

- Tytgat Institute for Liver and Intestinal Research, Department of Gastroenterology and Hepatology, Academic Medical Center, Amsterdam, Netherlands
Intestinal epithelial cells (IECs) are integral players in homeostasis of immunity and host defense in the gut and are under influence of the intestinal microbiome. Microbial metabolites and dietary components, including short chain fatty acids (acetate, propionate, and butyrate, SCFAs), have an impact on the physiology of IECs at multiple levels, including the inhibition of deacetylases affecting chromatin remodeling and global changes in transcriptional activity. The number and diversity of butyrate-producing bacteria is subject to factors related to age, disease, and to diet. At physiological levels, SCFAs are inhibitors of histone deacetylases (HDACs) which may explain the transcriptional effects of SCFAs on epithelial cells, although many effects of SCFAs on colonic mucosa can be ascribed to mechanisms beyond HDAC inhibition. Interference with this type of post-translational modification has great potential in cancer and different inflammatory diseases, because HDAC inhibition has anti-proliferative and anti-inflammatory effects in vitro, and in in vivo models of intestinal inflammation. Hence, the influence of dietary modulators on HDAC activity in epithelia is likely to be an important determinant of its responses to inflammatory and microbial challenges.
The Intestinal Epithelium Contributes to Intestinal Homeostasis
The intestinal epithelial cells (IECs) act as a physical barrier between the stromal cells and immune cells in the lamina propria and luminal antigens. IECs contribute to intestinal homeostasis via secretion of cytokines, metabolites, and anti-microbial peptides. The intestinal epithelial layer in fact comprises specialized epithelia of many different cell types: Paneth cells that secrete different anti-microbial peptides, mucin secreting goblet cells, enteroendocrine cells, enterocytes, and colonocytes outside the crypt and M-cells, located on top of intestinal lymphoid follicles. Interspersed with the IECs are different immune cells, including intra-epithelial γδ-T cells and specialized mucosal macrophages with the capacity to sampling antigen from the lumen.
Intestinal epithelial cells express different pattern recognition receptors, including Toll-like receptors and NOD-like receptors but the tolerogenic phenotype of epithelia is not readily understood. For example, IECs acquire endotoxin tolerance toward lipopolysaccharide despite the expression of TLR4 soon after birth (1), in some aspects comparable to the TLR hyporesponsiveness found in intestinal macrophages (2). In support of intestinal homeostasis, IECs secrete many factors that are needed for homeostasis of the mucosal compartment including transforming growth factor beta (TGF-beta), IL-10, thymic stromal lymphopoietin (TSLP), retinoic acid (RA), prostaglandin E2 (PGE2). Some of these factors have prominent autocrine effects. TGF-beta, for example, suppresses proliferation of rat IECs thereby preventing hyperproliferation and tumor formation (3). Cao et al. showed that diet supplemented with 20% pectin (dietary fiber) could increase TGF-beta/SMAD3 signaling in mouse jejunum, but not in colon (4). TGF-beta is also a regulator of immune cell polarization and function, but these effects have been discussed elsewhere (5–7).
Microbiome, Metabolites, and Epithelial Biology
Intestinal microbes have a mutualistic relationship with its host in part by dictating epithelial cell responses and intestinal barrier homeostasis. We coexist with our microbiota, but this relationship sometimes becomes pathological, and contributes to diseases such as inflammatory bowel disease (IBD). The impact of the gut microbiome on immune homeostasis has been identified around 60 years ago, but recent metagenomic analyses of microbiome changes associated with multiple diseases have advanced this field enormously (8–10). These recent screens have advanced our understanding of the changed microbiome in IBD (11, 12). A main outcome of microbiome sequencing is an underrepresentation of SCFA producing strains in IBD patients (13), corresponding with earlier observations of for instance decreased SCFA levels in feces of children with IBD (14).
Carbohydrates, resistant to breakdown in the stomach and small intestine, are subject to colonic fermentation to result in the production of SCFAs, fatty acids containing 1–6 carbon atoms. Anaerobic bacteria generate the major SCFAs acetate, propionate, and butyrate and the highest production is found in the proximal colon. SCFA concentrations are dependent on the availability and source of substrate, the microflora, and gut transit time. The estimated total amount of SCFAs in the proximal colon is higher than in the distal colon (from 70–140 mM in proximal to 20–70 mM distal part), mainly because of availability of carbohydrates and presence of water (15, 16). Oxidation of SCFAs accounts for 60–70% of the colonic epithelial energy need (16). Butyrate is through β-oxidation the main, and preferred, energy source for colonocytes and also has a variety of other physiological effects, including modulation of epithelial responses to cytokines, see for instance Figure 1 and a summary of previous work on this matter in Table 1.
