
 Mamoru Niikura
Mamoru Niikura
- Department of Infectious Diseases, Kyorin University School of Medicine, Mitaka, Tokyo, Japan
Malaria is caused by infection with Plasmodium parasites. Various studies with knockout mice have indicated that IFN-γ plays essential roles in protective immunity against blood-stage Plasmodium infection. However, after Plasmodium infection, increased IFN-γ production by various types of cells is involved not only in protective immunity, but also in immunopathology. Recent reports have shown that IFN-γ acts as a pro-inflammatory cytokine to induce not only the activation of macrophages, but also the generation of uncommon myelolymphoid progenitor cells after Plasmodium infection. However, the effects of IFN-γ on hematopoietic stem cells and progenitor cells are unclear. Therefore, the regulation of hematopoiesis by IFN-γ during Plasmodium infection remains to be clarified. Although there are conflicting reports concerning the significance of γδ T cells in protective immunity against Plasmodium infection, γδ T cells may respond to infection and produce IFN-γ as innate immune cells in the early phase of blood-stage malaria. Our recent studies have shown that γδ T cells express CD40 ligand and produce IFN-γ after Plasmodium infection, resulting in the enhancement of dendritic cell activation as part of the immune response to eliminate Plasmodium parasites. These data suggest that the function of γδ T cells is similar to that of NK cells. Although several reports suggest that γδ T cells have the potential to act as memory cells for various infections, it remains to be determined whether memory γδ T cells are generated by Plasmodium infection and whether memory γδ T cells can contribute to the host defense against re-infection with Plasmodium. Here, we summarize and discuss the effects of IFN-γ and the various functions of γδ T cells in blood-stage Plasmodium infection.
Malaria is one of the most serious public health problems worldwide, and the malaria-endemic areas include both tropical and subtropical regions. Plasmodium, the malaria-causing protozoan parasite, has a complex life cycle. Mammalian hosts, such as humans, monkeys, and mice, are inoculated with sporozoites into the host dermis of skin via a bite by a Plasmodium-infected Anopheles mosquito (1). Sporozoites then enter blood stream for invasion into hepatocytes and develop into merozoites with replication (liver-stage). Merozoites parasitize red blood cells (RBCs), replicate, and then rupture the RBCs, allowing for new infection of additional normal RBCs (blood-stage). In contrast to asymptomatic liver-stage malaria, blood-stage malaria is the symptomatic phase. Thus, protective immunity against blood-stage malaria is important for reducing the severity of disease (Figure 1).
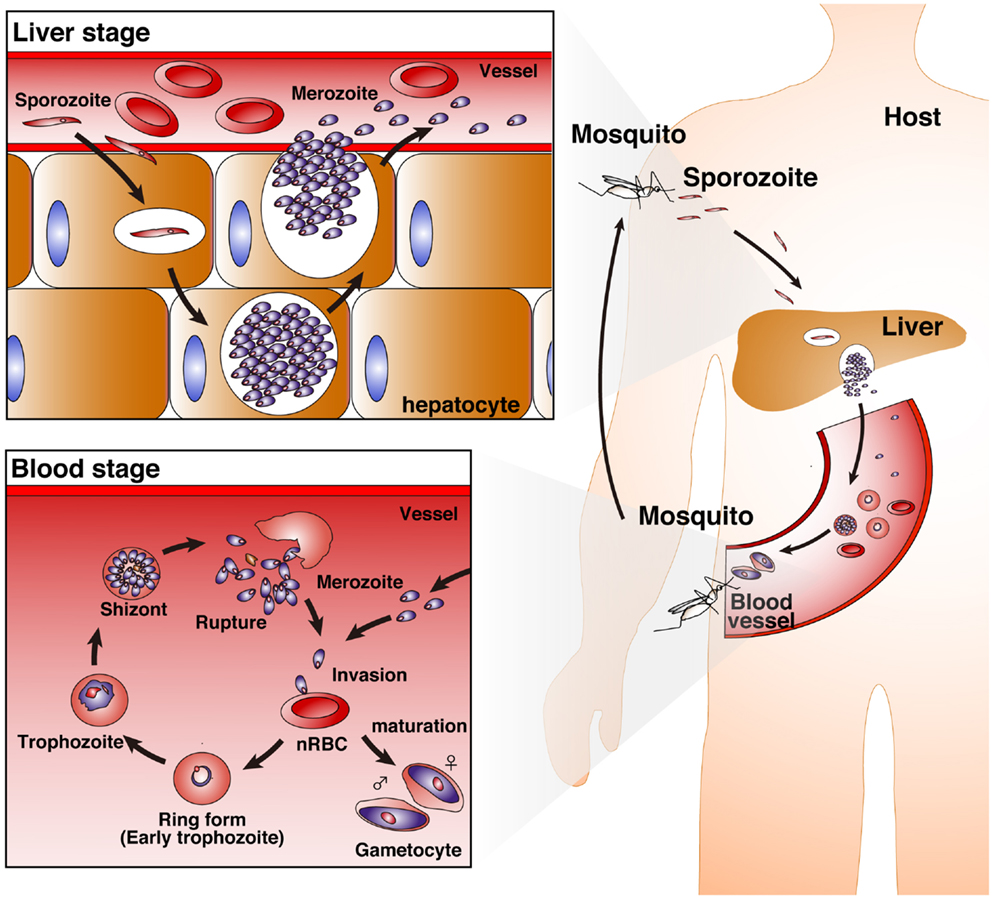
Figure 1. Life cycle of Plasmodium parasites. A bite from a Plasmodium-infected Anopheles mosquito inoculates human hosts with sporozoites in the dermis of skin. The sporozoites move into the bloodstream, and parasitize hepatocytes. Invaded sporozoites develop into merozoites with replication (liver-stage). Then, merozoites are released from hepatocytes. Merozoites parasitize red blood cells (RBCs), replicate, and then rupture the RBCs, leading to new infection of normal RBCs (nRBCs; blood-stage). Merozoites have several developmental phases: the ring form (early trophozoite), trophozoite, and schizont. Furthermore, some merozoites mature into female and male gametocytes.
