS Biol. 7:e51. doi:10.1371/journal.pbio.1000051
Baron, U., Floess, S., Wieczorek, G., Baumann, K., Grützkau, A., Dong, J., et al. (2007). DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur. J. Immunol. 37, 2378–2389. doi:10.1002/eji.200737594
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi:10.1016/j.cell.2009.01.002
Battaglia, M., Stabilini, A., Migliavacca, B., Horejs-Hoeck, J., Kaupper, T., and Roncarolo, M. G. (2006). Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J. Immunol. 177, 8338–8347.
Belkaid, Y. (2007). Regulatory T cells and infection: a dangerous necessity. Nat. Rev. Immunol. 7, 875–888. doi:10.1038/nri2189
Belkaid, Y., Piccirillo, C. A., Mendez, S., Shevach, E. M., and Sacks, D. L. (2002). CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420, 502–507. doi:10.1038/nature01152
Bennett, C. L., Christie, J., Ramsdell, F., Brunkow, M. E., Ferguson, P. J., Whitesell, L., et al. (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21. doi:10.1038/83713
Bensinger, S. J., Bandeira, A., Jordan, M. S., Caton, A. J., and Laufer, T. M. (2001). Major histocompatibility complex class II-positive cortical epithelium mediates the selection of CD4(+)25(+) immunoregulatory T cells. J. Exp. Med. 194, 427–438. doi:10.1084/jem.194.4.427
Benson, M. J., Pino-Lagos, K., Rosemblatt, M., and Noelle, R. J. (2007). All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 204, 1765–1774. doi:10.1084/jem.20070719
Beriou, G., Costantino, C. M., Ashley, C. W., Yang, L., Kuchroo, V. K., Baecher-Allan, C., et al. (2009). IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood 113, 4240–4249. doi:10.1182/blood-2008-10-183251
Bettelli, E., Carrier, Y., Gao, W., Korn, T., Strom, T. B., Oukka, M., et al. (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238. doi:10.1038/nature04753
Bettelli, E., Dastrange, M., and Oukka, M. (2005). Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc. Natl. Acad. Sci. U.S.A. 102, 5138–5143. doi:10.1073/pnas.0501675102
Bettini, M., Castellaw, A. H., Lennon, G. P., Burton, A. R., and Vignali, D. A. A. (2012). Prevention of autoimmune diabetes by ectopic pancreatic-cell expression of interleukin-35. Diabetes 61, 1519–1526. doi:10.2337/db11-0784
Blair, P. J., Bultman, S. J., Haas, J. C., Rouse, B. T., Wilkinson, J. E., and Godfrey, V. L. (1994). CD4+CD8 − T cells are the effector cells in disease pathogenesis in the scurfy (sf) mouse. J. Immunol. 153, 3764–3774.
Blazar, B. R., Murphy, W. J., and Abedi, M. (2012). Advances in graft-versus-host disease biology and therapy. Nat. Rev. Immunol. 12, 443–458. doi:10.1038/nri3212
Boks, M. A., Kager-Groenland, J. R., Haasjes, M. S. P., Zwaginga, J. J., van Ham, S. M., and ten Brinke, A. (2012). IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction – a comparative study of human clinical-applicable DC. Clin. Immunol. 142, 332–342. doi:10.1016/j.clim.2011.11.011
Bopp, T., Becker, C., Klein, M., Klein-Hessling, S., Palmetshofer, A., Serfling, E., et al. (2007). Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J. Exp. Med. 204, 1303–1310. doi:10.1084/jem.20062129
Bruder, D., Probst-Kepper, M., Westendorf, A. M., Geffers, R., Beissert, S., Loser, K., et al. (2004). Neuropilin-1: a surface marker of regulatory T cells. Eur. J. Immunol. 34, 623–630. doi:10.1002/eji.200324799
Brun, V., Bastian, H., Neveu, V., and Foussat, A. (2009). Clinical grade production of IL-10 producing regulatory Tr1 lymphocytes for cell therapy of chronic inflammatory diseases. Int. Immunopharmacol. 9, 609–613. doi:10.1016/j.intimp.2009.01.032
Brunkow, M. E., Jeffery, E. W., Hjerrild, K. A., Paeper, B., Clark, L. B., Yasayko, S. A., et al. (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27, 68–73. doi:10.1038/83784
Brüstle, A., Heink, S., Huber, M., Rosenplänter, C., Stadelmann, C., Yu, P., et al. (2007). The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 8, 958–966. doi:10.1038/ni1500
Burchill, M. A., Yang, J., Vang, K. B., Moon, J. J., Chu, H. H., Lio, C.-W. J., et al. (2008). Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity 28, 112–121. doi:10.1016/j.immuni.2007.11.022
Burchill, M. A., Yang, J., Vogtenhuber, C., Blazar, B. R., and Farrar, M. A. (2007). IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 178, 280–290.
Bushell, A., Jones, E., Gallimore, A., and Wood, K. (2005). The generation of CD25+ CD4+ regulatory T cells that prevent allograft rejection does not compromise immunity to a viral pathogen. J. Immunol. 174, 3290–3297.
Cao, X., Cai, S. F., Fehniger, T. A., Song, J., Collins, L. I., Piwnica-Worms, D. R., et al. (2007). Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 27, 635–646. doi:10.1016/j.immuni.2007.08.014
Cardone, J., Le Friec, G., Vantourout, P., Roberts, A., Fuchs, A., Jackson, I., et al. (2010). Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat. Immunol. 11, 862–871. doi:10.1038/ni.1917
Cedar, H., and Bergman, Y. (2011). Epigenetics of haematopoietic cell development. Nat. Rev. Immunol. 11, 478–488. doi:10.1038/nri2991
Chatila, T. A., Blaeser, F., Ho, N., Lederman, H. M., Voulgaropoulos, C., Helms, C., et al. (2000). JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J. Clin. Invest. 106, R75–R81. doi:10.1172/JCI11679
Chaturvedi, V., Collison, L. W., Guy, C. S., Workman, C. J., and Vignali, D. A. (2011). Cutting edge: human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J. Immunol. 186, 6661–6666. doi:10.4049/jimmunol.1100315
Chaudhry, A., Rudra, D., Treuting, P., Samstein, R. M., Liang, Y., Kas, A., et al. (2009). CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Sci. Signal. 326, 986. doi:10.1038/nrg1990
Chen, K., and Rajewsky, N. (2007). The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 8, 93–103. doi:10.1038/nrg1990
Chen, W., Jin, W., Hardegen, N., Lei, K.-J., Li, L., Marinos, N., et al. (2003). Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886. doi:10.1084/jem.20030152
Chen, W., Jin, W., and Wahl, S. M. (1998). Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor beta (TGF-beta) production by murine CD4(+) T cells. J. Exp. Med. 188, 1849–1857. doi:10.1084/jem.188.10.1849
Chen, X., Priatel, J. J., Chow, M. T., and Teh, H.-S. (2008). Preferential development of CD4 and CD8 T regulatory cells in RasGRP1-deficient mice. J. Immunol. 180, 5973–5982.
Chen, Y., Kuchroo, V. K., Inobe, J., Hafler, D. A., and Weiner, H. L. (1994). Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 265, 1237–1240. doi:10.1126/science.7520605
Chendrimada, T. P., Finn, K. J., Ji, X., Baillat, D., Gregory, R. I., Liebhaber, S. A., et al. (2007). MicroRNA silencing through RISC recruitment of eIF6. Nature 447, 823–828. doi:10.1038/nature05841
Cheng, G., Yu, A., Dee, M. J., and Malek, T. R. (2013). IL-2R signaling is essential for functional maturation of regulatory T cells during thymic development. J. Immunol. 190, 1567–1575. doi:10.4049/jimmunol.1201218
Chiang, Y. J., Kole, H. K., Brown, K., Naramura, M., Fukuhara, S., Hu, R. J., et al. (2000). Cbl-b regulates the CD28 dependence of T-cell activation. Nature 403, 216–220. doi:10.1038/35003235
Chong, M. M. W., Rasmussen, J. P., Rudensky, A. Y., Rundensky, A. Y., and Littman, D. R. (2008). The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J. Exp. Med. 205, 2005–2017. doi:10.1084/jem.20081219
Ciubotariu, R., Colovai, A. I., Pennesi, G., Liu, Z., Smith, D., Berlocco, P., et al. (1998). Specific suppression of human CD4+ Th cell responses to pig MHC antigens by CD8+CD28− regulatory T cells. J. Immunol. 161, 5193–5202.