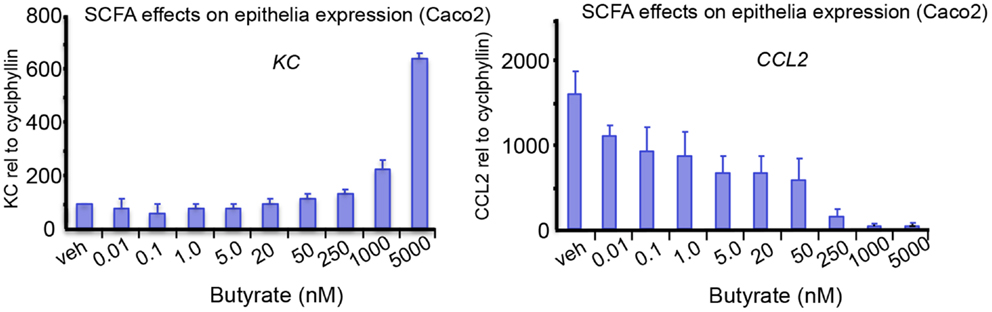
Figure 1. The effects of SCFA butyrate on epithelial cell responses in Caco-2 enterocyte-like cells. Cells were stimulated with 10 ng/mL of ILlb and tested against indicated butyrate concentrations added simultaneously. Expression levels of IL-8, and CCL2, were measured after 24 h treatment. Butyrate modulates the epithelial responses to cytokines at relevant concentrations. Tytgat Institute, 2013.
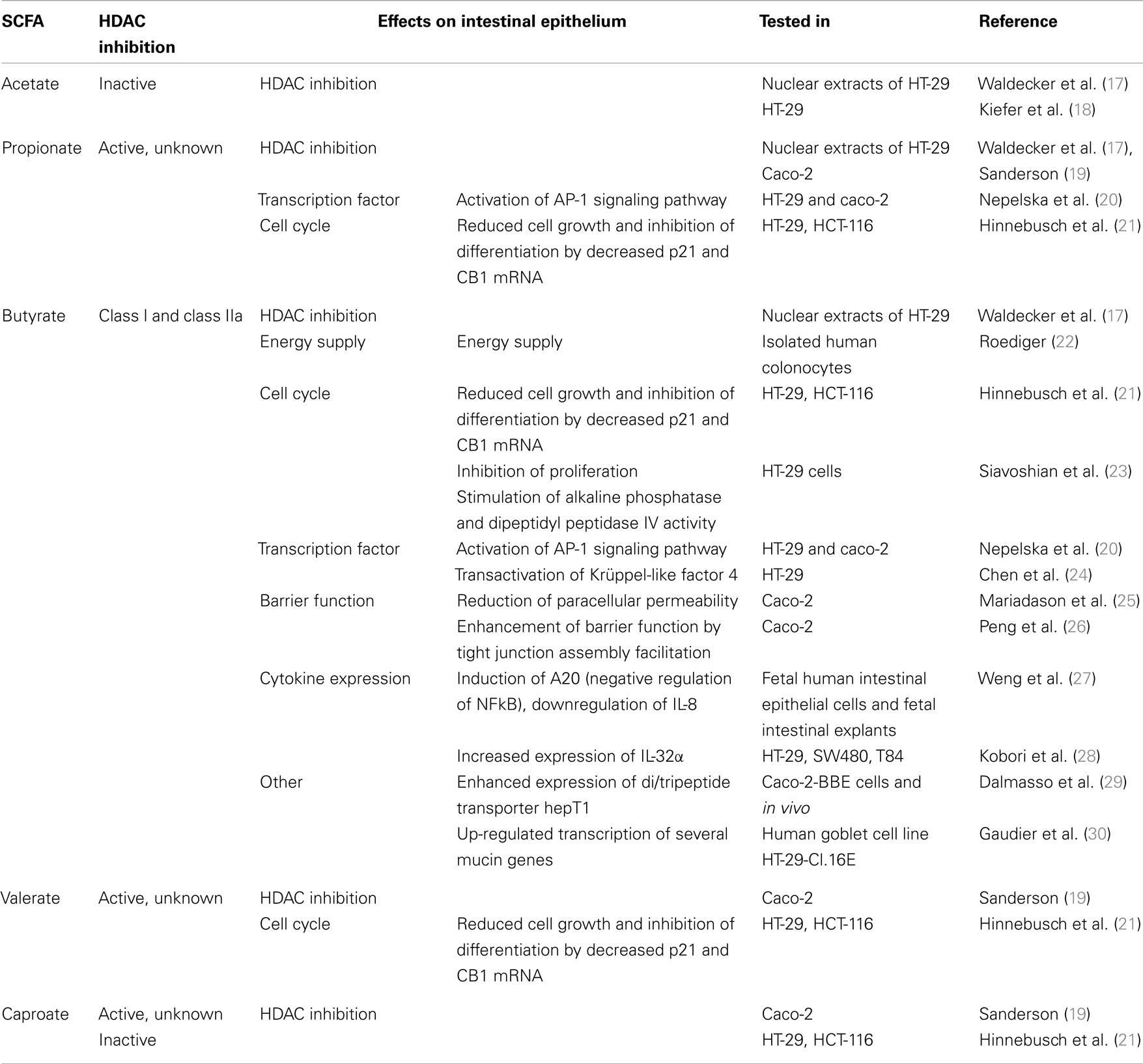
Table 1. Examples of SCFA-induced effects on intestinal epithelium.
