




- 1Department of Inflammation and Immunology, Humanitas Clinical and Research Center, Rozzano, Italy
- 2Department of Veterinary Science and Public Health, University of Milan, Milan, Italy
- 3Department of Biotechnology and Translational Medicine, University of Milan, Rozzano, Milan, Italy
Members of the IL-1 family play a key role in innate and adaptive immunity and in the pathogenesis of diverse diseases. Members of IL-1R like receptor (ILR) family include signaling molecules and negative regulators. The latter include decoy receptors (IL-1RII; IL-18BP) and “receptors” with regulatory function (TIR8/SIGIRR; IL-1RAcPb; DIGIRR). Structural considerations suggest that also TIGIRR-1 and IL-1RAPL may have regulatory function. The presence of multiple pathways of negative regulation of members of the IL-1/IL-1R family emphasizes the need for a tight control of members of this fundamental system.
Introduction
IL-1R like receptors (ILRs) belong, together with Toll-like receptors (TLRs), to a superfamily of phylogenetically conserved proteins involved in innate immunity and inflammation (1–5). The common characteristic of the members of this family is the presence of a conserved domain in the cytoplasmic region, called TIR domain, originally defined as the Toll/IL-1-resistance and now generally assumed as an acronym for Toll/IL-1R domain. The TIR domain is involved in the activation of an evolutionarily conserved signaling pathway leading to NF-kB translocation to the nucleus and activation of protein kinases such as p38, c-Jun N-terminal kinases (JNKs), extracellular signal-regulated kinases (ERKs), and mitogen-activated protein kinases (mAPKs) (6). The ILR subfamily includes the receptors and the accessory proteins (AcP) for IL-1α (IL-1F1) and IL-1β (IL-1F2), IL-18/IL-1F4, IL-33/IL-F11, and other IL-1 family members (IL-36α/IL-1F6, IL-36β/IL-1F8, and IL-36γ/IL-1F9), which are involved in the initiation of an amplification cascade of innate resistance and inflammation and contribute to the activation and orientation of adaptive immunity (7–9). Some members of the family remain orphan receptors with still unknown ligands and functions. For instance, in the IL-1R subfamily, TIR8/SIGIRR, TIGIRR-1, and IL-1RAPL have no characterized ligands so far (2, 10, 11).
The activation of the ILR-dependent signaling cascade is tightly regulated. Indeed, the deregulated activation of these receptors, which lead to the production of proteins related to inflammation and immunity, potentially mediates damaging local and systemic inflammatory reactions. Several pathological conditions depend, at least in part, on the inflammatory potential of the IL-1 family members mentioned above. For instance, the IL-1 system represents a relevant therapeutic target in arthritis, type 2 diabetes, psoriasis, sepsis, ischemia and reperfusion, atherosclerosis, graft rejection, cancer (12–15). The regulatory mechanisms identified so far in the IL-1 system (ligands, receptors, signaling pathway) act extracellularly or intracellularly (16, 17). IL-1R antagonists (IL-1Ra)/IL-1F3 and IL-36Ra/IL-1F5 are polypeptide antagonists competing with IL-1 and IL-36α/IL-1F6, IL-36β/IL-1F8, and IL-36γ/IL-1F9, respectively, for receptor binding (3, 7, 18–20). IL-1RII lacks a signaling domain and by binding IL-1 prevents its interaction with a signaling receptor complex and therefore acts as a decoy, dominant-negative molecule, and scavenger. The negative regulator of ILR and TLR signaling, TIR8 (also known as SIGIRR), acts intracellularly. IRAK-M and MyD88s are intracellular negative regulators of ILRs and TLRs signaling (21, 22). Finally, ILR or TLR signaling proteins or transcription factors are targets of miRNAs, such as miR-155, miR-21, miR-146a, miR-132, miR-9, and miR-147, whose transcription is induced by inflammatory mediators [lipopolysaccharide (LPS), TNFα, IL-1 β] through NF-kB (23–25).
Here, we summarize our current understanding of the structure and function of negative regulatory receptors of the ILR family, in particular IL-1RII, which has served to defining the decoy receptor paradigm, and TIR8/SIGIRR, focusing on their regulatory roles in different pathological disorders dependent on ILRs and TLRs activity, and finally describe other largely uncharacterized members of the family with a negative regulatory potential, TIGIRR-1, IL-1RAPL, IL-1RAPb.
The Decoy Receptor IL-1RII
Gene and Protein
The first IL-1R was cloned from murine and human T cells, whereas IL-1RII was identified soon after in B lymphocytes and myelomonocytic cells (26, 27). On the basis of their structures, IL-1RI and IL-1RII belong to the Ig-like superfamily of receptors, with the extracellular portion containing 3 Ig-like domains. The signaling IL-1R complex includes the type I IL-1 receptor (IL-1RI) and IL-1R AcP, which both have a cytoplasmic TIR domain (Figure 1).
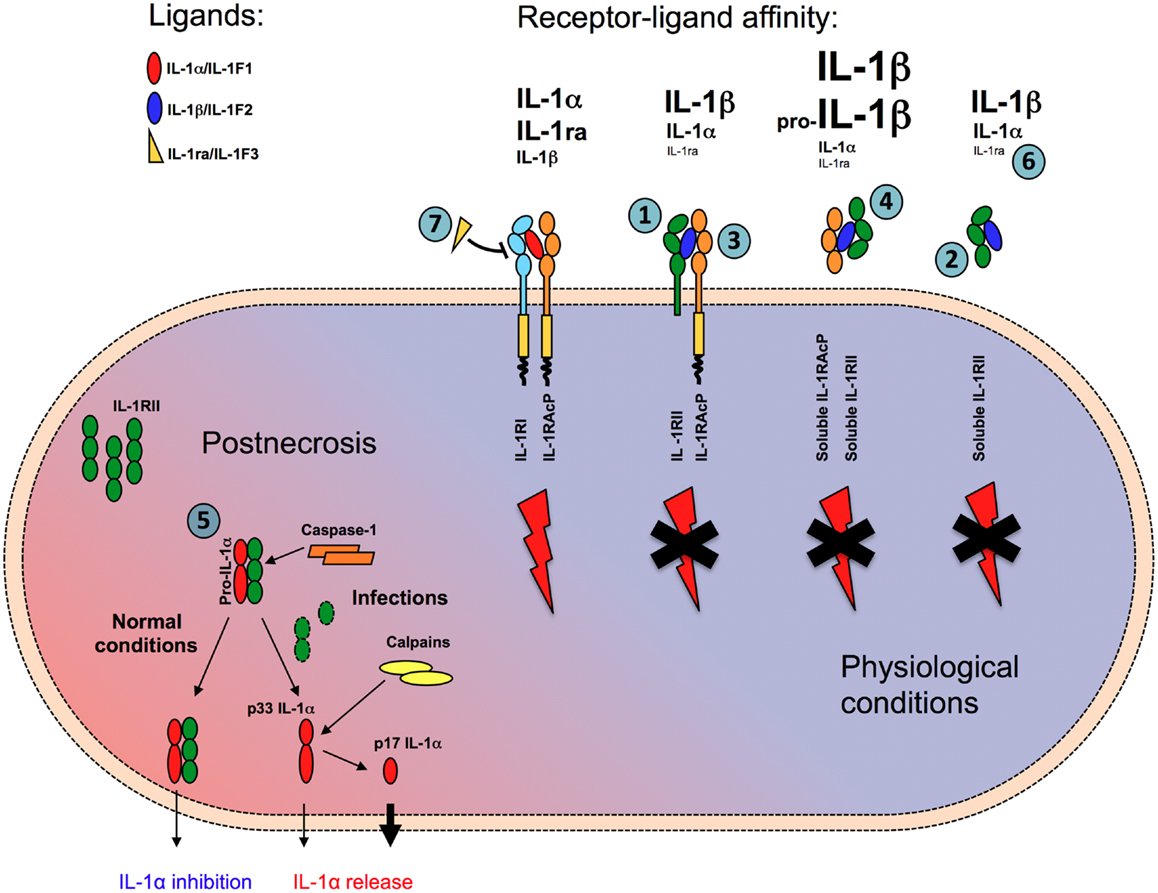
Figure 1. Mechanisms of negative regulation mediated by IL-1RII. IL-1RII negatively regulates IL-1 responses in multiple ways. In membrane-bound (1) or soluble form (2) IL-1RII acts as a decoy, capturing with high-affinity IL-1β and IL-1α, but not IL-1ra, and preventing their interaction with IL-1RI. IL-1RII acts as a dominant-negative influencing IL-1RI-IL-1RAcp signaling receptor complex formation (3). The interaction of ligand-bound soluble IL-1RII with soluble IL-1RAcP increases the affinity for both IL-1α and IL-1β by about two orders of magnitude (4). Finally, in cytosol soluble form, IL-1RII interacts with pro-IL-1α preventing its pro-inflammatory activity during necrosis, until caspase-1 cleaves IL-1RII and allows the secretion of IL-1α (5). Because of low affinity for IL-1RII, IL-1ra does not compete with IL-1α and IL-1β for the interaction with the decoy receptor (6), whereas it competes with IL-1α and IL-1β antagonizing their interaction with the signaling receptor IL-1RI (7).
The gene encoding IL-1RII is located on chromosome 2 (q12–22) in humans and in the centromeric region of chromosome 1 in mice (28), in cluster with IL-1RI and other members of the family (IL-33R, IL-18R, IL-36R). The type II receptor is highly conserved in evolution and is found in bony fish, where it functions to inhibit IL-1-induced inflammation (29). The third Ig domain of IL-1RII is homologous to the Ig domain of IL-18BP (30) and indeed, it has been suggested that IL-1RII and IL-18BP have a common ancestral gene and diverged at the level of fish (31). The IL-1RII locus spans about 38 kb of genomic DNA, of which about 21 kb contains the coding region. The exon structures of the extracellular portion of IL-1RII and IL-1RI receptors are identical and amino acid sequences share 28% homology. A single exon encodes the transmembrane region and a short cytoplasmic tail (29 amino acids) of IL-1RII, which has no TIR domain and does not signal (Figure 1). The human transcript encodes for a 386 amino acid glycosylated protein of 68 kDa, in contrast with IL-1RI which is a 80–85 kDa glycosylated protein and has a 213 amino acid cytoplasmic tail containing a TIR domain responsible for signaling (32).
IL-1RII can be proteolytically processed and released in a soluble form, via the actions of a metalloproteinase, A Disintegrin and Metalloprotease 17 (ADAM17, also know as TACE) (33, 34). In addition, IL-1RII can be processed in a manner similar to Amyloid β protein precursor (APP), by alpha-, beta-, and gamma-secretase: the ectodomain is shed in an alpha-secretase-like manner, whereas the IL-1RII C-terminal fragment undergoes further intramembrane proteolysis by gamma-secretase (35). Finally, the aminopeptidase regulator of TNFR1 shedding (ARTS-1) has been implicated in IL-1RII shedding in basal condition and upon cell stimulation with phorbol myristate acetate (PMA) (36).
