



- 1Nuffield Department of Medicine, The Jenner Institute (ORCRB), University of Oxford, Oxford, UK
- 2Infection and Immunity, School of Medicine, Henry Wellcome Building, Cardiff University, Cardiff, UK
CD4+Foxp3+ regulatory T cells (Tregs) have a fundamental role in maintaining immune balance by preventing autoreactivity and immune-mediated pathology. However this role of Tregs extends to suppression of anti-tumor immune responses and remains a major obstacle in the development of anti-cancer vaccines and immunotherapies. This feature of Treg activity is exacerbated by the discovery that Treg frequencies are not only elevated in the blood of cancer patients, but are also significantly enriched within tumors in comparison to other sites. These observations have sparked off the quest to understand the processes through which Tregs become elevated in cancer-bearing hosts and to identify the specific mechanisms leading to their accumulation within the tumor microenvironment. This manuscript reviews the evidence for specific mechanisms of intra-tumoral Treg enrichment and will discuss how this information may be utilized for the purpose of manipulating the balance of tumor-infiltrating T cells in favor of anti-tumor effector cells.
What are Tregs?
Regulatory T cells (Tregs) are suppressor cells that are necessary for maintaining immune homeostasis and immunological tolerance to self and which play a key role in limiting excessive and harmful immune responses (1). Several different types of suppressor T cells have been described including cells within both CD4+ and CD8+ populations (2). The most prominent of these express both CD4 and Foxp3 and can arise either in the thymus or in the periphery (1). A recent correspondence in Nature Immunology recommended the adoption of new nomenclature for Tregs (3). The authors suggested that thymus-derived Treg cells be called tTreg (rather than nTreg) to denote those that are thymus-derived and pTreg for those that differentiate in the periphery [therefore replacing the terms i(induced)Treg and a(adaptive)Treg]. This review will, as far as possible, adopt the recently recommended nomenclature. In addition the term Treg will only be used to describe cells where suppressor activity has been demonstrated either in vivo or in vitro and where suppressor function has not been confirmed, the cells will be termed Foxp3+CD4+ T cells.
Promotion of Tumor Progression by Tregs
There is an emerging consensus that effective anti-tumor immunity is characterized by a Thelper1 (Th1)/CD8+ T cell response (4). This type of response however, is susceptible to suppression by Tregs and several studies using mouse models have shown that partial or complete removal of this inhibitory influence uncovers anti-tumor immune responses capable of preventing tumor development and limiting tumor progression (5–7). Approaches aimed at modulating Foxp3+ T cell frequencies in patients with cancer have been shown to enhance vaccine-induced anti-tumor immune responses and even boost endogenous responses (8–11). These exciting findings underpin the importance of fully understanding the role of Tregs in cancer so that these cells can be manipulated in order to optimize cancer immunotherapy.
Mechanisms of Foxp3+ T Cell Enrichment within Tumors
Studies have shown that progressing mouse and human tumors can be associated with enhanced Tregs activity and escalating immune suppression (12, 13). Indeed Foxp3+ T cells manage to successfully pervade, and often dominate the tumor-specific immune response; Foxp3+ to Foxp3− T cell ratios in the range 0.5–1:1 have been described in some tumors (12, 14, 15). A few theories have been proposed to explain how Foxp3+ T cells become enriched in tumors and in the peripheral blood of tumor-bearing hosts. There may be preferential migration of Foxp3+ T cells to tumors in response to chemokines expressed by tumor cells and stroma. Foxp3+ T cells, preferentially attracted to the tumor microenvironment may use the same or additional cues to aid their retention within the tumor mass. In addition, tumor establishment may trigger production of a cocktail of factors that support increased Foxp3+ T cell proliferation and/or the conversion of conventional Foxp3−CD4+ T cells into Foxp3+ cells. Various lines of supporting evidence exist for these mechanisms of Foxp3+ T cell enrichment in tumors and will be discussed in this review.
Chemokine-Mediated Recruitment of Foxp3+ T Cells into Tumors
Migration of cells into peripheral tissues and sites of inflammation depends on their expression of various chemokine receptors, selectins (and selectin ligands), and integrins. Generally, effector-like inflammation-seeking T cells (including Tregs) express inflammatory chemokine receptors and adhesion molecules that enhance their capacity to migrate to inflamed tissues (16–24). Different tumors are characterized by unique albeit overlapping chemokine signatures. Tumor cells and surrounding stromal cells can express these chemokines, which serve to facilitate migration and accumulation of various leukocytes in the tumor (25–27). While some of these leukocytic infiltrates comprise macrophages (28) and myeloid derived suppressor cells (MDSCs) (29) which promote tumor progression and metastasis, a high frequency of infiltrating CD3+ T cells often correlates with improved clinical outcome, e.g., in ovarian and colorectal cancer (CRC) (30, 31). Whether or not the degree of CD3+ T cell infiltrate correlates with strong anti-tumor immunity may also depend on the frequency and suppressive capacity of tumor-infiltrating Tregs. Consequently, increased infiltration of Foxp3+ T cells is often associated with a poor prognosis and accelerated tumor progression (32).
Inflammatory Chemokines and Their Receptors
CCR4
CCR4 has been shown to be expressed on a greater proportion of Tregs than conventional T cells and to be important for guiding Tregs to sites of inflammation (24, 33). Several studies indicate that the tumor-expressed chemokines CCL22 and CCL17, which are ligands for CCR4, play a role in the recruitment and enrichment of Tregs. A study by Curiel and colleagues, clearly demonstrated a major role for CCL22 in recruitment of CCR4+ Tregs into human ovarian carcinomas (13). CCL22 alone, or in combination with CCL17, has been implicated in Treg recruitment to human breast (34, 35) and prostate (36) cancers. Increased levels of CCL17 and/or CCL22 are also associated with higher frequencies of CD4+Foxp3+ T cells in cerebral spinal fluid of patients with lymphomatous and carcinomatous meningitis (37), gastric (38), and esophageal squamous cell carcinomas (39). Using mouse models, several approaches, including the use of specific antibodies, antagonists, or siRNA, have been used to block the CCL22/CCL17 – CCR4 axis, resulting in reduction in Treg frequencies and a concomitant increase in anti-tumor activity (40–42).