Regulation of Epithelial Gene Expression through Acetylation and Deacetylation
In the large bowel, SCFAs butyrate and propionate reach concentrations that are able to inhibit the activity of an important class of epigenetic modifiers, histone deacetylases (HDACs). Mammalian cells contain 18 different HDACs, grouped into 4 different classes: 11 HDACs within classes I, II, and IV are Zn2+ dependent, while class III is comprised of 7 sirtuins which are NAD+ dependent. Class 1 HDACs (HDAC1, HDAC2, HDAC3, and HDAC8) are predominantly localized in the nucleus, have strong deacetylase activity and are expressed in every cell. Class II HDACs shuttle between nucleus and cytoplasm and can be subdivided into class IIa (HDAC4, HDAC5, HDAC7, and HDAC9) and class IIb (HDAC6 and HDAC10). Class IIa enzymes are found in multiprotein complexes and have weak deacetalyse activity, class IIb enzymes are active deacetylases (31).
Acetylation and deacetylation of lysines are post-translational modifications that occur on histone tails, but also on many other non-histone proteins. Choudhary et al. used high-resolution mass spectrometry to identify a total of 1750 acetylated proteins. Lysine acetylation sites overlapped between different cell lines for 60–70% and occurs preferentially in macromolecular complexes affecting the regulation of almost all nuclear, but also many cytoplasmic processes, including chaperone complexes and cytoskeleton remodeling (32).
Acetylation, particularly of histone H3 and histone H4 tails, has almost invariably been linked to a loosened chromatin structure and activation of transcription (33–35). Conversely, histone hypoacetylation correlates with condensed chromatin and transcriptional repression. Histone acetyl transferases (HATs) are enzymes that catalyze the addition of acetyl moieties to histones and other proteins. HAT activity is counter-acted by the activity of HDACs, which remove acetyl groups from lysines in histones and other proteins (36). The function of acetylation on non histone proteins, such as transcription factors, has been elucidated by identification of acetylation of individual proteins (e.g., different STATs, p53, NF-κB) (37–39). The effects of HAT and HDAC activity play direct and indirect roles in a variety of factors including mRNA stability, cell signaling, protein localization, binding, and function and can also prevent or increase proteasomal degradation (40).
Endogenous Deacetylase Inhibitors
Among SCFAs, butyrate induces the highest acetylation (41). It is generally thought that butyrate inhibits class I and class IIa HDACs but not class IIb (HDAC6 and HDAC10) and sirtuins, however supporting evidence is lacking. One possibility is that butyrate inhibits deacetylation, but not HDAC activity directly (42). However, earlier studies have addressed the potential of SCFAs to alter epigenetic marks in epithelial cells (43). In particular the SCFA butyrate inhibits all class I HDACs. Not only this, it also seems to affect many other epigenetic-related enzymes by regulating the expression of the respective genes encoding HDACs. In bovine epithelial cell cultures, RNA sequencing analyses revealed that the expression of HDACs was modulated by butyrate treatment. Whereas the expression of HCACs 7, 8, and 9 are down-regulated, HDACs 5 and 11 are up-regulated (43). It is to be determined why inhibition of enzymatic activities, seemingly regulates their own expression at the mRNA level. We have summarized earlier data on the effect of SCFA on HDAC activity in different epithelial cell assays in Table 1.
In addition to SCFAs, the microbiome metabolic activity can lead to secretion of metabolites that can interfere with HDAC activity. There are a variety of dietary HDAC inhibitors described earlier, either as dietary component or arising after metabolic activity in the microbiome. Lactate and pyruvate are two of these bacterial components. Lactate itself serves as a substrate for butyrate formation, but has the capacity to inhibit the activity of HDACs, but only in very high concentrations (IC50 of 40 mM) (44, 45). These levels of lactate are found in muscle cells only during intense exercise, so it is unlikely that lactate acts as a deacetylase inhibitor in the gut. As such, it is unlikely that the concentrations of SCFAs, besides butyrate and propionate, or bacterial components, like pyruvate and lactate, are high enough to significantly inhibit HDAC function in epithelia, although local fluctuations may render transient HDAC inhibiting properties.
Further examples are isothiocyanates and allyl sulfides, and the foods from which they are derived (46). Sulforaphane is another example, an isothiocyanate, derived from glucoraphanin in broccoli and broccoli sprouts, first identified as a potent inducer of phase 2 detoxification enzymes (46). A common denominator of these compounds is food constituents with chemical structures that contained a spacer “arm” that might fit the HDAC active site, and a functional group that could interact with the buried catalytic zinc atom (47).
Receptors for SCFAs
Over 90–95% of the produced SCFAs are taken up in the colon and the remaining 5–10% is secreted in the feces. Over 60% of this uptake happens by diffusion of SCFAs across the epithelial membrane, the rest is transported into the cell (16). In human colon, the G-protein coupled nicotinate receptor GPR109A is expressed at the apical membrane of the epithelial layer. Activation of GPR109A by butyrate and other ligands blocked basal and LPS-induced NF-κB activation (48). Butyrate concentration in the lumen is over 10 times higher than what is needed to half-maximal activate this receptor, 1.6 vs. 20 mM, respectively (48).
