 Elke S. Bergmann-Leitner1 Heather Hosie1 Jessica Trichilo2 Elizabeth DeRiso1 Ryan T. Ranallo3† Timothy Alefantis2† Tatyana Savranskaya4 Paul Grewal2 Christian F. Ockenhouse1 Malabi M. Venkatesan3 Vito G. DelVecchio2
Elke S. Bergmann-Leitner1 Heather Hosie1 Jessica Trichilo2 Elizabeth DeRiso1 Ryan T. Ranallo3† Timothy Alefantis2† Tatyana Savranskaya4 Paul Grewal2 Christian F. Ockenhouse1 Malabi M. Venkatesan3 Vito G. DelVecchio2 Evelina Angov1*
Evelina Angov1*
- 1Malaria Vaccine Branch, Walter Reed Army Institute of Research, Silver Spring, MD, USA
- 2Vital Probes, Inc., Mayfield, PA, USA
- 3Division of Bacterial and Rickettsial Diseases, WRAIR, Silver Spring, MD, USA
- 4Division of Entomology, WRAIR, Silver Spring, MD, USA
Genetically inactivated, Gram-negative bacteria that express malaria vaccine candidates represent a promising novel self-adjuvanting vaccine approach. Antigens expressed on particulate bacterial carriers not only target directly to antigen-presenting cells but also provide a strong danger signal thus circumventing the requirement for potent extraneous adjuvants. E. coli expressing malarial antigens resulted in the induction of either Th1 or Th2 biased responses that were dependent on both antigen and sub-cellular localization. Some of these constructs induced higher quality humoral responses compared to recombinant protein and most importantly they were able to induce sterile protection against sporozoite challenge in a murine model of malaria. In light of these encouraging results, two major Plasmodium falciparum pre-erythrocytic malaria vaccine targets, the Cell-Traversal protein for Ookinetes and Sporozoites (CelTOS) fused to the Maltose-binding protein in the periplasmic space and the Circumsporozoite Protein (CSP) fused to the Outer membrane (OM) protein A in the OM were expressed in a clinically relevant, attenuated Shigella strain (Shigella flexneri 2a). This type of live-attenuated vector has previously undergone clinical investigations as a vaccine against shigellosis. Using this novel delivery platform for malaria, we find that vaccination with the whole-organism represents an effective vaccination alternative that induces protective efficacy against sporozoite challenge. Shigella GeMI-Vax expressing malaria targets warrant further evaluation to determine their full potential as a dual disease, multivalent, self-adjuvanting vaccine system, against both shigellosis, and malaria.
Introduction
While traditional whole-cell, killed, or live-attenuated microorganism-based vaccines have been the most effective for disease prevention, a large number of subunit vaccines are currently in development and are being evaluated for their ability to elicit protective immune responses. These approaches include DNA vaccines, recombinant protein (with adjuvant or as conjugates), viral vectors, and bacteria expressing full-length or immunologically crucial antigens (1–11). Vaccine development for the three major human diseases, namely HIV, tuberculosis, and malaria, has been primarily impeded by host immune evasion mechanisms employed by pathogens, the poor immunogenicity of protective antigens and/or the inability to generate effective whole-organism vaccines. Poor immunogenicity can often be overcome by administering the vaccine with potent adjuvants (12, 13). However, the limited repertoire and availability of human-use adjuvants is hampering the clinical vaccine development pipeline (14). Using recombinant viruses or bacteria as vectors could overcome some of these limitations. Vaccination with live-attenuated organisms mimic the immune stimulation of natural infections and offer induction of a more natural and robust immune response. Although some concerns over their safety have prevented their universal acceptance and licensure; recent advances toward increased safety through detoxification and unique delivery routes may quell some of these concerns. Using bacteria as recombinant vectors to mount an immune response against xenogeneic transgenes has several advantages: (1) pathogen-associated molecular patterns (PAMPs) on the bacteria are recognized by specialized pattern recognition receptors (PRRs) of the host and lead to the activation of strong innate immune responses (15). This is the crucial first step in initiating an antigen-specific adaptive immune response. Depending on the type of PAMPs, distinctive types of adaptive immune responses are induced. (2) Complement factors MBL, C1q, and C3b also recognize PAMPs which results in proteolytic activation of the complement cascade. Binding with complement mediates the targeting of the bacteria to the complement receptors (CR) on antigen-presenting cells (APC) (16) therefore improving the antigen presentation of xenogeneic antigens. (3) In individuals with pre-existing immunity to the bacterium, cross-linking between antigen receptor and CR results in strong activation of the respective B cells (17, 18) thus improving humoral immune responses. (4) Opsonized pathogens targeted to CR on follicular dendritic cells ensure long-lasting immunity (19 –21). (5) Finally, using bacteria of clinical relevance as vectors for delivery of xenogeneic pathogen-derived antigens holds the potential for their application as dual-use vaccines. Currently, several virulence-attenuated bacteria are used as vaccines and are either commercially available or in clinical development, i.e., Salmonella enterica serovar Typhi Ty21a (22, 23), Vibrio cholera CVD 103-HgR (24, 25), Mycobacterium bovis BCG (26 –28), Shigella dysenteriae Type 1, Shigella flexneri 2a, and Shigella sonnei (29 –33).
These advantages warrant further evaluation of recombinant bacteria as vectors for delivering heterologous target antigens either by co-expression, adsorption, or encapsulation (28, 34 –37). Traditionally, microorganisms have been inactivated or killed using methods with strong denaturing conditions involving heat or chemical treatments such as formaldehyde or formalin. This process is meant to ensure the safety of the formulation but the harsh treatment can negatively affect the structure of the pathogen’s proteins and thus antigenicity of key protective antigens (38, 39). Molecular methods to sustain surface antigen functionality and integrity that circumvent these denaturing conditions include the controlled expression of PhiX174 gene E leading to the concept of Bacterial Ghosts (BGs) as a vaccine delivery platform (40, 41). A new approach to inactive bacteria not previously described uses genetic means to express inhibitors of key metabolic processes that disrupt cellular functions without significantly altering bacterial cell structure integrity. In the current study, we utilize this Gene-Mediated-Inactivation Vaccine (GeMI-Vax) process to generate inactivated Gram-negative bacteria carrying heterologous protein antigens. In GeMI-Vax production, a Gram-negative pathogen is transformed with plasmids containing a gene for an antigen of interest and the GeMI-Vax inactivation gene, ColE3, which encodes a colicin that degrades mRNA. GeMI-Vax bacteria serve as the antigen delivery system in the context of whole bacterial cells that are rendered non-replicating and non-viable through this type of genetic manipulation. Moreover, since these bacteria are not chemically modified, conformational epitopes on the recombinant antigens, and the bacterial derived PAMPs (such as lipopolysaccharide, lipoproteins, flagellin, and DNA) are unchanged allowing for the induction of potent immune responses. The advantage of using GeMI-Vax bacteria as delivery platform compared to traditional adjuvants is that a wide range of PAMPs can trigger distinct PRRs, both surface bound (e.g., TLR-4) and intracellular (e.g., TLR-9) thus resulting in the engagement of multiple signaling pathways.
Malaria caused by Plasmodium falciparum results in serious illness and often leads to death if left untreated. The development of an efficacious vaccine to prevent this global disease is of utmost importance. There is an urgent need to develop a highly efficacious, low cost, self-adjuvanting, pre-erythrocytic stage malaria vaccine from P. falciparum target antigens (sporozoite and liver stages) to protect populations in malaria endemic regions. In initial studies, Escherichia coli GeMI-Vax were co-transformed with plasmids expressing the malaria target antigen and the bacterial host inactivation gene product. The malaria targets used in the E. coli experiments was the rodent malaria Plasmodium berghei Circumsporozoite Protein (PbCSP) and the Cell-traversal protein of ookinetes and sporozoites (PbCelTOS). Three sub-cellular localizations of the target were evaluated; expression in the cytosol (Cyto), the periplasmic space (PPS), and the outer membrane (OM) of the bacterial carrier. Localization to the PPS and OM was accomplished through genetic fusions between the Maltose-Binding Protein (MBP) and the Outer Membrane Protein A (OmpA), forming chimeric proteins with the malaria targets. Culture conditions were optimized to maximize antigen expression, effective bacterial inactivation, and the isolation of whole, intact GeMI-Vax cells. Since no harsh physical or chemical agents are employed, proteins, and other immune-stimulating components of GeMI-Vax cells are intact and recognized by the immune system in the same manner as their live pathogenic counterpart. Thus they elicit a robust immune response without the need for additional adjuvants. GeMI-Vax cells were evaluated for their ability to induce protection against malaria challenge in murine challenge models, and both the humoral and cellular immune responses were evaluated.
Evidence of immunological potency and protection from studies performed using E. coli GeMI-Vax supported translation to the clinically more relevant whole-cell Shigella flexneri as the delivery platform. The GeMI-Vax platform was used to inactivate engineered Shigella flexneri 2a (15G strain) expressing P. falciparum CelTOS (PfCelTOS) in the PPS or P. falciparum CSP (PfCSP) on the OM. Similar to E. coli GeMI-Vax, the Shigella GeMI-Vax cells have none of the disadvantages of chemical killing or regulatory hurdles associated with live vaccines. The live-attenuated S. flexneri used in this study were previously shown to be safe and protective in an animal model when administered intranasally (42). Both applications of GeMI-Vax, E. coli, and S. flexneri 2a (15G strain), expressing malaria targets on the surface of bacteria elicited antigen-specific antibodies and IFN-γ producing T cells when expressed on the OMs. Malaria antigen localized to intracellular spaces, i.e., periplasmic and cytosol, skewed toward cellular responses with no significant levels of antibodies detected. Thus the protective efficacy of the different constructs supports GeMI-Vax as a vaccine vector system for delivery of target antigens. Additionally, expressing target antigens at different sites on bacteria influences the type of immune responses induced and allows for investigations into antigen-specific immune correlates of protection.
Materials and Methods
PbCSP in the Cytosol for E. coli Expression
The Plasmodium berghei (Pb) CSP gene coding sequence was obtained from the plasmid pET(K-) PbCSP-tr. The gene insert was synthesized by Retrogen Inc. (San Diego, CA, USA) using a WRAIR proprietary codon harmonized nucleotide sequence for optimal expression in E. coli (43). The PbCSP nucleotide sequence was truncated in the total number of repeat motifs for cloning purposes. The gene product was 572 bp long and contained a His tag at the 5′end. The final expressed product has a molecular weight of 21 kDa, but runs anomalously on gradient Tris-Glycine SDS-PAGE (Invitrogen, Carlsbad, CA, USA) due to the complex nature of its structure. The PbCSP-tr DNA from pET(K-)PbCSP-tr was used for constructing the OM-PbCSP and PPS-PbCSP GeMI-Vax plasmids as well as for expressing PbCSP in the cytosol of E. coli (Cyto-PbCSP).
PbCSP in the Outer Membrane for E. coli Expression
The PbCSP gene from pET(K-)PbCSP-tr was PCR amplified and cloned into the pVPIHS64K OM vector yielding an E. coli OmpA-PbCSP fusion protein. After the sequence was confirmed, the plasmid was used to transform NM522 E. coli cells. Positive clones were identified by colony PCR using vector specific primers. Expression was confirmed by induction of bacterial cultures with 1 mM isopropyl-thio-β-galactoside (IPTG) (Roche, Indianapolis, IN, USA), and analyzed by whole-cell extraction and SDS-PAGE Western blotting (Invitrogen) using an anti-His6 antibody (Sigma-Aldrich, St. Louis, MO, USA). Expression was detected at the expected molecular weight of 60 kDa for the fusion protein. The PbCelTOS codon harmonized nucleotide sequence was similarly constructed to be expressed in the OM and the PPS of E. coli (44). The details for growth, expression, and isolation are identical to those used for the PbCSP GeMI-Vax constructs.