CCR5
Disruption of CCR5/CCL5 signaling has also been shown in mouse models to impair intra-tumoral Treg accumulation and slow tumor progression (43). Similarly, CCL5 levels correlate with increased Treg frequencies and impaired CD8+ T cell responses in human colon cancer (44). Further evidence for CCR5-dependent Treg enrichment comes from a study exploring the potential mechanisms through which MDSCs inhibit anti-tumor immunity. MDSCs infiltrating mouse RMA-S lymphomas were shown to increase the levels of CCL3, CCL4, and CCL5, which in turn enhanced the recruitment of CD4+Foxp3+ T cells via CCR5 (45). CCR5 deficiency (demonstrated by use of CCR5−/− mice) or CCL5 blockade (using Met-RANTES) led to diminished CD4+Foxp3+ T cells numbers and slower tumor growth (45). However, apart from its ability to attract CD4+Foxp3+ T cells to tumors, the pivotal role for CCR5 in mediating recruitment and activation of conventional T cells dictates that CCR5 is also important for achieving strong anti-tumor immune responses and regression of established tumors (46–49). Thus, although the findings of some mouse models indicate that the CCR5 axis can be targeted to reduce Treg accumulation, the general utility of this approach is likely to be limited by the potential for concurrent effects on anti-tumor effector cells.
CXCR3
A similar situation applies to the chemokine receptor CXCR3. Intra-tumoral accumulation of CXCR3+Foxp3+ T cells has been reported in human ovarian, colorectal, and hepatocellular carcinomas (50, 51). However, like CCR5, CXCR3 is abundantly expressed on activated cells, binding the IFN-γ-induced chemokines, CXCL9, CXCL10, and CXCL11. Indeed, homing and migration of activated effector cells (CTL, NK, NKT, and T helper) is highly dependent on CXCL9/CXCL10/CXCL11 – CXCR3 signaling thereby limiting the utility of this pathway for targeted prevention of Treg recruitment. CXCR3 and CCR5 are often co-expressed by effector T cells. In a study of human colorectal carcinomas expressing CXCL10 and CCL5, the CD8+IFNγ+ T cell infiltrate comprised predominantly CXCR3+CCR5+ cells (52), concurrent with a favorable prognosis as previously described (30). Similarly, CXCL9, CXCL10, and CXCL11 expression by sporadic human renal cell carcinomas was associated with increased frequency of CXCR3+CCR5+ T cells and a favorable prognosis which was characterized by the absence of recurrences following curative surgery (53). Furthermore tumor-expressed CXCL9 was shown to be crucial for immune control of murine cutaneous fibrosarcomas (54) while CXCL11 secretion by genetically modified mouse T cell lymphoma cells (EL4) led to increased infiltration of CD8+CXCR3+ T cells and subsequent tumor rejection (55). Considering the body of evidence highlighting a favorable prognosis for cancers expressing these IFN-γ induced chemokines, disruption of the CXCR3 and/or CCR5 pathways to prevent Treg accumulation in tumors is unlikely to be effective for promoting tumor immunity.
Hypoxia-Induced Chemokines and Their Receptors
CCR10
Hypoxia and angiogenesis are both characteristic features of advanced solid tumors. Both of these features also serve to modulate the enrichment of intra-tumoral Tregs expressing CCR10. CCL28, a chemokine known to be upregulated by hypoxia has recently been shown to recruit CCR10+ Tregs to mouse ovarian cancers (56). These CCR10+ Tregs contributed to tumor progression by secreting vascular endothelial growth factor A (VEGF-A), thereby promoting angiogenesis. Understanding the nature of the relationship between VEGF-A and Tregs may prove important. VEGF-A blockade not only reduced angiogenesis but also has been shown to reduce the extent of Treg infiltration in mouse models resulting in enhanced vaccine-induced immune responses (57). Moreover, treatment of CRC patients with the anti-VEGF-A monoclonal antibody bevacizumab reversed Treg accumulation in patients’ blood (58) whilst VEGFR2+CD4+Foxp3+ cells are reportedly associated with poor prognosis in CRC (59) supporting the theory that angiogenic factors may be targeted for the purpose of modulating both angiogenesis and the anti-tumor immune response.
CXCR4
Vascular endothelial growth factor A has also been shown to work synergistically with CXCL12, a chemokine commonly expressed by tumors, to promote tumor angiogenesis (60). In a study of patients with basal-like breast cancers, infiltration with Foxp3+ cells was shown, as above, to correlate with tumor hypoxia (61). In this study however, a preferential accumulation of Foxp3+ cells expressing CXCR4, the receptor for CXCL12, was observed. The authors further showed that accumulation of these CXCR4+Foxp3+ cells was associated with a poor prognosis. Although CD8+ and Foxp3−CD4+ T cells can also express CXCR4, there are reports that CXCL12 preferentially attracts Tregs to human lung adenocarcinomas (62) and advanced cervical cancers (63), thereby indicating that targeting the CXCR4–CXCL12 axis may represent a useful means of selectively reducing the intra-tumoral Treg infiltrate. In support of this, using a mouse model of ovarian cancer, Righi and colleagues showed that administration of a specific CXCR4 antagonist, AMD3100 (64), was associated with several anti-tumor effects including increased tumor cell death, reduced dissemination and angiogenesis and better survival of the treated animals (65). Significantly, the authors also observed a selective reduction in the recruitment of Foxp3+ T cells in comparison with CD8+ T cells (65).
The picture that emerges from these reports is that tumors with high levels of Tregs, recruited in response to hypoxia (via CCR10 and/or CXCR4), are rich in VEGF-A and therefore, serve to drive neovascularization. Such a pathway implies that angiogenesis and the recruitment and activity of Tregs work side-by-side, facilitating tumor growth directly through neovascularization and indirectly through promoting immune suppression. With this in mind, it may prove useful to further explore potential synergy between therapies targeting angiogenesis and those targeting Tregs.
Lymphoid-Associated Chemokines and Their Receptors
CCR7
The role of CCL21/CCR7 signaling in the recruitment and accumulation of Tregs in tumors has been described in one study using B16 melanomas engineered to express higher levels of CCL21. These tumors recruited high numbers of Tregs and progressed more rapidly compared to tumors expressing normal or lower CCL21 levels (66). In contrast, other studies indicate that the CCL21/CCR7 pathway promotes increased tumor control as a result of increased recruitment of effector immune cells (67). Furthermore, intra-tumoral expression of CCL21 boosted CTL responses after DNA vaccination of mice and induced regression of B16 melanomas (68). In another study, intra-tumoral delivery of CCL21 inhibited lung cancer growth in mice. Inhibition of tumor growth was associated with reduced frequencies of Tregs and MDSC but enhanced recruitment of CCR7+Foxp3− T cells (69). Moreover, a recent study of patients with metastatic CRC indicated that tumor infiltration with CCR7+ T cells was associated with a more favorable prognosis (70). Given the plethora of studies highlighting the important role of CCL21 in recruitment of immune effector cells and subsequent tumor immunity and the paucity of studies to support enhanced Treg recruitment to the tumor via CCL21/CCR7, it is highly unlikely that selective targeting of this pathway as a means to prevent Treg recruitment will be of clinical benefit in cancer patients.