Wang et al. (37) solved the structure of IL-1 and IL-1RAcP in complex with the extracellular domain of IL-1RII and showed that the mode of interaction among IL-1β, IL-1RII, and IL-1RAcP and the overall structure are extremely similar to those of the signaling ligand-receptor complex (IL-1, IL-1R1, IL-1AcP).
Mechanisms of Negative Regulation
Several lines of evidence are consistent with the view that the IL-1RII is a bonafide IL-1 decoy.
A first level of control is represented by the differential affinity of the signaling and decoy receptors for agonist or antagonist ligands of the IL-1 family (Figure 1). IL-1RI binds IL-1α with higher affinity than IL-1β (Kd ≈ 10−10 and 10−9 M, respectively) and IL-1ra with an affinity similar to that for IL-1α. By contrast, IL-1RII binds IL-1β and IL-1α with high affinity (Kd ≈ 10−9–10−10 and 10−8 M, respectively), but it binds IL-1ra at least 100 times less efficiently (38). Plasmon resonance analysis revealed that IL-1β has a slow off-rate from IL-1RII, whereas IL-1ra rapidly dissociates from IL-1RII but not from IL-1RI (39), in agreement with the need that the two regulators of IL-1 do not bind each other self defeating and frustrating their regulatory activity. By binding agonist ligands with high affinity without inducing signaling, IL-1RII acts as a molecular trap for IL-1 inhibiting its activity (27, 40) (Figure 1).
Second, IL-1RII also forms a complex with IL-1 and the IL-1RAcP. It therefore exerts a dominant-negative effect on the formation of a signaling receptor complex, by sequestering AcP, which is essential for signal transduction (41, 42) (Figure 1).
In addition, IL-1RII is also found in a soluble form, released from cells via the actions of a metalloproteinase (see above). Soluble IL-1RII is found in normal blood relatively at high concentrations, in the order of nanogram per milliliter. Cell-surface shedding is the major mechanism responsible of soluble IL-1RII generation, but in addition an alternatively spliced transcript encoding a soluble version of IL-1RII has been described (43). Soluble, but not cell-associated, IL-1RII binds pro-IL-1β and blocks its processing by IL-1-converting enzyme (ICE)/caspase-1 (38) (Figure 1). Soluble AcP, encoded by an alternatively spliced mRNA (44) and found at high levels in the circulation (300 ng/ml in humans), can interact with ligand-bound soluble IL-1RII, enhancing the latter’s affinity for IL-1α and IL-1β by two orders of magnitude, while not affecting the very low affinity for IL-1Ra (40, 45) (Figure 1). In mouse and monkey, the interaction between AcP and IL-1RII is required for high-affinity binding of IL-1β and effective inhibition (45). Thus, the interaction with AcP renders IL-1RII a much more effective inhibitor of IL-1.
Finally, a further mechanism of negative control of IL-1α by IL-1RII during necrosis has recently been proposed (46). The soluble form of IL-1RII has been detected in the cytosol in large amounts, possibly because the IL-1RII signal peptide is short and relatively weak. In line with previous reports on systemic sclerosis fibroblasts (47), in this cytosolic form, soluble IL-1RII interacts with pro-IL-1α (Figure 1). This interaction protects pro-IL-1α from cleavage by different enzymes (calpain, granzyme B, chymase, and elastase) normally involved in the generation of the active form (48, 49) and prevents IL-1α activity (46). This blockade would be abrogated by active caspase-1 (for instance during infections), which specifically cleaves IL-1RII, causing dissociation from IL-1α, calpain processing, and complete restoration of IL-1α activity after necrosis or during regulated secretion (Figure 1). Since IL-1RII is expressed by a limited set of cell types, in contrast with IL-1RI, which is widely expressed, this mechanism of negative regulation would be cell type specific. Thus, the activity of IL-1α during necrosis and sterile inflammation would be somehow restricted to cell types which do not express IL-1RII. For instance, the high inflammatory profile of vascular smooth muscle cells to necrosis, which is IL-1α-dependent (50), would be in agreement with low levels of IL-1RII. These findings would explain the tissue specificity of inflammatory damage during necrosis.
The anti-inflammatory role of IL-1RII has been demonstrated in different pathological conditions in animal models. Gene-targeted mice overexpressing IL-1RII under the control of the human keratin gene promoter were resistant to PMA-induced chronic skin inflammation (51). Recombinant IL-1RII delivered via implanted human keratinocytes overexpressing soluble IL-1RII played a protective role in a mouse model of collagen-induced arthritis (52) and intravenous administration of soluble IL-1RII significantly reduced joint swelling and erosion in a model of arthritis in rabbit (53). Gene transfer of a soluble IL-1RII-Ig fusion protein reduced allograft rejection and prolonged graft survival in a rat model of heart transplantation, reduced infiltrating macrophages, and CD4+ T cells, and lowered levels of TNF-α and TGF-β (54). Similarly, IL-1RII ameliorated experimental autoimmune myocarditis by blocking IL-1 and inhibiting production of the cytokines [IL-6, transforming growth factor-β, retinoic acid-related orphan nuclear receptor (RORγt) and IL-17] involved in the polarization of Th17 cells (55). Finally, in a mouse model of endometriosis, consisting of human endometrial tissue implanted in nude mice, human soluble IL-1-RII administered intraperitoneally reduced the growth and dissemination of endometrial implants and the expression of IL-1β-dependent inflammatory, angiogenic, and cell growth mediators (56).
In support of the view that IL-1RII is a professional anti-IL-1 molecule, Pox viruses have acquired and retained a soluble version of type II IL-1R, that plays a key role in the regulation of pathogenicity (57).
Thus, IL-1RII negatively regulates IL-1 responses in multiple complementary ways. In membrane-bound or soluble form IL-1RII acts as a decoy, capturing with high affinity IL-1, and preventing it from interacting with IL-1RI. It acts as a dominant-negative influencing IL-1RI-IL-1RAcp signaling receptor complex formation. The interaction of ligand-bound soluble IL-1RII with soluble IL-1RAcP increases the affinity for both IL-1α and IL-1β by about two orders of magnitude and makes IL-1RII a powerful inhibitor for both agonists. Finally, in cytosol soluble form, IL-1RII interacts with pro-IL-1α preventing its pro-inflammatory activity during necrosis.
Expression
In contrast with IL-1RI, which is expressed by a large variety of cell types, IL-1RII is expressed by a limited set of cell types, which also often express IL-1RI: among leukocytes, IL-1RII is the predominant IL-1-binding protein found in monocytes, neutrophils, and B cells (26, 27, 40, 58). Monocyte differentiation to macrophages, in particular M2 or M2-like macrophages, is associated to increased expression of IL-1RII (58, 59). IL-1RII is also expressed by microglial cells, in particular upon stimulation with LPS and has been shown to regulate IL-1β actions by binding excess levels of this cytokine during brain inflammation (60). In addition, noradrenaline has been reported to upregulate IL-1RII in mixed microglia via β-adrenoceptor activation and downstream activation of protein kinase A and ERK, thus preventing IL-1β-induced neurotoxicity (61). Other stimuli involved in IL-1RII upregulation in the CNS include cerebral ischemia, kainic acid administration, and central administration of IL-1β (62).
T regulatory cells (Tregs) have been shown to express surface and soluble functional IL-1RII, as well as IL-1Ra mRNA. This property has been exploited for the purification of activated human FOXP3+ regulatory T cells from expansion cultures (63). Activated human Tregs rapidly up-regulated IL-1RII and were able to neutralize IL-1β, which suggests a physiological significance for the expression of IL-1 decoy receptor on Tregs (64).
Differential levels of IL-1RII have been described in osteoclasts. In particular, lower expression of IL-1RII has been detected in large osteoclasts compared to small osteoclasts, and this is in line with increased resorptive activity of large osteoclasts in response to IL-1 (65). IL-1RII is also expressed by basal epithelial cells of the skin (66), epithelium of endometrium (67), vagina and urethra, and chondrocytes. Endothelial cells and fibroblasts generally express only IL-1RI and AcP.
Surface and soluble IL-1RII expression is strongly enhanced by anti-inflammatory signals. Glucocorticoid hormones (GCs), prostaglandins, the anti-inflammatory T helper 2 (Th2) cytokines (IL-4 and IL-13), and IL-27 induced augmented surface expression and release of IL-1RII in vitro, in particular in myelomonocytic cells, and in vivo (27, 58, 68–72). In particular, IL-4 and dexamethasone, by inducing IL-1RII, antagonized the prosurvival effect of IL-1 in neutrophils in vitro (27). IL-10 increased circulating soluble IL-1RII levels in vivo in mice. Aspirin increased IL-1RII release from mononuclear cell cultures in vitro and in vivo (73). IL-27 inhibited IL-1β-induced signaling in human macrophages by downregulating the expression of the signaling receptor IL-1RI, inducing expression of the receptor antagonist IL-1Ra, and by upregulating the expression of the decoy receptor IL-1RII (72). These data suggest that induction of IL-1RII contributes to the anti-inflammatory effect of these mediators.
In contrast, pro-inflammatory molecules inhibit IL-1RII expression. For instance, bacterial LPS caused a rapid shedding of surface IL-1RII in monocytes, followed by down-regulation of expression, whereas it stimulated the expression of IL-1RI, AcP and the adapter protein MyD88 (74). Interferon γ (IFN-γ) inhibited IL-1RII expression and release in myelomonocytic cells and counteracted IL-4-dependent upregulation of IL-1RII (71). In addition to LPS, chemoattractants such as formyl Meth-Leu-Phe (fMLP), reactive oxygen intermediates (ROI), TNF, and PMA caused rapid shedding of IL-1RII (33, 75, 76). PMA also induced alternatively spliced soluble IL-1RAcP (44). Thus, shedding of IL-1RII by circulating phagocytes and generation of alternatively spliced soluble IL-1RAcP induced by chemoattractants in the early steps of recruitment, could prepare cells to respond to IL-1 once they enter tissues.
Acetylated low density (ac-LDL) and very low density (VLDL) lipoprotein intracellular accumulation caused decreased IL-1RII mRNA and protein expression in macrophages in vitro. In agreement with these in vitro data, patients with familial combined hyperlipidemia showed decreased expression of IL-1RII in monocytes. Finally, IL-1RII expression in human atherosclerotic vessels was defective compared to non-atherosclerotic arteries (77).
Naturally circulating levels of soluble IL-1R type II are in the range of 5–10 ng/ml, although these can rise in certain chronic (78) or acute (79) inflammatory settings (see below), in part reflecting the activation of negative circuits of regulation of the cytokine action.
IL-1RII in Human Pathological Conditions: Diagnostic and Therapeutic Implications
High levels of soluble IL-1RII are normally present in plasma of healthy individuals. Defective or increased expression of tissue or body fluid levels of soluble IL-1RII have been described in diverse pathological conditions, ranging from critical conditions to autoimmune diseases, neuroinflammatory diseases and tumors.