Cobb, B. S., Hertweck, A., Smith, J., O’Connor, E., Graf, D., Cook, T., et al. (2006). A role for Dicer in immune regulation. J. Exp. Med. 203, 2519–2527. doi:10.1084/jem.20061692
Collison, L. W., Chaturvedi, V., Henderson, A. L., Giacomin, P. R., Guy, C., Bankoti, J., et al. (2010). IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 11, 1093–1101. doi:10.1038/ni.1952
Collison, L. W., Pillai, M. R., Chaturvedi, V., and Vignali, D. A. A. (2009). Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J. Immunol. 182, 6121–6128. doi:10.4049/jimmunol.0803646
Collison, L. W., Workman, C. J., Kuo, T. T., Boyd, K., Wang, Y., Vignali, K. M., et al. (2007). The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450, 566–569. doi:10.1038/nature06306
Constant, S. L., and Bottomly, K. (1997). Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu. Rev. Immunol. 15, 297–322. doi:10.1146/annurev.immunol.15.1.297
Coombes, J. L., Siddiqui, K. R. R., Arancibia-Cárcamo, C. V., Hall, J., Sun, C.-M., Belkaid, Y., et al. (2007). A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764. doi:10.1084/jem.20070590
Curotto de Lafaille, M. A., Kutchukhidze, N., Shen, S., Ding, Y., Yee, H., and Lafaille, J. J. (2008). Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity 29, 114–126. doi:10.1016/j.immuni.2008.05.010
De Jong, E., Suddason, T., and Lord, G. (2010). Translational mini review series on Th17 cells: development of mouse and human T helper 17 cells. Clin. Exp. Immunol. 159, 148–158. doi:10.1111/j.1365-2249.2009.04041.x
Deaglio, S., Dwyer, K. M., Gao, W., Friedman, D., Usheva, A., Erat, A., et al. (2007). Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265. doi:10.1084/jem.20062512
Desreumaux, P., Foussat, A., Allez, M., Beaugerie, L., Hébuterne, X., Bouhnik, Y., et al. (2012). Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 143, 1207.e1–2–12017.e1–2. doi:10.1053/j.gastro.2012.07.116
Dhodapkar, M. V., Steinman, R. M., Krasovsky, J., Munz, C., and Bhardwaj, N. (2001). Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 193, 233–238. doi:10.1084/jem.193.2.233
Di Ianni, M., Falzetti, F., Carotti, A., Terenzi, A., Castellino, F., Bonifacio, E., et al. (2011). Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 117, 3921–3928. doi:10.1182/blood-2010-10-311894
DiPaolo, R. J., Brinster, C., Davidson, T. S., Andersson, J., Glass, D., and Shevach, E. M. (2007). Autoantigen-specific TGFbeta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J. Immunol. 179, 4685–4693.
E, X. Q., Meng, H. X., Cao, Y., Zhang, S. Q., Bi, Z. G., and Yamakawa, M. (2012). Distribution of regulatory T cells and interaction with dendritic cells in the synovium of rheumatoid arthritis. Scand. J. Rheumatol. 41, 413–420. doi:10.3109/03009742.2012.696135
Elias, K. M., Laurence, A., Davidson, T. S., Stephens, G., Kanno, Y., Shevach, E. M., et al. (2007). Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111, 1013–1020. doi:10.1182/blood-2007-06-096438
Fayyad-Kazan, H., Rouas, R., Merimi, M., Zein El, N., Lewalle, P., Jebbawi, F., et al. (2010). Valproate treatment of human cord blood CD4-positive effector T cells confers on them the molecular profile (microRNA signature and FOXP3 expression) of natural regulatory CD4-positive cells through inhibition of histone deacetylase. J. Biol. Chem. 285, 20481–20491. doi:10.1074/jbc.M110.119628
Feng, T., Cao, A. T., Weaver, C. T., Elson, C. O., and Cong, Y. (2011). Interleukin-12 converts Foxp3+ regulatory T cells to interferon-γ-producing Foxp3+ T cells that inhibit colitis. Gastroenterology 140, 2031–2043. doi:10.1053/j.gastro.2011.03.009
Ferrara, N., Gerber, H.-P., and LeCouter, J. (2003). The biology of VEGF and its receptors. Nat. Med. 9, 669–676. doi:10.1038/nm0603-669
Floess, S., Freyer, J., Siewert, C., Baron, U., Olek, S., Polansky, J., et al. (2007). Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 5:e38. doi:10.1371/journal.pbio.0050038
Fontenot, J. D., Dooley, J. L., Farr, A. G., and Rudensky, A. Y. (2005a). Developmental regulation of Foxp3 expression during ontogeny. J. Exp. Med. 202, 901–906. doi:10.1084/jem.20050784
Fontenot, J. D., Rasmussen, J. P., Gavin, M. A., and Rudensky, A. Y. (2005b). A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 6, 1142–1151. doi:10.1038/ni1263
Fontenot, J. D., Rasmussen, J. P., Williams, L. M., Dooley, J. L., Farr, A. G., and Rudensky, A. Y. (2005c). Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22, 329–341. doi:10.1016/j.immuni.2005.01.016
Fontenot, J. D., Gavin, M. A., and Rudensky, A. Y. (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336. doi:10.1038/ni904
Gagliani, N., Magnani, C. F., Huber, S., Gianolini, M. E., Pala, M., Licona-Limon, P., et al. (2013). Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 19, 739–746. doi:10.1038/nm.3179
Gaidot, A., Landau, D. A., Martin, G. H., Bonduelle, O., Grinberg-Bleyer, Y., Matheoud, D., et al. (2011). Immune reconstitution is preserved in hematopoietic stem cell transplantation coadministered with regulatory T cells for GVHD prevention. Blood 117, 2975–2983. doi:10.1182/blood-2010-08-299974
Gavin, M. A., Rasmussen, J. P., Fontenot, J. D., Vasta, V., Manganiello, V. C., Beavo, J. A., et al. (2007). Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445, 771–775. doi:10.1038/nature05543
Gavin, M. A., Torgerson, T. R., Houston, E., DeRoos, P., Ho, W. Y., Stray-Pedersen, A., et al. (2006). Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc. Natl. Acad. Sci. U.S.A. 103, 6659–6664. doi:10.1073/pnas.0509484103
Gilliet, M., and Liu, Y.-J. (2002). Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J. Exp. Med. 195, 695–704. doi:10.1084/jem.20011603
Glinka, Y., and Prud’Homme, G. J. (2008). Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J. Leukoc. Biol. 84, 302–310. doi:10.1189/jlb.0208090
Golovina, T. N., Mikheeva, T., Brusko, T. M., Blazar, B. R., Bluestone, J. A., and Riley, J. L. (2011). Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS ONE 6:e15868. doi:10.1371/journal.pone.0015868
Gondek, D. C., Lu, L.-F., Quezada, S. A., Sakaguchi, S., and Noelle, R. J. (2005). Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol. 174, 1783–1786.