Whether chemokines lead to the preferential enrichment of Tregs in tumors is as yet unclear although there is evidence that Treg recruitment to tumors may be selectively inhibited through chemokine receptor blockade: the most notable candidates being CCR4, CXCR4, and CCR10. Such strategies may not however, impinge on the existing pool of tumor-infiltrating Tregs. Chemokines may perform functions other than to attract Tregs to tumors. It is highly likely for example, that chemokines, expressed intra-tumorally, serve to retain Treg cells, perhaps preferentially, within the tumor mass. If this is the case, selective retention of Tregs in the tumor microenvironment could significantly influence their fate compared to that of conventional T cells, with clear immunosuppressive consequences. Although the key role of chemokines is to act as chemoattractants, a role for CCL5 in promoting T cell activation has been demonstrated; in these studies CCL5 was shown to induce signaling events in T cells in antigen-independent fashion (71, 72). This finding raises the intriguing possibility that chemokines present within the tumor microenvironment may influence T cell activity, including the activity of Tregs.
Induction of Tregs in the Tumor Microenvironment
The possibility that conversion of conventional T cells into Tregs represents a mechanism of Treg enrichment in tumors has been explored. In adoptive transfer experiments, purified CD4+CD25− T cells transferred into tumor bearing mice have been shown to convert into Foxp3+CD4+CD25+ cells within the tumor microenvironment (73, 74). In studies of patients with melanoma, Fourcade and colleagues demonstrated that CD4+CD25− T cells and Foxp3+CD4+ T cells could recognize the same peptide and moreover, clonotypic analyses of these cells revealed a common T cell receptor (TCR) Vβ usage (75). These findings are compatible with the hypothesis that conventional tumor-specific T cells can convert into Tregs. Whilst the potential for conversion of conventional T cells into Tregs is undoubtedly demonstrated in these types of studies, the extent to which this contributes to what is a significant intra-tumoral enrichment of Foxp3+ T cells is unclear. Addressing this question directly has been hampered by reports that Foxp3 can be transiently upregulated on activated T cells without necessarily conferring suppressor functions and a lack of definitive markers to discriminate tTregs from pTregs. The “best” markers are the transcription factor, Helios, and the type 1 transmembrane protein, neuropilin 1 (Nrp1), which, according to some reports, are expressed mainly by tTregs (76, 77). In the case of renal cell cancer patients, the significant increase in Foxp3+ T cells observed in both untreated and IL-2-treated patients are helios+ suggesting that tumors drive expansion of tTreg and not pTregs (78). The same observation has been made in studies of patients with glioblastoma multiforme (GBM) and parallel studies of orthotopic mouse models of brain tumors (79). Studies of Nrp1 expression have resulted in mixed findings where in some mouse tumors Nrp1+Foxp3+ T cells predominate whereas in others they do not (80, 81). The validity of both helios and Nrp1 as true discriminators of tTregs versus pTregs has however been disputed, thus no definitive conclusions can be drawn from the studies described above (82, 83).
Working on the premise that pTregs and conventional T cells share the same TCRs, we used a mouse model of carcinogen-induced tumors to compare the TCR repertoires of tumor-infiltrating Foxp3− and Foxp3+CD4+ T cells in order to determine the extent of TCR overlap between the two populations following their recovery from the tumor microenvironment (15). The data clearly indicated that the TCR repertoires of tumor-infiltrating Foxp3− and Foxp3+CD4+ T cells are distinct, implying that at least in the case of carcinogen-induced tumors, conversion of conventional T cells is not a significant cause of intra-tumoral Treg enrichment. This finding was confirmed in a similar analysis of CD4+Foxp3+ T cells recovered from TRAMP mice in which prostate cancer is driven by transgenic expression of SV40 large T antigen (84). In this study, thymic development of the CD4+Foxp3+ T cells was Aire-dependent and the cells appeared to be specific for a prostate-associated self-antigen. Overall, the results of this study support enrichment of intra-tumoral tTregs as the main mechanism of Treg accumulation in tumors rather than conversion of conventional T cells to pTregs.
Collectively, evidence to support conversion as a major mechanism of Treg enrichment in tumors is currently weak. Most of the direct evidence for a limited role for conversion has however, come from mouse models. There are reports that human CD4+Foxp3− cells can convert into CD4+Foxp3+/lo Tregs in vitro and that the phenotypic characteristics of these cells can resemble CD4+ T cell sub-populations isolated from tumors (85). Whilst these data do not provide definitive answers relating to the relationship between different tumor-infiltrating T cell subpopulation (such as that gained from TCR clonotyping), it remains possible that in human cancers, there is some enrichment of pTregs.
Superior Proliferation of tTregs within the Tumor Microenvironment
Given the evidence that Foxp3+CD4+ T cells gain an edge in accessing the tumor microenvironment through a combination of differential chemokine receptor expression and an increased capacity to migrate in response to hypoxia-induced chemokines and VEGF-A, it is reasonable to speculate that migration does contribute to their observed enrichment within the tumor microenvironment. Higher frequencies of Foxp3+CD4+ T cells are however also observed in spleen/blood and draining lymph of tumor-bearing mice and patients with cancer compared to non-tumor-bearing controls. In carcinogen-induced tumors, enhanced proliferation of CD4+CD25+ T cells has been reported (86). Studies examining proliferation of intra-tumoral Foxp3+ T cells in brain tumors imply that the majority of proliferating cells are helios+; for example in mouse models of glioblastoma, it has been reported that the majority of tumor-infiltrating Foxp3+ T cells express helios and are highly proliferative, significantly more so than helios−Foxp3+ and Foxp3−CD4+ T cells (79). Moreover, if these highly proliferative CD4+Foxp3+helios+ cells are, as this study suggests, suppressive within the tumor microenvironment then the available evidence favors intra-tumoral expansion of tTreg as a major mechanism of Treg enrichment in tumors.