Infection with Plasmodium falciparum causes the most severe malaria in humans. The predominant symptoms are anemia, splenomegaly, and fever. Cerebral malaria, liver dysfunction, acute renal failure, acidosis, hypoglycemia, respiratory distress, and edema are also observed as complications in malaria patients, although these symptoms do not always appear. Certain Plasmodium species can specifically infect rodents and cause malaria. A rodent malaria model is very useful not only for the development of anti-malarial drugs and vaccines, but also in research studies into the protective and pathologic immune responses during malaria. The lethality of the Plasmodium infection is dependent upon combinations of Plasmodium species and inbred mouse strains. Here, we review experimental studies on Plasmodium infections in the mouse and human studies with P. falciparum, focusing on protective immunity against Plasmodium infections.
Interferon-γ (IFN-γ) is produced mainly by lymphocytes, such as αβ T cells, natural killer (NK) cells, NKT cells, and γδ T cells (2–5). Some myeloid cells have also been reported to have the potential to produce IFN-γ (6–10). IFN-γ is an important pro-inflammatory cytokine and a mediator of immune responses against intracellular bacteria and viruses (11–14). Furthermore, it plays a protective role against infection by protozoan parasites (15–18). It enhances phagocyte activity, resulting in the elimination of extracellular bacteria and protozoan parasites, and its production by CD4+ helper T cells, CD8+ killer T cells, and NK cells is greatly induced by IL-12 and IL-18 from antigen-presenting cells (APCs), such as dendritic cells (DCs) (19–22). Moreover, some reports have shown that IFN-γ production from APCs is regulated by IL-12, IL-15, and IL-18 (23–25).
γδ T cells work as innate lymphocytes, the first line of defense against infectious pathogens. On the other hand, αβ T cells, such as CD4+ helper T cells and CD8+ killer T cells, are typically related to adaptive immunity. γδ T cells play critical roles in protective immune responses against protozoan parasites, bacteria, and viruses that are associated with various infectious diseases (26–32). This review focuses on the protective abilities of γδ T cells and IFN-γ in the response against malaria infection.
IFN-γ Mediates Protective Immunity Against Blood-Stage Plasmodium Parasites
Mice (on a C57BL/6 or CBA background) that are genetically IFN-γ-deficient or IFN-γ receptor (IFN-γR)-deficient or that are treated with anti-IFN-γ antibody and infected with blood-stage P. berghei are unable to control the infecting parasite (33–35). In the cases of infection with blood-stage P. chabaudi and P. yoelii parasites, genetically IFN-γ-deficient or IFN-γ receptor (IFN-γR)-deficient mice or anti-IFN-γ antibody-treated mice on a C57BL/6 or CBA background show delayed elimination of the parasites (36–39). These experimental malaria models demonstrate that IFN-γ is a key pro-inflammatory cytokine for controlling blood-stage Plasmodium parasites (Table 1). IFN-γ is produced by many cell types and involved in many steps of immune responses. αβ T cells, NK cells, NKT cells, and γδ T cells have been shown to produce IFN-γ after infection with Plasmodium parasites. The contributions of producers of IFN-γ to protective immunity against Plasmodium parasites are complicated (Figure 2). Thus, more detailed experiments using IFN-γ signaling-deficient models should be performed to determine the mechanism(s) underlying the involvement of IFN-γ in immune protection against Plasmodium parasites.