Gregori, S., Tomasoni, D., Pacciani, V., Scirpoli, M., Battaglia, M., Magnani, C. F., et al. (2010). Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood 116, 935–944. doi:10.1182/blood-2009-07-234872
Grigoriadis, G., Vasanthakumar, A., Banerjee, A., Grumont, R., Overall, S., Gleeson, P., et al. (2011). c-Rel controls multiple discrete steps in the thymic development of Foxp3+ CD4 regulatory T cells. PLoS ONE 6:e26851. doi:10.1371/journal.pone.0026851
Groux, H., O’Garra, A., Bigler, M., Rouleau, M., Antonenko, S., de Vries, J. E., et al. (1997). A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389, 737–742. doi:10.1038/39614
Hamano, K., Rawsthorne, M. A., Bushell, A. R., Morris, P. J., and Wood, K. J. (1996). Evidence that the continued presence of the organ graft and not peripheral donor microchimerism is essential for maintenance of tolerance to alloantigen in vivo in anti-CD4 treated recipients. Transplantation 62, 856–860. doi:10.1097/00007890-199609270-00026
Han, J., Lee, Y., Yeom, K.-H., Nam, J.-W., Heo, I., Rhee, J.-K., et al. (2006). Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125, 887–901. doi:10.1016/j.cell.2006.03.043
Hansen, W., Hutzler, M., Abel, S., Alter, C., Stockmann, C., Kliche, S., et al. (2012). Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med. 209, 2001–2016. doi:10.1084/jem.20111497
Harada, Y., Harada, Y., Elly, C., Ying, G., Paik, J.-H., DePinho, R. A., et al. (2010). Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J. Exp. Med. 207, 1381–1391. doi:10.1084/jem.20100004
Haxhinasto, S., Mathis, D., and Benoist, C. (2008). The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 205, 565–574. doi:10.1084/jem.20071477
Hill, J. A., Feuerer, M., Tash, K., Haxhinasto, S., Perez, J., Melamed, R., et al. (2007). Foxp3 transcription-factor-dependent and-independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800. doi:10.1016/j.immuni.2007.09.010
Hill, J. A., Hall, J. A., Sun, C.-M., Cai, Q., Ghyselinck, N., Chambon, P., et al. (2008). Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi cells. Immunity 29, 758–770. doi:10.1016/j.immuni
Hippen, K. L., Riley, J. L., June, C. H., and Blazar, B. R. (2011). Clinical perspectives for regulatory T cells in transplantation tolerance. Semin. Immunol. 23, 462–468. doi:10.1016/j.smim.2011.07.008
Hirahara, K., Vahedi, G., Ghoreschi, K., Yang, X.-P., Nakayamada, S., Kanno, Y., et al. (2011). Helper T-cell differentiation and plasticity: insights from epigenetics. Immunology 134, 235–245. doi:10.1111/j.1365-2567.2011.03483.x
Hori, S. (2010). Developmental plasticity of Foxp3+ regulatory T cells. Curr. Opin. Immunol. 22, 575–582. doi:10.1016/j.coi
Hori, S. (2011). Regulatory T cell plasticity: beyond the controversies. Trends Immunol. 32, 295–300. doi:10.1016/j.it.2011.04.004
Hori, S., Nomura, T., and Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061. doi:10.1126/science.1079490
Hsieh, C.-S., Liang, Y., Tyznik, A. J., Self, S. G., Liggitt, D., and Rudensky, A. Y. (2004). Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity 21, 267–277. doi:10.1016/j.immuni.2004.07.009
Hsieh, C.-S., Zheng, Y., Liang, Y., Fontenot, J. D., and Rudensky, A. Y. (2006). An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat. Immunol. 7, 401–410. doi:10.1038/ni1318
Huang, C.-T., Workman, C. J., Flies, D., Pan, X., Marson, A. L., Zhou, G., et al. (2004). Role of LAG-3 in regulatory T cells. Immunity 21, 503–513. doi:10.1016/j.immuni.2004.08.010
Inobe, J.-I., Slavin, A. J., Komagata, Y., Chen, Y., Liu, L., and Weiner, H. L. (1998). IL-4 is a differentiation factor for transforming growth factor-β secreting Th3 cells and oral administration of IL-4 enhances oral tolerance in experimental allergic encephalomyelitis. Eur. J. Immunol. 28, 2780–2790. doi:10.1002/(SICI)1521-4141(199809)28:09<2780::AID-IMMU2780>3.0.CO;2-J
Ishikawa, H., Tanaka, K., Maeda, Y., Aiba, Y., Hata, A., Tsuji, N. M., et al. (2008). Effect of intestinal microbiota on the induction of regulatory CD25+ CD4+ T cells. Clin. Exp. Immunol. 153, 127–135. doi:10.1111/j.1365-2249
Isomura, I., Palmer, S., Grumont, R. J., Bunting, K., Hoyne, G., Wilkinson, N., et al. (2009). c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J. Exp. Med. 206, 3001–3014. doi:10.1084/jem.20091411
Issa, F., Chandrasekharan, D., and Wood, K. J. (2011). Regulatory T cells as modulators of chronic allograft dysfunction. Curr. Opin. Immunol. 23, 648–654. doi:10.1016/j.coi.2011.06.005
Ivanov, I. I., Frutos, R., de, L., Manel, N., Yoshinaga, K., Rifkin, D. B., et al. (2008). Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349. doi:10.1016/j.chom.2008.09.009
Ivanov, I. I., McKenzie, B. S., Zhou, L., Tadokoro, C. E., Lepelley, A., Lafaille, J. J., et al. (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133. doi:10.1016/j.cell.2006.07.035
Izcue, A., Coombes, J. L., and Powrie, F. (2006). Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol. Rev. 212, 256–271. doi:10.1111/j.0105-2896.2006.00423.x
Janson, P. C. J., Winerdal, M. E., Marits, P., Thörn, M., Ohlsson, R., and Winqvist, O. (2008). FOXP3 promoter demethylation reveals the committed Treg population in humans. PLoS ONE 3:e1612. doi:10.1371/journal.pone.0001612
Jeker, L. T., Zhou, X., Blelloch, R., and Bluestone, J. A. (2013). DGCR8-mediated production of canonical micrornas is critical for regulatory T cell function and stability. PLoS ONE 8:e66282. doi:10.1371/journal.pone.0066282
Jeker, L. T., Zhou, X., Gershberg, K., de Kouchkovsky, D., Morar, M. M., Stadthagen, G., et al. (2012). MicroRNA 10a marks regulatory T cells. PLoS ONE 7:e36684. doi:10.1371/journal.pone.0036684
Jiang, S., Tugulea, S., Pennesi, G., Liu, Z., Mulder, A., Lederman, S., et al. (1998). Induction of MHC-class I restricted human suppressor T cells by peptide priming in vitro. Hum. Immunol. 59, 690–699. doi:10.1016/S0198-8859(98)00073-1
Johansson-Lindbom, B., Svensson, M., Pabst, O., Palmqvist, C., Marquez, G., Förster, R., et al. (2005). Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J. Exp. Med. 202, 1063–1073. doi:10.1084/jem.20051100
Jonuleit, H., Schmitt, E., Schuler, G., Knop, J., and Enk, A. H. (2000). Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J. Exp. Med. 192, 1213–1222. doi:10.1084/jem.192.9.1213
Jordan, M. S., Boesteanu, A., Reed, A. J., Petrone, A. L., Holenbeck, A. E., Lerman, M. A., et al. (2001). Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2, 301–306. doi:10.1038/86302
Josefowicz, S. Z., Lu, L.-F., and Rudensky, A. Y. (2012). Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 30, 531–564. doi:10.1146/annurev.immunol.25.022106.141623
Josefowicz, S. Z., and Rudensky, A. (2009). Control of regulatory T cell lineage commitment and maintenance. Immunity 30, 616–625. doi:10.1016/j.immuni.2009.04.009
Josefowicz, S. Z., Wilson, C. B., and Rudensky, A. Y. (2009). Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J. Immunol. 182, 6648–6652. doi:10.4049/jimmunol.0803320
Kanangat, S., Blair, P., Reddy, R., Daheshia, M., Godfrey, V., Rouse, B. T., et al. (1996). Disease in the scurfy (sf) mouse is associated with overexpression of cytokine genes. Eur. J. Immunol. 26, 161–165. doi:10.1002/eji.1830260125
Kemper, C., Chan, A. C., Green, J. M., Brett, K. A., Murphy, K. M., and Atkinson, J. P. (2003). Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature 421, 388–392. doi:10.1038/nature01315
Khattri, R., Cox, T., Yasayko, S.-A., and Ramsdell, F. (2003). An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4, 337–342. doi:10.1038/ni909
Kim, H.-P., and Leonard, W. J. (2007). CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J. Exp. Med. 204, 1543–1551.