Increased blood levels of soluble IL-1RII have been detected in critically ill patients with infectious conditions such as sepsis, acute meningococcal infection, experimental endotoxemia, operative trauma, or necrotizing enterocolitis in preterm infants (73, 80, 81). In critically ill patients, IL-1RII levels were elevated especially in severe, systemic infection and culture-positive infections. In patients with a marked systemic inflammatory response syndrome, further pronounced increase of circulating IL-1RII levels was observed in patients developing sepsis (80). Treatment with glucocorticoids further increased IL-1RII levels, suggesting that it potentially behaves as a biomarker for the activation of anti-inflammatory pathways or for responsiveness to anti-inflammatory agents. In acute meningococcal infections, increased soluble IL-1RII levels correlated with disease severity, in particular with endotoxemia, complement-activation, and shock (82). Increased IL-1RII levels were also observed in patients upon treatment with aspirin (73).
IL-1Ra and/or IL-1RII increased levels were also detected in sera of multiple sclerosis patients after steroid treatment for relapse (83) and in the cerebrospinal fluid of patients with Alzheimer’s disease, where it may be a marker of disease progression (84).
In psoriasis, IL-1ra and IL-1RII were both significantly overexpressed in the suprabasal and basal compartment, respectively, and inversely correlated with the expressions of IL-1α (66). Increased levels of soluble IL-1RII have been found in the synovial fluid (39) and plasma of individuals with RA (78), and these negatively correlated with severity of disease, suggesting IL-1RII acts as natural antagonist of IL-1-driven joint destruction. In contrast, plasma levels of IL-1Ra correlated positively with disease progression, possibly reflecting disease exacerbation (78). These data are in line with experimental in vitro and in vivo data showing that overexpression of IL-1RII in chondrocytes protected them from IL-1 stimulation (85), or that the transfer of cells overexpressing and releasing IL-1RII resulted in the inhibition of collagen-induced arthritis (52). These results, as well as the binding properties of IL-1RII (high affinity for IL-1, low affinity for IL-1ra), supported the development of IL-1RII as therapeutic molecule in rheumatoid arthritis (see below). The expression of both receptors for IL-1 was demonstrated by immunostaining and laser confocal microscopy in sarcolemma from human muscle tissue samples, at higher levels in patients with polymyositis and dermatomyositis as compared with healthy individuals, together with increased expression of IL-1α, IL-1β, and IL-1Ra (86).
IL-1RII is upregulated in some tumors, including pancreatic ductal adenocarcinoma (87), prostatic cancer and benign prostatic hyperplasia (88), and ovarian cancer, where it provides a powerful distinction between primary and recurrent tumors (89).
In contrast to these conditions associated to upregulation of IL-1RII, in other contexts, defective expression of IL-1RII has been associated to the pathogenesis of the disease. For instance, gene-array analysis of osteoarthritic lesions indicated a lack of expression of IL-1RII and IL-1ra (85), suggesting that defective expression of negative regulators of the IL-1 system contributes to pathogenesis. Similarly, endometriosis and endometrioid ovarian cancer are associated with lower levels of serum and local IL-1RII and with IL-1RII polymorphisms (90– 92). In the context of atherosclerosis, it has been proposed that, since macrophages from hyperlipidemic patients have decreased IL-1RII mRNA and protein expression, IL-1-dependent inflammation could be relatively unchecked during atheroma formation (77). Genome-wide association studies identified several candidate genes potentially involved in inflammatory bowel disease (IBD) pathogenesis, including IL-1RII (91).
Autoimmune inner ear disease is characterized by recurring episodes of sudden or progressive sensorineural hearing loss. Defective responsiveness to corticosteroid in this disease has been correlated to the low induction of IL-1RII in peripheral blood mononuclear cells (93).
Secretion of embryonic IL-1β is one of the first responses of the blastocyst to the receptive endometrium. IL-1β is involved in inducing molecular changes that are essential for attachment of the blastocyst, such as immunomodulation, angiogenesis, and endometrial tissue remodeling. In this context, it has been proposed that these IL-1 activities are regulated by chorionic gonadotropin, which acts directly on endometrial epithelial cells to down-regulate the synthesis and release of IL-1RII (94).
The IL-1 decoy receptor IL-1RII was originally tested as a therapeutic by Amgen in arthritis, based on the promising results in this context (45), but no clinical development of this agent has been reported. Recently, the soluble IL-1RI (Rilonacept) was introduced as therapeutic and approved by the FDA for selected autoinflammatory diseases, in particular cryopyrin-associated periodic syndromes (familial cold autoinflammatory syndrome and Muckle–Wells Syndrome) (95). The drug consists in a fusion protein containing the extracellular domains of IL-1R1 and IL-1RAcP coupled to the Fc region of human IgG1. Rilonacept acts similarly to soluble IL-1RII, as a decoy, by binding IL-1β and IL-1α with higher affinity than IL-1Ra (96).
The Negative Regulator TIR8/SIGIRR
Gene and Protein
TIR8/SIGIRR gene is localized on human chromosome 11 and on murine chromosome 7 (97). The 410 amino acid-long protein is constituted by a single Ig extracellular domain, a transmembrane domain, an intracellular conserved TIR domain, and a 95 amino acid-long tail at the C-terminal, reminiscent of the intracellular tails of few ILR/TLR family members, in particular IL-1AcPb and TIGIRR (see below) (Figure 2). Both in human and mouse, TIR8/SIGIRR has several N- and O-glycosylation sites in the extracellular domain (97, 98). The sequence and pattern of expression of TIR8/SIGIRR is conserved among vertebrates, from chicken to humans (99). In particular, human and mouse protein sequences share 82% homology. TIR8/SIGIRR is expressed in several tissues, particularly in kidney, digestive tract, liver, lung, and lymphoid organs (97, 100).
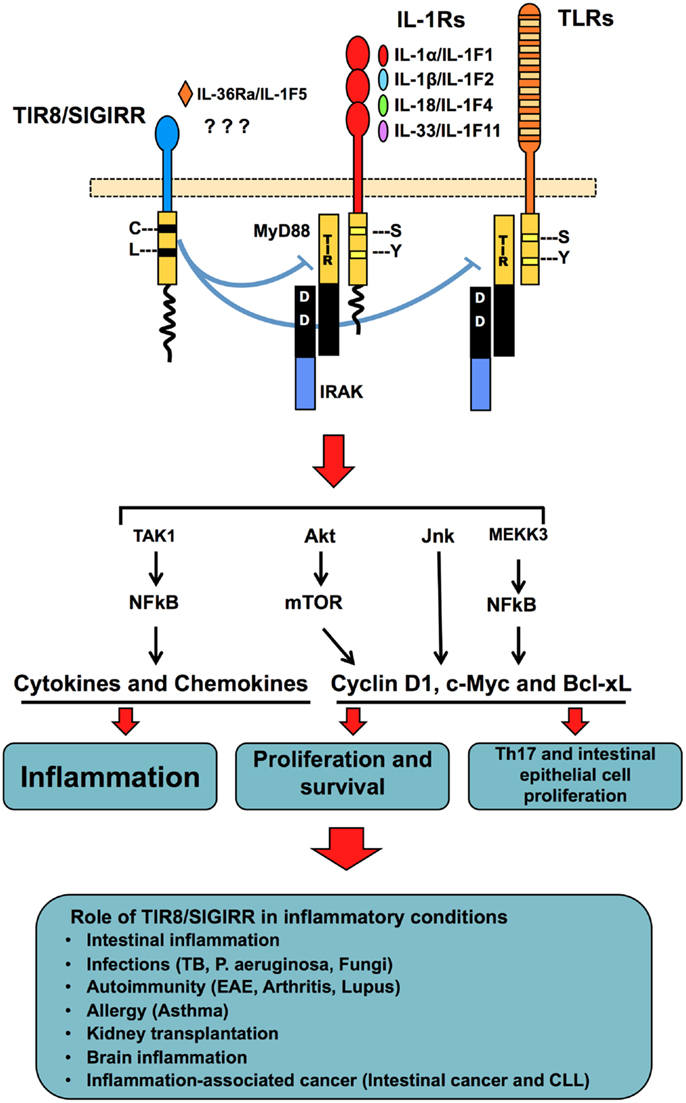
Figure 2. TIR8/SIGIRR, a negative regulatory receptor of ILR and TLR signaling. TIR8/SIGIRR is composed by a single extracellular Ig domain, a transmembrane domain, an intracellular conserved TIR domain and a long intracellular tail. Two replaced amino acid in the TIR domain (Cys 222, Leu 305) are potentially involved in non-conventional activation. TIR8/SIGIRR is an orphan receptor, but IL-36Ra has been proposed as a TIR8/SIGIRR ligand in glial cells. TIR8/SIGIRR inhibits ILR and TLR signaling by competing with MyD88 and IRAK recruitment at the TIR domain thus dampening the signaling pathway leading to NF-kB activation. In T cells and epithelial cells, TIR8/SIGIRR inhibits IL-1-dependent activation of the Akt- mTOR pathway and of JNK, thus controlling cell proliferation and survival. In vivo studies in gene-targeted mice demonstrate that Tir8/Sigirr acts as a non-redundant negative regulator in different inflammatory conditions dependent on ILRs or TLRs.
TIR8/SIGIRR proximal promoter has a binding site for SP1, which enhances its transcription in basal conditions (101). LPS stimulation reduces SP1 binding to TIR8/SIGIRR promoter, possibly explaining the TIR8/SIGIRR down-regulation in inflammatory conditions (LPS administration, ulcerative colitis, lung and urinary infections,infestations) (100–106). Recent studies demonstrated lower expression of TIR8/SIGIRR in fetal human enterocytes providing a reasonable explanation to the excessive inflammatory response in the immature intestine (107).
In contrast with these studies, TIR8/SIGIRR up-regulation was shown in human monocytes during sepsis and sterile systemic inflammation (108). Th2-lymphocytes expressed higher levels of TIR8/SIGIRR compared to Th1 polarized or non-differentiated lymphocytes (109). Pseudomonas aeruginosa infected mice showed up-regulation of Tir8/Sigirr in the cornea, macrophages, and Langerhans cells through the activity of vasoactive intestinal peptide (110). Lactobacillus jensenii, a probiotic microorganism, induced up-regulation of TIR8/SIGIRR in porcine Payer’s patch antigen presenting cells through activation of TLR2 (111). Similarly, LPS-induced Tir8/Sigirr in murine Payer’s patch DCs, but not in spleen DCs (112). These data suggest that Payer’s patch DCs use Tir8/Sigirr to tune TLRs signaling.
Mechanisms of Negative Regulation
The function of TIR8/SIGIRR consists in the specific inhibition of NF-kB and JNK activation following stimulation of ILR or TLR family members (102, 113). TIR8 can modulate the signal transduction activated by IL-1RI, IL-18R, T1/ST2, TLR1/2, TLR3, TLR4, TLR7, and TLR9 (98, 102, 109, 113–115) (Figure 2).
The extracellular Ig-like domain of TIR8/SIGIRR has been show to interfere with the dimerization of IL-1RI and IL-1RAcP. The cytoplasmic TIR domain binds TIR-containing adaptor molecules, which are no more available for signaling, whereas the cytoplasmic tail is not involved in the inhibitory activity (102, 114). A computational approach suggests a three-dimensional model for the interaction among the TIR domains of TLR4, TLR7, MyD88, and TIR8/SIGIRR. In this model, TIR8/SIGIRR binds TLR4 and TLR7 through its BB-loop region preventing their dimerization and MyD88 recruitment (116).