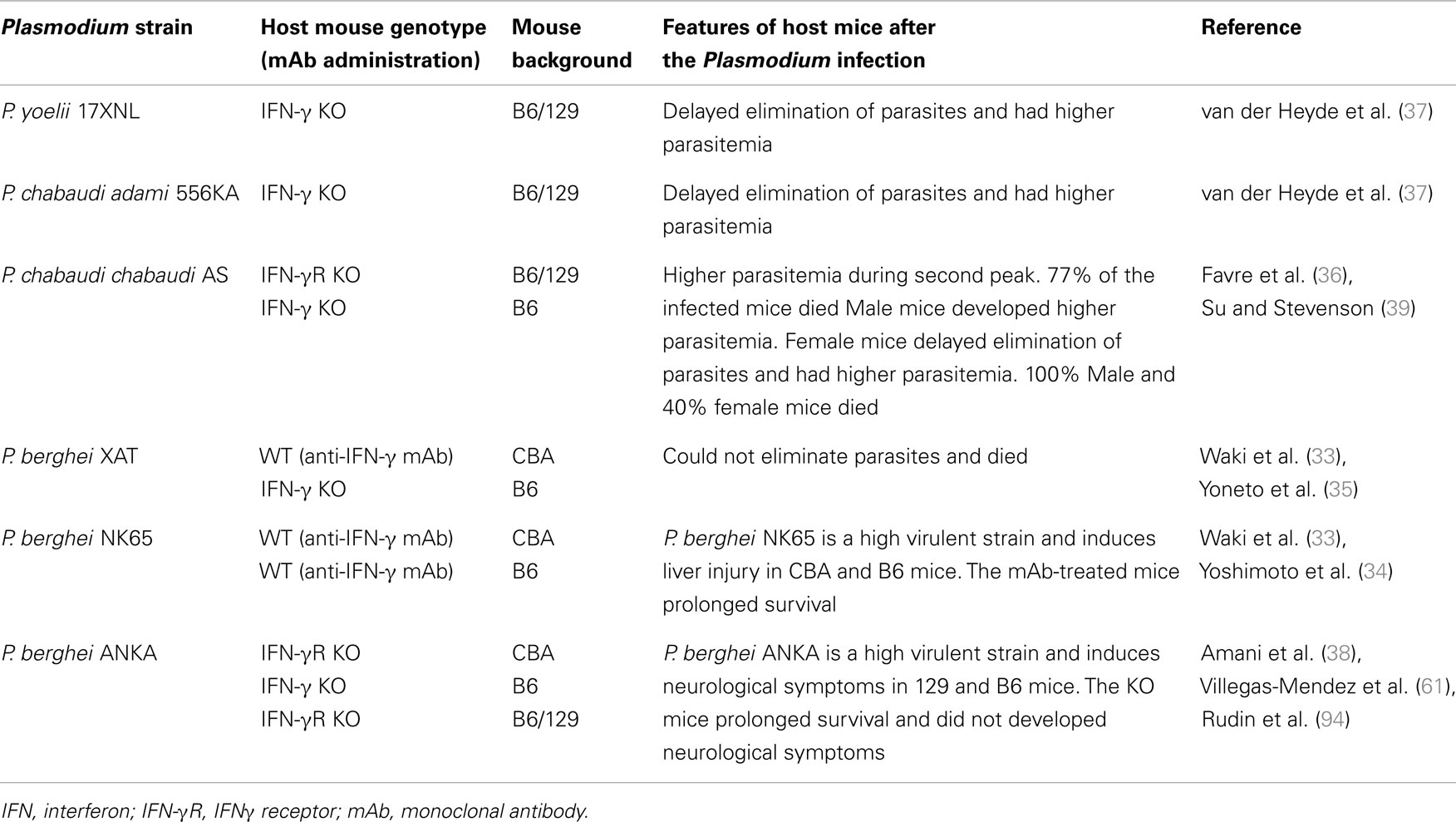
Table 1. Influence of IFN-γ-signal deficiency on control of Plasmodium parasites in mice.