PbCSP in the Periplasmic Space for E. coli Expression
The PbCSP gene coding sequence was obtained from plasmid pET(K-)PbCSP-tr. The gene was PCR amplified and cloned into the p2XK MBP vector, yielding an E. coli MBP-PbCSP fusion protein. After the sequence was confirmed, the plasmid was used to transform NM522 cells. Positive clones were identified by colony PCR using vector specific primers. Expression was confirmed by induction of bacterial cultures with 1 mM IPTG; and analyzed by whole-cell extraction and SDS-PAGE Western blotting using an anti-His6 antibody. Expression was detected at the expected molecular weight of 64 kDa for the fusion protein.
Preparation of E. coli GeMI-Vax Cells
E. coli NM522 cells were grown to an OD600 of 0.3 at 30°C in Hyperbroth (Accurate Chemicals & Scientific Corp., Westbury, NY, USA) with 2% Glucose Nutrient Mixture™ (Accurate Chemicals & Scientific Corp.) media containing 40 μg/mL kanamycin (Sigma-Aldrich) and 20 μg/mL gentamicin (Sigma-Aldrich). Key steps in the GeMI-Vax expression and isolation are outlined in Table 1. Briefly, E. coli cells were induced with 1 mM IPTG to express the malaria target antigen for 1 h. Induction of colE3 gene, which encodes a colicin that degrades mRNA, was used for cell inactivation. The colE3 inactivation gene was cloned behind the araBAD promoter in a pBAD vector (Invitrogen) (modified for gentamicin selection) using AflII and NcoI restriction sites and expression was induced by the addition of 2.5% L-(+)-arabinose (≥99%, Sigma-Aldrich) for 1.5 h. During cell inactivation, 100 μg/mL gentamicin was added to guarantee plasmid retention. As a final step to ensure 100% cell inactivation, 2 mg/mL solid streptomycin (Sigma-Aldrich) was added to cultures for 2 h. Cells were collected by centrifugation at 5,000 rpm for 15 min, pellets were resuspended in 50 mL sterile water, and the suspension was stored with 800 μg/mL of streptomycin overnight at 4°C. To determine final cell suspension sterility, 100 μL of cells were plated onto antibiotic-free Luria Broth agar plates and also inoculated into 10 mL liquid culture media with no antibiotics. LB plates and culture tubes were incubated at 37°C overnight or for 37°C for 5 days, respectively.

Table 1. Summary steps for production of GeMI-Vax cells.
Washing and Lyophilization
For final storage, the cell pellets were washed extensively by sequential centrifugation and resuspension with four exchanges of sterile water to remove any residual antibiotics. After the final wash, cells were aliquoted into cryotubes, snap-frozen in liquid nitrogen and kept at −80°C overnight. Frozen cells were lyophilized using Labconco-Fast Freeze Flasks (Labconco, Kansas City, MO, USA) and a Flexi-Dry™ Microprocessor (μP) Control-Corrosion Resistant Freeze Dryer (FTF Systems, Stone Ridge, NY, USA). Lyophilization was completed over a two day period at a temperature of −70°C under vacuum (600 mTorr). The final lyophilized vials were stored at 4°C.
PfCelTOS in the Periplasmic Space for Shigella Expression
A plasmid containing the Plasmodium falciparum (Pf) CelTOS gene served as the template for PCR amplification. Briefly, the parent plasmid included a 522 bp DNA fragment of PfCelTOS 3D7 designed by using the codon-harmonization algorithm (43, 45) and cloned into a modified pET(K−) expression vector (Novagen, Madison, WI, USA). Oligonucleotides were designed for cloning the PfCelTOS sequence into the p2xK periplasmic vector yielding a fusion protein with the MBP. After sequence confirmation, the plasmid was transformed into S. flexneri 2a (15G strain) (ATCC, Manassas, VA, USA). Positive clones were identified by colony PCR using vector specific primers. Expression testing was accomplished by induction of bacterial cultures with IPTG followed by whole-cell extraction and SDS-PAGE Western blot using an anti-MBP antibody (New England BioLabs, Ipswich, MA, USA). Expression was confirmed at the expected molecular weight of 61 kDa for the fusion protein.
PfCSP in the Outer Membrane for Shigella Expression
The Plasmodium falciparum (Pf) CSP gene coding sequence was synthesized by DNA 2.0 (Menlo Park, CA, USA). DNA 2.0 adjusted the coding sequence for optimized expression in E. coli. The gene was 807 bp long and a His tag was added to the 5′end. The highly repetitive central repeat region was retained in its entirety. The gene was PCR amplified and cloned into the pVPIHS64K OM vector, yielding an E. coli OmpA-PfCSP fusion protein. After the sequence was confirmed, the plasmid was used to transform S. flexneri 2a (15G strain). Positive clones were identified by colony PCR using vector specific primers. Expression testing was performed by induction of bacterial cultures with IPTG followed by whole-cell extraction and SDS-PAGE Western blot using an anti-His6 antibody (Sigma). Expression was detected at the expected molecular weight of 68.5 kDa.
Construction of pλColE3 Plasmid
A plasmid synthesized by DNA2.0 (Menlo Park) designated GV001λVPI was used to engineer the GeMI-Vax inactivation gene ColE3 behind the lambda promoter. This plasmid contained the ColE3 gene under control of the araBAD promoter (ColE3 cassette), a malaria gene under control of the λ promoter (a temperature sensitive promoter), the gentamicin resistance marker, and the pUC origin of replication and was used as the template for constructing the pλColE3 plasmid. The ColE3 cassette was removed by digesting the plasmid with PmeI and SfiI. After gel purification the vector backbone was self ligated and transformed into E. coli and plated onto gentamicin plates. DNA plasmid purification was performed. The malaria gene was removed by digesting with NdeI and NotI and the backbone vector was gel purifying. The ColE3 gene was amplified by PCR using primers that incorporated NdeI and NotI recognition sites. The amplicon was digested and then ligated behind the λ promoter, in place of the malaria gene. The newly constructed “GV200” plasmid was transformed into E. coli. Sequence confirmation was performed at Genewiz Inc (South Plainfield, NJ). Expression of the ColE3 gene was also confirmed by expression profile.
Co-Expression of Malaria Target and Inactivation Genes on Same Plasmid for Shigella Expression
A single plasmid was created which contained either of the malaria antigens as well as the GeMI-Vax inactivation gene, ColE3. The malaria antigens were controlled by the IPTG inducible pTac promoter as described above. The ColE3 inactivation gene was controlled by the lambda promoter, also described above. The malaria antigen cassette, consisting of the lac repressor, the pTac promoter, the malaria antigen, and the terminator, was amplified by PCR using primers which contained restriction enzyme tails. Once amplification was confirmed by agarose gel electrophoresis, restriction enzyme digestion was performed. The cassette was then ligated into the GV200 vector, which contained the GeMI-Vax inactivation gene ColE3 under the control of the lambda promoter, which was then linearized with the same restriction enzymes. Each of the malaria target antigens was then subcloned into the GV200 vector described above resulting in the double target GV200-p2xkPfCelTOS and GV200-pHS64PfCSP plasmids, respectively. Positive clones were obtained and the inserted sequences were verified by DNA sequencing. Cell inactivation for these constructs was achieved by a temperature shift from 30 to 40°C resulting in the denaturation of the c1857 repressor and induction of expression of the inactivation gene. Upon sequence confirmation the plasmids were transformed into Shigella and antigen expression and inactivation studies were performed.
Preparation of S. flexneri 15G-GeMI-Vax Cells
S. flexneri 15G is an asd mutant strain derived from the parent 2457T strain that is non-replicating in LB medium in the absence of added diaminopimelic acid (DAP) or within eukaryotic cells that lack an endogenous source of DAP. Bacteria cells that are asd mutants have an obligate requirement for DAP, an essential constituent of the cell wall. APF Luria Broth (Athena ES, Baltimore, MD, USA) supplemented with 2% glucose, 20 μg/mL gentamicin, 100 μg/mL DAP, was inoculated with 0.15 OD595 of stationary phase culture S. flexneri 2a 15G. The cultures were grown at 30°C with shaking at 250 RPM to an OD595 of 0.3 and induced with 1 mM of IPTG. One hour following IPTG induction, an additional 20 μg/mL of gentamicin was added and the temperature was raised to 40°C for an additional 90 min to induce ColE3 induction and cell inactivation. 100 μg/mL of streptomycin was added after the 90 min incubation and the cells continued to incubate for an additional 2.5 h. Cells were collected by centrifugation at 4,000 rpm for 20 min at 4°C. Pellets were resuspended in cold, sterile 1 x PBS (Quality Biological, Gaithersburg, MD, USA). This step was repeated several times. After a fourth centrifugation, the pellet was resuspended in 1 × PBS with 100 μg/mL of streptomycin and incubated with gentle rocking at 4°C overnight. Streptomycin was removed by washing the cell pellet with 1 × PBS with at least four exchanges. Antigen expression was monitored by whole-cell extraction, SDS-PAGE, and Western blot.
Washing and Lyophilization
Shigella GeMI-Vax cell pellets were washed extensively by the process of sequential centrifugation and resuspension with four exchanges of sterile 1 × PBS to remove any residual antibiotics. All subsequent steps are identical to those described above.
Immunizations
Mice used for immunizations were either 6- to 8-week-old BALB/c-J or C57BL6 (Jackson Laboratories, Bar Harbor, ME, USA) female mice (n = 10 for challenges and n = 5 for cellular assays). For all PbCSP and PbCelTOS based immunization, BALB/c were immunized three times in the scruff of the neck at 3 week intervals subcutaneously (sc) with either 200 μL of 1.7 × 109 GeMI-Vax cells or 1 μg rPbCSP-tr/ISA 720 (Seppic, Inc.). For PfCelTOS-based immunizations, BALB/C were immunized three times at 3 week intervals in the scruff of the neck subcutaneously (sc) with either 1.7 × 109 GeMI-Vax cells delivered in two 100 μL aliquots or with 10 μg rPfCelTOS/ISA 720 (Seppic, Inc.). All recombinant proteins used had endotoxin levels below the limits of detection (Pyrochrome LAL Kit, Cape Cod Associates, MA, USA). For PfCSP based immunizations, C57BL6 mice were immunized four times with 1.7 × 109 GeMI-Vax cells delivered in two 100 μL aliquots in the scruff of the neck. Sera were collected one day prior to each immunization and four weeks after the last immunization, prior to challenge with either live wild-type P. berghei sporozoites or live P. berghei P. falciparum CSP transgenic sporozoites (46).
Challenge
Thirty days after the final immunization, mice were challenged with either wild-type Plasmodium berghei sporozoites dissected from infected mosquito salivary glands (4,000 sporozoites for BALB/c) by subcutaneous inoculation (into the inguinal region) (47) or (5,000 Pb-PfCSP-transgenic sporozoites for C57BL6) by intravenous inoculation. Infection was determined by the presence of blood stage parasites in Giemsa-stained thin blood smears on days 6, 8, 10, and 14 after challenge. Mice that were aparasitemic on day 14 were scored as protected (i.e., sterile protection).
ELISA
The ELISA was performed as previously described (45, 48). Briefly, 96-well plates (Immunolon 2 HB, Thermo Milford, MA, USA) were coated by overnight incubation with either 50 μL of rPbCSP-tr at 0.05 μg or 100 μL PfCelTOS and PfCSP at 0.025 and 0.035 μg per well, respectively, in 1 × PBS at 4°C. Sera were serially diluted in blocking buffer and incubated at 37°C for 2 h. Antibody concentration was determined by establishing a standard curve (run with each assay) with purified mouse IgG. For each serum, we determined a concentration that was within the linear portion of the reaction curve and used this dilution to extrapolate the actual antibody concentration in the assay wells. Antibody avidity measurements were done using the chaotropic agent sodium isothiocyanate (NaSCN) as previously described (48). Briefly, antigen-specific interactions were disrupted by the addition of varying amounts of the chaotropic agent NaSCN. Results are reported as the effective concentration of NaSCN required to release 50% of antisera (ED50) and are used to evaluate antibody avidities for the different vaccination groups.