Why might CD4+Foxp3+ Tregs demonstrate enhanced proliferation in the tumor microenvironment compared to CD4+Foxp3− T cells? Evidence suggests that the Treg population contains a higher number of cells that respond to self-antigens compared to Tconv cells (87, 88). Thus, in the case of tumors, Tregs may receive stronger antigen-driven signals than conventional T cells, promoting their expansion in tumors. Using a mouse model of melanoma (B16), Ghiringhelli and colleagues showed that tumors can license dendritic cells (DCs) to promote the proliferation of Tregs through the production of TGF-β (89). Another study, also utilizing the B16 tumor cell line showed that plasmacytoid DCs promoted Treg activation in an indoleamine 2,3-dioxygenase (IDO)-dependent manner (90). Whilst both studies assessed Treg activity in tumor-draining lymph nodes, it is possible similar signals serve to further promote Treg cell proliferation and survival within the tumor microenvironment. Of note, IDO production by human monocyte-derived DCs has also been shown to drive proliferation of highly suppressive CD4+Foxp3+ T cells (91). In addition, to these pathways, it has recently been shown that VEGFR+ Tregs, purified from tumor-bearing mice proliferated in response to VEGF. The same study also demonstrated reduced Treg frequencies in the peripheral blood of CRC patients treated with the VEGF-A blocking antibody, bevacuzimab (58).
Any signal that serves to promote Treg activity, also therefore serves to indirectly suppress the activities of conventional T cells, one effect of which is to reduce local production of IL-2. Through expression of high levels of CD25, Tregs may out-compete conventional T cells for the limited supply of IL-2. Thus, within the tumor microenvironment Tregs may gain superiority by utilizing the available IL-2 to support their own proliferation and moreover, to further promote their immunosuppressive capability (92).
Foxp3+ Tregs – The Tip of the Iceberg
Although most available evidence indicates that the bulk of tumor-infiltrating Foxp3+ Tregs are tTreg, this does not preclude a significant immunosuppressive role for pTreg or indeed Foxp3− cells. With the possible exception of melanoma, there is a distinct paucity of publications detailing the phenotypic and functional characteristics of tumor-infiltrating T cells, particularly tumor-infiltrating CD4+ T cells. There are however suggestions that Foxp3+ Tregs, whether pTregs or tTregs, are not the only suppressive CD4+ T cell sub-population found in tumors. Using a transgenic mouse model of prostate cancer, Donkor et al. showed that TGF-β-blockade in Foxp3− T cells resulted in heightened CTL responses and better immune-mediated control of primary and metastatic tumors (93). In human studies, elevated frequencies of CD4+ T cells expressing latency associated peptide (LAP) have been observed in blood of CRC patients compared to healthy controls. Interestingly, many of these did not express Foxp3 but could suppress proliferation of LAP− cells in a TGF-β-dependent fashion (94). Moreover the LAP+CD4+ sub-population cells were also found in colorectal tumors where their proportions within the CD4+ tumor-infiltrating T cell pool increased with disease progression (94). Similarly, in a study of patients with hepatocellular carcinoma, elevated frequencies of intrahepatic CD4+Foxp3− cells were observed in cancer patients compared to hepatitis C virus infected individuals with chronic liver disease; these CD4+Foxp3− T cells expressed IL-10 and were suppressive in vitro (95). Collectively the data thus far, support a major role for the immunosuppressive cytokines IL-10 and TGFβ in mediating the suppressive effects of CD4+Foxp3− T cells. Whether Foxp3− suppressor T cells arise through sustained but inadequate activation of conventional T cells and/or through mechanisms of infectious tolerance is unknown. It is extremely important however, to determine whether or not Foxp3+ Tregs are responsible, directly or indirectly, for driving the acquisition of suppressor functions of tumor-infiltrating Foxp3−CD4+ T cells. This information will reveal whether or not modulating Foxp3+ Tregs will be sufficient for overcoming the influence of intra-tumoral suppressor T cells or whether multiple suppressor T cell subsets will need to be independently targeted (Figure 1).
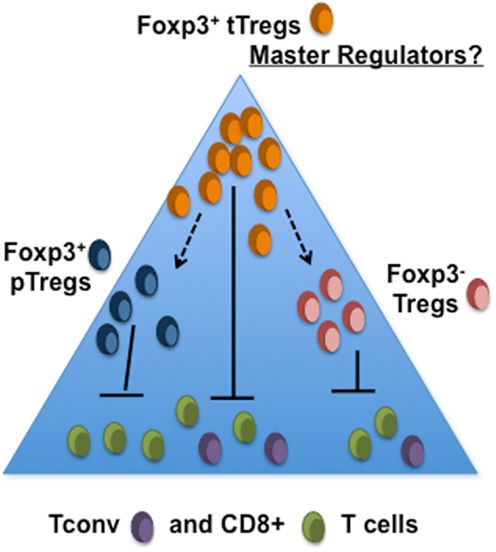
Figure 1. Are intra-tumoral Foxp3+ tregs simply the tip of the iceberg? The tumor microenvironment may consist of several subsets of Treg that serve to suppress the activities of tumor-specific CD4+ and CD8+ T cells. It is not yet known whether development of pTreg or suppressor activity within CD4+Foxp3− T population requires the presence of tTreg.
Implications for Cancer Immunotherapy
Strategies aimed at non-specifically targeting pathways of tolerance induction have proven extremely informative and potentially useful methods of cancer immunotherapy. Boosting conventional T cell activity through use of CTLA4-blocking antibodies can be highly effective in the treatment of metastatic melanoma (96, 97). Similarly, early findings with PD-1- or PDL1-blockade has shown clinical efficacy in melanoma patients without the toxicities observed with anti-CTLA4 antibody treatment (98). These differential toxic effects of CTLA4- versus PD-1-blockade reflect the phenotypes described for CTLA4- and PD-1-deficient mice. Whereas mice lacking CTLA4 exhibit systemic T cell proliferation (99, 100), those lacking PD-1 exhibit milder symptoms (101). This difference may be due to the ability of CTLA4 blockade to induce global T cell activation whereas PD-1 blockade serves to promote effector T cell responses. Differential effects of CTLA4- and PD-1-blockade on Tregs are also likely to contribute.
Has our understanding thus far of intra-tumoral Foxp3+ T cell-enrichment identified mechanisms through which their potential influence on the anti-tumor immune response can be modulated and used to improve current T cell-orientated treatments (Figure 2)? Blockade of recruitment may be possible; administration of methyl gallate has been shown to inhibit recruitment of CD4+Foxp3+ cells through modulating expression of CCR4 whilst AMD3100 can antagonize the CXCR4–CXCL12 interaction (41, 65). VEGF-A blockade may also reduce the numbers of tumor-infiltrating Tregs through effects on both recruitment (57, 80) and proliferation (58). Moreover, this approach may also serve to enhance homing of anti-tumor T cells, possibly due the effects of its blockade on normalization of tumor blood vessels (102, 103). It may also be the case that targeting blood vessels can alter the composition of the intra-tumoral T cell pool. Recently we found that carcinogen-induced tumors were controlled in a proportion of mice in which Tregs had been largely ablated. The tumors of these mice, unlike progressing tumors, were distinguished by the presence of high endothelial venules (HEV); specialized blood vessels normally found only in lymph nodes that when present in tumors facilitated entry of anti-tumor effector cells (5). Thus, disabling Tregs can, directly or indirectly, impact on blood vessel differentiation, promoting access of anti-tumor T cells.