SCFAs can be transported into the cell by monocarboxylate transporters (MCT), which are coupled to H+ transport. Because there is almost no H+ gradient over the luminal membrane, this type of transport does not seem to be very active. An alternative transport which is more likely to be the main type of transport is mediated through SLC5A8 (or sodium-coupled MCT, SMCT), which is coupled to Na+ over the electrochemical Na+ gradient. SLC5A8 is expressed in colonic epithelium at the apical membrane and transports substrates lactate, pyruvate, acetate, propionate, and butyrate (49).
Two additional epithelial receptors for SCFAs were identified. FFAR3 (GPR41) and FFAR2 (GPR43), both G-protein coupled receptors, are activated by high micromolar or millimolar SCFA concentrations, but have different substrate activation potencies (50–52). FFAR2 is expressed in immune cells: mast cells, B lymphocytes, monocytes, eosinophils, and also neutrophils, in which FFAR2 is the only functional receptor for SCFAs in mice. Expression of FFAR2 on immune cells seems to be important in the regulation of immune responses, since mice lacking FFAR2 develop exacerbated inflammation in models for colitis, asthma, and arthritis due to higher production of inflammatory mediators and increased recruitment of immune cells (43, 53). The expression of FFAR2 is also needed for the SCFA-mediated regulation of size and function of the colonic regulatory T-cell pool (54). In addition, FFAR2 functions as a chemotactic receptor, moving neutrophils, and probably other immune cells expressing FFAR2, toward sources of SCFAs (55). FFAR3 mRNA is expressed in many different tissues, but high expression was found primarily in adipose tissue (50, 51). Tazoe et al. identified FFAR3 protein expression in enterocytes and peptide YY containing enteroendocrine cells (56), which also express FFAR2 (57).
How SCFAs are transported from the colonocytes into the circulation is not entirely clear yet. One possibility is by transport via the MCT. This transporter is expressed at the basolateral membrane, which is in line with this hypothesis (58), see Figure 2.
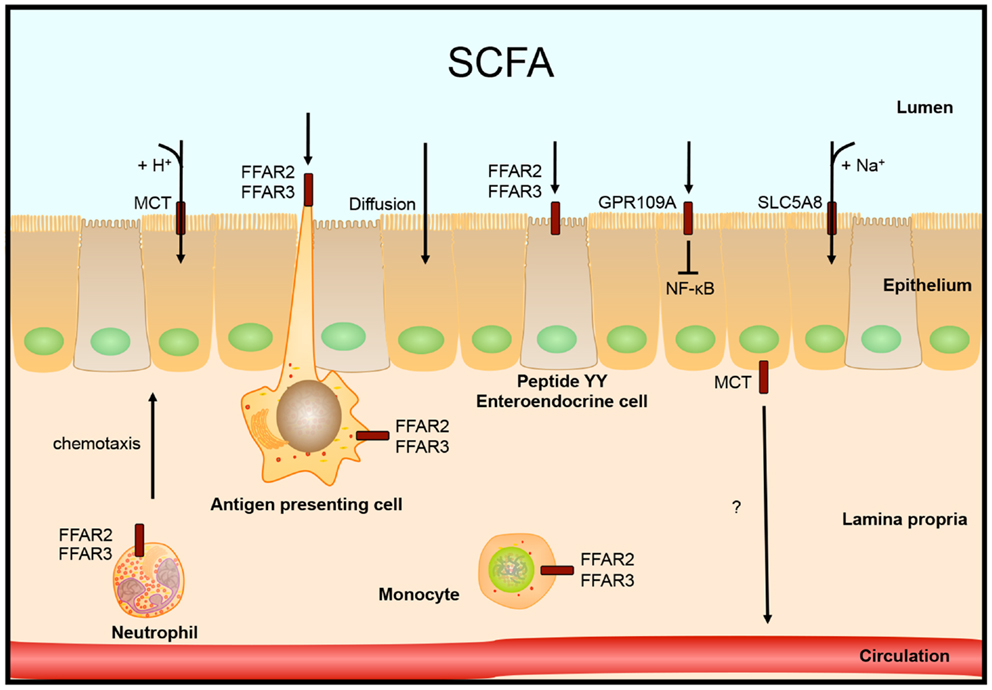
Figure 2. Intestinal SCFA receptors and transporters. SCFAs are taken up by the epithelial cells by diffusion, H+ coupled transport by monocarboxylate transporters (MCT) or by Na+ coupled transport by SLC5A8. Other receptors that are activated by SCFA are localized on colonocytes, peptide YY expressing enteroendocrine cells, or different immune cells. Receptor FFAR2 is involved in neutrophil chemotaxis toward sources of SCFA.