Immunofluoresence Assay
Immunofluoresence assay (IFA) were performed as previously described using salivary gland P. berghei or P. falciparum sporozoites (45).
ELISpot Assays
Cells were stimulated with various concentrations of recombinant protein or 1 μg/mL P. berghei CSP peptides CS57-70 (49), CS58-65 (50), CS260-279 (51), and CS252-260 (52). IFN-γ or IL-4 specific ELISpot assays (R&D Systems, Minneapolis, MN, USA) were performed as previously described (45).
Western Blot Analysis
Protein expression was determined by separating whole-cell extracts of GeMI-Vax cells (OD600 = 0.3) on 4–20% Tris-Glycine SDS-PAGE. Proteins were transferred to 0.2 μm nitrocellulose membranes (Invitrogen) and blocked with 1 × PBS pH 7.4, 0.1% Tween 20. Blots were probed with PbCSP-specific mAb 4B10 (generously provided by Dr. Robert Wirtz, CDC), or PfCSP-specific mAbs (clones WR3 and WR7, WRAIR, Malaria Vaccine Branch) or PfCelTOS-specific mAbs (clones 3D11.E2, 3C3.C2) or Ag-specific polyclonal antibodies. Membranes were washed after 1 h incubation and then incubated with alkaline-phosphatase-conjugated secondary antibodies (Promega, Madison, WI, USA), with either goat-anti-mouse IgG (1:10,000), goat-anti-human IgG (1:10,000), or goat-anti-rabbit IgG (1:20,000). Blots were washed and then developed for 10 min at RT with NBT/BCIP in 0.1 M NaCl, 5 mM MgCl2, and 0.1 M Tris-HCl pH 9.0. For detection of PbCSP, membranes were probed with HRP-conjugated mouse mAb 4B10 (1:20,000) for 2 h. Membranes were washed with 1 × PBS, 0.05% Tween 20 prior to applying the Bio-Rad Immun-Star™ WesternC™ reagent (Bio-Rad, Laboratories, Hercules, CA, USA). Bio-Rad’s VersaDoc Imaging system was used to measure the band intensity after 60 s exposures.
Statistical Analysis
The protective effect of vaccination against P. berghei sporozoite challenge was evaluated using the Fisher’s exact test comparing differences between the GeMI-Vax expressing malaria antigens and empty cells; adjuvant control groups and recombinant protein vaccine groups, respectively. Statistical significance of the serological data and cellular responses was tested using ANOVA and Student T-tests (two-sided), respectively, employing the SigmaPlot v12 (Systat Software, Inc., San Jose, CA, USA). Data not meeting normality testing was subsequently tested using Kruskal–Wallace One-way analysis of variance on ranks.
Results
E. coli Gene-Mediated-Inactivation Vaccine Expressing Malaria Antigens
Gram-negative E. coli were selected as the bacterial vector system to evaluate the GeMI-Vax approach for malaria. These studies were designed to investigate: (1) the relationship between antigen-specific dosage levels, (2) effect of sub-cellular localization of antigen, (3) the type of immune responses induced, and (4) the efficacy of the GeMI-Vax vaccine platform in E. coli. Prior to embarking on immunological studies, bacterial growth, expression, localization, and inactivation conditions were established.
Malaria target sequences used for co-expression were codon harmonized for optimal expression and protein folding in the bacterial host. The approach relies on codon frequency matching between the antigen-“donor” species (in this case Plasmodium) and the “recipient” species (in this case E. coli) yielding regulated co-translational folding (43).
Malaria targets such as Plasmodium CSP are characterized by a central, highly repetitive region, which contains varying numbers of repeat motifs. These repeat motifs have made it technically difficult to synthesize full-length CSP genes. Thus, for this purpose, a reduced number of central repeats were incorporated into the PbCSP sequence. Several plasmid constructs were generated to express PbCSP on the OM, to the PPS or in the cytosol of recombinant bacteria (Figure 1). Fusing pbcsp with the ompa gene resulted in insertion into the OM while fusing the CSP gene with the MBP gene yielded expression to the PPS. Removal of the MBP fusion partner from the latter construct resulted in the expression of CSP to the cytosol (Cyto). Despite the engineering to truncate the central repeat region and expression as fusion proteins, expressed CS proteins were recognized by PbCSP-repeat specific monoclonal antibody 4B10 (Figure 2) and by PbCSP-specific polyclonal antibodies induced in mice with recombinant PbCSP/Montanide ISA 720 (data not shown). These results confirm the cellular expression and immune-recognition of the CS proteins. High levels of heterologous expression were measured in the OM (Figure 2A) and the PPS (Figure 2B), while cytosolic expression was only detectable after IPTG induction and lost after inactivation and lyophilization (Figure 2C). The cytosolic expression likely resulted in intracellular proteolysis in the absence of a fusion partner. Immunostaining of E. coli GeMI-Vax using a PbCSP-specific mAb verified the localization and distribution of fusion proteins (Figure 3). Staining of unfixed OM-PbCSP showed a circumferential staining pattern consistent with its surface expression through fusion with the OM protein (Figure 3A); while surface staining of the unfixed PPS-PbCSP cells was negative (Figure 3C) and required permeabilization of the cells to detect the intracellularly expressed PPS-PbCSP (Figure 3D). PPS-PbCSP staining in the PPS appeared uniform throughout the bacterium. No surface staining was observed of OM-PbCSP GeMI-Vax probed with an irrelevant mAb (Figure 3B).
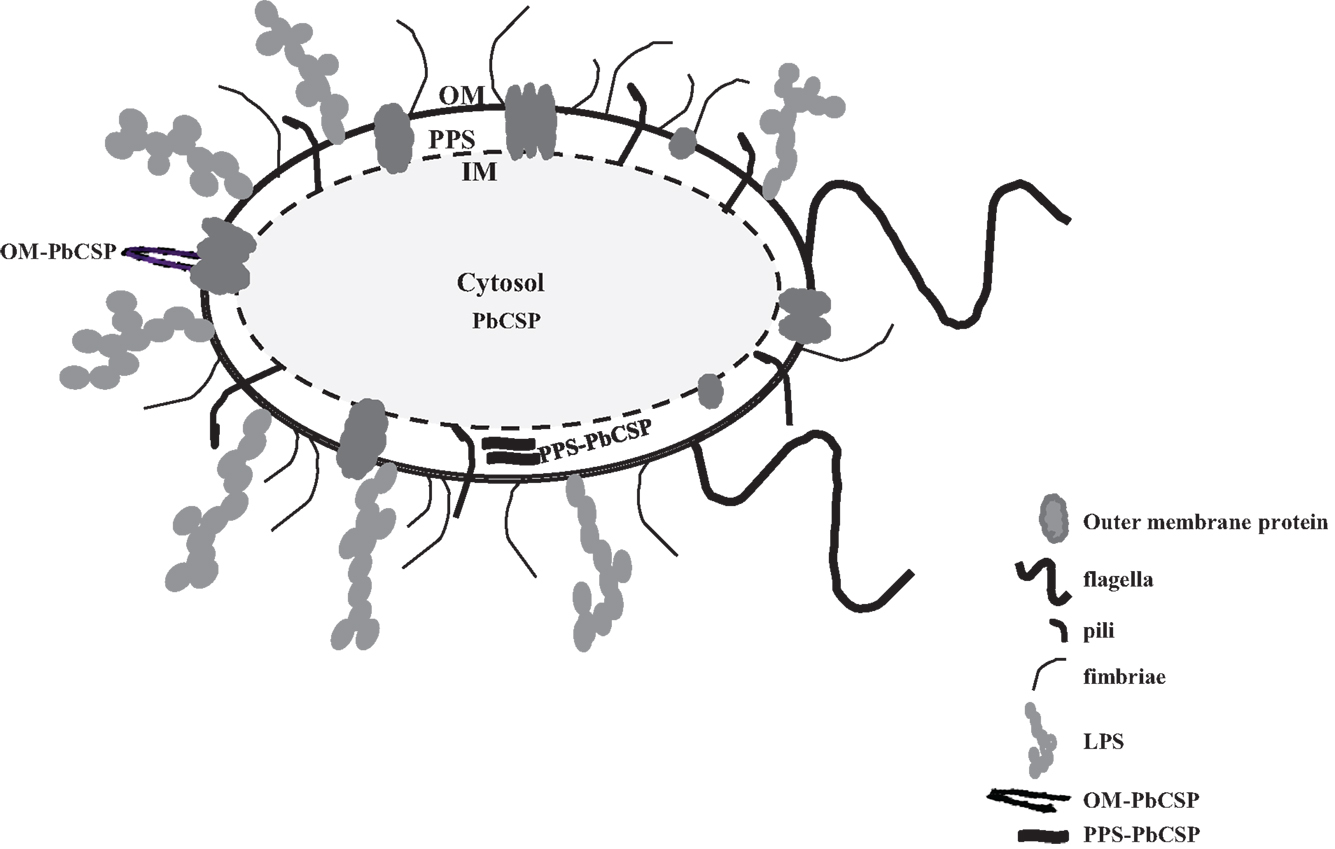
Figure 1. Diagram of Gram-negative bacterium representing the outer membrane, periplasmic space, and the cytosol. PbCSP expression is represented as either a fusion with outer membrane protein OmpA or in the periplasmic space with MBP Inner membrane (IM). Cartoons of various cell surface molecules are included but not drawn to scale, for instance pili, flagella, LPS, and transmembrane proteins.
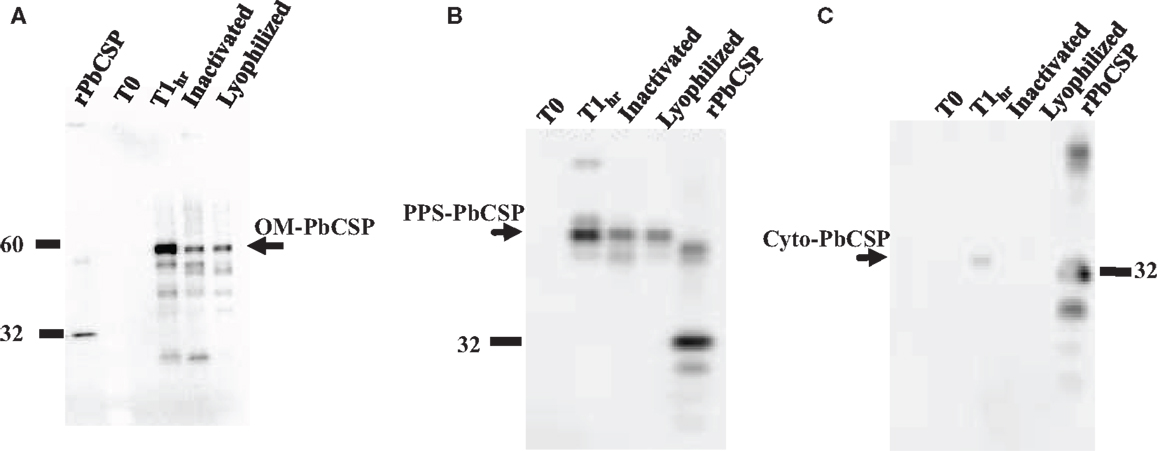
Figure 2. Western blot expression profiles of GeMI-Vax PbCSP from different sub-cellular localizations. (A) Targeting PbCSP to the outer membrane, OM-PbCSP. Lanes: 5 ng rPbCSP; un-induced cells (T0), 1 h post IPTG induction (T1h), post ColE3 inactivation, post lyophilization; (B) Targeting PbCSP to the periplasmic space, PPS-PbCSP. Lanes: un-induced cells (T0), 1 h post IPTG induction (T1h), post ColE3 inactivation, post lyophilization; 1 μg rPbCSP. (C) Expression of PbCSP to the cytoplasmic space. Lanes: un-induced cells (T0), 1 h post IPTG induction (T1h), post ColE3 inactivation, post lyophilization, 1 μg rPbCSP. Arrow points to rPbCSP which migrates at ∼32 kDa. Final lyophilized product loaded at 1.2 × 107 cells per lane.