Kim, J. M., and Rudensky, A. (2006). The role of the transcription factor Foxp3 in the development of regulatory T cells. Immunol. Rev. 212, 86–98. doi:10.1111/j.0105-2896.2006.00426.x
Koch, M. A., Tucker-Heard, G., Perdue, N. R., Killebrew, J. R., Urdahl, K. B., and Campbell, D. J. (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602. doi:10.1038/ni.1731
Koenen, H. J. P. M., Smeets, R. L., Vink, P. M., van Rijssen, E., Boots, A. M. H., and Joosten, I. (2008). Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 112, 2340–2352. doi:10.1182/blood-2008-01-133967
Kojima, A., Taguchi, O., and Nishizuka, Y. (1980). Experimental production of possible autoimmune castritis followed by macrocytic anemia in athymic nude mice. Lab. Invest. 42, 387–395.
Kojima, A., Tanaka-Kojima, Y., Sakakura, T., and Nishizuka, Y. (1976). Spontaneous development of autoimmune thyroiditis in neonatally thymectomized mice. Lab. Invest. 34, 550–557.
Kretschmer, K., Apostolou, I., Hawiger, D., Khazaie, K., Nussenzweig, M. C., and Boehmer von, H. (2005). Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 6, 1219–1227. doi:10.1038/ni1265
Kwan, W.-H., van der Touw, W., Paz-Artal, E., Li, M. O., and Heeger, P. S. (2013). Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J. Exp. Med. 210, 257–268. doi:10.1084/jem.20121525
Lathrop, S. K., Santacruz, N. A., Pham, D., Luo, J., and Hsieh, C.-S. (2008). Antigen-specific peripheral shaping of the natural regulatory T cell population. J. Exp. Med. 205, 3105–3117. doi:10.1084/jem.20081359
Laurence, A., Tato, C. M., Davidson, T. S., Kanno, Y., Chen, Z., Yao, Z., et al. (2007). Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26, 371–381. doi:10.1016/j.immuni.2007.02.009
Le Friec, G., Sheppard, D., Whiteman, P., Karsten, C. M., Shamoun, S. A.-T., Laing, A., et al. (2012). The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat. Immunol. 13, 1213–1221. doi:10.1038/ni.2454
Lee, Y., Kim, M., Han, J., Yeom, K.-H., Lee, S., Baek, S. H., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060. doi:10.1038/sj.emboj.7600385
Levings, M. K., Sangregorio, R., Galbiati, F., Squadrone, S., de Waal-Malefyt, R., and Roncarolo, M. G. (2001). IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells. J. Immunol. 166, 5530–5539.
Li, J., Liu, Z., Jiang, S., Cortesini, R., Lederman, S., and Suciu-Foca, N. (1999). T suppressor lymphocytes inhibit NF-kappa B-mediated transcription of CD86 gene in APC. J. Immunol. 163, 6386–6392.
Li, M. O., Sanjabi, S., and Flavell, R. A. (2006). Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 25, 455–471. doi:10.1016/j.immuni.2006.07.011
Lio, C.-W. J., and Hsieh, C.-S. (2008). A two-step process for thymic regulatory T cell development. Immunity 28, 100–111. doi:10.1016/j.immuni.2007.11.021
Liston, A., Lu, L.-F., O’Carroll, D., Tarakhovsky, A., and Rudensky, A. Y. (2008a). Dicer-dependent microRNA pathway safeguards regulatory T cell function. J. Exp. Med. 205, 1993–2004. doi:10.1084/jem.20081062
Liston, A., Nutsch, K. M., Farr, A. G., Lund, J. M., Rasmussen, J. P., Koni, P. A., et al. (2008b). Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc. Natl. Acad. Sci. U.S.A. 105, 11903–11908. doi:10.1073/pnas.0801506105
Liu, W., Putnam, A. L., Xu-Yu, Z., Szot, G. L., Lee, M. R., Zhu, S., et al. (2006). CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 203, 1701–1711. doi:10.1084/jem.20060772
Liu, Y., Zhang, P., Li, J., Kulkarni, A. B., Perruche, S., and Chen, W. (2008). A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 9, 632–640. doi:10.1038/ni.1607
Lodish, H. F., Zhou, B., Liu, G., and Chen, C.-Z. (2008). Micromanagement of the immune system by microRNAs. Nat. Rev. Immunol. 8, 120–130. doi:10.1038/nri2252
Lombardi, G., Sagoo, P., Scotta, C., Fazekasova, H., Smyth, L., Tsang, J., et al. (2011). Cell therapy to promote transplantation tolerance: a winning strategy? Immunotherapy 3, 28–31. doi:10.2217/imt.11.42
Long, M., Park, S.-G., Strickland, I., Hayden, M. S., and Ghosh, S. (2009). Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 31, 921–931. doi:10.1016/j.immuni.2009.09.022
Lu, L.-F., Boldin, M. P., Chaudhry, A., Lin, L.-L., Taganov, K. D., Hanada, T., et al. (2010). Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 142, 914–929. doi:10.1016/j.cell.2010.08.012
Lu, L.-F., Thai, T.-H., Calado, D. P., Chaudhry, A., Kubo, M., Tanaka, K., et al. (2009). Foxp3-dependent MicroRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity 30, 80–91. doi:10.1016/j.immuni.2008.11.010
Lykken, E. A., and Li, Q.-J. (2010). microRNAs at the regulatory frontier: an investigation into how microRNAs impact the development and effector functions of CD4 T cells. Immunol. Res. 49, 87–96. doi:10.1007/s12026-010-8196-4
Magnani, C. F., Alberigo, G., Bacchetta, R., Serafini, G., Andreani, M., Roncarolo, M. G., et al. (2011). Killing of myeloid APCs via HLA class I, CD2 and CD226 defines a novel mechanism of suppression by human Tr1 cells. Eur. J. Immunol. 41, 1652–1662. doi:10.1002/eji.201041120
Malek, T. R., Yu, A., Vincek, V., Scibelli, P., and Kong, L. (2002). CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity 17, 167–178. doi:10.1016/S1074-7613(02)00367-9
Maloy, K. J., Salaun, L., Cahill, R., Dougan, G., Saunders, N. J., and Powrie, F. (2003). CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 197, 111–119. doi:10.1084/jem.20021345
Mandapathil, M., Szczepanski, M. J., Szajnik, M., Ren, J., Jackson, E. K., Johnson, J. T., et al. (2010). Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J. Biol. Chem. 285, 27571–27580. doi:10.1074/jbc.M110.127100
Mangan, P. R., Harrington, L. E., O’Quinn, D. B., Helms, W. S., Bullard, D. C., Elson, C. O., et al. (2006). Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441, 231–234. doi:10.1038/nature04754
Mantel, P.-Y., Ouaked, N., Rückert, B., Karagiannidis, C., Welz, R., Blaser, K., et al. (2006). Molecular mechanisms underlying FOXP3 induction in human T cells. J. Immunol. 176, 3593–3602.