TIR8/SIGIRR can also regulate mTOR kinase activity in Th17 lymphocytes (117) and in intestinal epithelial cells (118) (Figure 2). These results are in agreement with the role of TIR8/SIGIRR in autoimmune diseases and in tumor suppression (see below).
Role of TIR8/SIGIRR in Vivo
Infection-associated inflammation
Tir8/Sigirr-deficient mice are more susceptible than wild type mice in several infections, such as tuberculosis, candidiasis, aspergillosis, P. aeruginosa infection, in terms of mortality and tissue damage due to an exaggerated inflammatory response (103, 106, 119, 120) (Figure 2). Results obtained with IL-1-blocking antibodies and IL-1RI-deficient mice indicated that in some of these infectious conditions (tuberculosis and P. aeruginosa lung infection), TIR8/SIGIRR played a major role in dampening inflammation induced by IL-1R activation.
Similarly, in a colitis mouse model, Tir8/Sigirr-deficient mice developed a more severe gut inflammation compared to wild type mice (113, 121). Commensal microflora activates enterocyte TLRs and consequently induces survival of epithelial cells and maintains gut homeostasis (122, 123). Lack of Tir8/Sigirr in colon epithelial cells was shown to be associated to constitutive NF-kB and JNK activation and up-regulated expression of Cyclin D1 and Bcl-xL in homeostatic conditions, which returned to the control level after depletion of commensal bacteria (121) (Figure 2).
Excessive systemic inflammation was observed in Tir8/Sigirr-deficient mice upon LPS challenge, and reduced inflammation and mortality were described in Tir8/Sigirr overexpressing mice in LPS-dependent acute lung injury model (102, 124). However, these phenotypes possibly depend on the genetic background since excessive systemic or local inflammatory reactions to LPS were not confirmed in other studies (102, 113).
In contrast with these data, in a urinary tract infection model, Tir8/Sigirr inhibited an effective host response against uropathogenic E. coli, as indicated by lower renal bacterial load and dysfunction in TIR8-deficient mice, associated to increased circulating and intrarenal neutrophils at the early phase of infection (125).
Sterile inflammatory conditions
Recent data suggest that TIR8/SIGIRR plays a direct role in inhibiting different IL-1-dependent signaling pathways, including IL-1R-mTOR, in Th17 lymphocytes, thus tuning initial Th17 differentiation and preventing Th17 cell-mediated pathogenic effects (117) (Figure 2). This effect was particularly evident in the control of CNS autoimmune inflammation in a model of experimental autoimmune encephalomyelitis (117). Tir8/Sigirr deficiency was also associated to increased susceptibility to develop autoimmunity in a model of systemic lupus erythematosus (B6lpr/lpr), as well as in a model of lupus nephritis induced by hydrocarbon oil (pristane) (115, 126). In the lpr/lpr model, Tir8/Sigirr deficiency was responsible for massive lymphoproliferation, peribronchial inflammation, and mesangio-proliferative glomerulonephritis, due to B and dendritic cell hyper-activation in TLR7- and TLR9-dependent response to autoantigens and nucleosomes (115). Tir8/Sigirr-deficient mice were also more susceptible than wild type mice to both zymosan-induced and collagen antibody-induced arthritis models, because of excessive inflammation at least in part dependent on IL-1 (127) (Figure 2).
In agreement with the results obtained in autoimmunity mouse models, TIR8/SIGIRR was down modulated together with other anti-inflammatory genes in psoriatic patients (128).
Studies on allergic inflammatory responses showed that Tir8/Sigirr plays an important role also in controlling the axis IL-33 – ST2 which is involved in Th2 cell polarization and Th2 cytokine production (109) (Figure 2).
DAMPs generated during renal ischemia/reperfusion are responsible of the activation of intrarenal DCs, macrophages, and neutrophils via TLRs and IL-1R, which are potentially involved in post ischemic renal failure. In models of renal ischemia/reperfusion or kidney transplantation, Tir8/Sigirr-deficient mice showed increased renal injury or severe graft rejection, respectively, associated to excessive cytokine and chemokine production and consequently, leukocyte recruitment and amplified adaptive immune response against donor antigens (129, 130) (Figure 2).
Finally, in agreement with the expression in neurons, microglia, and astrocytes (131), TIR8/SIGIRR was shown to be a modulator of microglia activation by LPS, and of neuroinflammation (132). Furthermore, Tir8/Sigirr-deficient mice showed impaired cognitive and synaptic functions associated to up-regulated IL-1R1 and TLR4 signaling in hippocampal tissue in response to IL-1α and high mobility group box 1 (133). Studies on the anti-inflammatory activity of IL-36Ra in the brain demonstrated at least a partial involvement of TIR8/SIGIRR in down modulating glial cell inflammatory responses through the production of IL-4 (18).
Cancer-related inflammation
Chronic inflammation is associated with promotion of malignancy and tumor progression and several studies in animals have shown the protumoral role of IL-1 in this context (134, 135). Along the same line, in different murine models, TIR8/SIGIRR has been demonstrated to play a key protective role in the pathogenesis of cancer-related inflammation. In a model of colitis-associated cancer (CAC), a colorectal disease that arises in patients suffering from chronic IBD, Tir8/Sigirr-deficient mice were highly susceptible to both inflammation and carcinogenesis in terms of number, size, and severity of lesions (121, 136) (Figure 2). The mechanism proposed suggests that TIR8/SIGIRR plays a protective role probably by modulating the signaling activated by commensal bacteria through TLRs in the epithelial cells and consequently, downstream events, including production of inflammatory mediators and factors involved in cell survival and proliferation, leukocyte recruitment, and angiogenesis (137). Moreover, Tir8/Sigirr deficiency in the Apcmin/+ mouse model was associated to increased intestinal lesion development due to higher Akt-mTOR activity, a crucial tumorigenic pathway (118, 138). The data suggest that Tir8/Sigirr exerts a tumor suppressor activity by controlling IL-1- and TLR-induced mTOR-mediated cell cycle progression and consequent genetic instability (118).
In Chronic Lymphocytic Leukemia (CLL), human malignant B cells express lower levels of TIR8/SIGIRR mRNA than normal B cells (139, 140). Similar results were found in the mouse where CD19+ cells express lower levels of Tir8 messenger compared to CD19+ cells isolated from a transgenic mouse model of CLL (TCL1 mice) (141). In CLL, both genetic (e.g., MyD88 mutations) and micro environmental factors concur to the development, expansion, and progression of the disease (139, 142). In a murine CLL model, the absence of Tir8/Sigirr led to a more severe and earlier appearance of monoclonal B-cell expansions and to shortened life span. The disease mimicked the aggressive variant of human CLL, characterized by the appearance of prolymphocytes (141) (Figure 2), suggesting that TIR8/SIGIRR acts as an inhibitor of CLL progression through a still unclear molecular mechanism.
Other ILR with Negative Regulatory Properties
IL-18 Binding Protein
IL-18 binding protein (IL-18BP) is a secreted high affinity IL-18 binding molecule, which acts as a potent inhibitor of IL-18 and a modulator of Th1 response. It is constituted by only one Ig-like domain and it is structurally and functionally similar to IL-1RII (30). Indeed, phylogenetic analysis suggests that IL-18BP and IL-1RII had a common ancestral gene and diverged at the level of fish (31). Recombinant IL-1F7 also binds to the IL-18BP, further increasing the ability of IL-18BP to neutralize IL-18 activity (143).
Proteins homolog to IL-18BP have been found in poxviruses (Ectromelia), which are responsible of neutralization of human IL-18 during the viral infection and of dampening the inflammatory response associated to the infection (144).
Further information about this molecule is available in the review by Dinarello et al. in this issue.
IL-1RAcPb
The IL-1RAcP is the receptor subunit of the IL-1RI complex, and it is also used by IL-36α/IL-1F6, IL-36β/IL-1F8 and IL-36γ/IL-1F9, and IL-33 receptors. It has been shown that an alternative form of AcP, called AcPb, can be generated by alternative splicing, in which the prototypical AcP C-terminal exon 12 is skipped and an alternative exon 12b is used (145, 146). Smith et al. (146) characterized this molecule and showed its regulatory properties in the brain. The C-terminus encoded by these two alternative exons has 35% amino acid identity, which includes conserved motifs of the TIR domain. Moreover, the exon 12b encodes a sequence of approximately 140 additional amino acids in the C-terminal of the TIR domain that has no homology to other protein sequences and is of unknown function. The general structure of AcPb is similar to that of AcP and suggests that the AcPb cytoplasmic domain is similar to AcP TIR domain. However, there is one area of substantial difference because of changed configuration in the DD loop and aD helix regions of the AcPb TIR domain, which remembers modification of TIR8/SIGIRR TIR domain, and altered charge distribution pattern on its surface. It has been proposed that these modifications could affect interaction with adapter and signaling molecules. Indeed, AcPb is capable of forming a ligand-dependent complex with IL-1R, but it does not lead to the recruitment of the adaptor molecules MyD88 and IRAK4 after stimulation with IL-1, and is unable to mediate specific IL-1 responses. In both human and mouse, the expression of the AcPb is restricted to the CNS (whole brain, fetal brain, cerebellum, and spinal cord). AcP and AcPb are coexpressed in the same cells, but AcPb is the more abundant isoform. It has been proposed that AcPb could also be recruited to other AcP-utilizing receptors, such as ST2 and IL-1Rrp2/IL-36R, which are expressed in the CNS, once they have bound their ligands (IL-33 and IL-36α,β,γ, respectively). In a model of LPS challenge in the CNS, AcPb-deficiency was associated to neuronal loss suggesting that AcPb may dampen the neurotoxic effects of IL-1 by modulating the intracellular signaling and gene expression response to LPS-induced IL-1, or possibly to other cytokines acting through AcP (146). The inhibitory effect of AcPb could depend on the failure to recruit MyD88 and IRAK4, on the competition with AcP in a IL-1 receptor complex containing multiple IL-1R and AcP molecules, or on unknown functions mediated by the C-terminal tail.
TIGIRR-1 and IL-1RAPL
TIGIRR-1 and IL-1RAPL (also named TIGIRR-2) are localized on the X chromosome and share between 22 and 48% overall identity to other ILR family members. IL-1RAPL and TIGIRR-1 exons are spread out over a very large segment of genomic DNA (more than 1500 kb for IL-1RAPL and 380 kb for TIGIRR-1). Both TIGIRR-1 and IL-1RAPL contain a signal peptide, three predicted extracellular Ig domains, a single transmembrane domain, and a highly conserved cytoplasmic region containing a C-terminal cytoplasmic extension reminiscent of the Drosophila Toll family, TIR8/SIGIRR, and AcPb cytoplasmic domains.