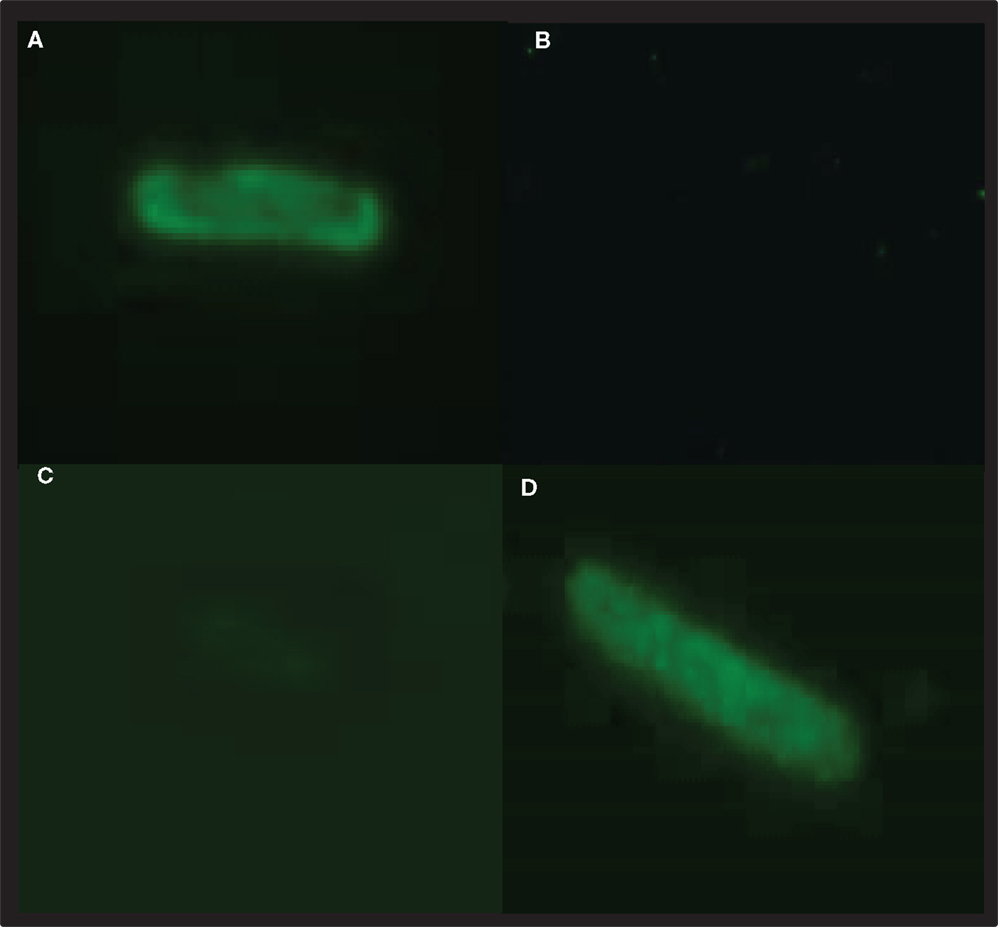
Figure 3. PbCSP-specific antibodies react with recombinant expressing PbCSP GeMI-Vax bacteria. Bacteria expressing PbCSP in the outer membrane (A,B) or periplasmic space (C,D) were stained with the PbCSP-specific mAb 4B10 (A,C,D) or control mAb (2C6.7 MSP119 specific mAb) (B). Bacteria were stained for surface expression (A,C) or after permeabilization for intracellular expression (D). Representative images were taken at 1,000× magnification.
CSP Expression to Different Cellular Compartments on E. coli Dictates Immune Responses
To evaluate the immune potential of bacterial vectors to induce malaria-specific responses, mice were immunized with E. coli GeMI-Vax cells. Based on protein expression estimates made from Western blots, the total antigen doses delivered were 0.5 μg/dose for OM-PbCSP, 0.1 μg/dose for the PPS-PbCSP and below detection limit for the Cyto-PbCSP. The number of cells delivered was based on experiments performed to determine tolerability in BALB/c mice, thus the maximum number of cells delivered for all constructs was 1.7 × 109 cells/dose. OM-PbCSP immunized mice and a reference group (mice immunized with recombinant 1 μg PbCSP/Montanide ISA-720) showed the highest titers of PbCSP-specific antibodies (Figure 4A). However, in all cases, the OM-Pbf CSP induced CSP-specific antibody responses that were significantly greater than for any other GeMI-Vax group (Kruskal–Wallace One-way analysis of variance on ranks, p < 0.001). PPS- and Cyto-PbCSP immunizations induced responses that could not be boosted and were not distinguishable from background, “empty” GeMI-Vax cells. Antibodies to E. coli were measured against an E. coli lysate by ELISA and did not vary by GeMI-Vax groups (Figure 4B). For GeMI-Vax groups, only the third immunization with OM-PbCSP boosted PbCSP-specific responses, however they were not statistically different compared with responses induce by the recombinant protein PbCSP/ISA 720 on Day 56, just prior to sporozoite challenge (ANOVA, p = 0.06). All GeMI-Vax groups had similar levels of anti-E. coli vector background responses by time of challenge. To characterize whether the PbCSP-specific antibodies recognize native antigen, IFAs with P. berghei sporozoites were performed (Figure 5). Only sera from mice immunized with either OM-PbCSP or the recombinant PbCSP had IFA-reactive antibodies. The mean endpoint titer of the OM-PbCSP was 1:64,000, while the mean endpoint titer of the recombinant PbCSP was 1:125,000. Sera from other GeMI-Vax groups failed to react with sporozoites above the background level of the empty GeMI-Vax group. Lack of reactivity to sporozoites could be due to: (a) GeMI-Vax expressing the PbCSP-induced epitope specificities not present or accessible in the native CSP on sporozoites or (b) the concentration or avidity of the antibodies was too low to detect binding or (c) the inability of the intracellular PbCSP delivery to significantly mount humoral responses at the doses of antigen delivered. Avidity measurements were performed using the chaotropic agent NaSCN in ELISAs to compare the OM-PbCSP and soluble protein PbCSP/ISA 720 responses. OM-PbCSP-induced antibody responses with enhanced avidity compared to the conventional antigen/adjuvant approach (ED50 NaSCN (mol/L) for OM-PbCSP and PbCSP/ISA 720, 2.0 versus 1.0, respectively).
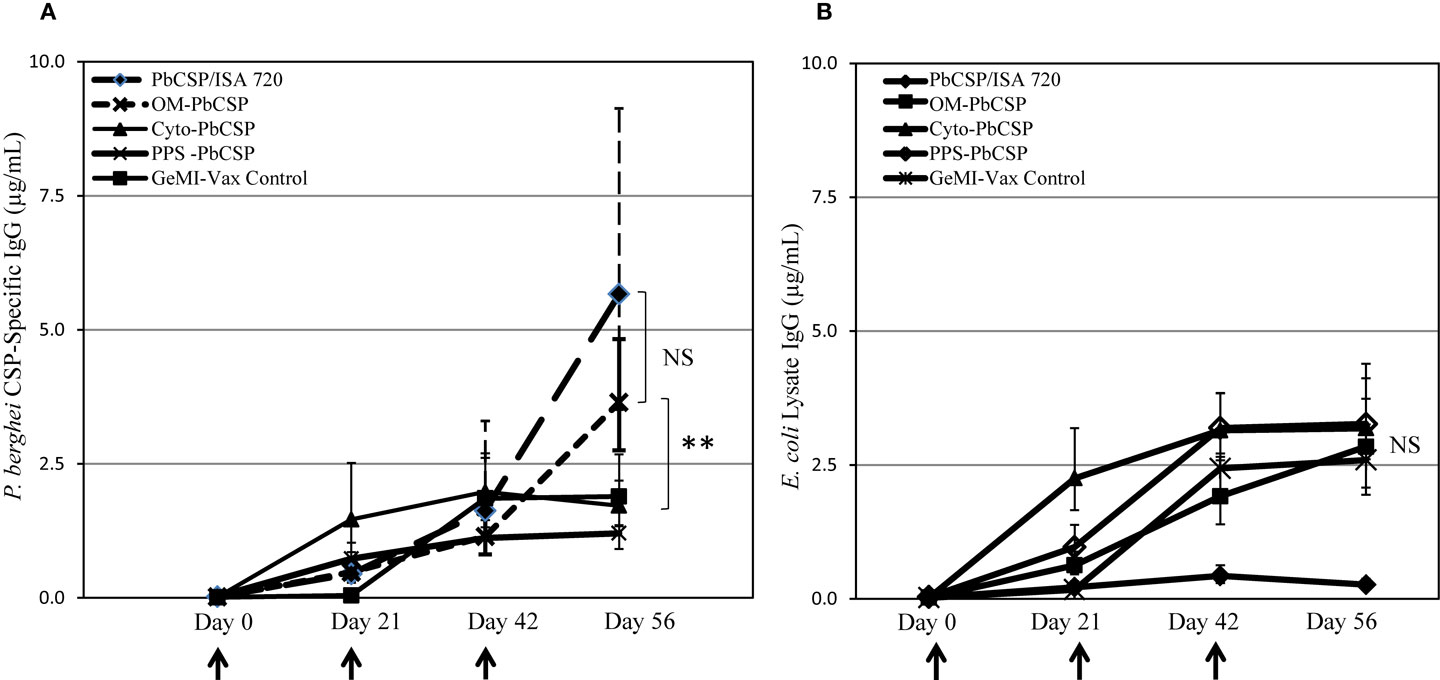
Figure 4. OM-PbCSP GeMI-Vax bacteria induce strong humoral immune responses. Sera from mice immunized with either OM-PbCSP, PPS-PbCSP, Cyto-PbCSP, or recombinant PbCSP adjuvanted with Montanide ISA-720 were collected two days prior to each immunization and tested for reactivity against rPbCSP (A) or on E. coli lysates (B). Data are expressed as geometric mean in μg/mL of antigen-specific IgG (n = 15 mice/group), and the error bars indicate the 95% confidence interval. NS denotes not statistically significant. Asterisk indicates statistical significance, **p < 0.001; Kruskal–Wallace One-Way Analysis of Variance on Ranks.
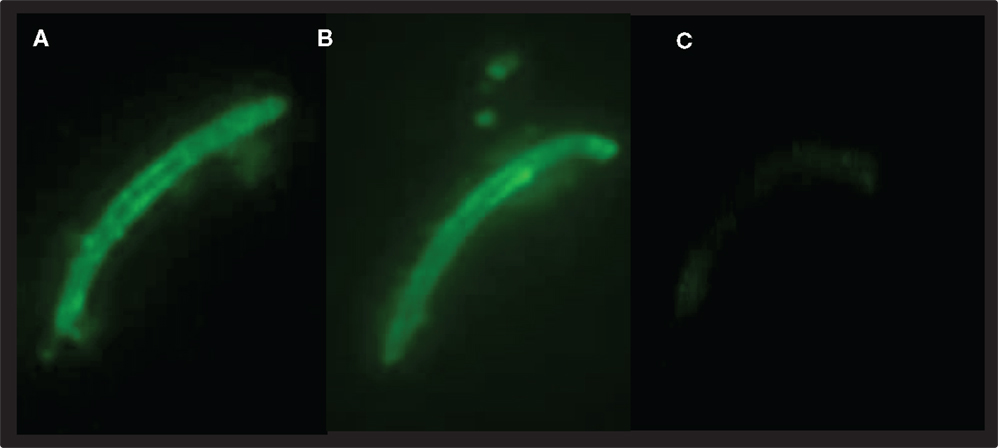
Figure 5. OM-PbCSP-GeMI-Vax (A) and PbCSP/ISA-720 (B) induce antibodies that react with P. berghei sporozoites by indirect immunofluorescence. The reactivity was specific as sera from mice immunized with empty GeMI-Vax did not react with sporozoites (C). Representative images were taken at 1,000 × magnification, staining at 1:10,000 dilutions.