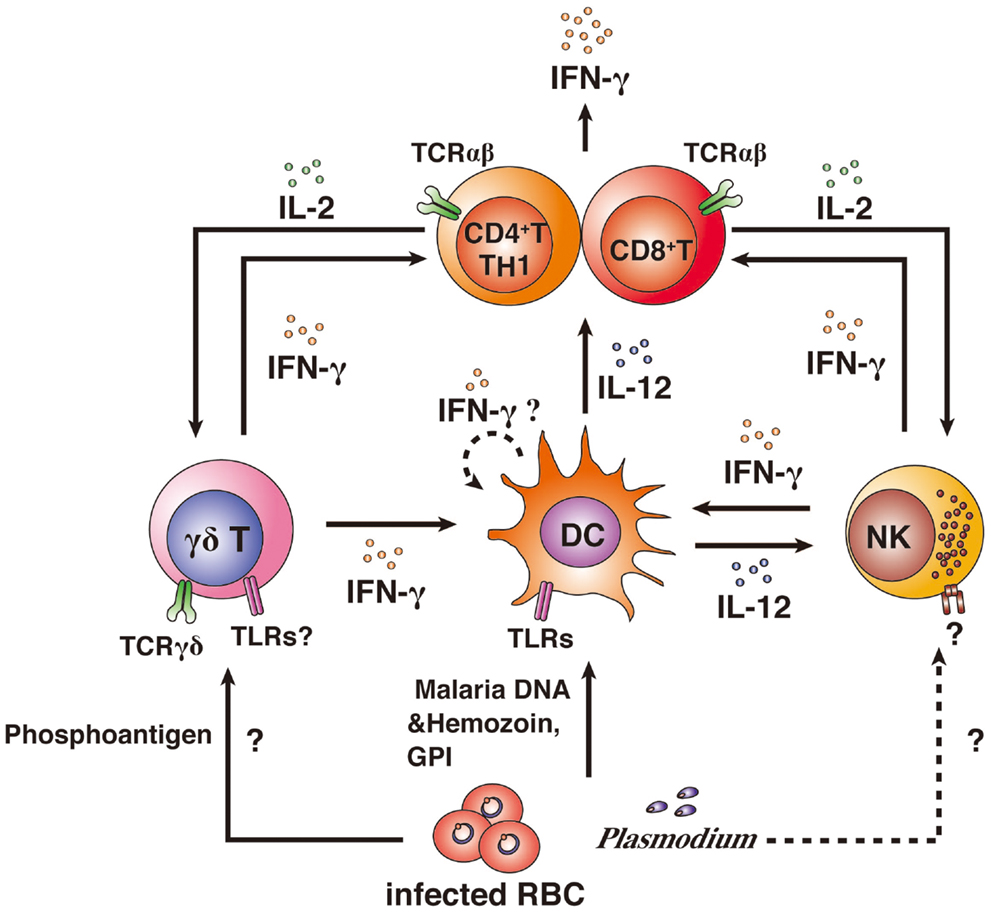
Figure 2. IFN-γ producers and their activation. CD4+ and CD8+ αβ T cells, NK cells, and γδ T cells have been shown to produce IFN-γ after infection with Plasmodium parasites. Innate immune cells can directly recognize Plasmodium antigens. γδ T cells can recognize phosphoantigens by MHC II-independent pathways via TCRγδ, resulting in the activation of DCs and IFN-γ production. Whether γδ T cells can recognize malarial antigens via TLR is unknown. DCs phagocytose iRBCs and recognize malarial DNA and hemozoin or glycosylphosphatidylinositol (GPI) anchors via TLRs, resulting in the activation of DCs. DCs are thought to have the potential to produce IFN-γ. However, it remains unclear as to whether IFN-γ production from DCs is induced by malarial antigens. Activated DCs produce IL-12 and present antigens to αβ T cells. αβ T cells are strongly activated by IL-12 and antigen presentation. IL-12 also activates NK cells. The malarial antigen-recognition receptor on NK cells remains to be determined. Activation of αβ T cells leads to high IFN-γ production. Furthermore, activated αβ T cells produce IL-2, resulting in the proliferation of γδ T cells and NK cells and the αβ T cells themselves.