A negative regulatory role has not yet been reported for these two receptors. However, in in vitro studies performed with chimeric molecules, the cytoplasmic domains of TIGIRR-1 and IL-1RAPL fused to the extracellular and transmembrane domains of IL-1RI or AcP could not induce NF-κB, similarly to TIR8/SIGIRR, and in contrast with the cytoplasmic domains of other members of the ILR family (10). Other functional studies showed that IL-1RAPL can activate JNK but not the ERK or the p38 MAP kinases, whereas TIGIRR-1 cannot activate JNK. Deletion mutagenesis studies showed that the activation of JNK by IL-1RAPL does not depend on the integrity of its TIR domain, suggesting a distinct mechanism of signaling through this receptor (147).
TIGIRR-1 is highly conserved in human and mouse (94.5% identical at the amino acid level) and it is expressed in skin, liver, placenta, and fetal brain. IL-1RAPL, whose crystal structure has been determined (147), is expressed in heart, brain, ovary, skin, and to a lesser extent in tonsil, fetal liver, prostate, testis, small intestine, placenta, and colon. IL-1RAPL was identified as the gene responsible for hereditary non-syndromic mental retardation and autism linked to chromosome region Xp22.1–21.3 (148, 149). It is expressed in brain structures involved in the hippocampal memory system, and it has a role in brain development and function (150). No information are available about a potential role of IL-1RAPL in inflammation and defense, however, its C-terminal extension is reported to interact with neuronal calcium sensor-1 and regulate neurite outgrowth (150–152).
DIGIRR
Recently, a new member called DIGIRR was added to the ILR family (153). DIGIRR was discovered in teleost fish and showed high homology with TIR8/SIGIRR. DIGIRR is characterized by an extracellular portion comprising two Ig-like domains, a transmembrane domain and TIR domain carrying two amino acid substitutions (Arg419-Tyr420), which are responsible for the loss of signaling. The DIGIRR mRNA was found expressed in several tissues and in leukocytes and was upregulated by LPS, oppositely to TIR8/SIGIRR, suggesting a different mechanism of response to inflammatory stimuli between the two molecules. At the subcellular level, DIGIRR showed a peculiar distribution within the Golgi apparatus.
Different lines of evidence suggest that DIGIRR acts as negative regulator of LPS- and IL-1β-induced inflammation. Indeed, siRNA knock down of DIGIRR lead to increased production of IL-β-induced pro-inflammatory cytokines in liver, kidney, and leukocytes. Moreover, in vitro administration of DIGIRR to zebrafish embryos significantly inhibited LPS- and IL-1β-induced activation of NF-kB.
The discovery of DIGIRR could help to understand the evolution of ILR family members. Indeed, the authors suggest the hypothesis that DIGIRR and TIR8/SIGIRR derive from a common ancestral molecule that lost respectively one or two Ig-like extracellular domains, and Ser or Arg-Tyr- amino acids in the TIR domain. DIGIRR might represent an evolutionary intermediate molecule between IL-1R and TIR8/SIGIRR, demonstrating a shift from a potent receptor to a negative regulator.
Concluding Remarks
Studies conducted in the early 1990s suggesting that the non-signaling IL-1RII acts as a molecular trap for the agonist and the AcP, led to the formulation of the decoy paradigm, which has then been extended to other cytokine families and chemokines. Decoy receptors are now recognized as a general strategy to tune the actions of primary inflammatory cytokines and chemokines.
The list of ILR/TLR family receptors acting as negative regulators now includes TIR8/SIGIRR, which acts by modulating ILRs- or TLRs-dependent signaling. In addition to these molecules, soluble forms of signaling receptors or AcP act as decoys or negative regulators by trapping the ligands. For instance, T1/ST2 exists also as a soluble isoform obtained by differential mRNA processing, which acts as an antagonistic decoy receptor for IL-33 (154), and has been proposed in the therapy of arthritis (155). Similarly, soluble IL-1AcP, generated by alternative splicing, forms a complex with IL-1β and IL-1RII playing a protective role in arthritis (156) and has been pharmacologically exploited.
For several of these molecules further studies have to be performed to unequivocally define their role in disease and their potential as therapeutic targets. For instance, unfortunately there are no genetic evidence on the consequence of IL-1RII-gene deficiency or data supporting the relevance of TIR8/SIGIRR in human disease. In addition, the clinical development of IL-RII pharmacological targeting has not been reported. Finally, pharmacological approaches targeting TIR8/SIGIRR functions have not been developed yet and they will be necessary to assess whether TIR8/SIGIRR might be a therapeutic target in inflammatory conditions.
However, the existence of IL-1RII, together with IL-1Ra, TIR8/SIGIRR, brain AcPb, and other soluble receptors acting as molecular traps emphasizes the need for tight control of the IL-1 system, which mediates potentially devastating local and systemic inflammatory reactions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The contributions of the European Commission (European Research Council project HIIS, MUGEN LSHG-CT-2005-005203, MUVAPRED LSH-CT-2003-503240 and TIMER HEALTH F4-2011-281608), Ministero dell’Istruzione, dell’Università e della Ricerca [progetto FIRB RBLA039LSF (www.miur.it)], Associazione Italiana per la Ricerca sul Cancro (AIRC), Fondazione CARIPLO, and the Italian Cystic Fibrosis Research Foundation (http://www.fibrosicisticaricerca.it) are gratefully acknowledged.
References
1. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol (2003) 21:335–76. doi:10.1146/annurev.immunol.21.120601.141126
2. O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev (2008) 226:10–8. doi:10.1111/j.1600-065X.2008.00701.x
3. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol (2009) 27:519–50. doi:10.1146/annurev.immunol.021908.132612
4. Dinarello CA. Anti-inflammatory agents: present and future. Cell (2010) 140:935–50. doi:10.1016/j.cell.2010.02.043
5. van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol (2011) 32:110–6. doi:10.1016/j.it.2011.01.003
6. Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science (2003) 300:1524–5. doi:10.1126/science.1085536
7. Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem (2004) 279:13677–88. doi:10.1074/jbc.M400117200
8. Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev (2008) 223:20–38. doi:10.1111/j.1600-065X.2008.00624.x
9. Dinarello C, Arend W, Sims J, Smith D, Blumberg H, O’Neill L, et al. IL-1 family nomenclature. Nat Immunol (2010) 11:973. doi:10.1038/ni1110-973
10. Born TL, Smith DE, Garka KE, Renshaw BR, Bertles JS, Sims JE. Identification and characterization of two members of a novel class of the interleukin-1 receptor (IL-1R) family. Delineation of a new class of IL-1R-related proteins based on signaling. J Biol Chem (2000) 275:29946–54. doi:10.1074/jbc.M004077200
11. Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, et al. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol (2005) 174:2942–50.
12. Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res (2007) 67:1062–71. doi:10.1158/0008-5472.CAN-06-2956
13. Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol (2010) 10:103–10. doi:10.1038/nri2692
14. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood (2011) 117:3720–32. doi:10.1182/blood-2010-07-273417
15. Towne JE, Sims JE. IL-36 in psoriasis. Curr Opin Pharmacol (2012) 12:486–90. doi:10.1016/j.coph.2012.02.009
16. Mantovani A, Locati M, Polentarutti N, Vecchi A, Garlanda C. Extracellular and intracellular decoys in the tuning of inflammatory cytokines and Toll-like receptors: the new entry TIR8/SIGIRR. J Leukocyte Biol (2004) 75:738–42. doi:10.1189/jlb.1003473
17. Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol (2005) 5:446–58. doi:10.1038/nri1630
18. Costelloe C, Watson M, Murphy A, McQuillan K, Loscher C, Armstrong ME, et al. IL-1F5 mediates anti-inflammatory activity in the brain through induction of IL-4 following interaction with SIGIRR/TIR8. J Neurochem (2008) 105:1960–9. doi:10.1111/j.1471-4159.2008.05304.x
19. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, Van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med (2009) 360:2426–37. doi:10.1056/NEJMoa0807865
20. Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med (2009) 360:2438–44. doi:10.1056/NEJMoa0809568
21. Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr., Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell (2002) 110:191–202. doi:10.1016/S0092-8674(02)00827-9
22. Janssens S, Burns K, Vercammen E, Tschopp J, Beyaert R. MyD88S, a splice variant of MyD88, differentially modulates NF-kappaB- and AP-1-dependent gene expression. FEBS Lett (2003) 548:103–7. doi:10.1016/S0014-5793(03)00747-6
23. Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci U S A (2009) 106:5282–7. doi:10.1073/pnas.0810909106
24. Nahid MA, Satoh M, Chan EK. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol Immunol (2011) 8:388–403. doi:10.1038/cmi.2011.26
25. Quinn SR, O’Neill LA. A trio of microRNAs that control Toll-like receptor signalling. Int Immunol (2011) 23:421–5. doi:10.1093/intimm/dxr034
26. McMahan CJ, Slack JL, Mosley B, Cosman D, Lupton SD, Brunton LL, et al. A novel IL-1 receptor, cloned from B cells by mammalian expression, is expressed in many cell types. EMBO J (1991) 10:2821–32.
27. Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, et al. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science (1993) 261:472–5. doi:10.1126/science.8332913
28. Copeland NG, Silan CM, Kingsley DM, Jenkins NA, Cannizzaro LA, Croce CM, et al. Chromosomal location of murine and human IL-1 receptor genes. Genomics (1991) 9:44–50. doi:10.1016/0888-7543(91)90219-5
29. Morrison RN, Young ND, Nowak BF. Description of an Atlantic salmon (Salmo salar L.) type II interleukin-1 receptor cDNA and analysis of interleukin-1 receptor expression in amoebic gill disease-affected fish. Fish Shellfish Immunol (2012) 32:1185–90. doi:10.1016/j.fsi.2012.03.005
30. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity (1999) 10:127–36. doi:10.1016/S1074-7613(00)80013-8
31. Watanabe M, Goto N, Watanabe Y, Nishiguchi S, Shimada K, Yasunga T, et al. Evolution of interleukin-18 binding proteins and interleukin-1 receptor, type II proteins. Int J Mol Med (2005) 15:561–6.
32. Mantovani A, Muzio M, Ghezzi P, Colotta C, Introna M. Regulation of inhibitory pathways of the interleukin-1 system. Ann N Y Acad Sci (1998) 840:338–51. doi:10.1111/j.1749-6632.1998.tb09573.x
33. Orlando S, Sironi M, Bianchi G, Drummond AH, Boraschi D, Yabes D, et al. Role of metalloproteases in the release of the IL-1 type II decoy receptor. J Biol Chem (1997) 272:31764–9. doi:10.1074/jbc.272.50.31764
34. Lorenzen I, Lokau J, Dusterhoft S, Trad A, Garbers C, Scheller J, et al. The membrane-proximal domain of A Disintegrin and Metalloprotease 17 (ADAM17) is responsible for recognition of the interleukin-6 receptor and interleukin-1 receptor II. FEBS Lett (2012) 586:1093–100. doi:10.1016/j.febslet.2012.03.012
35. Kuhn PH, Marjaux E, Imhof A, De Strooper B, Haass C, Lichtenthaler SF. Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J Biol Chem (2007) 282:11982–95. doi:10.1074/jbc.M700356200
36. Cui X, Rouhani FN, Hawari F, Levine SJ. Shedding of the type II IL-1 decoy receptor requires a multifunctional aminopeptidase, aminopeptidase regulator of TNF receptor type 1 shedding. J Immunol (2003) 171:6814–9.