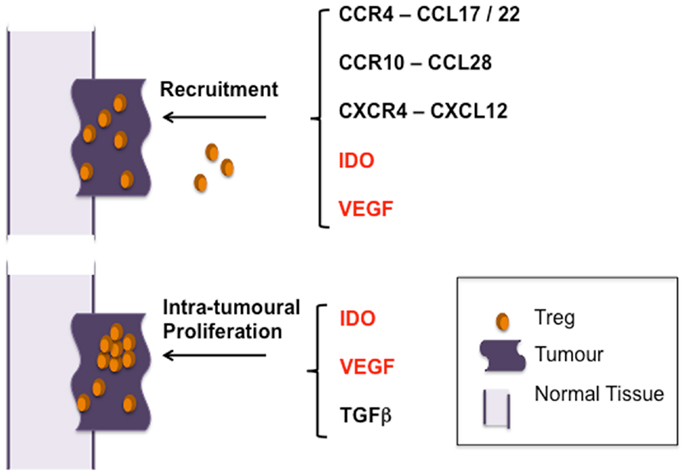
Figure 2. Pathways of Treg enrichment in tumors. Studies thus far indicate that selective migration of Treg and preferential proliferation of tTreg result in their accumulation in tumors – the main pathways identified thus far are indicated. Mechanisms promoting both recruitment and proliferation are indicated in red.
As well as VEGF-A, IDO has been implicated in both the recruitment and activation of Tregs, underpinning the potential for targeting these molecules for modulating Treg numbers within tumors (57, 80, 104) (Figure 2). Thus, inhibition of IDO, shown to be successful in promoting tumor-immunity in many mouse models, may prove a useful therapeutic target (105).
The enhanced proliferative activity of CD4+Foxp3+ T cells can also be exploited as a means of targeting Tregs with chemotherapeutic drugs such as cyclophosphamide. The findings of a recent study suggest that modulating Treg numbers in this way can be successfully combined with vaccination strategies aimed at inducing or boosting anti-tumor effector T cells (11). It was demonstrated, in a phase 2 trial involving patients with renal cell carcinoma, that a single dose of cyclophosphamide reduced Treg numbers and promoted immune responses to a peptide-based vaccine. These immune responses were associated with longer overall survival (11). Collectively, the data described herein point to the importance of exploring immunotherapeutic strategies aimed at modulating Treg numbers, boosting anti-tumor T cell responses through vaccination and influencing blood vessel differentiation for the purpose of facilitating access of effective anti-tumor T cells to the tumor microenvironment.
As discussed above, it appears that in terms of T cell-mediated immunosuppression in the tumor microenvironment, Foxp3+ Tregs are just one subpopulation of suppressor T cell. It is likely that the tumor-infiltrating CD4+ T cell pool is highly heterogeneous comprising both Foxp3+ and Foxp3− suppressor cells. It is not surprising therefore that even in mouse models whereby Foxp3+ Treg cells can be specifically and almost completely ablated that effects on tumor growth are often modest and in the majority of cases, despite the systemic autoreactivity induced by Foxp3+ Treg depletion, tumors continue to grow (5–7). It is important therefore to determine whether ablation of Foxp3+ T cells also reduces or removes the immunosuppressive influence of Foxp3− Tregs. Moreover, the nature of anti-tumor T cell responses is not completely understood. Whilst it is clear that Th1/CD8+ T cell responses can exert potent anti-tumor activities, some reports also suggest that Th17 cells can also participate in limiting tumor progression (106, 107). As reported recently in a study of patients with pancreatic cancer, Th17 cells may also represent relevant targets for suppression by Tregs (108). With these questions in mind, it is imperative that we continue to characterize tumor-infiltrating T cell pools with respect to deciphering the origins, specificities, and phenotypes of both Foxp3+ and Foxp3− Tregs cells and their targets. Such studies may reveal new means of disabling intra-tumoral Tregs.
Overall, our current knowledge of Tregs indicates that there is room for optimism. Preclinical and clinical studies will continue to use current and new findings to examine both benefits and toxicities of combination therapies (e.g., immune modulation, blood vessel normalization, vaccination) aimed at redressing the balance between tolerance and immunity within the tumor microenvironment. Modulating Treg numbers and activity is likely to represent an integral part of this process.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Awen Gallimore is supported by a University Award from The Wellcome Trust (086983).