To determine the effect of antigen localization on the induction of cellular responses, spleens from immunized mice were stimulated ex vivo with recombinant PbCSP as well as PbCSP-derived peptides representing immunodominant CD4 or CD8 epitopes. T cell responses were measured by the number of IFN-γ and IL-4 producing cells responding to antigen stimulation in ELISpot assays (Figures 6A,B). The responses to the recombinant protein and peptides for the rPbCSP/ISA 720 group were significantly higher than for any GeMI-Vax group (p < 0.03, p < 0.001, respectively, Student’s T-test). Interestingly, the magnitude of the T cell response did not parallel the magnitude of the antibody response since both the PPS-PbCSP and Cyto-PbCSP groups had similar numbers of IFN-γ spot-forming cells (SFC) as the OM-PbCSP group. The IFN-γ responses measured in the GeMI-Vax groups were significantly higher than those observed in the empty GeMI-Vax (control group) demonstrating antigen-specificity (p < 0.001, ANOVA). In contrast to the antibody responses, the differences in antigen localization and doses delivered by the different bacterial vectors did not appear to affect the overall magnitude of the cellular responses. Neither the GeMI-Vax groups nor the PbCSP/ISA 720 group mounted a significant Th2 type cellular response, as indicated by the overall lack of IL-4 production (Figure 6B).
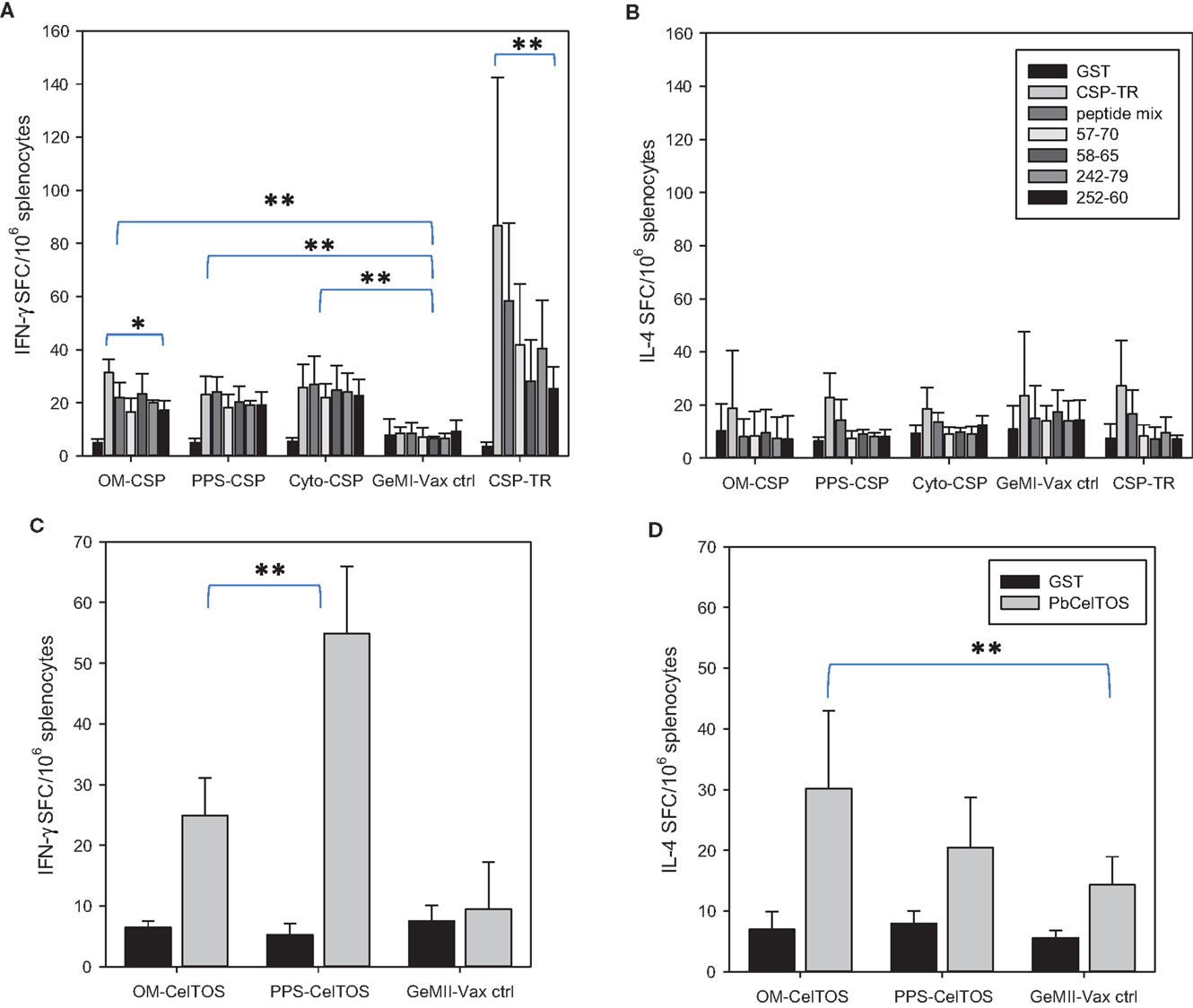
Figure 6. E. coli GeMI-Vax induce Th1 but not Th2 responses. GeMI-Vax constructs (indicated on the X-axis) were tested for their ability to mount antigen-specific IFN-γ responses (A,C), and IL-4 responses (B,D) after antigen-specific stimulation. (A,B) Splenocytes stimulated ex vivo with either recombinant PbCSP (10 μg/mL) or PbCSP-derived peptides (peptide mix = pool of all four peptides, peptides CS 57-70 and CS 242-279 represent I-Ad restricted CD4+ epitopes, peptides CS 58-65 and CS 252-60 represents H2Kd-restricted CD8+ epitopes). GST = negative control. (C,D) Splenocytes stimulated ex vivo with either recombinant PbCelTOS or GST protein (30 μg/mL). Data are expressed as the mean and standard error of the mean (SEM) number of spot-forming cells (SFC) from n = 5 mice/group. Asterisk indicates statistical significance (Students T-test): *p < 0.05, **p < 0.001.
To further explore the role of cellular localization and the modulation of immune responses, similar constructs to those describe for PbCSP were generated for expressing the PbCelTOS antigen (i.e., OM-PbCelTOS and PPS-PbCelTOS). CelTOS is a secreted, micronemal protein expressed from salivary gland sporozoites through early liver cell infection stages. Preclinical experiments utilizing recombinant protein revealed that CelTOS-based immunity is highly conserved and results in protection against sporozoite challenge in a homologous (44) and a heterologous (45) challenge model and that the protection is dependent on both cellular and humoral immunity (44). The observed cross-species protection is a unique feature of the CelTOS antigen and allows for evaluation of P. falciparum CelTOS in the P. berghei murine model. Unlike for OM-PbCSP, mice immunized with OM-PbCelTOS did not induce significant levels of antibodies above background (data not shown); however, a significant cellular response that was skewed toward Th1 was evident (Figures 6C,D). In this case, localization of CelTOS to the surface or the PPS yielded IFN-γ responses that were significantly different (p < 0.001, T-test). Only the OM-PbCelTOS construct induced IL-4 responses that were significantly different from the empty GeMI-Vax control (p < 0.01, T-test).
To evaluate the in vivo efficacy, BALB/c mice were challenged by injecting 4,000 salivary gland P. berghei sporozoites subcutaneously (SQ). We have previously shown that the SQ route better mimics the natural infection route than does the intravenous route (IV) by engaging humoral immune mechanisms (47). Mice that remained aparasitemic on day 14 after challenge were considered sterilely protected (Table 2). Sterile protection was observed in mice immunized with the OM-PbCSP, while PPS- and Cyto-PbCSP were only able to induce partial protection. Some mice were re-challenged 10 weeks after the initial challenge to determine (1) whether protection was long-lasting, and (2) whether the exposure to the parasites during the first challenge edited the immune response (48). Only mice immunized with OM-PbCSP achieved complete protection (7/7 mice) upon re-challenge (Table 2). Although low numbers of animals were re-challenged, these results suggest the GeMI-Vax platform is capable of inducing long-lasting immunity.
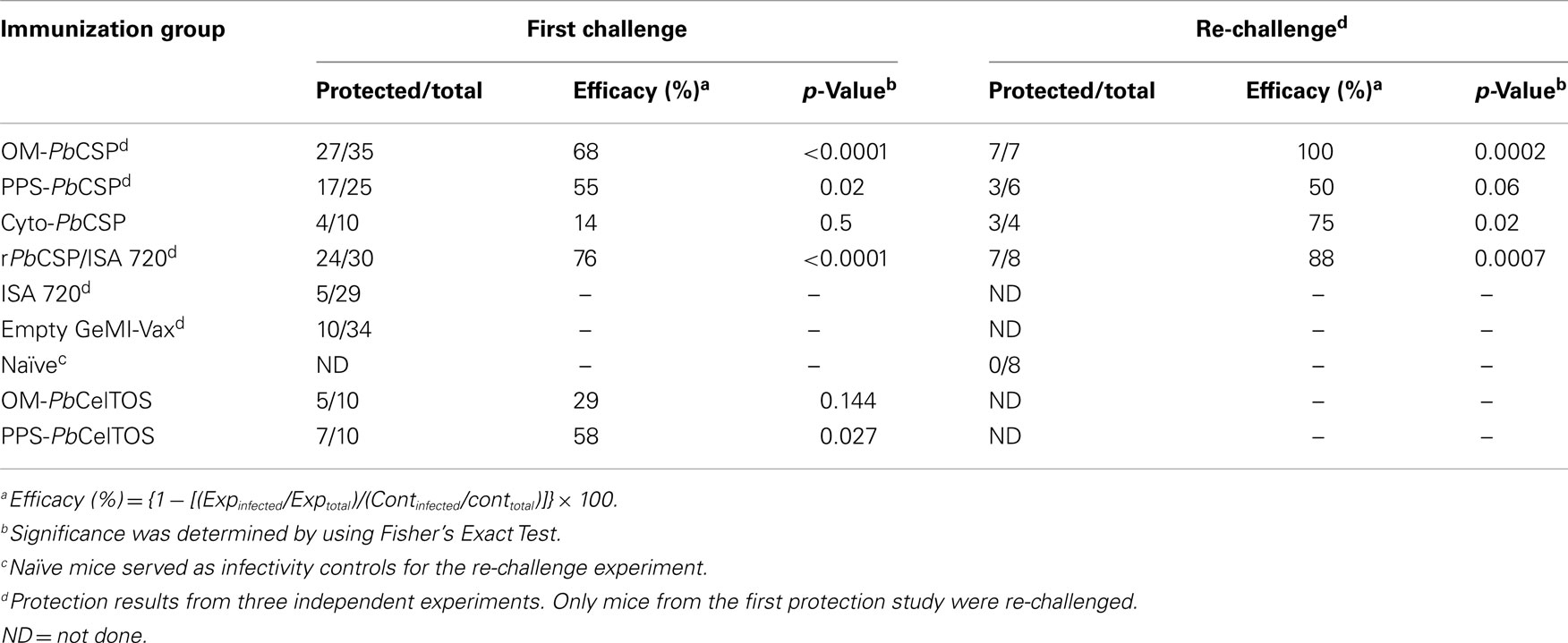
Table 2. GeMI-Vax induce long-lasting protective immunity that varies by sub-cellular localization.