37. Wang D, Zhang S, Li L, Liu X, Mei K, Wang X. Structural insights into the assembly and activation of IL-1beta with its receptors. Nat Immunol (2010) 11:905–11. doi:10.1038/ni.1925
38. Symons JA, Young PR, Duff GW. Soluble type II interleukin 1 (IL-1) receptor binds and blocks processing of IL-1 beta precursor and loses affinity for IL-1 receptor antagonist. Proc Natl Acad Sci U S A (1995) 92:1714–8. doi:10.1073/pnas.92.5.1714
39. Arend WP, Malyak M, Smith MF Jr., Whisenand TD, Slack JL, Sims JE, et al. Binding of IL-1 alpha, IL-1 beta, and IL-1 receptor antagonist by soluble IL-1 receptors and levels of soluble IL-1 receptors in synovial fluids. J Immunol (1994) 153:4766–74.
40. Re F, Sironi M, Muzio M, Matteucci C, Introna M, Orlando S, et al. Inhibition of interleukin-1 responsiveness by type II receptor gene transfer: a surface “receptor” with anti-interleukin-1 function. J Exp Med (1996) 183:1841–50. doi:10.1084/jem.183.4.1841
41. Lang D, Knop J, Wesche H, Raffetseder U, Kurrle R, Boraschi D, et al. The type II IL-1 receptor interacts with the IL-1 receptor accessory protein: a novel mechanism of regulation of IL-1 responsiveness. J Immunol (1998) 161:6871–7.
42. Malinowsky D, Lundkvist J, Laye S, Bartfai T. Interleukin-1 receptor accessory protein interacts with the type II interleukin-1 receptor. FEBS Lett (1998) 429:299–302. doi:10.1016/S0014-5793(98)00467-0
43. Liu C, Hart RP, Liu XJ, Clevenger W, Maki RA, De Souza EB. Cloning and characterization of an alternatively processed human type II interleukin-1 receptor mRNA. J Biol Chem (1996) 271:20965–72. doi:10.1074/jbc.271.34.20965
44. Jensen LE, Muzio M, Mantovani A, Whitehead AS. IL-1 signaling cascade in liver cells and the involvement of a soluble form of the IL-1 receptor accessory protein. J Immunol (2000) 164:5277–86.
45. Smith DE, Hanna R, Della F, Moore H, Chen H, Farese AM, et al. The soluble form of IL-1 receptor accessory protein enhances the ability of soluble type II IL-1 receptor to inhibit IL-1 action. Immunity (2003) 18:87–96. doi:10.1016/S1074-7613(02)00514-9
46. Zheng Y, Humphry M, Maguire JJ, Bennett MR, Clarke MC. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity (2013) 38:285–95. doi:10.1016/j.immuni.2013.01.008
47. Kawaguchi Y, Nishimagi E, Tochimoto A, Kawamoto M, Katsumata Y, Soejima M, et al. Intracellular IL-1alpha-binding proteins contribute to biological functions of endogenous IL-1alpha in systemic sclerosis fibroblasts. Proc Natl Acad Sci U S A (2006) 103:14501–6. doi:10.1073/pnas.0603545103
48. Kobayashi Y, Yamamoto K, Saido T, Kawasaki H, Oppenheim JJ, Matsushima K. Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1 alpha. Proc Natl Acad Sci U S A (1990) 87:5548–52. doi:10.1073/pnas.87.14.5548
49. Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Luthi AU, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol Cell (2011) 44:265–78. doi:10.1016/j.molcel.2011.07.037
50. Clarke MC, Talib S, Figg NL, Bennett MR. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res (2010) 106:363–72. doi:10.1161/CIRCRESAHA.109.208389
51. Rauschmayr T, Groves RW, Kupper TS. Keratinocyte expression of the type 2 interleukin 1 receptor mediates local and specific inhibition of interleukin 1-mediated inflammation. Proc Natl Acad Sci U S A (1997) 94:5814–9. doi:10.1073/pnas.94.11.5814
52. Bessis N, Guery L, Mantovani A, Vecchi A, Sims JE, Fradelizi D, et al. The type II decoy receptor of IL-1 inhibits murine collagen-induced arthritis. Eur J Immunol (2000) 30:867–75. doi:10.1002/1521-4141(200003)30:3<867::AID-IMMU867>3.0.CO;2-M
53. Dawson J, Engelhardt P, Kastelic T, Cheneval D, MacKenzie A, Ramage P. Effects of soluble interleukin-1 type II receptor on rabbit antigen-induced arthritis: clinical, biochemical and histological assessment. Rheumatology (1999) 38:401–6. doi:10.1093/rheumatology/38.5.401
54. Simeoni E, Dudler J, Fleury S, Li J, Pagnotta M, Pascual M, et al. Gene transfer of a soluble IL-1 type 2 receptor-Ig fusion protein improves cardiac allograft survival in rats. Eur J Cardiothorac Surg (2007) 31:222–8. doi:10.1016/j.ejcts.2006.10.042
55. Chang H, Wang Y, Wu W, Li G, Hanawa H, Zou J. Hydrodynamics-based delivery of an interleukin-1 receptor II fusion gene ameliorates rat autoimmune myocarditis by inhibiting IL-1 and Th17 cell polarization. Int J Mol Med (2013) 31:833–40. doi:10.3892/ijmm.2013.1276
56. Khoufache K, Bondza PK, Harir N, Daris M, Leboeuf M, Mailloux J, et al. Soluble human IL-1 receptor type 2 inhibits ectopic endometrial tissue implantation and growth: identification of a novel potential target for endometriosis treatment. Am J Pathol (2012) 181:1197–205. doi:10.1016/j.ajpath.2012.06.022
57. Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Immunol Today (2000) 21:447–55. doi:10.1016/S0167-5699(00)01699-6
58. Colotta F, Saccani S, Giri JG, Dower SK, Sims JE, Introna M, et al. Regulated expression and release of the IL-1 decoy receptor in human mononuclear phagocytes. J Immunol (1996) 156:2534–41.
59. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol (2006) 177:7303–11.
60. Pinteaux E, Parker LC, Rothwell NJ, Luheshi GN. Expression of interleukin-1 receptors and their role in interleukin-1 actions in murine microglial cells. J Neurochem (2002) 83:754–63. doi:10.1046/j.1471-4159.2002.01184.x
61. McNamee EN, Ryan KM, Kilroy D, Connor TJ. Noradrenaline induces IL-1ra and IL-1 type II receptor expression in primary glial cells and protects against IL-1beta-induced neurotoxicity. Eur J Pharmacol (2010) 626:219–28. doi:10.1016/j.ejphar.2009.09.054
62. Docagne F, Campbell SJ, Bristow AF, Poole S, Vigues S, Guaza C, et al. Differential regulation of type I and type II interleukin-1 receptors in focal brain inflammation. Eur J Neurosci (2005) 21:1205–14. doi:10.1111/j.1460-9568.2005.03965.x
63. Tran DQ, Andersson J, Hardwick D, Bebris L, Illei GG, Shevach EM. Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human FOXP3+ regulatory T cells allows for their purification from expansion cultures. Blood (2009) 113:5125–33. doi:10.1182/blood-2009-01-199950
64. Mercer F, Kozhaya L, Unutmaz D. Expression and function of TNF and IL-1 receptors on human regulatory T cells. PLoS ONE (2010) 5:e8639. doi:10.1371/journal.pone.0008639
65. Trebec DP, Chandra D, Gramoun A, Li K, Heersche JN, Manolson MF. Increased expression of activating factors in large osteoclasts could explain their excessive activity in osteolytic diseases. J Cell Biochem (2007) 101:205–20. doi:10.1002/jcb.21171
66. Debets R, Hegmans JP, Croughs P, Troost RJ, Prins JB, Benner R, et al. The IL-1 system in psoriatic skin: IL-1 antagonist sphere of influence in lesional psoriatic epidermis. J Immunol (1997) 158:2955–63.
67. Akoum A, Jolicoeur C, Kharfi A, Aube M. Decreased expression of the decoy interleukin-1 receptor type II in human endometriosis. Am J Pathol (2001) 158:481–9. doi:10.1016/S0002-9440(10)63990-9
68. Spriggs MK, Nevens PJ, Grabstein K, Dower SK, Cosman D, Armitage RJ, et al. Molecular characterization of the interleukin-1 receptor (IL-1R) on monocytes and polymorphonuclear cells. Cytokine (1992) 4:90–5. doi:10.1016/1043-4666(92)90042-P
69. Colotta F, Re F, Muzio M, Polentarutti N, Minty A, Caput D, et al. Interleukin-13 induces expression and release of interleukin-1 decoy receptor in human polymorphonuclear cells. J Biol Chem (1994) 269:12403–6.
70. Re F, Muzio M, De Rossi M, Polentarutti N, Giri JG, Mantovani A, et al. The type II “receptor” as a decoy target for interleukin 1 in polymorphonuclear leukocytes: characterization of induction by dexamethasone and ligand binding properties of the released decoy receptor. J Exp Med (1994) 179:739–43. doi:10.1084/jem.179.2.739
71. Dickensheets HL, Donnelly RP. IFN-gamma and IL-10 inhibit induction of IL-1 receptor type I and type II gene expression by IL-4 and IL-13 in human monocytes. J Immunol (1997) 159:6226–33.
72. Kalliolias GD, Gordon RA, Ivashkiv LB. Suppression of TNF-alpha and IL-1 signaling identifies a mechanism of homeostatic regulation of macrophages by IL-27. J Immunol (2010) 185:7047–56. doi:10.4049/jimmunol.1001290
73. Daun JM, Ball RW, Burger HR, Cannon JG. Aspirin-induced increases in soluble IL-1 receptor type II concentrations in vitro and in vivo. J Leukocyte Biol (1999) 65:863–6.
74. Penton-Rol G, Orlando S, Polentarutti N, Bernasconi S, Muzio M, Introna M, et al. Bacterial lipopolysaccharide causes rapid shedding, followed by inhibition of mRNA expression, of the IL-1 type II receptor, with concomitant up-regulation of the type I receptor and induction of incompletely spliced transcripts. J Immunol (1999) 162:2931–8.
75. Colotta F, Orlando S, Fadlon EJ, Sozzani S, Matteucci C, Mantovani A. Chemoattractants induce rapid release of the interleukin 1 type II decoy receptor in human polymorphonuclear cells. J Exp Med (1995) 181:2181–6. doi:10.1084/jem.181.6.2181
76. Orlando S, Matteucci C, Fadlon EJ, Buurman WA, Bardella MT, Colotta F, et al. TNF-alpha, unlike other pro- and anti-inflammatory cytokines, induces rapid release of the IL-1 type II decoy receptor in human myelomonocytic cells. J Immunol (1997) 158:3861–8.