References
1. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi:10.1016/j.cell.2008.05.009
2. Smith TR, Kumar V. Revival of CD8+ Treg-mediated suppression. Trends Immunol (2008) 29(7):337–42. doi:10.1016/j.it.2008.04.002
3. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14(4):307–8. doi:10.1038/ni.2554
4. Fridman WH, Galon J, Dieu-Nosjean MC, Cremer I, Fisson S, Damotte D, et al. Immune infiltration in human cancer: prognostic significance and disease control. Curr Top Microbiol Immunol (2011) 344:1–24. doi:10.1007/82_2010_46
5. Hindley JP, Jones E, Smart K, Bridgeman H, Lauder SN, Ondondo B, et al. T-cell trafficking facilitated by high endothelial venules is required for tumor control after regulatory T-cell depletion. Cancer Res (2012) 72(21):5473–82. doi:10.1158/0008-5472.CAN-12-1912
6. Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MW, Ngiow SF, et al. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res (2010) 70(20):7788–99. doi:10.1158/0008-5472.CAN-10-1736
7. Teng MW, Ngiow SF, von Scheidt B, McLaughlin N, Sparwasser T, Smyth MJ. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res (2010) 70(20):7800–9. doi:10.1158/0008-5472.CAN-10-1681
8. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother (2007) 56(5):641–8. doi:10.1007/s00262-006-0225-8
9. Schuetze SM, Zhao L, Chugh R, Thomas DG, Lucas DR, Metko G, et al. Results of a phase II study of sirolimus and cyclophosphamide in patients with advanced sarcoma. Eur J Cancer (2012) 48(9):1347–53. doi:10.1016/j.ejca.2012.03.022
10. Vermeij R, Leffers N, Hoogeboom BN, Hamming IL, Wolf R, Reyners AK, et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: a single-arm phase II study. Int J Cancer (2012) 131(5):E670–80. doi:10.1002/ijc.27388
11. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med (2012) 18(8):1254–61. doi:10.1038/nm.2883
12. Betts G, Jones E, Junaid S, El-Shanawany T, Scurr M, Mizen P, et al. Suppression of tumour-specific CD4(+) T cells by regulatory T cells is associated with progression of human colorectal cancer. Gut (2012) 61(8):1163–71. doi:10.1136/gutjnl-2011-300970
13. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi:10.1038/nm1093
14. Betts G, Twohig J, Van den Broek M, Sierro S, Godkin A, Gallimore A. The impact of regulatory T cells on carcinogen-induced sarcogenesis. Br J Cancer (2007) 96(12):1849–54. doi:10.1038/sj.bjc.6603824
15. Hindley JP, Ferreira C, Jones E, Lauder SN, Ladell K, Wynn KK, et al. Analysis of the T-cell receptor repertoires of tumor-infiltrating conventional and regulatory T cells reveals no evidence for conversion in carcinogen-induced tumors. Cancer Res (2011) 71(3):736–46. doi:10.1158/0008-5472.CAN-10-1797
16. Iellem A, Colantonio L, D’Ambrosio D. Skin-versus gut-skewed homing receptor expression and intrinsic CCR4 expression on human peripheral blood CD4+CD25+ suppressor T cells. Eur J Immunol (2003) 33(6):1488–96. doi:10.1002/eji.200323658
17. Bystry RS, Aluvihare V, Welch KA, Kallikourdis M, Betz AG. B cells and professional APCs recruit regulatory T cells via CCL4. Nat Immunol (2001) 2(12):1126–32. doi:10.1038/ni735
18. Annunziato F, Cosmi L, Liotta F, Lazzeri E, Manetti R, Vanini V, et al. Phenotype, localization, and mechanism of suppression of CD4(+)CD25(+) human thymocytes. J Exp Med (2002) 196(3):379–87. doi:10.1084/jem.20020110
19. Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, Su L, et al. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T cells during acute graft-versus-host disease. Blood (2005) 106(9):3300–7. doi:10.1182/blood-2005-04-1632
20. Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, Piccirillo CA. CCR5-dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. J Exp Med (2006) 203(11):2451–60. doi:10.1084/jem.20060956
21. Kallikourdis M, Andersen KG, Welch KA, Betz AG. Alloantigen-enhanced accumulation of CCR5+ “effector” regulatory T cells in the gravid uterus. Proc Natl Acad Sci U S A (2007) 104(2):594–9. doi:10.1073/pnas.0604268104
22. Hirahara K, Liu L, Clark RA, Yamanaka K, Fuhlbrigge RC, Kupper TS. The majority of human peripheral blood CD4+CD25highFoxp3+ regulatory T cells bear functional skin-homing receptors. J Immunol (2006) 177(7):4488–94.
23. Menning A, Hopken UE, Siegmund K, Lipp M, Hamann A, Huehn J. Distinctive role of CCR7 in migration and functional activity of naive- and effector/memory-like Treg subsets. Eur J Immunol (2007) 37(6):1575–83. doi:10.1002/eji.200737201
24. Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med (2001) 194(6):847–53. doi:10.1084/jem.194.6.847
25. Negus RP, Stamp GW, Hadley J, Balkwill FR. Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am J Pathol (1997) 150(5):1723–34.
26. Scotton C, Milliken D, Wilson J, Raju S, Balkwill F. Analysis of CC chemokine and chemokine receptor expression in solid ovarian tumours. Br J Cancer (2001) 85(6):891–7. doi:10.1054/bjoc.2001.2020
27. Milliken D, Scotton C, Raju S, Balkwill F, Wilson J. Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin Cancer Res (2002) 8(4):1108–14.
28. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol (2002) 196(3):254–65. doi:10.1002/path.1027
29. Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett (2007) 252(1):86–92. doi:10.1016/j.canlet.2006.12.012
30. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi:10.1126/science.1129139
31. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003) 348(3):203–13. doi:10.1056/NEJMoa020177
32. Gallimore A, Godkin A. Regulatory T cells and tumour immunity – observations in mice and men. Immunology (2008) 123(2):157–63. doi:10.1111/j.1365-2567.2007.02748.x
33. Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med (2007) 204(6):1335–47. doi:10.1084/jem.20070081
34. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res (2009) 69(5):2000–9. doi:10.1158/0008-5472.CAN-08-2360
35. Faget J, Biota C, Bachelot T, Gobert M, Treilleux I, Goutagny N, et al. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res (2011) 71(19):6143–52. doi:10.1158/0008-5472.CAN-11-0573
36. Miller AM, Lundberg K, Ozenci V, Banham AH, Hellstrom M, Egevad L, et al. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol (2006) 177(10):7398–405.
37. Haas J, Schopp L, Storch-Hagenlocher B, Fritzsching B, Jacobi C, Milkova L, et al. Specific recruitment of regulatory T cells into the CSF in lymphomatous and carcinomatous meningitis. Blood (2008) 111(2):761–6. doi:10.1182/blood-2007-08-104877
38. Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer (2008) 122(10):2286–93. doi:10.1002/ijc.23392
39. Maruyama T, Kono K, Izawa S, Mizukami Y, Kawaguchi Y, Mimura K, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to infiltration of regulatory T cells in esophageal squamous cell carcinoma. Dis Esophagus (2010) 23(5):422–9.
40. Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S, et al. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res (2006) 66(11):5716–22. doi:10.1158/0008-5472.CAN-06-0261
41. Lee H, Lee H, Kwon Y, Lee JH, Kim J, Shin MK, et al. Methyl gallate exhibits potent antitumor activities by inhibiting tumor infiltration of CD4+CD25+ regulatory T cells. J Immunol (2010) 185(11):6698–705. doi:10.4049/jimmunol.1001373
42. Pere H, Montier Y, Bayry J, Quintin-Colonna F, Merillon N, Dransart E, et al. A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood (2011) 118(18):4853–62. doi:10.1182/blood-2011-01-329656
43. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol (2009) 182(3):1746–55.