HDAC Inhibiting Agents in Inflammatory Bowel Disease
Because of the anti-inflammatory and anti-tumor properties of HDAC inhibition, deacetylase inhibitors have potential for treatment of IBD and IBD associated colorectal cancer, further reviewed in Refs. (59, 60). Increased intake of SCFAs has the capacity to ameliorate colitis disease parameters (53), possibly via the impact on HDAC activity. Glauben et al. tested HDAC inhibitors and SAHA in innate (DSS) and T-cell driven (TNBS) colitis models. In DSS and TNBS models, oral intake of both SAHA and valproic acid reduced several disease parameters. Treatment with VPA in DSS conditions increased acetylation, which was found locally in lamina propria mononuclear cells, but not in liver homogenate or splenocytes (61). A clear impact of HDAC inhibitors is described on the generation of regulatory T cells, an effect that may well contribute to the efficacy of HDAC inhibitors to colitis (62).
Histone Deacetylases, Colonic Cell Differentiation, and Cancer
Given the ability of HDAC inhibitors to induce growth arrest, maturation, and apoptosis of colon cancer cell lines, it is likely that HDACs themselves play a physiological role in the maintenance of cell proliferation and survival and suppression of IEC maturation. In comparison with HATs, HDACs are of particular interest in medical research because of the ability to inhibit their deacetylase activity. Inhibition of deacetylases leads to hyperacetylation and results in a block of proliferation in tumor cells, by a variety of mechanisms, including the induction of differentiation, apoptosis, and transcriptional upregulation of tumor suppressors (63). Two chemical HDAC inhibitors, vorinostat and romidepsin, have been approved by the FDA for treatment of cutaneous T-cell lymphoma (46). HDAC inhibitors also negatively effect tumorigenesis by affecting angiogenesis and modulating immune responses (64). In addition, SAHA has prominent effects on the inflammatory response (65). This gives rise to potential use in a range of other diseases, including chronic inflammatory disease like IBD (66).
The modulating effects of HDAC inhibition on epithelial cell differentiation can have major effects on the development of colon cancers. Functional genetic screenings to elucidate potential pathways targeted by HDAC inhibitors were described earlier (67). In these screens overexpression of genes involved in RA signaling selectively rendered tumor cells resistant to treatment with HDAC inhibitors. In these resistant cells, overexpression of two genes allowed cells to bypass proliferation arrest and apoptosis imposed by HDAC inhibitors: RA receptor alpha (RARα) and preferentially expressed antigen of melanoma (67). The latter gene encodes a tumor antigen and has been identified as a repressor of RA signaling (68). Hence, HDAC inhibitor treatment caused de-repression of RA target genes in these cells, suggesting a role for RA signaling in the anti-cancer effects induced by HDAC inhibitors.
This is particularly interesting in the light of effects of butyrate on epithelial modulation of cell proliferation, survival, and apoptosis (16, 43). In particular HDAC3 was put forward because overexpression of HDAC overcomes the butyrate repression of p21, a potent cyclin-dependent kinase inhibitor that functions as a regulator of cell cycle progression at G1 (69, 70). Additionally, protein expression of HDAC3 was significantly up-regulated in a panel of human colon tumors compared with adjacent normal mucosa and in small intestinal adenomas derived from APC mutant mice epithelia, establishing a link between HDAC3 expression and intestinal cell transformation. From these findings it is expected that aberrant expression of HDAC3 and other class I HDACs play a role in the progression of colon cancer. HDAC3 activity may be dependent on the activity of class IIa HDAC4 protein, as Sp1-dependent targeting of HDAC4 to the proximal p21 promoter in colon cancer cells was shown (70). Hence, repression of the activity of the proximal p21 promoter is likely mediated by HDAC4 through association with the catalytically active HDAC3, within the N-CoR/SMRT co-repressor complex (69, 71). Furthermore, it has been shown that HDAC4 acts as a “scaffold” protein with the HDAC3–NCo-R/SMRT complex without contributing to the overall deacetylase activity of the complex, consistent with the weak catalytic activity of HDAC4 and other class IIa HDACs (71, 72).
Future Perspectives
The human intestinal flora has an important role in homeostasis in health and disease, not only because the help to digest food, but also because of the secretion of many metabolites. Differences in diversity were found between feces from healthy individuals and feces from patients with various disease states, obesity, type 2 diabetes, IBD, allergies (11). It remains unknown whether the shifts in flora are consequential of disease or whether they also have a causative nature. Interestingly, in feces from IBD patients, the diversity of butyrate-producing bacteria is lower than in feces of healthy controls (13, 14). Even though fecal samples do not reflect the actual intestinal flora, such a shift has the potential to affect the intestine considerably by the altering metabolite and SCFA secretion (73).
In line with this, it will also be interesting to see to what extend the SCFA levels and HDAC-mediated processes can be modulated by administration of prebiotics and probiotics (74–76). In patients with chronic pouchitis and ulcerative colitis patients in remission, probiotic treatment was effective in the prevention of relapse. However, additional controlled clinical trials are needed to fully appreciate the potential of prebiotic or probiotic administration in IBD (77–79).