77. Pou J, Martinez-Gonzalez J, Rebollo A, Rodriguez C, Rodriguez-Calvo R, Martin-Fuentes P, et al. Type II interleukin-1 receptor expression is reduced in monocytes/macrophages and atherosclerotic lesions. Biochim Biophys Acta (2011) 1811:556–63. doi:10.1016/j.bbalip.2011.05.014
78. Jouvenne P, Vannier E, Dinarello CA, Miossec P. Elevated levels of soluble interleukin-1 receptor type II and interleukin-1 receptor antagonist in patients with chronic arthritis: correlations with markers of inflammation and joint destruction. Arthritis Rheum (1998) 41:1083–9. doi:10.1002/1529-0131(199806)41:6<1083::AID-ART15>3.0.CO;2-9
79. Giri JG, Wells J, Dower SK, McCall CE, Guzman RN, Slack J, et al. Elevated levels of shed type II IL-1 receptor in sepsis. Potential role for type II receptor in regulation of IL-1 responses. J Immunol (1994) 153:5802–9.
80. Muller B, Peri G, Doni A, Perruchoud AP, Landmann R, Pasqualini F, et al. High circulating levels of the IL-1 type II decoy receptor in critically ill patients with sepsis: association of high decoy receptor levels with glucocorticoid administration. J Leukoc Biol (2002) 72:643–9.
81. Chan KY, Leung FW, Lam HS, Tam YH, To KF, Cheung HM, et al. Immunoregulatory protein profiles of necrotizing enterocolitis versus spontaneous intestinal perforation in preterm infants. PLoS ONE (2012) 7:e36977. doi:10.1371/journal.pone.0036977
82. van Deuren M, Van Der Ven-Jongekrijg J, Vannier E, Van Dalen R, Pesman G, Bartelink AK, et al. The pattern of interleukin-1beta (IL-1beta) and its modulating agents IL-1 receptor antagonist and IL-1 soluble receptor type II in acute meningococcal infections. Blood (1997) 90:1101–8.
83. Dujmovic I, Mangano K, Pekmezovic T, Quattrocchi C, Mesaros S, Stojsavljevic N, et al. The analysis of IL-1 beta and its naturally occurring inhibitors in multiple sclerosis: the elevation of IL-1 receptor antagonist and IL-1 receptor type II after steroid therapy. J Neuroimmunol (2009) 207:101–6. doi:10.1016/j.jneuroim.2008.11.004
84. Garlind A, Brauner A, Hojeberg B, Basun H, Schultzberg M. Soluble interleukin-1 receptor type II levels are elevated in cerebrospinal fluid in Alzheimer’s disease patients. Brain Res (1999) 826:112–6. doi:10.1016/S0006-8993(99)01092-6
85. Attur MG, Dave M, Cipolletta C, Kang P, Goldring MB, Patel IR, et al. Reversal of autocrine and paracrine effects of interleukin 1 (IL-1) in human arthritis by type II IL-1 decoy receptor. Potential for pharmacological intervention. J Biol Chem (2000) 275:40307–15. doi:10.1074/jbc.M002721200
86. Grundtman C, Salomonsson S, Dorph C, Bruton J, Andersson U, Lundberg IE. Immunolocalization of interleukin-1 receptors in the sarcolemma and nuclei of skeletal muscle in patients with idiopathic inflammatory myopathies. Arthritis Rheum (2007) 56:674–87. doi:10.1002/art.22388
87. Ruckert F, Dawelbait G, Winter C, Hartmann A, Denz A, Ammerpohl O, et al. Examination of apoptosis signaling in pancreatic cancer by computational signal transduction analysis. PLoS ONE (2010) 5:e12243. doi:10.1371/journal.pone.0012243
88. Ricote M, Garcia-Tunon I, Bethencourt FR, Fraile B, Paniagua R, Royuela M. Interleukin-1 (IL-1alpha and IL-1beta) and its receptors (IL-1RI, IL-1RII, and IL-1Ra) in prostate carcinoma. Cancer (2004) 100:1388–96. doi:10.1002/cncr.20142
89. Laios A, O’Toole SA, Flavin R, Martin C, Ring M, Gleeson N, et al. An integrative model for recurrence in ovarian cancer. Mol Cancer (2008) 7:8. doi:10.1186/1476-4598-7-8
90. Akoum A, Al-Akoum M, Lemay A, Maheux R, Leboeuf M. Imbalance in the peritoneal levels of interleukin 1 and its decoy inhibitory receptor type II in endometriosis women with infertility and pelvic pain. Fertil Steril (2008) 89:1618–24. doi:10.1016/j.fertnstert.2007.06.019
91. Keita M, Ainmelk Y, Pelmus M, Bessette P, Aris A. Endometrioid ovarian cancer and endometriotic cells exhibit the same alteration in the expression of interleukin-1 receptor II: to a link between endometriosis and endometrioid ovarian cancer. J Obstet Gynaecol Res (2011) 37:99–107. doi:10.1111/j.1447-0756.2010.01320.x
92. Chun S, Kim H, Ku SY, Suh CS, Kim SH, Kim JG. The association between endometriosis and polymorphisms in the interleukin-1 family genes in Korean women. Am J Repr Immunol (2012) 68:154–63. doi:10.1111/j.1600-0897.2012.01136.x
93. Vambutas A, Devoti J, Goldofsky E, Gordon M, Lesser M, Bonagura V. Alternate splicing of interleukin-1 receptor type II (IL1R2) in vitro correlates with clinical glucocorticoid responsiveness in patients with AIED. PLoS ONE (2009) 4:e5293. doi:10.1371/journal.pone.0005293
94. Herrmann-Lavoie C, Rao CV, Akoum A. Chorionic gonadotropin down-regulates the expression of the decoy inhibitory interleukin 1 receptor type II in human endometrial epithelial cells. Endocrinology (2007) 148:5377–84. doi:10.1210/en.2007-0368
95. Gillespie J, Mathews R, McDermott MF. Rilonacept in the management of cryopyrin-associated periodic syndromes (CAPS). J Inflamm Res (2010) 3:1–8.
96. Dinarello CA, Simon A, Van Der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov (2012) 11:633–52. doi:10.1038/nrd3800
97. Thomassen E, Renshaw BR, Sims JE. Identification and characterization of SIGIRR, a molecule representing a novel subtype of the IL-1R superfamily. Cytokine (1999) 11:389–99. doi:10.1006/cyto.1998.0452
98. Lech M, Garlanda C, Mantovani A, Kirschning CJ, Schlondorff D, Anders HJ. Different roles of TiR8/Sigirr on toll-like receptor signaling in intrarenal antigen-presenting cells and tubular epithelial cells. Kidney Int (2007) 72:182–92. doi:10.1038/sj.ki.5002293
99. Riva F, Polentarutti N, Tribbioli G, Mantovani A, Garlanda C, Turin L. The expression pattern of TIR8 is conserved among vertebrates. Vet Immunol Immunopathol (2009) 131:44–9. doi:10.1016/j.vetimm.2009.03.009
100. Polentarutti N, Rol GP, Muzio M, Bosisio D, Camnasio M, Riva F, et al. Unique pattern of expression and inhibition of IL-1 signaling by the IL-1 receptor family member TIR8/SIGIRR. Eur Cytokine Netw (2003) 14:211–8.
101. Kadota C, Ishihara S, Aziz MM, Rumi MA, Oshima N, Mishima Y, et al. Down-regulation of single immunoglobulin interleukin-1R-related molecule (SIGIRR)/TIR8 expression in intestinal epithelial cells during inflammation. Clin Exp Immunol (2010) 162:348–61. doi:10.1111/j.1365-2249.2010.04254.x
102. Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol (2003) 4:920–7. doi:10.1038/ni968
103. Huang X, Hazlett LD, Du W, Barrett RP. SIGIRR promotes resistance against Pseudomonas aeruginosa keratitis by down-regulating type-1 immunity and IL-1R1 and TLR4 signaling. J Immunol (2006) 177:548–56.
104. Ragnarsdottir B, Samuelsson M, Gustafsson MC, Leijonhufvud I, Karpman D, Svanborg C. Reduced toll-like receptor 4 expression in children with asymptomatic bacteriuria. J Infect Dis (2007) 196:475–84. doi:10.1086/518893
105. Gopal R, Birdsell D, Monroy FP. Regulation of toll-like receptors in intestinal epithelial cells by stress and Toxoplasma gondii infection. Parasite Immunol (2008) 30:563–76. doi:10.1111/j.1365-3024.2008.01055.x
106. Veliz Rodriguez T, Moalli F, Polentarutti N, Paroni M, Bonavita E, Anselmo A, et al. Role of Toll interleukin-1 receptor (IL-1R) 8, a negative regulator of IL-1R/Toll-like receptor signaling, in resistance to acute Pseudomonas aeruginosa lung infection. Infect Immun (2012) 80:100–9. doi:10.1128/IAI.05695-11
107. Nanthakumar N, Meng D, Goldstein AM, Zhu W, Lu L, Uauy R, et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS ONE (2011) 6:e17776. doi:10.1371/journal.pone.0017776
108. Adib-Conquy M, Adrie C, Fitting C, Gattolliat O, Beyaert R, Cavaillon JM. Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit Care Med (2006) 34:2377–85. doi:10.1097/01.CCM.0000233875.93866.88
109. Bulek K, Swaidani S, Qin J, Lu Y, Gulen MF, Herjan T, et al. The essential role of single Ig IL-1 receptor-related molecule/Toll IL-1R8 in regulation of Th2 immune response. J Immunol (2009) 182:2601–9. doi:10.4049/jimmunol.0802729
110. Jiang X, McClellan SA, Barrett RP, Zhang Y, Hazlett LD. Vasoactive intestinal peptide downregulates proinflammatory TLRs while upregulating anti-inflammatory TLRs in the infected cornea. J Immunol (2012) 189:269–78. doi:10.4049/jimmunol.1200365
111. Villena J, Suzuki R, Fujie H, Chiba E, Takahashi T, Tomosada Y, et al. Immunobiotic Lactobacillus jensenii modulates the Toll-like receptor 4-induced inflammatory response via negative regulation in porcine antigen-presenting cells. Clin Vaccine Immunol (2012) 19:1038–53. doi:10.1128/CVI.00199-12
112. Davies JM, MacSharry J, Shanahan F. Differential regulation of Toll-like receptor signalling in spleen and Peyer’s patch dendritic cells. Immunology (2010) 131:438–48. doi:10.1111/j.1365-2567.2010.03317.x
113. Garlanda C, Riva F, Polentarutti N, Buracchi C, Sironi M, De Bortoli M, et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc Natl Acad Sci U S A (2004) 101:3522–6. doi:10.1073/pnas.0308680101
114. Qin J, Qian Y, Yao J, Grace C, Li X. SIGIRR inhibits interleukin-1 receptor- and toll-like receptor 4-mediated signaling through different mechanisms. J Biol Chem (2005) 280:25233–41. doi:10.1074/jbc.M501363200
115. Lech M, Kulkarni OP, Pfeiffer S, Savarese E, Krug A, Garlanda C, et al. Tir8/Sigirr prevents murine lupus by suppressing the immunostimulatory effects of lupus autoantigens. J Exp Med (2008) 205:1879–88. doi:10.1084/jem.20072646
116. Gong J, Wei T, Stark RW, Jamitzky F, Heckl WM, Anders HJ, et al. Inhibition of Toll-like receptors TLR4 and 7 signaling pathways by SIGIRR: a computational approach. J Struct Biol (2010) 169:323–30. doi:10.1016/j.jsb.2009.12.007
117. Gulen MF, Kang Z, Bulek K, Youzhong W, Kim TW, Chen Y, et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity (2010) 32:54–66. doi:10.1016/j.immuni.2009.12.003
118. Xiao H, Yin W, Khan MA, Gulen MF, Zhou H, Sham HP, et al. Loss of single immunoglobulin interlukin-1 receptor-related molecule leads to enhanced colonic polyposis in Apc(min) mice. Gastroenterology (2010) 139:574–85. doi:10.1053/j.gastro
119. Garlanda C, Di Liberto D, Vecchi A, La Manna MP, Buracchi C, Caccamo N, et al. Damping excessive inflammation and tissue damage in Mycobacterium tuberculosis infection by Toll IL-1 receptor 8/single Ig IL-1-related receptor, a negative regulator of IL-1/TLR signaling. J Immunol (2007) 179:3119–25.