44. Chang LY, Lin YC, Mahalingam J, Huang CT, Chen TW, Kang CW, et al. Tumor-derived chemokine CCL5 enhances TGF-beta-mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res (2012) 72(5):1092–102. doi:10.1158/0008-5472.CAN-11-2493
45. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol (2012) 189(12):5602–11. doi:10.4049/jimmunol.1201018
46. Nakazaki Y, Hase H, Inoue H, Beppu Y, Meng XK, Sakaguchi G, et al. Serial analysis of gene expression in progressing and regressing mouse tumors implicates the involvement of RANTES and TARC in antitumor immune responses. Mol Ther (2006) 14(4):599–606. doi:10.1016/j.ymthe.2006.04.014
47. Gough M, Crittenden M, Thanarajasingam U, Sanchez-Perez L, Thompson J, Jevremovic D, et al. Gene therapy to manipulate effector T cell trafficking to tumors for immunotherapy. J Immunol (2005) 174(9):5766–73.
48. Uekusa Y, Yu WG, Mukai T, Gao P, Yamaguchi N, Murai M, et al. A pivotal role for CC chemokine receptor 5 in T-cell migration to tumor sites induced by interleukin 12 treatment in tumor-bearing mice. Cancer Res (2002) 62(13):3751–8.
49. Gonzalez-Martin A, Gomez L, Lustgarten J, Mira E, Manes S. Maximal T cell-mediated antitumor responses rely upon CCR5 expression in both CD4(+) and CD8(+) T cells. Cancer Res (2011) 71(16):5455–66. doi:10.1158/0008-5472.CAN-11-1687
50. Redjimi N, Raffin C, Raimbaud I, Pignon P, Matsuzaki J, Odunsi K, et al. CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res (2012) 72(17):4351–60. doi:10.1158/0008-5472.CAN-12-0579
51. Yang S, Wang B, Guan C, Wu B, Cai C, Wang M, et al. Foxp3+IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. J Leukoc Biol (2011) 89(1):85–91. doi:10.1189/jlb.0910506
52. Musha H, Ohtani H, Mizoi T, Kinouchi M, Nakayama T, Shiiba K, et al. Selective infiltration of CCR5(+)CXCR3(+) T lymphocytes in human colorectal carcinoma. Int J Cancer (2005) 116(6):949–56. doi:10.1002/ijc.21135
53. Kondo T, Nakazawa H, Ito F, Hashimoto Y, Osaka Y, Futatsuyama K, et al. Favorable prognosis of renal cell carcinoma with increased expression of chemokines associated with a Th1-type immune response. Cancer Sci (2006) 97(8):780–6. doi:10.1111/j.1349-7006.2006.00231.x
54. Gorbachev AV, Kobayashi H, Kudo D, Tannenbaum CS, Finke JH, Shu S, et al. CXC chemokine ligand 9/monokine induced by IFN-gamma production by tumor cells is critical for T cell-mediated suppression of cutaneous tumors. J Immunol (2007) 178(4):2278–86.
55. Hensbergen PJ, Wijnands PG, Schreurs MW, Scheper RJ, Willemze R, Tensen CP. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J Immunother (2005) 28(4):343–51. doi:10.1097/01.cji.0000165355.26795.27
56. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475(7355):226–30. doi:10.1038/nature10169
57. Li B, Lalani AS, Harding TC, Luan B, Koprivnikar K, Huan Tu G, et al. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin Cancer Res (2006) 12(22):6808–16. doi:10.1158/1078-0432.CCR-06-1558
58. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res (2013) 73(2):539–49. doi:10.1158/0008-5472.CAN-12-2325
59. Suzuki H, Onishi H, Morisaki T, Tanaka M, Katano M. Intratumoral FOXP3+VEGFR2+ regulatory T cells are predictive markers for recurrence and survival in patients with colorectal cancer. Clin Immunol (2013) 146(1):26–33. doi:10.1016/j.clim.2012.10.007
60. Kryczek I, Lange A, Mottram P, Alvarez X, Cheng P, Hogan M, et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res (2005) 65(2):465–72.
61. Yan M, Jene N, Byrne D, Millar EK, O’Toole SA, McNeil CM, et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res (2011) 13(2):R47. doi:10.1186/bcr2869
62. Wald O, Izhar U, Amir G, Avniel S, Bar-Shavit Y, Wald H, et al. CD4+CXCR4highCD69+ T cells accumulate in lung adenocarcinoma. J Immunol (2006) 177(10):6983–90.
63. Jaafar F, Righi E, Lindstrom V, Linton C, Nohadani M, Van Noorden S, et al. Correlation of CXCL12 expression and FoxP3+ cell infiltration with human papillomavirus infection and clinicopathological progression of cervical cancer. Am J Pathol (2009) 175(4):1525–35. doi:10.2353/ajpath.2009.090295
64. Liles WC, Broxmeyer HE, Rodger E, Wood B, Hubel K, Cooper S, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood (2003) 102(8):2728–30. doi:10.1182/blood-2003-02-0663
65. Righi E, Kashiwagi S, Yuan J, Santosuosso M, Leblanc P, Ingraham R, et al. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res (2011) 71(16):5522–34. doi:10.1158/0008-5472.CAN-10-3143
66. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science (2010) 328(5979):749–52. doi:10.1126/science.1185837
67. Turnquist HR, Lin X, Ashour AE, Hollingsworth MA, Singh RK, Talmadge JE, et al. CCL21 induces extensive intratumoral immune cell infiltration and specific anti-tumor cellular immunity. Int J Oncol (2007) 30(3):631–9.
68. Igoucheva O, Grazzini M, Pidich A, Kemp DM, Larijani M, Farber M, et al. Immunotargeting and eradication of orthotopic melanoma using a chemokine-enhanced DNA vaccine. Gene Ther (2013). doi:10.1038/gt.2013.17 [Epub ahead of print].
69. Kar UK, Srivastava MK, Andersson A, Baratelli F, Huang M, Kickhoefer VA, et al. Novel CCL21-vault nanocapsule intratumoral delivery inhibits lung cancer growth. PLoS ONE (2011) 6(5):e18758. doi:10.1371/journal.pone.0018758
70. Correale P, Rotundo MS, Botta C, Del Vecchio MT, Tassone P, Tagliaferri P. Tumor infiltration by chemokine receptor 7 (CCR7)(+) T-lymphocytes is a favorable prognostic factor in metastatic colorectal cancer. Oncoimmunology (2012) 1(4):531–2. doi:10.4161/onci.19404
71. Bacon KB, Premack BA, Gardner P, Schall TJ. Activation of dual T cell signaling pathways by the chemokine RANTES. Science (1995) 269(5231):1727–30. doi:10.1126/science.7569902
72. Bacon KB, Szabo MC, Yssel H, Bolen JB, Schall TJ. RANTES induces tyrosine kinase activity of stably complexed p125FAK and ZAP-70 in human T cells. J Exp Med (1996) 184(3):873–82. doi:10.1084/jem.184.3.873
73. Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, et al. Tumor evasion of the immune system by converting CD4+CD25- T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol (2007) 178(5):2883–92.