Marek-Trzonkowska, N., Mysliwiec, M. L., Dobyszuk, A., Grabowska, M., Techmanska, I., Juscinska, J., et al. (2012). Administration of CD4+ CD25highCD127- regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care 35, 1817–1820. doi:10.2337/dc12-0038
Marie, J. C., Letterio, J. J., Gavin, M., and Rudensky, A. Y. (2005). TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med. 201, 1061–1067. doi:10.1084/jem.20042276
Marson, A., Kretschmer, K., Frampton, G. M., Jacobsen, E. S., Polansky, J. K., MacIsaac, K. D., et al. (2007). Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 445, 931–935. doi:10.1038/nature05478
Mayack, S. R., and Berg, L. J. (2006). Cutting edge: an alternative pathway of CD4+ T cell differentiation is induced following activation in the absence of gamma-chain-dependent cytokine signals. J. Immunol. 176, 2059–2063.
Maynard, C. L., Hatton, R. D., Helms, W. S., Oliver, J. R., Stephensen, C. B., and Weaver, C. T. (2009). Contrasting roles for all-trans retinoic acid in TGF-beta-mediated induction of Foxp3 and Il10 genes in developing regulatory T cells. J. Exp. Med. 206, 343–357. doi:10.1084/jem.20080950
Meiler, F., Zumkehr, J., Klunker, S., Rückert, B., Akdis, C. A., and Akdis, M. (2008). In vivo switch to IL-10-secreting T regulatory cells in high dose allergen exposure. J. Exp. Med. 205, 2887–2898. doi:10.1084/jem.20080193
Miyara, M., Yoshioka, Y., Kitoh, A., Shima, T., Wing, K., Niwa, A., et al. (2009). Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 30, 899–911. doi:10.1016/j.immuni.2009.03.019
Morrissey, P. J., Charrier, K., Braddy, S., Liggitt, D., and Watson, J. D. (1993). CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J. Exp. Med. 178, 237–244. doi:10.1084/jem.178.1.237
Mucida, D., Kutchukhidze, N., Erazo, A., Russo, M., Lafaille, J. J., and Curotto de Lafaille, M. A. (2005). Oral tolerance in the absence of naturally occurring Tregs. J. Clin. Invest. 115, 1923–1933. doi:10.1172/JCI24487
Mucida, D., Park, Y., Kim, G., Turovskaya, O., Scott, I., Kronenberg, M., et al. (2007). Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317, 256–260. doi:10.1126/science.1145697
Muraro, P. A., Vergelli, M., Kalbus, M., Banks, D. E., Nagle, J. W., Tranquill, L. R., et al. (1997). Immunodominance of a low-affinity major histocompatibility complex-binding myelin basic protein epitope (residues 111-129) in HLA-DR4 (B1∗0401) subjects is associated with a restricted T cell receptor repertoire. J. Clin. Invest. 100, 339–349. doi:10.1172/JCI119539
Nagar, M., Vernitsky, H., Cohen, Y., Dominissini, D., Berkun, Y., Rechavi, G., et al. (2008). Epigenetic inheritance of DNA methylation limits activation-induced expression of FOXP3 in conventional human CD25-CD4+ T cells. Int. Immunol. 20, 1041–1055. doi:10.1093/intimm/dxn062
Nakamura, K., Kitani, A., and Strober, W. (2001). Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J. Exp. Med. 194, 629–644. doi:10.1084/jem.194.5.629
Nakayamada, S., Takahashi, H., Kanno, Y., and O’Shea, J. J. (2012). Helper T cell diversity and plasticity. Curr. Opin. Immunol. 24, 297–302. doi:10.1016/j.coi.2012.01.014
Naranjo-Gómez, M., Raïch-Regué, D., Oñate, C., Grau-López, L., Ramo-Tello, C., Pujol-Borrell, R., et al. (2011). Comparative study of clinical grade human tolerogenic dendritic cells. J. Transl. Med. 9, 89. doi:10.1186/1479-5876-9-89
Ng, W. F., Duggan, P. J., Ponchel, F., Matarese, G., Lombardi, G., Edwards, A. D., et al. (2001). Human CD4(+)CD25(+) cells: a naturally occurring population of regulatory T cells. Blood 98, 2736–2744. doi:10.1182/blood.V98.9.2736
Nishizuka, Y., and Sakakura, T. (1969). Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science 166, 753–755. doi:10.1126/science.166.3906.753
Ohkura, N., Hamaguchi, M., Morikawa, H., Sugimura, K., Tanaka, A., Ito, Y., et al. (2012). T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 37, 785–799. doi:10.1016/j.immuni.2012.09.010
Olson, B. M., Jankowska-Gan, E., Becker, J. T., Vignali, D. A. A., Burlingham, W. J., and McNeel, D. G. (2012). Human prostate tumor antigen-specific CD8+ regulatory T cells are inhibited by CTLA-4 or IL-35 blockade. J. Immunol. 189, 5590–5601. doi:10.4049/jimmunol.1201744
O’Shea, J. J., and Paul, W. E. (2010). Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102. doi:10.1126/science.1178334
Östman, S., Rask, C., Wold, A. E., Hultkrantz, S., and Telemo, E. (2006). Impaired regulatory T cell function in germ-free mice. Eur. J. Immunol. 36, 2336–2346. doi:10.1002/eji.200535244
Ouyang, W., Beckett, O., Ma, Q., Paik, J.-H., DePinho, R. A., and Li, M. O. (2010). Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat. Immunol. 11, 618–627. doi:10.1038/ni.1884
Paiva, R. S., Lino, A. C., Bergman, M.-L., Caramalho, I., Sousa, A. E., Zelenay, S., et al. (2013). Recent thymic emigrants are the preferential precursors of regulatory T cells differentiated in the periphery. Proc. Natl. Acad. Sci. U.S.A. 110, 6494–6499. doi:10.1073/pnas.1221955110
Pandiyan, P., Zheng, L., Ishihara, S., Reed, J., and Lenardo, M. J. (2007). CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 8, 1353–1362. doi:10.1038/ni1536
Papiernik, M., de Moraes, M. L., Pontoux, C., Vasseur, F., and Pénit, C. (1998). Regulatory CD4 T cells: expression of IL-2R alpha chain, resistance to clonal deletion and IL-2 dependency. Int. Immunol. 10, 371–378. doi:10.1093/intimm/10.4.371
Passerini, L., Di Nunzio, S., Gregori, S., Gambineri, E., Cecconi, M., Seidel, M. G., et al. (2011). Functional type 1 regulatory T cells develop regardless of FOXP3 mutations in patients with IPEX syndrome. Eur. J. Immunol. 41, 1120–1131. doi:10.1002/eji.201040909
Pennington, D. J., Silva-Santos, B., Silberzahn, T., Escórcio-Correia, M., Woodward, M. J., Roberts, S. J., et al. (2006). Early events in the thymus affect the balance of effector and regulatory T cells. Nature 444, 1073–1077. doi:10.1038/nature06051
Pesenacker, A. M., Bending, D., Ursu, S., Wu, Q., Nistala, K., and Wedderburn, L. R. (2013). CD161 defines the subset of FoxP3+ T cells capable of producing proinflammatory cytokines. Blood 121, 2647–2658. doi:10.1182/blood-2012-08-443473
Picca, C. C., and Caton, A. J. (2005). The role of self-peptides in the development of CD4+ CD25+ regulatory T cells. Curr. Opin. Immunol. 17, 131–136. doi:10.1016/j.coi.2005.01.003
Pillai, R. S., Bhattacharyya, S. N., and Filipowicz, W. (2007). Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 17, 118–126. doi:10.1016/j.tcb.2006.12.007
Polansky, J. K., Kretschmer, K., Freyer, J., Floess, S., Garbe, A., Baron, U., et al. (2008). DNA methylation controls Foxp3 gene expression. Eur. J. Immunol. 38, 1654–1663. doi:10.1002/eji.200838105
Powrie, F., Carlino, J., Leach, M. W., Mauze, S., and Coffman, R. L. (1996). A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med. 183, 2669–2674. doi:10.1084/jem.183.6.2669
Puccetti, P., and Grohmann, U. (2007). IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat. Rev. Immunol. 7, 817–823. doi:10.1038/nri2163
Read, S., Greenwald, R., Izcue, A., Robinson, N., Mandelbrot, D., Francisco, L., et al. (2006). Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 177, 4376–4383.