Translation to Clinically Relevant Shigella flexneri 15G-GeMI-Vax Expressing Malarial Antigens
After the initial success using E. coli, the GeMI-Vax platform was adapted to an asd mutant of S. flexneri 2a (15G strain). The rationale for switching to Shigella was based on safety concerns related to immunization with a commensal E. coli present in the host gut. Attenuated derivatives of the S. flexneri 2a have previously been tested as oral vaccines to prevent shigellosis (30, 53, 54) and used in animal studies as potential multivalent mucosal vaccine candidates for delivery of bacterial antigens and eukaryotic genes (55). An asd mutant of Shigella is unable to grow in the absence of DAP, an essential peptidoglycan component of the bacterial cell wall. Since mammalian cells lack DAP, Shigella asd mutants can readily invade epithelial cells but are unable to replicate thus rendering them highly attenuated. Facile transition from E. coli to Shigella was achieved due to their genetic similarities thus allowing identical approaches for establishing the GeMI-Vax Shigella concept. The malaria targets were derived from the lethal Plasmodium spp., P. falciparum, and were engineered for cellular localization as described for the P. berghei CSP and CelTOS. Based on results from E. coli GeMI-Vax, the PfCSP was targeted to the OM while the CelTOS antigen was targeted to the PPS of Shigella. Because of its native surface localization, the objective for the OM-PfCSP was to achieve strong antibody responses while the intracellular PPS-PfCelTOS was intended to induce primarily T cell responses. Since in these experiments Shigella GeMI-Vax was delivered by parenteral routes the immune responses were focused on the malaria antigens and not the shigella delivery vehicle.
Expression levels of the S. flexneri 15G encoding either OM-PfCSP or PPS-PfCelTOS were confirmed by Western blotting with antigen-specific mAbs (Figure 7). OM-PfCSP was detected by polyclonal mouse antisera as well as mAbs specific for the C-terminus (clone WR7) or the central repeat, NANP region (clone WR3) (Figure 7A) of the P. falciparum CSP. Both the C-terminal and the repeat region-specific mAbs recognized the OM-PbCSP fusion protein (68.5 kDa) and the recombinant full-length protein, rPfCSP (∼32 kDa). For immunizations, the amount of OM-PfCSP delivered was estimated to be 0.05 μg (low dose) and 5 μg (high dose) per dose based on immunoreactivities on Western blots. Expression of PPS-PfCelTOS was confirmed by Western blots probed with CelTOS specific mAbs and polyclonal antibodies (Figure 7B). The fusion protein was confirmed to have a size of ∼61 kDa. Fine specificity of the PfCelTOS-specific mAbs indicate that anti-PfCelTOS mAb 3C3.C2 binds primarily to linear C-terminal epitopes while mAb 3D11.E2 binds primarily to the recombinant PfCelTOS protein indicating specificity for a conformational epitope. The lack of immunoreactivity by the mAb 3C3.C2 to the PPS-PfCelTOS protein suggests that the binding epitope for this mAb may not be accessible due to folding constraints. Both mAbs 3C3.C2 and 3D11.E2 and the polyclonal immune sera recognized the reference protein, rPfCelTOS (19 kDa). Based on Western blot probing the amount of PPS-PfCelTOS delivered during immunizations was estimated to be ∼0.01 μg (low dose) and 1 μg (high dose) per dose.
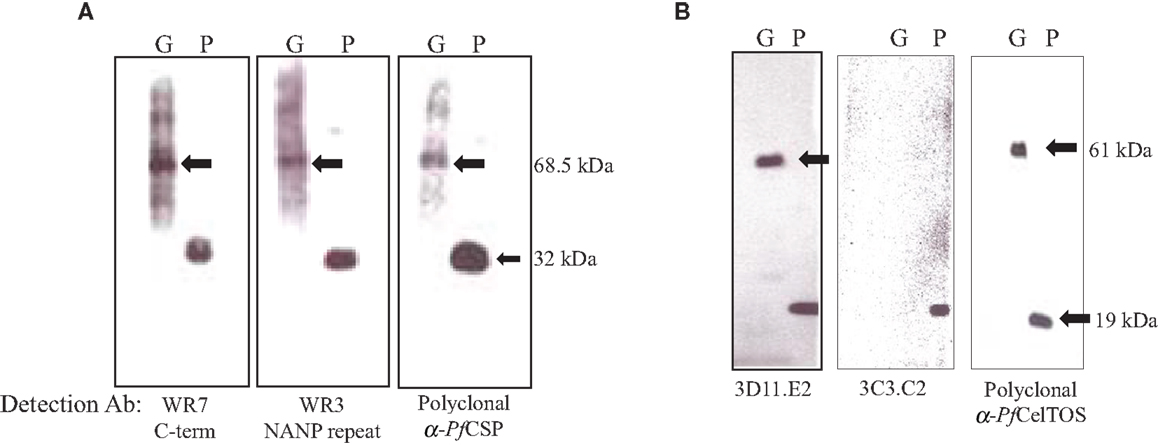
Figure 7. Analysis of expression levels of malarial antigens from the different sub-cellular compartments on Shigella GeMI-Vax. Lysates from GeMI-Vax expressing OM-PfCSP (A) or PPS-PfCelTOS (B) were probed with antigen-specific mAbs and polyclonal Abs: clone WR7 = PfCSP C-terminus specific mAb; clone WR3 = PfCSP repeat region-specific mAb, polyclonal = serum pooled from mice immunized with recombinant PfCSP; 3D11.E2 = PfCelTOS-specific, conformation-dependent mAb; 3C3.C2 = PfCelTOS-specific, conformation-independent mAb; polyclonal = serum pool from rabbits immunized with recombinant PfCelTOS/ISA 720. Arrows indicate fusion protein in molecular mass. G is GeMI-Vax lysate; P is recombinant protein. Molecular weight of the rPfCSP is 34 kDa and the rPfCelTOS is 19.1 kDa.
Induction of PfCelTOS or PfCSP-Specific Antibody Responses by Shigella GeMI-Vax Depends on Antigen Localization
The immunogenicity of GeMI-Vax expressing PfCelTOS or PfCSP was evaluated by immunizing mice with a high dose (1 μg) or low dose (0.01 μg) of PPS-PfCelTOS or high dose (5 μg) or low dose (0.05 μg) of OM-PfCSP. The doses were selected based on the initial experiments performed with E. coli where 1.7 × 109 cells/mouse/dose was the highest tolerated dose. The kinetics of the antibody response in the immunized mice confirmed the earlier observation that targeting the antigen to the OM results in more robust humoral responses (Figure 8). Due to issues related to the availability of P. berghei Pf CSP-transgenic sporozoites for challenge it was necessary to include a fourth immunization of the OM-PfCSP GeMI-Vax. Delivering a higher dose of GeMI-Vax cells yielded statistically significant higher antigen-specific antibody responses at all time points (ANOVA, p < 0.05). Based on antibody kinetics, the fourth immunization did not significantly boost the response in the group receiving the high dose of OM-PfCSP. In contrast, the PPS-PfCelTOS groups did not show any significant antigen-specific antibodies above the background empty GeMI-Vax control cells (data not shown). IFA-results using P. falciparum sporozoites paralleled and confirmed the results from the ELISA in that immunization with OM-PfCSP led to high titers of CSP antibodies that are sporozoite-reactive, while the titers induced by the PPS-PfCelTOS were negligible (data not shown).
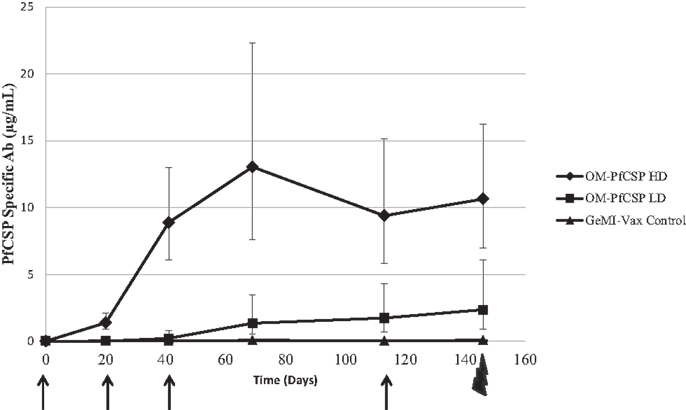
Figure 8. Kinetics of the humoral immune responses induced by Shigella GeMI-Vax. Sera from mice immunized with OM-PfCSP HD (high dose) and OM-PfCSP LD (low dose) were collected prior to, 3 weeks after each immunization and 1 day prior to the challenge and were analyzed using a quantitative ELISA. Data are expressed as the geometric mean PfCSP-specific antibody concentration (μg/mL) and the error bars represent the 95% confidence interval of n = 10 mice/group. Arrows indicate the days of immunizations, Day-1, 20, 41, and 113, and the bolt indicates time of challenge. Mice were immunized with either high dose = 1.7 × 109 cells or low dose = 1.7 × 107 number of cells. At each time point following the primary immunization, PfCSP-specific antibody responses for the OM-PfCSP HD group were significantly higher than for the OM-PfCSP LD group (ANOVA, p < 0.05).
T Cell Responses Differed by Targeting to Cellular Components
Immunization with OM-PfCSP induced PfCSP antigen-specific IFN-γ and IL-4 producing T cells with an apparent bias toward Th1-type responses compared to stimulations with irrelevant protein (GST as negative control) (Figure 9A). In contrast to the IFN-γ responses, the magnitude of the IL-4 responses was dependent on the immunization dose, however, only splenocytes stimulated with 30 μg/mL PfCSP yielding a significant difference between the two dosage groups (Student’s t-test; p < 0.05). Immunization with the PPS-PfCelTOS induced a relatively more balanced T cell response that did not significantly change as a function of the immunization dose, except for the low dose PPS-PfCelTOS splenocytes stimulated with 3 μg/mL of recombinant CelTOS, which were significantly higher than high dose (Students t-test; p < 0.05) (Figure 9B). The ability of the PPS-construct to induce antigen-specific T cells while failing to induce significant antibodies supports the earlier finding that localization to the periplasm leads to predominantly cellular responses.
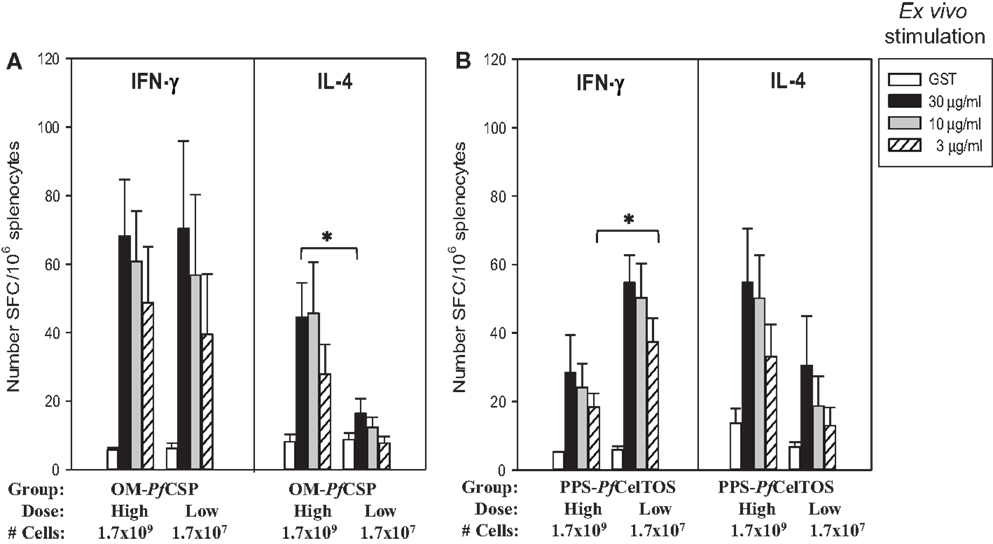
Figure 9. Shigella GeMI-Vax-induced a balanced T cell response that is dose-independent. Splenocytes from mice immunized with either OM-PfCSP (A) or PPS-PfCelTOS (B) were stimulated ex vivo with the respective recombinant protein (either rPfCSP or rPfCelTOS) or GST (negative control). Data are expressed as the mean and standard error from the mean (SEM) number of spot-forming cells (SFC) from n = 5 mice/group. Mice were immunized with either High dose = 1.7 × 109 or Low dose = 1.7 × 107 number of cells. Asterisk indicates statistical significance (Students T-test; *p < 0.05).