Other promising studies that are ongoing are fecal microbiota transplantation studies performed in different diseased settings, including obesity, metabolic syndrome, IBD, and in recurrent Clostridium difficile infections (80). In a recent trail with patients with recurrent C. difficile infections, 81.3% of the patients cured from infection after a single infusion of donor feces, which was substantially higher than the 30% with standard antibiotic treatment. Microbiota diversity of the patients increased 2 weeks after infusion to reach levels of donors (81). In another study, intestinal microbiota transfer was used to increase insulin sensitivity in patients with metabolic syndrome. Again, microbial diversity was increased after infusion, among them were different strains of butyrate-producing bacteria (82). Careful analysis of these therapies can possible give an indication whether the altered microbiota composition is the consequence of chronic ongoing inflammation or whether it has a more causative nature.
Conflict of Interest Statement
W. J. de Jonge receives funding from Mead Johnson Nutrition and GlaxoSmithKline Research.
Acknowledgments
R. Schilderink and W. J. de Jonge are funded by a grant from the Dutch Organization for Scientific Research (NWO-VIDI).
References
1. Lotz M, Gütle D, Walther S, Ménard S, Bogdan C, Hornef MW. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J Exp Med (2006) 203:973–84. doi:10.1084/jem.20050625
2. Smythies LE, Shen R, Bimczok D, Novak L, Clements RH, Eckhoff DE, et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J Biol Chem (2010) 285:19593–604. doi:10.1074/jbc.M109.069955
3. Barnard JA, Beauchamp RD, Coffey RJ, Moses HL. Regulation of intestinal epithelial cell growth by transforming growth factor type beta. Proc Natl Acad Sci U S A (1989) 86:1578–82. doi:10.1073/pnas.86.5.1578
4. Cao Y, Gao X, Zhang W, Zhang G, Nguyen AK, Liu X, et al. Dietary fiber enhances TGF-β signaling and growth inhibition in the gut. Am J Physiol Gastrointest Liver Physiol (2011) 301:G156–64. doi:10.1152/ajpgi.00362.2010
5. Li MO, Flavell RA. TGF-β: a master of all T cell trades. Cell (2008) 134:392–404. doi:10.1016/j.cell.2008.07.025
6. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol (2010) 10:554–67. doi:10.1038/nri2808
7. Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest (2004) 115:66–75. doi:10.1172/JCI200519229
8. Dubos RJ, Schaedler RW. The effect of diet on the fecal bacterial flora of mice and on their resistance to infection. J Exp Med (1962) 115:1161–72. doi:10.1084/jem.115.6.1161
9. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature (2010) 464:59–65. doi:10.1038/nature08821
10. Human Microbiome Project Consortium. A framework for human microbiome research. Nature (2012) 486:215–21. doi:10.1038/nature11209
11. Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell (2012) 148:1258–70. doi:10.1016/j.cell.2012.01.035
12. Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci U S A (2012) 109:594–9. doi:10.1073/pnas.1116053109
13. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A (2007) 104:13780–5. doi:10.1073/pnas.0706625104
14. Treem WR, Ahsan N, Shoup M, Hyams JS. Fecal short-chain fatty acids in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr (1994) 18:159–64. doi:10.1097/00005176-199402000-00007
15. Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev (2001) 81:1031–64.
16. Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol (2006) 40:235–43. doi:10.1097/00004836-200603000-00015
17. Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem (2008) 19:7–7. doi:10.1016/j.jnutbio.2007.08.002
18. Kiefer J, Beyer-Sehlmeyer G, Pool-Zobel BL. Mixtures of SCFA, composed according to physiologically available concentrations in the gut lumen, modulate histone acetylation in human HT29 colon cancer cells. Br J Nutr (2007) 96(5):803–10.
19. Sanderson IR. Short chain fatty acid regulation of signaling genes expressed by the intestinal epithelium. J Nutr (2004) 134:2450S–4.
20. Nepelska M, Cultrone A, Béguet-Crespel F, Le Roux K, Doré J, Arulampalam V, et al. Butyrate produced by commensal bacteria potentiates phorbol esters induced AP-1 response in human intestinal epithelial cells. PLoS ONE (2012) 7:e52869. doi:10.1371/journal.pone.0052869
21. Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr (2002) 132:1012–7.
22. Roediger WE. The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet (1980) 2:712–5. doi:10.1016/S0140-6736(80)91934-0
23. Siavoshian S, Segain JP, Kornprobst M, Bonnet C, Cherbut C, Galmiche JP, et al. Butyrate and trichostatin A effects on the proliferation/differentiation of human intestinal epithelial cells: induction of cyclin D3 and p21 expression. Gut (2000) 46:507–14. doi:10.1136/gut.46.4.507
24. Chen ZY, Rex S, Tseng CC. Krüppel-like factor 4 is transactivated by butyrate in colon cancer cells. J Nutr (2004) 134:792–8.
25. Mariadason JM, Barkla DH, Gibson PR. Effect of short-chain fatty acids on paracellular permeability in Caco-2 intestinal epithelium model. Am J Physiol (1997) 272:G705–12.
26. Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr (2009) 139:1619–25. doi:10.3945/jn.109.104638
27. Weng M, Walker WA, Sanderson IR. Butyrate regulates the expression of pathogen-triggered IL-8 in intestinal epithelia. Pediatr Res (2007) 62:542–6. doi:10.1203/PDR.0b013e318155a422
28. Kobori A, Bamba S, Imaeda H, Ban H, Tsujikawa T, Saito Y, et al. Butyrate stimulates IL-32alpha expression in human intestinal epithelial cell lines. World J Gastroenterol (2010) 16:2355–61. doi:10.3748/wjg.v16.i19.2355
29. Dalmasso G, Nguyen HT, Yan Y, Charrier-Hisamuddin L, Sitaraman SV, Merlin D. Butyrate transcriptionally enhances peptide transporter PepT1 expression and activity. PLoS ONE (2008) 3:e2476. doi:10.1371/journal.pone.0002476
30. Gaudier E, Jarry A, Blottière HM, de Coppet P, Buisine MP, Aubert JP, et al. Butyrate specifically modulates MUC gene expression in intestinal epithelial goblet cells deprived of glucose. Am J Physiol Gastrointest Liver Physiol (2004) 287:G1168–74. doi:10.1152/ajpgi.00219.2004
31. Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol (2008) 9:206–18. doi:10.1038/nrm2346
32. Choudhary C, Kumar C, Gnad F, Nielsen M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science (2009) 325(5942):834–40. doi:10.1126/science.1175371
33. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in regulation of RNA synthesis. Proc Natl Acad Sci U S A (1964) 51:786–94. doi:10.1073/pnas.51.5.786
34. Cheung WL, Briggs SD, Allis CD. Acetylation and chromosomal functions. Curr Opin Cell Biol (2000) 12:326–33. doi:10.1016/S0955-0674(00)00096-X
35. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature (1997) 389:349–52. doi:10.1038/38664
36. Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem (2001) 70:81–120. doi:10.1146/annurev.biochem.70.1.81
37. Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene (2005) 363:15–23. doi:10.1016/j.gene.2005.09.010
38. Yuan Z, Guan Y, Chatterjee D. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science (2005) 307(5707):269–73. doi:10.1126/science.1105166
39. Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev (2000) 64:435–59. doi:10.1128/MMBR.64.2.435-459.2000
40. Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol (2008) 41:185–98. doi:10.1016/j.biocel.2008.08.027
41. Cousens LS, Gallwitz D, Alberts BM. Different accessibilities in chromatin to histone acetylase. J Biol Chem (1979) 254:1716–23.
42. Corfe BM. Hypothesis: butyrate is not an HDAC inhibitor, but a product inhibitor of deacetylation. Mol Biosyst (2012) 8:1609–12. doi:10.1039/c2mb25028d
43. Wu S, Li RW, Li W, Li CJ. Transcriptome characterization by RNA-seq unravels the mechanisms of butyrate-induced epigenomic regulation in bovine cells. PLoS ONE (2012) 7:e36940. doi:10.1371/journal.pone.0036940
44. Belenguer A, Holtrop G, Duncan SH, Anderson SE, Calder AG, Flint HJ, et al. Rates of production and utilization of lactate by microbial communities from the human colon. FEMS Microbiol Ecol (2011) 77:107–19. doi:10.1111/j.1574-6941.2011.01086.x
45. Latham T, Mackay L, Sproul D, Karim M, Culley J, Harrison DJ, et al. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res (2012) 40:4794–803. doi:10.1093/nar/gks066
46. Rajendran P, Williams DE, Ho E, Dashwood RH. Metabolism as a key to histone deacetylase inhibition. Crit Rev Biochem Mol Biol (2011) 46:181–99. doi:10.3109/10409238.2011.557713
47. Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem (2005) 96:293–304. doi:10.1002/jcb.20532
48. Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res (2009) 69:2826–32. doi:10.1158/0008-5472.CAN-08-4466
49. Paroder V, Spencer SR, Paroder M, Arango D, Schwartz S Jr, Mariadason JM, et al. Na+/monocarboxylate transport (SMCT) protein expression correlates with survival in colon cancer: molecular characterization of SMCT. Proc Natl Acad Sci U S A (2006) 103:7270–5. doi:10.1073/pnas.0602365103
50. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem (2003) 278:11312–9. doi:10.1074/jbc.M211609200
51. Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem (2003) 278:25481–9. doi:10.1074/jbc.M301403200
52. Ulven TT. Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front Endocrinol (Lausanne) (2011) 3:111. doi:10.3389/fendo.2012.00111
53. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature (2009) 461:1282–6. doi:10.1038/nature08530
54. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013). doi:10.1126/science.1241165
55. Vinolo MA, Ferguson GJ, Kulkarni S, Damoulakis G, Anderson K, Bohlooly-Y M, et al. SCFAs induce mouse neutrophil chemotaxis through the GPR43 Receptor. PLoS ONE (2011) 6:e21205. doi:10.1371/journal.pone.0021205
56. Tazoe H, Otomo Y, Karaki S, Kato I, Fukami Y, Terasaki M, et al. Expression of short-chain fatty acid receptor GPR41 in the human colon. Biomed Res (2009) 30:149–56. doi:10.2220/biomedres.30.149
57. Karaki S, Tazoe H, Hayashi H, Kashiwabara H, Tooyama K, Suzuki Y, et al. Expression of the short-chain fatty acid receptor, GPR43, in the human colon. J Mol Histol (2007) 39:135–42. doi:10.1007/s10735-007-9145-y
58. Iwanaga T, Takebe K, Kato I, Karaki SI, Kuwahara A. Cellular expression of monocarboxylate transporters (MCT) in the digestive tract of the mouse, rat, and humans, with special reference to slc5a8. Biomed Res (2006) 27:243–54. doi:10.2220/biomedres.27.243
59. Edwards AJ, Pender SL. Histone deacetylase inhibitors and their potential role in inflammatory bowel diseases. Biochem Soc Trans (2011) 39:1092–5. doi:10.1042/BST0391092
60. Glauben R, Siegmund B. Inhibition of histone deacetylases in inflammatory bowel diseases. Mol Med (2011) 17:426–33. doi:10.2119/molmed.2011.00069
61. Glauben R, Batra A, Fedke I, Zeitz M, Lehr HA, Leoni F, et al. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol (2006) 176:5015–22.