Rengarajan, J., Mowen, K. A., McBride, K. D., Smith, E. D., Singh, H., and Glimcher, L. H. (2002). Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J. Exp. Med. 195, 1003–1012. doi:10.1084/jem.20011128
Robertson, S. J., and Hasenkrug, K. J. (2006). The role of virus-induced regulatory T cells in immunopathology. Springer Semin. Immunopathol. 28, 51–62. doi:10.1007/s00281-006-0019-2
Romagnoli, P., Hudrisier, D., and van Meerwijk, J. P. M. (2002). Preferential recognition of self antigens despite normal thymic deletion of CD4(+)CD25(+) regulatory T cells. J. Immunol. 168, 1644–1648.
Rouas, R., Fayyad hyphen Kazan, H., Zein, E. N., Lewalle, P., Roth eacute, F. C. O., Simion, A., et al. (2009). Human natural Treg microRNA signature: role of microRNA & hyphen; 31 and microRNA & hyphen; 21 in FOXP3 expression. Eur. J. Immunol. 39, 1608–1618.
Roychoudhuri, R., Hirahara, K., Mousavi, K., Clever, D., Klebanoff, C. A., Bonelli, M., et al. (2013). BACH2 represses effector programs to stabilize Treg-mediated immune homeostasis. Nature doi:10.1038/nature12199. [Epub ahead of print].
Ruan, Q., Kameswaran, V., Tone, Y., Li, L., Liou, H.-C., Greene, M. I., et al. (2009). Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 31, 932–940. doi:10.1016/j.immuni.2009.10.006
Rubtsov, Y. P., Niec, R. E., Josefowicz, S., Li, L., Darce, J., Mathis, D., et al. (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671. doi:10.1126/science.1191996
Russell, W. L., Russell, L. B., and Gower, J. S. (1959). Exceptional inheritance of a sex-linked gene in the mouse explained on the basis that the X/O sex-chromosome constitution is female. Proc. Natl. Acad. Sci. U.S.A. 45, 554–560. doi:10.1073/pnas.45.4.554
Sadlack, B., Merz, H., Schorle, H., Schimpl, A., Feller, A. C., and Horak, I. (1993). Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 75, 253–261. doi:10.1016/0092-8674(93)80067-O
Safinia, N., Leech, J., Hernandez-Fuentes, M., Lechler, R., and Lombardi, G. (2013). Promoting transplantation tolerance; adoptive regulatory T cell therapy. Clin. Exp. Immunol. 172, 158–168. doi:10.1111/cei.12052
Sakaguchi, S. (2005). Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 6, 345–352. doi:10.1038/ni1178
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M., and Toda, M. (1995). Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164.
Sakaguchi, S., Takahashi, T., and Nishizuka, Y. (1982). Study on cellular events in post-thymectomy autoimmune oophoritis in mice. II. Requirement of Lyt-1 cells in normal female mice for the prevention of oophoritis. J. Exp. Med. 156, 1577–1586. doi:10.1084/jem.156.6.1565
Sakaguchi, S., Toda, M., Asano, M., Itoh, M., Morse, S. S., and Sakaguchi, N. (1996). T cell-mediated maintenance of natural self-tolerance: its breakdown as a possible cause of various autoimmune diseases. J. Autoimmun. 9, 211–220. doi:10.1006/jaut.1996.0026
Sakaguchi, S., Wing, K., Onishi, Y., Prieto-Martin, P., and Yamaguchi, T. (2009). Regulatory T cells: how do they suppress immune responses? Int. Immunol. 21, 1105–1111. doi:10.1093/intimm/dxp095
Sakaguchi, S., Yamaguchi, T., Nomura, T., and Ono, M. (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. doi:10.1016/j.cell.2008.05.009
Salomon, B., Lenschow, D. J., Rhee, L., Ashourian, N., Singh, B., Sharpe, A., et al. (2000). B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12, 431–440. doi:10.1016/S1074-7613(00)80195-8
Samon, J. B., Champhekar, A., Minter, L. M., Telfer, J. C., Miele, L., Fauq, A., et al. (2008). Notch1 and TGF 1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood 112, 1813–1821. doi:10.1182/blood-2008-03-144980
Samstein, R. M., Arvey, A., Josefowicz, S. Z., Peng, X., Reynolds, A., Sandstrom, R., et al. (2012). Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166. doi:10.1016/j.cell.2012.06.053
Sarris, M., Andersen, K. G., Randow, F., Mayr, L., and Betz, A. G. (2008). Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity 28, 402–413. doi:10.1016/j.immuni.2008.01.012
Sauer, S., Bruno, L., Hertweck, A., Finlay, D., Leleu, M., Spivakov, M., et al. (2008). T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. U.S.A. 105, 7797–7802. doi:10.1073/pnas.0800928105
Saurwein-Teissl, M., Lung, T. L., Marx, F., Gschösser, C., Asch, E., Blasko, I., et al. (2002). Lack of antibody production following immunization in old age: association with CD8(+)CD28(−) T cell clonal expansions and an imbalance in the production of Th1 and Th2 cytokines. J. Immunol. 168, 5893–5899.
Schmidt-Supprian, M., Courtois, G., Tian, J., Coyle, A. J., Israël, A., Rajewsky, K., et al. (2003). Mature T cells depend on signaling through the IKK complex. Immunity 19, 377–389. doi:10.1016/S1074-7613(03)00237-1
Schmidt-Supprian, M., Tian, J., Grant, E. P., Pasparakis, M., Maehr, R., Ovaa, H., et al. (2004). Differential dependence of CD4+CD25+ regulatory and natural killer-like T cells on signals leading to NF-kappaB activation. Proc. Natl. Acad. Sci. U.S.A. 101, 4566–4571. doi:10.1073/pnas.0400885101
Schubert, L. A., Jeffery, E., Zhang, Y., Ramsdell, F., and Ziegler, S. F. (2001). Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. J. Biol. Chem. 276, 37672–37679. doi:10.1074/jbc.M104521200
Schuster, M., Glauben, R., Plaza-Sirvent, C., Schreiber, L., Annemann, M., Floess, S., et al. (2012). IκB(NS) protein mediates regulatory T cell development via induction of the Foxp3 transcription factor. Immunity 37, 998–1008. doi:10.1016/j.immuni.2012.08.023
Scotta, C., Esposito, M., Fazekasova, H., Fanelli, G., Edozie, F. C., Ali, N., et al. (2012). Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4+CD25+FOXP3+ Treg subpopulations. Haematologica doi:10.3324/haematol.2012.074088. [Epub ahead of print].
Scotto, L., Naiyer, A. J., Galluzzo, S., Rossi, P., Manavalan, J. S., Kim-Schulze, S., et al. (2004). Overlap between molecular markers expressed by naturally occurring CD4+CD25+ regulatory T cells and antigen specific CD4+CD25+ and CD8+CD28− T suppressor cells. Hum. Immunol. 65, 1297–1306. doi:10.1016/j.humimm.2004.09.004
Seder, R. A., Marth, T., Sieve, M. C., Strober, W., Letterio, J. J., Roberts, A. B., et al. (1998). Factors involved in the differentiation of TGF-beta-producing cells from naive CD4+ T cells: IL-4 and IFN-gamma have opposing effects, while TGF-beta positively regulates its own production. J. Immunol. 160, 5719–5728.