Shigella GeMI-Vax Cells Induced Protective Immunity Mediated by PfCelTOS in the Periplasm
To evaluate the protective efficacy of the PPS-PfCelTOS, mice were challenged with wild-type P. berghei parasites (45) while the OM-PfCSP mice were challenged with PfCSP-transgenic P. berghei parasites (46). A summary of the Shigella GeMI-Vax-induced protection is shown in Table 3. The level of protection induced by PPS-PfCelTOS was comparable to previously reported results using 10 μg PfCelTOS/Montanide ISA-720 (45). The effective antigen dose delivered by Shigella PPS-PfCelTOS was 10-fold lower than for soluble protein/adjuvant. When equivalent low doses of recombinant protein were delivered as soluble protein/adjuvant, no protection was observed (45), suggesting that the PPS-PfCelTOS GeMI-Vax approach is antigen dose sparing. Although the high dose of S. flexneri GeMI-Vax OM-PfCSP also induced a dose-dependent, antigen-specific antibody response, no significant protection was observed in contrast to the observations with the OM-PbCSP in E. coli.
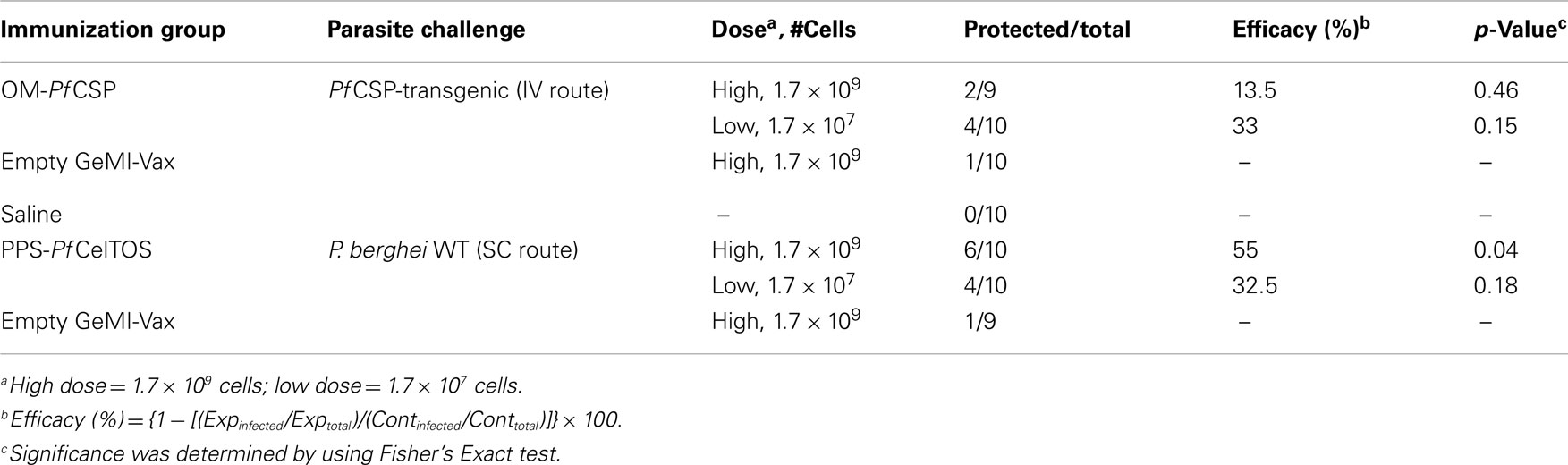
Table 3. Immunization with high dose PPS-PfCelTOS protects mice against challenge with wild-type P. berghei sporozoites.
Discussion
New and improved methods for the simultaneous presentation of pathogenic antigens and immune stimulation are needed for the development of efficacious vaccines. Platforms that present antigen to the host immune system in a particulate manner can mimic the structure of a natural pathogen leading to improved immunogenicity. GeMI-Vax are intact, Gram-negative bacteria, that are inactivated through genetic means and provide a delivery platform for presenting xenogeneic expressed target antigens with intrinsic danger signals to the host immune system. Although immunostimulatory, and from a safety standpoint, the endotoxin constituent of Gram-negative OMs does not limit their use as vaccine vectors primarily due to their minimal toxicity when presented as cell-associated lipopolysaccharides compared to the free-soluble forms (56).
In this study, GeMI-Vax served to evaluate the induction of antigen-specific humoral and cellular immune responses by expressing malaria proteins to different sub-cellular localizations. Localization of the PbCSP in E. coli GeMI-Vax to different cellular compartments induced distinct immune responses. PbCSP fused to the OM induced both humoral and Th1 cellular responses that induced sterile protection against homologous sporozoite challenge. Immunostaining of OM-PbCSP GeMI-Vax verified the surface localization and revealed a punctate and circumferential staining pattern that mimicked the authentic conformational display of CSP on sporozoites. Interestingly, none of the intracellular PbCSP localizations whether to the PPS or to the cytosol, induced high levels of PbCSP-specific antibodies. However, sub-cellular localizations of PbCSP led to the induction of IFN-γ responses by both CD4 and CD8 T cells, recognizing peptide epitopes throughout PbCSP (Figure 6). In contrast to PbCSP, OM localization of the PbCelTOS did not lead to pronounced levels of antibody. These results point to both cellular localization and the nature of the target antigen in influencing the induced immunity. Thus the key immunological findings are (1) protection was highest in the groups that mounted significant antibody responses, (2) PbCSP-antigen delivered by GeMI-Vax from different sub-cellular localizations does not influence the magnitude of the IFN-γ responses; and (3) none of the PbCSP-GeMI-Vax constructs induced significant PbCSP-specific IL-4 responses. These findings are consistent with other studies where PbCSP-specific antibodies were shown to play an important role in protection against a homologous sporozoite challenge (48). In the current study, the longevity of the protective response and its robustness against parasitic editing was measured by re-challenging the protected mice 10 weeks after the first challenge (Table 2). Only the OM-PbCSP immunized mice which notably had the highest antibody response and a concomitant Th1-type cellular response were completely protected. Currently, it is unclear whether the surface presentation on bacteria led to extended persistence of the antigen on follicular dendritic cells as described for other targeted vaccines (48). Another explanation for the sustained protection could be an increase in the precursor frequency of high-avidity B cells induced by lower antigen doses in the context of a potent activator of the innate immune system, a hypothesis supported by the finding that immune sera induced by OM-PbCSP GeMI-Vax had higher avidities than did the soluble protein/adjuvant. Thus presentation of PbCSP on the bacterial surface likely resembled the natural surface display on sporozoites and the adjuvanticity of the bacterium led to robust humoral responses despite the lower antigen dose. Concomitantly, surface display of PPS-PbCelTOS did not generate significant levels of antibodies and the protection seen was primarily due to a potent Th1-type IFN-γ T cell response. The present study emphasizes that the nature of the antigen; and both the quantity and quality of vaccine-induced immune responses, have a role in the potency of this vaccine platform.
The efficacy of Shigella GeMI-Vax OM-PfCSP differed from the results obtained in E. coli since the OM-PfCSP did not lead to significant protection against sporozoite challenge. One difference between the two experiments was the challenge route: the PbCSP E. coli GeMI-Vax construct were evaluated using the subcutaneous injection with wild-type P. berghei sporozoites which more closely mimics the natural challenge route (mosquito bite) (47). In contrast, the PfCSP Shigella GeMI-Vax constructs were evaluated using intravenous injection of PfCSP-transgenic P. berghei sporozoites. By injecting sporozoites directly IV, effector antibodies may have had little chance to contribute to protection (57). In this case, the partial protection may be attributed solely to the cellular responses induced by this vaccine. While the periplasmic location of the PbCSP did not induce statistically significant protective immunity, the opposite was seen with PfCelTOS localized to the PPS. These findings further confirm that the protection is not solely a function of the sub-cellular localization but that (a) the nature of the antigen and (b) the antigen-specific dosage levels may contribute to protection.
These results reveal that GeMI-Vax expressing the two major pre-erythrocytic targets, CSP or CelTOS, can elicit protective efficacy against homologous sporozoite challenge in murine models. In comparison with other vaccine technologies, the GeMI-Vax platform has several advantages: (a) ease and low cost of manufacturing; (b) highly stable final lyophilized product well suited for cold chain-free transport to remote field sites; (c) self-adjuvanting; (d) induces high-avidity antibodies known to be more efficacious in mediating protection; (e) triggers distinct immune responses by the choice of cellular localization of the target antigen; (f) integration with Gram-negative human pathogens such as Shigella GeMI-Vax holds the possibility for a dual-use vaccine to mediate protection against shigellosis and xenogeneic antigen(s) expressed by the bacteria. Finally, based on practical issues, GeMI-Vax may be suitable for use in the early immunological characterization of novel target antigens identified from high throughput antigen discovery projects. Using relatively standard molecular engineering, novel targets can be expressed and delivered to assess their immune potential in murine models without time consuming protein purification and process development. GeMI-Vax is also suitable for high density display of target antigens and can be easily integrated into a multi-epitope, multi-stage malaria vaccine system.
Disclaimer
The authors’ views are private and are not to be construed as official policy of the Department of Defense or the U.S. Army. Research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 1996 edition.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Ms. Katharine Boyle and Ms. Elizabeth Duncan for their technical assistance on the animal studies and immunological assays, Dr. Sheetij Dutta for providing recombinant P. falciparum circumsporozoite protein used in ELISAs and Western blots. This work was supported by the United States Army Medical Research and Materiel Command through ARMY SBIR funding to Vital Probes Inc. under contract #W81XWH-10-C-0003, through the Military Infectious Diseases and Research Program (MIDRP) and the ARMY In-house Laboratory Independent Research (ILIR) program.
References
1. Ben-Yedidia T, Arnon R. Design of peptide and polypeptide vaccines. Curr Opin Biotechnol (1997) 8:442–8.
2. Liljeqvist S, Stahl S. Production of recombinant subunit vaccines: protein immunogens, live delivery systems and nucleic acid vaccines. J Biotechnol (1999) 73:1–33.
3. Arnon R. Synthetic vaccines based on peptides, oligonucleotides and conjugate antigens. Behring Inst Mitt (1997) (98):184–90.
4. Hansson M, Nygren PA, Stahl S. Design and production of recombinant subunit vaccines. Biotechnol Appl Biochem (2000) 32(Pt 2):95–107.
5. Lesinski GB, Westerink MA. Vaccines against polysaccharide antigens. Curr Drug Targets Infect Disord (2001) 1:325–34.
7. Kotton CN, Hohmann EL. Enteric pathogens as vaccine vectors for foreign antigen delivery. Infect Immun (2004) 72:5535–47.
8. Clark TG, Cassidy-Hanley D. Recombinant subunit vaccines: potentials and constraints. Dev Biol (Basel) (2005) 121:153–63.