120. Bozza S, Zelante T, Moretti S, Bonifazi P, Deluca A, D’Angelo C, et al. Lack of Toll IL-1R8 exacerbates Th17 cell responses in fungal infection. J Immunol (2008) 180:4022–31.
121. Xiao H, Gulen MF, Qin J, Yao J, Bulek K, Kish D, et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity (2007) 26:461–75. doi:10.1016/j.immuni.2007.02.012
122. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118:229–41. doi:10.1016/j.cell.2004.07.002
123. Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell (2006) 124:823–35. doi:10.1016/j.cell.2006.02.016
124. Chen X, Zhao Y, Wu X, Qian G. Enhanced expression of single immunoglobulin IL-1 receptor-related molecule ameliorates LPS-induced acute lung injury in mice. Shock (2011) 35:198–204. doi:10.1097/SHK.0b013e3181f226f3
125. Leemans JC, Butter LM, Teske GJ, Stroo I, Pulskens WP, Florquin S. The toll interleukin-1 receptor (IL-1R) 8/single Ig domain IL-1R-related molecule modulates the renal response to bacterial infection. Infect Immunity (2012) 80:3812–20. doi:10.1128/IAI.00422-12
126. Lech M, Skuginna V, Kulkarni OP, Gong J, Wei T, Stark RW, et al. Lack of SIGIRR/TIR8 aggravates hydrocarbon oil-induced lupus nephritis. J Pathol (2010) 220:596–607. doi:10.1002/path.2678
127. Drexler SK, Kong P, Inglis J, Williams RO, Garlanda C, Mantovani A, et al. SIGIRR/TIR-8 is an inhibitor of Toll-like receptor signaling in primary human cells and regulates inflammation in models of rheumatoid arthritis. Arthritis Rheum (2010) 62:2249–61. doi:10.1002/art.27517
128. Batliwalla FM, Li W, Ritchlin CT, Xiao X, Brenner M, Laragione T, et al. Microarray analyses of peripheral blood cells identifies unique gene expression signature in psoriatic arthritis. Mol Med (2005) 11:21–9. doi:10.2119/2006-00003.Gulko
129. Lech M, Avila-Ferrufino A, Allam R, Segerer S, Khandoga A, Krombach F, et al. Resident dendritic cells prevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J Immunol (2009) 183:4109–18. doi:10.4049/jimmunol.0900118
130. Noris M, Cassis P, Azzollini N, Cavinato R, Cugini D, Casiraghi F, et al. The Toll-IL-1R member Tir8/SIGIRR negatively regulates adaptive immunity against kidney grafts. J Immunol (2009) 183:4249–60. doi:10.4049/jimmunol.0803549
131. Andre R, Lerouet D, Kimber I, Pinteaux E, Rothwell NJ. Regulation of expression of the novel IL-1 receptor family members in the mouse brain. J Neurochem (2005) 95:324–30. doi:10.1111/j.1471-4159.2005.03364.x
132. Watson MB, Costello DA, Carney DG, McQuillan K, Lynch MA. SIGIRR modulates the inflammatory response in the brain. Brain Behav Immun (2010) 24:985–95. doi:10.1016/j.bbi.2010.04.002
133. Costello DA, Watson MB, Cowley TR, Murphy N, Murphy Royal C, Garlanda C, et al. Interleukin-1alpha and HMGB1 mediate hippocampal dysfunction in SIGIRR-deficient mice. J Neurosci (2011) 31:3871–9. doi:10.1523/JNEUROSCI.6676-10.2011
134. Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A (2003) 100:2645–50. doi:10.1073/pnas.0437939100
135. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis (2009) 30:1073–81. doi:10.1093/carcin/bgp127
136. Garlanda C, Riva F, Veliz T, Polentarutti N, Pasqualini F, Radaelli E, et al. Increased susceptibility to colitis-associated cancer of mice lacking TIR8, an inhibitory member of the interleukin-1 receptor family. Cancer Res (2007) 67:6017–21. doi:10.1158/0008-5472.CAN-07-0560
137. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature (2006) 441:431–6. doi:10.1038/nature04870
138. Aoki K, Tamai Y, Horiike S, Oshima M, Taketo MM. Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Delta716 Cdx2+ compound mutant mice. Nat Genet (2003) 35:323–30. doi:10.1038/ng1265
139. Muzio M, Scielzo C, Bertilaccio MT, Frenquelli M, Ghia P, Caligaris-Cappio F. Expression and function of toll like receptors in chronic lymphocytic leukaemia cells. Br J Haematol (2009) 144:507–16. doi:10.1111/j.1365-2141.2008.07475.x
140. Arvaniti E, Ntoufa S, Papakonstantinou N, Touloumenidou T, Laoutaris N, Anagnostopoulos A, et al. Toll-like receptor signaling pathway in chronic lymphocytic leukemia: distinct gene expression profiles of potential pathogenic significance in specific subsets of patients. Haematologica (2011) 96:1644–52. doi:10.3324/haematol.2011.044792
141. Bertilaccio MT, Simonetti G, Dagklis A, Rocchi M, Rodriguez TV, Apollonio B, et al. Lack of TIR8/SIGIRR triggers progression of chronic lymphocytic leukemia in mouse models. Blood (2011) 118:660–9. doi:10.1182/blood-2011-01-329870
142. Muzio M, Bertilaccio MT, Simonetti G, Frenquelli M, Caligaris-Cappio F. The role of toll-like receptors in chronic B-cell malignancies. Leuk Lymphoma (2009) 50:1573–80. doi:10.1080/10428190903115410
143. Bufler P, Azam T, Gamboni-Robertson F, Reznikov LL, Kumar S, Dinarello CA, et al. A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc Natl Acad Sci U S A (2002) 99:13723–8. doi:10.1073/pnas.212519099
144. Xiang Y, Moss B. Correspondence of the functional epitopes of poxvirus and human interleukin-18-binding proteins. J Virol (2001) 75:9947–54. doi:10.1128/JVI.75.20.9947-9954.2001
145. Lu HL, Yang CY, Chen HC, Hung CS, Chiang YC, Ting LP. A novel alternatively spliced interleukin-1 receptor accessory protein mIL-1RAcP687. Mol Immunol (2008) 45:1374–84. doi:10.1016/j.molimm.2007.09.002
146. Smith DE, Lipsky BP, Russell C, Ketchem RR, Kirchner J, Hensley K, et al. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity (2009) 30:817–31. doi:10.1016/j.immuni.2009.03.020
147. Khan JA, Brint EK, O’Neill LA, Tong L. Crystal structure of the Toll/interleukin-1 receptor domain of human IL-1RAPL. J Biol Chem (2004) 279:31664–70. doi:10.1074/jbc.M403434200
148. Ferrante MI, Ghiani M, Bulfone A, Franco B. IL1RAPL2 maps to Xq22 and is specifically expressed in the central nervous system. Gene (2001) 275:217–21. doi:10.1016/S0378-1119(01)00659-X
149. Tabolacci E, Pomponi MG, Pietrobono R, Terracciano A, Chiurazzi P, Neri G. A truncating mutation in the IL1RAPL1 gene is responsible for X-linked mental retardation in the MRX21 family. Am J Med Genet (2006) 140:482–7. doi:10.1002/ajmg.a.31107
150. Gao X, Xi G, Niu Y, Zhang S, Fu R, Zheng Z, et al. A study on the correlation between IL1RAPL1 and human cognitive ability. Neurosci Lett (2008) 438:163–7. doi:10.1016/j.neulet.2008.03.084
151. Bahi N, Friocourt G, Carrie A, Graham ME, Weiss JL, Chafey P, et al. IL1 receptor accessory protein like, a protein involved in X-linked mental retardation, interacts with Neuronal Calcium Sensor-1 and regulates exocytosis. Hum Mol Genet (2003) 12:1415–25. doi:10.1093/hmg/ddg147
152. Piton A, Michaud JL, Peng H, Aradhya S, Gauthier J, Mottron L, et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum Mol Genet (2008) 17:3965–74. doi:10.1093/hmg/ddn300
153. Gu YF, Fang Y, Jin Y, Dong WR, Xiang LX, Shao JZ. Discovery of the DIGIRR gene from teleost fish: a novel Toll-IL-1 receptor family member serving as a negative regulator of IL-1 signaling. J Immunol (2011) 187:2514–30. doi:10.4049/jimmunol.1003457
154. Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem (2007) 282:26369–80. doi:10.1074/jbc.M704916200
155. Leung BP, Xu D, Culshaw S, McInnes IB, Liew FY. A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J Immunol (2004) 173:145–50.
156. Smeets RL, Joosten LA, Arntz OJ, Bennink MB, Takahashi N, Carlsen H, et al. Soluble interleukin-1 receptor accessory protein ameliorates collagen-induced arthritis by a different mode of action from that of interleukin-1 receptor antagonist. Arthritis Rheum (2005) 52:2202–11. doi:10.1002/art.21108
Keywords: cytokine, interleukin-1, inflammation, decoy receptor
Citation: Garlanda C, Riva F, Bonavita E, Gentile S and Mantovani A (2013) Decoys and regulatory “receptors” of the IL-1/Toll-like receptor superfamily. Front. Immunol. 4:180. doi: 10.3389/fimmu.2013.00180
Received: 03 May 2013; Accepted: 22 June 2013;
Published online: 09 July 2013.
Edited by:
Pietro Ghezzi, Brighton and Sussex Medical School, UKReviewed by:
Nick Gay, University of Cambridge, UKCem Gabay, University Hospitals of Geneva, Switzerland
Copyright: © 2013 Garlanda, Riva, Bonavita, Gentile and Mantovani. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Cecilia Garlanda, Laboratory of Experimental Immunopathology, Istituto Clinico Humanitas, Via Manzoni 113, 20089 Rozzano, Italy e-mail:Y2VjaWxpYS5nYXJsYW5kYUBodW1hbml0YXNyZXNlYXJjaC5pdA==