Selvaraj, R. K., and Geiger, T. L. (2007). A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J. Immunol. 178, 7667–7677.
Shevach, E. M. (2000). Regulatory T cells in autoimmmunity. Annu. Rev. Immunol. 18, 423–449. doi:10.1146/annurev.immunol.18.1.423
Shimizu, J., Yamazaki, S., and Sakaguchi, S. (1999). Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J. Immunol. 163, 5211–5218.
Shimizu, J., Yamazaki, S., Takahashi, T., Ishida, Y., and Sakaguchi, S. (2002). Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 3, 135–142. doi:10.1038/ni759
Shimo, Y., Yanai, H., Ohshima, D., Qin, J., Motegi, H., Maruyama, Y., et al. (2011). TRAF6 directs commitment to regulatory T cells in thymocytes. Genes Cells 16, 437–447. doi:10.1111/j.1365-2443.2011.01500.x
Siewert, C., Lauer, U., Cording, S., Bopp, T., Schmitt, E., Hamann, A., et al. (2008). Experience-driven development: effector/memory-like alphaE+Foxp3+ regulatory T cells originate from both naive T cells and naturally occurring naive-like regulatory T cells. J. Immunol. 180, 146–155.
Singer, A., Adoro, S., and Park, J.-H. (2008). Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat. Rev. Immunol. 8, 788–801. doi:10.1038/nri2416
Strainic, M. G., Shevach, E. M., An, F., Lin, F., and Medof, M. E. (2012). Absence of signaling into CD4+ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3+ regulatory T cells. Nat. Immunol. 14, 162–171. doi:10.1038/ni.2499
Strauch, U. G., Obermeier, F., Grunwald, N., Gürster, S., Dunger, N., Schultz, M., et al. (2005). Influence of intestinal bacteria on induction of regulatory T cells: lessons from a transfer model of colitis. Gut 54, 1546–1552. doi:10.1136/gut.2004.059451
Sun, C.-M., Hall, J. A., Blank, R. B., Bouladoux, N., Oukka, M., Mora, J. R., et al. (2007). Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204, 1775–1785. doi:10.1084/jem.20070602
Suri-Payer, E., Amar, A. Z., Thornton, A. M., and Shevach, E. M. (1998). CD4+CD25+ T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J. Immunol. 160, 1212–1218.
Suzuki, H., Kündig, T. M., Furlonger, C., Wakeham, A., Timms, E., Matsuyama, T., et al. (1995). Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science 268, 1472–1476. doi:10.1126/science.7770771
Szabo, S. J., Kim, S. T., Costa, G. L., Zhang, X., Fathman, C. G., and Glimcher, L. H. (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. doi:10.1016/S0092-8674(00)80702-3
Szabo, S. J., Sullivan, B. M., Stemmann, C., Satoskar, A. R., Sleckman, B. P., and Glimcher, L. H. (2002). Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 295, 338–342. doi:10.1126/science.1065543
Taguchi, O., and Nishizuka, Y. (1981). Experimental autoimmune orchitis after neonatal thymectomy in the mouse. Clin. Exp. Immunol. 46, 425–434.
Tai, X., Cowan, M., Feigenbaum, L., and Singer, A. (2005). CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol. 6, 152–162. doi:10.1038/ni1160
Takahashi, H., Kanno, T., Nakayamada, S., Hirahara, K., Sciumè, G., Muljo, S. A., et al. (2012). TGF-β and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat. Immunol. 13, 587–595. doi:10.1038/ni.2286
Takahashi, T., Kuniyasu, Y., Toda, M., Sakaguchi, N., Itoh, M., Iwata, M., et al. (1998). Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10, 1969–1980. doi:10.1093/intimm/10.12.1969
Tang, Q., Bluestone, J. A., and Kang, S.-M. (2012). CD4(+)Foxp3(+) regulatory T cell therapy in transplantation. J. Mol. Cell Biol. 4, 11–21. doi:10.1093/jmcb/mjr047
Tang, Y., Luo, X., Cui, H., Ni, X., Yuan, M., Guo, Y., et al. (2009). MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 60, 1065–1075. doi:10.1002/art.24436
Thornton, A. M., Korty, P. E., Tran, D. Q., Wohlfert, E. A., Murray, P. E., Belkaid, Y., et al. (2010). Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 184, 3433–3441. doi:10.4049/jimmunol.0904028
Thornton, A. M., and Shevach, E. M. (1998). CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188, 287–296. doi:10.1084/jem.188.2.287
Tone, Y., Furuuchi, K., Kojima, Y., Tykocinski, M. L., Greene, M. I., and Tone, M. (2008). Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 9, 194–202. doi:10.1038/ni1549
Tresoldi, E., Dell’Albani, I., Stabilini, A., Jofra, T., Valle, A., Gagliani, N., et al. (2011). Stability of human rapamycin-expanded CD4+CD25+ T regulatory cells. Haematologica 96, 1357–1365. doi:10.3324/haematol
Trzonkowski, P., Bieniaszewska, M., Juscinska, J., Dobyszuk, A., Krzystyniak, A., Marek, N., et al. (2009). First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127−T regulatory cells. Clin. Immunol. 133, 22–26. doi:10.1016/j.clim
Veldhoen, M., Hocking, R. J., Atkins, C. J., Locksley, R. M., and Stockinger, B. (2006). TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189. doi:10.1016/j.immuni.2006.01.001
Venuprasad, K., Huang, H., Harada, Y., Elly, C., Subramaniam, M., Spelsberg, T., et al. (2008). The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat. Immunol. 9, 245–253. doi:10.1038/ni1564
Vieira, P. L., Christensen, J. R., Minaee, S., O’Neill, E. J., Barrat, F. J., Boonstra, A., et al. (2004). IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J. Immunol. 172, 5986–5993.
Waldmann, H., Adams, E., Fairchild, P., and Cobbold, S. (2006). Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol. Rev. 212, 301–313. doi:10.1111/j.0105-2896.2006.00406.x
Walker, M. R., Kasprowicz, D. J., Gersuk, V. H., Benard, A., van Landeghen, M., Buckner, J. H., et al. (2003). Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25− T cells. J. Clin. Invest. 112, 1437–1443. doi:10.1172/JCI200319441
Wan, Y. Y., and Flavell, R. A. (2005). Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc. Natl. Acad. Sci. U.S.A. 102, 5126–5131. doi:10.1073/pnas.0501701102
Wang, J., Ioan-Facsinay, A., van der Voort, E. I. H., Huizinga, T. W. J., and Toes, R. E. M. (2007). Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur. J. Immunol. 37, 129–138. doi:10.1002/eji.200636435
Wei, G., Wei, L., Zhu, J., Zang, C., Hu-Li, J., Yao, Z., et al. (2009). Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167. doi:10.1016/j.immuni.2008.12.009
Wei, J., Duramad, O., Perng, O. A., Reiner, S. L., Liu, Y.-J., and Qin, F. X.-F. (2007). Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 104, 18169–18174. doi:10.1073/pnas.0703642104
Weiner, H. L. (2001). Induction and mechanism of action of transforming growth factorβ secreting Th3 regulatory cells. Immunol. Rev. 182, 207–214. doi:10.1034/j.1600-065X.2001.1820117.x
Weiss, J. M., Bilate, A. M., Gobert, M., Ding, Y., Curotto de Lafaille, M. A., Parkhurst, C. N., et al. (2012). Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J. Exp. Med. 209, 1723–1742. doi:10.1084/jem.20120914
Weng, N.-P., Akbar, A. N., and Goronzy, J. (2009). CD28- T cells: their role in the age-associated decline of immune function. Trends Immunol. 30, 306–312. doi:10.1016/j.it.2009.03.013
Wildin, R. S., Ramsdell, F., Peake, J., Faravelli, F., Casanova, J. L., Buist, N., et al. (2001). X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 27, 18–20. doi:10.1038/83707
Willerford, D. M., Chen, J., Ferry, J. A., Davidson, L., Ma, A., and Alt, F. W. (1995). Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3, 521–530. doi:10.1016/1074-7613(95)90180-9
Willoughby, J. E., Costello, P. S., Nicolas, R. H., Robinson, N. J., Stamp, G., Powrie, F., et al. (2007). Raf signaling but not the ERK effector SAP-1 is required for regulatory T cell development. J. Immunol. 179, 6836–6844.