9. Detmer A, Glenting J. Live bacterial vaccines – a review and identification of potential hazards. Microb Cell Fact (2006) 5:23.
10. Tabrizi CA, Walcher P, Mayr UB, Stiedl T, Binder M, Mcgrath J, et al. Bacterial ghosts – biological particles as delivery systems for antigens, nucleic acids and drugs. Curr Opin Biotechnol (2004) 15:530–7.
11. Moyle PM, Toth I. Modern subunit vaccines: development, components, and research opportunities. ChemMedChem (2013) 8(3):360–76.
13. Levitz SM, Golenbock DT. Beyond empiricism: informing vaccine development through innate immunity research. Cell (2012) 148:1284–92.
15. Medzhitov R, Janeway CA Jr. Decoding the patterns of self and nonself by the innate immune system. Science (2002) 296:298–300.
16. Bergmann-Leitner ES, Leitner WW, Tsokos GC. Complement 3d: from molecular adjuvant to target of immune escape mechanisms. Clin Immunol (2006) 121:177–85.
17. Mongini PK, Tolani S, Fattah RJ, Inman JK. Antigen receptor triggered upregulation of CD86 and CD80 in human B cells: augmenting role of the CD21/CD19 co-stimulatory complex and Il-4. Cell Immunol (2002) 216:50–64.
18. Hjelm F, Carlsson F, Getahun A, Heyman B. Antibody-mediated regulation of the immune response. Scand J Immunol (2006) 64:177–84.
19. EL Shikh ME, EL Sayed RM, Sukumar S, Szakal AK, Tew JG. Activation of B cells by antigens on follicular dendritic cells. Trends Immunol (2010) 31:205–11.
20. Wu Y, Sukumar S, EL Shikh ME, Best AM, Szakal AK, Tew JG. Immune complex-bearing follicular dendritic cells deliver a late antigenic signal that promotes somatic hypermutation. J Immunol (2008) 180:281–90.
21. Aydar Y, Sukumar S, Szakal AK, Tew JG. The influence of immune complex-bearing follicular dendritic cells on the IgM response, Ig class switching, and production of high affinity IgG. J Immunol (2005) 174:5358–66.
22. Germanier R, Fuer E. Isolation and characterization of Gal E mutant Ty 21a of Salmonella typhi: a candidate strain for a live, oral typhoid vaccine. J Infect Dis (1975) 131:553–8.
23. Sztein MB. Cell-mediated immunity and antibody responses elicited by attenuated Salmonella enterica Serovar Typhi strains used as live oral vaccines in humans. Clin Infect Dis (2007) 45(Suppl 1):S15–9.
24. Lagos R, San Martin O, Wasserman SS, Prado V, Losonsky GA, Bustamante C, et al. Palatability, reactogenicity and immunogenicity of engineered live oral cholera vaccine CVD 103-HgR in Chilean infants and toddlers. Pediatr Infect Dis J (1999) 18:624–30.
25. Taylor DN, Sanchez JL, Castro JM, Lebron C, Parrado CM, Johnson DE, et al. Expanded safety and immunogenicity of a bivalent, oral, attenuated cholera vaccine, CVD 103-HgR plus CVD 111, in United States military personnel stationed in Panama. Infect Immun (1999) 67:2030–4.
26. Checkley AM, Mcshane H. Tuberculosis vaccines: progress and challenges. Trends Pharmacol Sci (2011) 32:601–6.
27. Mcshane H. Tuberculosis vaccines: beyond bacille Calmette-Guerin. Philos Trans R Soc Lond B Biol Sci (2011) 366:2782–9.
28. Roland KL, Tinge SA, Killeen KP, Kochi SK. Recent advances in the development of live, attenuated bacterial vectors. Curr Opin Mol Ther (2005) 7:62–72.
29. Collins TA, Barnoy S, Baqar S, Ranallo RT, Nemelka KW, Venkatesan MM. Safety and colonization of two novel VirG(IcsA)-based live Shigella sonnei vaccine strains in rhesus macaques (Macaca mulatta). Comp Med (2008) 58:88–94.
30. Mckenzie R, Venkatesan MM, Wolf MK, Islam D, Grahek S, Jones AM, et al. Safety and immunogenicity of WRSd1, a live attenuated Shigella dysenteriae type 1 vaccine candidate. Vaccine (2008) 26:3291–6.
31. Kaminski RW, Oaks EV. Inactivated and subunit vaccines to prevent shigellosis. Expert Rev Vaccines (2009) 8:1693–704.
32. Ranallo RT, Fonseka S, Boren TL, Bedford LA, Kaminski RW, Thakkar S, et al. Two live attenuated Shigella flexneri 2a strains WRSf2G12 and WRSf2G15: a new combination of gene deletions for 2nd generation live attenuated vaccine candidates. Vaccine (2012) 30:5159–71.
33. Simon JK, Maciel M Jr., Weld ED, Wahid R, Pasetti MF, Picking WL, et al. Antigen-specific IgA B memory cell responses to Shigella antigens elicited in volunteers immunized with live attenuated Shigella flexneri 2a oral vaccine candidates. Clin Immunol (2011) 139:185–92.
34. Tartz S, Russmann H, Kamanova J, Sebo P, Sturm A, Heussler V, et al. Complete protection against P. berghei malaria upon heterologous prime/boost immunization against circumsporozoite protein employing Salmonella type III secretion system and Bordetella adenylate cyclase toxoid. Vaccine (2008) 26:5935–43.
35. Nganou-Makamdop K, Van Roosmalen ML, Audouy SA, Van Gemert GJ, Leenhouts K, Hermsen CC, et al. Bacterium-like particles as multi-epitope delivery platform for Plasmodium berghei circumsporozoite protein induce complete protection against malaria in mice. Malar J (2012) 11:50.
36. Jiang S, Rasmussen RA, Nolan KM, Frankel FR, Lieberman J, Mcclure HM, et al. Live attenuated Listeria monocytogenes expressing HIV Gag: immunogenicity in rhesus monkeys. Vaccine (2007) 25:7470–9.
37. Loeffler DI, Schoen CU, Goebel W, Pilgrim S. Comparison of different live vaccine strategies in vivo for delivery of protein antigen or antigen-encoding DNA and mRNA by virulence-attenuated Listeria monocytogenes. Infect Immun (2006) 74:3946–57.
38. Cranage MP, Mcbride BW, Rud EW. The simian immunodeficiency virus transmembrane protein is poorly immunogenic in inactivated virus vaccine. Vaccine (1995) 13:895–900.
39. Rossio JL, Esser MT, Suryanarayana K, Schneider DK, Bess JW Jr., Vasquez GM, et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J Virol (1998) 72:7992–8001.
40. Eko FO, Witte A, Huter V, Kuen B, Furst-Ladani S, Haslberger A, et al. New strategies for combination vaccines based on the extended recombinant bacterial ghost system. Vaccine (1999) 17:1643–9.
41. Langemann T, Koller VJ, Muhammad A, Kudela P, Mayr UB, Lubitz W. The Bacterial Ghost platform system: production and applications. Bioeng Bugs (2010) 1:326–36.
42. Fennelly GJ, Khan SA, Abadi MA, Wild TF, Bloom BR. Mucosal DNA vaccine immunization against measles with a highly attenuated Shigella flexneri vector. J Immunol (1999) 162:1603–10.
43. Angov E, Hillier CJ, Kincaid RL, Lyon JA. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE (2008) 3:e2189.
44. Bergmann-Leitner ES, Legler PM, Savranskaya T, Ockenhouse CF, Angov E. Cellular and humoral immune effector mechanisms required for sterile protection against sporozoite challenge induced with the novel malaria vaccine candidate CelTOS. Vaccine (2011) 29:5940–9.
45. Bergmann-Leitner ES, Mease RM, DE LA, Vega P, Savranskaya T, Polhemus M, et al. Immunization with pre-erythrocytic antigen CelTOS from Plasmodium falciparum elicits cross-species protection against heterologous challenge with Plasmodium berghei. PLoS ONE (2010) 5:e12294.
46. Tewari R, Spaccapelo R, Bistoni F, Holder AA, Crisanti A. Function of region I and II adhesive motifs of Plasmodium falciparum circumsporozoite protein in sporozoite motility and infectivity. J Biol Chem (2002) 277:47613–8.
47. Leitner WW, Bergmann-Leitner ES, Angov E. Comparison of Plasmodium berghei challenge models for the evaluation of pre-erythrocytic malaria vaccines and their effect on perceived vaccine efficacy. Malar J (2010) 9:145.
48. Bergmann-Leitner ES, Scheiblhofer S, Weiss R, Duncan EH, Leitner WW, Chen D, et al. C3d binding to the circumsporozoite protein carboxy-terminus deviates immunity against malaria. Int Immunol (2005) 17:245–55.
49. Migliorini P, Betschart B, Corradin G. Malaria vaccine: immunization of mice with a synthetic T cell helper epitope alone leads to protective immunity. Eur J Immunol (1993) 23:582–5.
50. Leitner WW, Ying H, Restifo NP. DNA and RNA-based vaccines: principles, progress and prospects. Vaccine (1999) 18:765–77.
51. Romero PJ, Tam JP, Schlesinger D, Clavijo P, Gibson H, Barr PJ, et al. Multiple T helper cell epitopes of the circumsporozoite protein of Plasmodium berghei. Eur J Immunol (1988) 18:1951–7.
52. Romero P, Corradin G, Luescher IF, Maryanski JL. H-2Kd-restricted antigenic peptides share a simple binding motif. J Exp Med (1991) 174:603–12.
53. Ranallo RT, Thakkar S, Chen Q, Venkatesan MM. Immunogenicity and characterization of WRSF2G11: a second generation live attenuated Shigella flexneri 2a vaccine strain. Vaccine (2007) 25:2269–78.
54. Venkatesan MM, Ranallo RT. Live-attenuated Shigella vaccines. Expert Rev Vaccines (2006) 5:669–86.
55. Sizemore DR, Branstrom AA, Sadoff JC. Attenuated Shigella as a DNA delivery vehicle for DNA-mediated immunization. Science (1995) 270:299–302.
56. Mader HJ, Szostak MP, Hensel A, Lubitz W, Haslberger AG. Endotoxicity does not limit the use of bacterial ghosts as candidate vaccines. Vaccine (1997) 15:195–202.
Keywords: E. coli, Shigella, malaria, self-adjuvanting, cellular targeting, bacterial vaccine vector, CelTOS, CSP
Citation: Bergmann-Leitner ES, Hosie H, Trichilo J, DeRiso E, Ranallo RT, Alefantis T, Savranskaya T, Grewal P, Ockenhouse CF, Venkatesan MM, DelVecchio VG and Angov E (2013) Self-adjuvanting bacterial vectors expressing pre-erythrocytic antigens induce sterile protection against malaria. Front. Immunol. 4:176. doi: 10.3389/fimmu.2013.00176
Received: 02 March 2013; Accepted: 18 June 2013;
Published online: 04 July 2013.
Edited by:
Lorne A. Babiuk, University of Alberta, CanadaReviewed by:
Yasmin Thanavala, Roswell Park Cancer Institute, USAOlivier Silvie, Institut National de la Santé et de la Recherche Médicale, France
Copyright: © 2013 Bergmann-Leitner, Hosie, Trichilo, DeRiso, Ranallo, Alefantis, Savranskaya, Grewal, Ockenhouse, Venkatesan, DelVecchio and Angov. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Evelina Angov, Malaria Vaccine Branch, Department of Molecular Parasitology, Walter Reed Army Institute of Research, Silver Spring, MD 20910, USA e-mail:ZXZlbGluYS5hbmdvdi5jaXZAbWFpbC5taWw=
†Present address: Ryan T. Ranallo, Division of Microbiology and Infectious Diseases, National Institutes of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA; Timothy Alefantis, Sanofi Pasteur Biologics, LLC, Cambridge, MA, USA.