62. Tao R, de Zoeten EF, Özkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med (2007) 13:1299–307. doi:10.1038/nm1652
63. Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol (2005) 45:495–528. doi:10.1146/annurev.pharmtox.45.120403.095825
64. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov (2006) 5:769–84. doi:10.1038/nrd2133
65. Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A (2002) 99(5):2995–3000. doi:10.1073/pnas.052702999
66. Dinarello CA, Fossati G, Mascagni P. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol Med (2011) 17(5–6):333–52. doi:10.2119/molmed.2011.00116
67. Epping MT, Wang L, Plumb JA, Lieb M, Gronemeyer H, Brown R, et al. A functional genetic screen identifies retinoic acid signaling as a target of histone deacetylase inhibitors. Proc Natl Acad Sci U S A (2007) 104:17777–82. doi:10.1073/pnas.0702518104
68. Epping MT, Wang L, Edel MJ, Carlée L, Hernandez M, Bernards R. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell (2005) 122:13–13. doi:10.1016/j.cell.2005.07.003
69. Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem (2006) 281(19):13548–58. doi:10.1074/jbc.M510023200
70. Wilson AJ, Byun DS, Nasser S, Murray LB, Ayyanar K, Arango D, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell (2008) 19:4062–75. doi:10.1091/mbc.E08-02-0139
71. Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, et al. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell (2002) 9:45–57. doi:10.1016/S1097-2765(01)00429-4
72. Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A (2007) 104:17335–40. doi:10.1073/pnas.0706487104
73. Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans AD, de Vos WM. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol (2002) 68:3401–7. doi:10.1128/AEM.68.7.3401-3407.2002
74. Quigley EM, Quera R. Small intestinal bacterial overgrowth: roles of antibiotics, prebiotics, and probiotics. Gastroenterology (2006) 130:S78–90. doi:10.1053/j.gastro.2005.11.046
75. Wollowski I, Rechkemmer G, Pool-Zobel BL. Protective role of probiotics and prebiotics in colon cancer. Am J Clin Nutr (2001) 73:451S–5.
76. Licciardi PV, Wong SS, Tang ML, Karagiannis TC. Epigenome targeting by probiotic metabolites. Gut Pathog (2010) 2:24. doi:10.1186/1757-4749-2-24
77. Gionchetti P, Rizzello F, Venturi A, Brigidi P, Matteuzzi D, Bazzocchi G, et al. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: a double-blind, placebo-controlled trial. Gastroenterology (2000) 119:305–9. doi:10.1053/gast.2000.9370
78. Kruis W, Fric P, Pokrotnieks J, Lukás M, Fixa B, Kascák M, et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut (2004) 53:1617–23. doi:10.1136/gut.2003.037747
79. Sartor RB. Efficacy of probiotics for the management of inflammatory bowel disease. Gastroenterol Hepatol (N Y) (2011) 7:606–8.
80. Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol (2010) 9:88–96. doi:10.1038/nrgastro.2011.244
81. van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med (2013) 368(5):407–15. doi:10.1056/NEJMoa1205037
Keywords: HDAC, intestinal epithelium, inflammatory bowel diseases, short chain fatty acids, butyrate
Citation: Schilderink R, Verseijden C and de Jonge WJ (2013) Dietary inhibitors of histone deacetylases in intestinal immunity and homeostasis. Front. Immunol. 4:226. doi: 10.3389/fimmu.2013.00226
Received: 12 May 2013; Accepted: 18 July 2013;
Published online: 01 August 2013.
Edited by:
Cecil Czerkinsky, Göteborg University, SwedenReviewed by:
Emma Slack, ETH Zürich, SwitzerlandJ. Rodrigo Mora, Massachusetts General Hospital, USA
Copyright: © 2013 Schilderink, Verseijden and de Jonge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: W. J. de Jonge, Tytgat Institute for Liver and Intestinal Research, Academic Medical Center, Meibergdreef 69-71, 1105BK Amsterdam, Netherlands e-mail:dy5qLmRlam9uZ2VAYW1jLnV2YS5ubA==