Wilson, C. B., Rowell, E., and Sekimata, M. (2009). Epigenetic control of T-helper-cell differentiation. Nat. Rev. Immunol. 9, 91–105. doi:10.1038/nri2487
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., et al. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275. doi:10.1126/science.1160062
Wohlfert, E., and Belkaid, Y. (2008). Role of endogenous and induced regulatory T cells during infections. J. Clin. Immunol. 28, 707–715. doi:10.1007/s10875-008-9248-6
Wohlfert, E. A., Gorelik, L., Mittler, R., Flavell, R. A., and Clark, R. B. (2006). Cutting edge: deficiency in the E3 ubiquitin ligase Cbl-b results in a multifunctional defect in T cell TGF-beta sensitivity in vitro and in vivo. J. Immunol. 176, 1316–1320.
Wong, J., Mathis, D., and Benoist, C. (2007). TCR-based lineage tracing: no evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J. Exp. Med. 204, 2039–2045. doi:10.1084/jem.20070822
Wood, K. J., and Sakaguchi, S. (2003). Regulatory T cells in transplantation tolerance. Nat. Rev. Immunol. 3, 199–210. doi:10.1038/nri1027
Xiao, S., Jin, H., Korn, T., Liu, S. M., Oukka, M., Lim, B., et al. (2008). Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J. Immunol. 181, 2277–2284.
Xu, L., Kitani, A., Fuss, I., and Strober, W. (2007). Cutting edge: regulatory T cells induce CD4+CD25-Foxp3− T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J. Immunol. 178, 6725–6729.
Xu, L., Kitani, A., Stuelten, C., McGrady, G., Fuss, I., and Strober, W. (2010). Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity 33, 313–325. doi:10.1016/j.immuni.2010.09.001
Yadav, M., Louvet, C., Davini, D., Gardner, J. M., Martínez-Llordella, M., Bailey-Bucktrout, S., et al. (2012). Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med. 209, 1713–1722. doi:10.1084/jem.20120822
Yamamoto, M., Kondo, E., Takeuchi, M., Harashima, A., Otani, T., Tsuji-Takayama, K., et al. (2011). miR-155, a Modulator of FOXO3a Protein Expression, Is Underexpressed and Cannot Be Upregulated by Stimulation of HOZOT, a Line of Multifunctional Treg. PLoS ONE 6:e16841. doi:10.1371/journal.pone.0016841
Yamazaki, S., Dudziak, D., Heidkamp, G. F., Fiorese, C., Bonito, A. J., Inaba, K., et al. (2008). CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J. Immunol. 181, 6923–6933.
Yang, X. O., Nurieva, R., Martinez, G. J., Kang, H. S., Chung, Y., Pappu, B. P., et al. (2008). Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56. doi:10.1016/j.immuni.2008.05.007
Yao, Z., Kanno, Y., Kerenyi, M., Stephens, G., Durant, L., Watford, W. T., et al. (2007). Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood 109, 4368–4375. doi:10.1182/blood-2006-11-055756
Yi, R., Qin, Y., Macara, I. G., and Cullen, B. R. (2003). Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 17, 3011–3016. doi:10.1101/gad.1158803
Yu, A., Zhu, L., Altman, N. H., and Malek, T. R. (2009). A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity 30, 204–217. doi:10.1016/j.immuni.2008.11.014
Zelenay, S., Bergman, M.-L., Paiva, R. S., Lino, A. C., Martins, A. C., Duarte, J. H., et al. (2010). Cutting edge: intrathymic differentiation of adaptive Foxp3+ regulatory T cells upon peripheral proinflammatory immunization. J. Immunol. 185, 3829–3833. doi:10.4049/jimmunol.1001281
Zheng, S. G., Wang, J., and Horwitz, D. A. (2008). Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. J. Immunol. 180, 7112–7116.
Zheng, S. G., Wang, J. H., Stohl, W., Kim, K. S., Gray, J. D., and Horwitz, D. A. (2006). TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J. Immunol. 176, 3321–3329.
Zheng, W., and Flavell, R. A. (1997). The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89, 587–596. doi:10.1016/S0092-8674(00)80240-8
Zheng, Y., Chaudhry, A., Kas, A., DeRoos, P., Kim, J. M., Chu, T.-T., et al. (2009). Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356. doi:10.1038/nature07674
Zheng, Y., Josefowicz, S., Chaudhry, A., Peng, X. P., Forbush, K., and Rudensky, A. Y. (2010). Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812. doi:10.1038/nature08750
Zheng, Y., Josefowicz, S. Z., Kas, A., Chu, T.-T., Gavin, M. A., and Rudensky, A. Y. (2007). Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445, 936–940. doi:10.1038/nature05563
Zhou, L., Chong, M. M. W., and Littman, D. R. (2009a). Plasticity of CD4+ T cell lineage differentiation. Immunity 30, 646–655. doi:10.1016/j.immuni.2009.05.001
Zhou, X., Bailey-Bucktrout, S. L., Jeker, L. T., Penaranda, C., Martínez-Llordella, M., Ashby, M., et al. (2009b). Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10, 1000–1007. doi:10.1038/ni.1774
Zhou, L., Lopes, J. E., Chong, M. M. W., Ivanov, I. I., Min, R., Victora, G. D., et al. (2008a). TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453, 236–240. doi:10.1038/nature06878
Zhou, X., Jeker, L. T., Fife, B. T., Zhu, S., Anderson, M. S., McManus, M. T., et al. (2008b). Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J. Exp. Med. 205, 1983–1991. doi:10.1084/jem.20080707
Zhu, J., Yamane, H., and Paul, W. E. (2010). Differentiation of effector CD4 T cell populations(*). Annu. Rev. Immunol. 28, 445–489. doi:10.1146/annurev-immunol-030409-101212
Ziegler, S. F. (2006). FOXP3: of mice and men. Annu. Rev. Immunol. 24, 209–226. doi:10.1146/annurev.immunol.24.021605.090547
Zorn, E., Nelson, E. A., Mohseni, M., Porcheray, F., Kim, H., Litsa, D., et al. (2006). IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 108, 1571–1579. doi:10.1182/blood-2006-02-004747
Keywords: regulatory T cells, Foxp3, Tr1, Th3, iTr35, interleukin-17, epigenetics, microRNA
Citation: Povoleri GAM, Scottà C, Nova-Lamperti EA, John S, Lombardi G and Afzali B (2013) Thymic versus induced regulatory T cells – who regulates the regulators? Front. Immunol. 4:169. doi: 10.3389/fimmu.2013.00169
Received: 17 May 2013; Paper pending published: 11 June 2013;
Accepted: 13 June 2013; Published online: 27 June 2013.
Edited by:
Eyad Elkord, United Arab Emirates University, UAE; University of Salford and University of Manchester, UKReviewed by:
Jocelyne Demengeot, Instituto Gulbenkian de Ciencia, PortugalRichard DiPaolo, Saint Louis University School of Medicine, USA
Theresa L. Whiteside, University of Pittsburgh Cancer Institute, USA
Copyright: © 2013 Povoleri, Scottà, Nova-Lamperti, John, Lombardi and Afzali. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Behdad Afzali, Immunoregulation Laboratory, MRC Centre for Transplantation, 5th Floor Bermondsey Wing, Guy’s Hospital, London SE1 9RT, UK e-mail:YmVoZGFkLmFmemFsaUBrY2wuYWMudWs=