74. Valzasina B, Piconese S, Guiducci C, Colombo MP. Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25- lymphocytes is thymus and proliferation independent. Cancer Res (2006) 66(8):4488–95. doi:10.1158/0008-5472.CAN-05-4217
75. Fourcade J, Sun Z, Kudela P, Janjic B, Kirkwood JM, El-Hafnawy T, et al. Human tumor antigen-specific helper and regulatory T cells share common epitope specificity but exhibit distinct T cell repertoire. J Immunol (2010) 184(12):6709–18. doi:10.4049/jimmunol.0903612
76. Bruder D, Probst-Kepper M, Westendorf AM, Geffers R, Beissert S, Loser K, et al. Neuropilin-1: a surface marker of regulatory T cells. Eur J Immunol (2004) 34(3):623–30. doi:10.1002/eji.200324799
77. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol (2010) 184(7):3433–41. doi:10.4049/jimmunol.0904028
78. Elkord E, Sharma S, Burt DJ, Hawkins RE. Expanded subpopulation of FoxP3+ T regulatory cells in renal cell carcinoma co-express Helios, indicating they could be derived from natural but not induced Tregs. Clin Immunol (2011) 140(3):218–22. doi:10.1016/j.clim.2011.04.014
79. Wainwright DA, Sengupta S, Han Y, Lesniak MS. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol (2011) 13(12):1308–23. doi:10.1093/neuonc/nor134
80. Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med (2012) 209(11):2001–16. doi:10.1084/jem.20111497
81. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med (2012) 209(10):1723–42, S1.
82. Gottschalk RA, Corse E, Allison JP. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol (2012) 188(3):976–80. doi:10.4049/jimmunol.1102964
83. Milpied P, Renand A, Bruneau J, Mendes-da-Cruz DA, Jacquelin S, Asnafi V, et al. Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur J Immunol (2009) 39(6):1466–71. doi:10.1002/eji.200839040
84. Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science (2013) 339(6124):1219–24. doi:10.1126/science.1233913
85. Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res (2007) 67(18):8865–73. doi:10.1158/0008-5472.CAN-07-0767
86. Bui JD, Uppaluri R, Hsieh CS, Schreiber RD. Comparative analysis of regulatory and effector T cells in progressively growing versus rejecting tumors of similar origins. Cancer Res (2006) 66(14):7301–9. doi:10.1158/0008-5472.CAN-06-0556
87. Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity (2004) 21(2):267–77. doi:10.1016/j.immuni.2004.07.009
88. Pacholczyk R, Kern J, Singh N, Iwashima M, Kraj P, Ignatowicz L. Nonself-antigens are the cognate specificities of Foxp3+ regulatory T cells. Immunity (2007) 27(3):493–504. doi:10.1016/j.immuni.2007.07.019
89. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med (2005) 202(7):919–29. doi:10.1084/jem.20050463
90. Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest (2007) 117(9):2570–82. doi:10.1172/JCI31911
91. Chung DJ, Rossi M, Romano E, Ghith J, Yuan J, Munn DH, et al. Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood (2009) 114(3):555–63. doi:10.1182/blood-2008-11-191197
92. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi:10.1038/ni1536
93. Donkor MK, Sarkar A, Savage PA, Franklin RA, Johnson LK, Jungbluth AA, et al. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-beta1 cytokine. Immunity (2011) 35(1):123–34. doi:10.1016/j.immuni.2011.04.019
94. Mahalingam J, Lin YC, Chiang JM, Su PJ, Fang JH, Chu YY, et al. LAP+CD4+ T cells are suppressors accumulated in the tumor sites and associated with the progression of colorectal cancer. Clin Cancer Res (2012) 18(19):5224–33. doi:10.1158/1078-0432.CCR-12-0211
95. Kakita N, Kanto T, Itose I, Kuroda S, Inoue M, Matsubara T, et al. Comparative analyses of regulatory T cell subsets in patients with hepatocellular carcinoma: a crucial role of CD25(-) FOXP3(-) T cells. Int J Cancer (2012) 131(11):2573–83. doi:10.1002/ijc.27535
96. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
97. Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A (2003) 100(14):8372–7. doi:10.1073/pnas.1533209100
98. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28(19):3167–75. doi:10.1200/JCO.2009.26.7609
99. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi:10.1016/1074-7613(95)90125-6
100. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270(5238):985–8. doi:10.1126/science.270.5238.985
101. Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol (1998) 10(10):1563–72. doi:10.1093/intimm/10.10.1563
102. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A (2012) 109(43):17561–6. doi:10.1073/pnas.1215397109
103. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res (2010) 70(15):6171–80. doi:10.1158/0008-5472.CAN-10-0153
104. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res (2012) 18(22):6110–21. doi:10.1158/1078-0432.CCR-12-2130
105. Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed) (2012) 4:734–45. doi:10.2741/414
106. Benchetrit F, Ciree A, Vives V, Warnier G, Gey A, Sautes-Fridman C, et al. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood (2002) 99(6):2114–21. doi:10.1182/blood.V99.6.2114
107. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity (2009) 31(5):787–98. doi:10.1016/j.immuni.2009.09.014
108. Amedei A, Niccolai E, Benagiano M, Della Bella C, Cianchi F, Bechi P, et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother (2013) 62(7):1249–60. doi:10.1007/s00262-013-1429-3
Keywords: tumor immunology, regulatory T cells, intra-tumoral proliferation, chemokines, immunotherapy of cancer
Citation: Ondondo B, Jones E, Godkin A and Gallimore A (2013) Home sweet home: the tumor microenvironment as a haven for regulatory T cells. Front. Immunol. 4:197. doi: 10.3389/fimmu.2013.00197
Received: 24 May 2013; Paper pending published: 11 June 2013;
Accepted: 03 July 2013; Published online: 16 July 2013.
Edited by:
Eyad Elkord, United Arab Emirates University, UAE; University of Salford, UK; University of Manchester, UKReviewed by:
Bin Li, Chinese Academy of Sciences, ChinaAmedeo Amedei, University of Florence, Italy
Copyright: © 2013 Ondondo, Jones, Godkin and Gallimore. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Awen Gallimore, Infection and Immunity, School of Medicine, Henry Wellcome Building, University Cardiff, Cardiff CF14 4XN, UK e-mail:Z2FsbGltb3JlYW1AY2YuYWMudWs=