IFN-γ Producers and Their Activation in Plasmodium Infection
CD4+ αβ T Cells and CD8+ αβ T Cells
Given that MHC class II-deficient mice on a C57BL/6 background are unable to control P. yoelii- and P. chabaudi-infected RBCs (iRBCs), CD4+ αβ T cells respond to Plasmodium-iRBCs after priming with malarial antigens in an MHC class II context on DCs (40). CD4+ αβ T cells strongly increase their ability to produce IFN-γ after infection with P. berghei (32). High proportions of CD4+ αβ T cells from P. falciparum-infected human subjects respond to iRBCs and produce IFN-γ, compared to CD4+ αβ T cells from naïve human subjects (4). Moreover, some studies have shown that CD4+ αβ T cell-depleted C57BL/6 or CBA mice treated with anti-CD4 antibody are unable to control blood-stage P. chabaudi or P. berghei (32, 33, 41, 42). These lines of evidence suggest that CD4+ αβ T cells are major sources of IFN-γ and are required for protective immunity against Plasmodium infection, resulting in complete clearance of the parasites. In contrast, the results for Plasmodium-infected mice with CD8+ αβ T-cell depletion are comparable to those for control mice (33, 41, 42). Moreover, CD8+ αβ T cells from highly immunized mice may play critical roles in protective immunity against infection with a lethal P. yoelii strain in C57BL/6 mice (41–43), although CD8+ T cells are not required for the resolution of primary infection with P. chabaudi or P. yoelii parasites in C57BL/6 mice. Interestingly, involvement of perforin molecules is only partial in protective immunity involving CD8+ αβ T cells. IFN-γ and macrophages are key molecular and cellular factors in protective immunity involving CD8+ αβ T cells (43). These data suggest that the role of the CD8+ αβ T cells is redundant in relation to the role of CD4+ αβ T cells in protective immunity in repeatedly immunized mice. A repeat infection with Plasmodium parasites may induce CD4+ αβ T-cell exhaustion (44). Cytokine production by T cells is inhibited by this exhaustion via the signaling of inhibitory receptors, such as PD-1 and LAG3. Since CD4+ αβ T cells are still needed to exert protective immunity against re-infection with the Plasmodium parasites, only partial exhaustion of CD4+ αβ T cells may occur. Therefore, both CD4+ αβ T cells and CD8+ αβ T cells may need to produce adequate quantities of IFN-γ to ensure clearance of the Plasmodium parasites.
Dendritic Cells
Several reports have shown that IFN-γ is also produced by DCs (8–10). The IFN-γ-producing DCs are important for priming lymphocytes (10), although it remains to be determined whether IFN-γ production by DCs enhances protective immunity against Plasmodium parasites, and whether stimulation of Toll-like receptors (TLRs) by parasite components induces IFN-γ production by DCs (45–48).
γδ T Cells and NK Cells
γδ T cells and NK cells are considered to be important IFN-γ producers in blood-stage malaria infections and to be associated with the control of malarial parasites (2, 3, 5). In the early stages of malaria infection, γδ T cells directly recognize the pathogen through MHC-independent mechanisms that involve the γδTCR, and high levels of IFN-γ production and proliferation are induced (49–51). Several reports have suggested that proliferation of γδ T cells depends on IL-2 (52, 53). Human and murine γδ T cells usually express TLRs, and the expression levels of TLRs are enhanced by γδTCR stimulation (54–56). Therefore, TLR is another candidate receptor for γδ T-cell responses to malaria antigens (46, 57). Although studies have suggested that γδ T cells have the potential to react to malarial antigens via TLRs, TLR signaling in myeloid cells may be more important for the induction of protective immunity against malaria (58).
A study of experimental P. falciparum infection showed that, in contrast to γδ T cells, NK cells were minor IFN-γ producers in response to iRBCs before and after P. falciparum infection (5). A longitudinal study of children in Papua New Guinea indicated that IFN-γ-producing responses of malaria antigen-specific γδ T cells, but not those of NK cells, were important for protective immunity against P. falciparum infection (59). In contrast, several in vitro culture studies have shown that NK cells rapidly induce IFN-γ production in response to P. falciparum-iRBCs via IL-12 and IL-18 signaling. These reports suggest that NK cells have the potential to produce IFN-γ in response to P. falciparum.
IFN-γ Induces Pathologic Effects in Severe Malaria
Interferon-γ is not only a key factor in protection against Plasmodium infection, but also a pathogenicity factor in severe malaria symptoms (33, 38, 60, 61, 94). It is well-known that P. berghei ANKA infection leads to the development of experimental cerebral malaria (ECM), and P. berghei NK65 infection induces liver injury. Those P. berghei-infected mice are useful models for studies of severe malarial symptoms. IFN-γR-deficient mice on a C57BL/6 or 129 background do not develop ECM. Furthermore, P. berghei NK65-infected mice on a C57BL/6 or CBA background treated with anti-IFN-γ antibody live longer than those treated with control-IgG. These reports suggest that IFN-γ is important for the development of severe malarial symptoms (Table 1) (33, 34, 38, 94). ECM also does not develop in P. berghei ANKA-infected mice after depletion of CD4+ T cells or CD8+ T cells (62). Cytotoxic CD8+ αβ T cells, which express perforin or granzyme B, have important roles in the development of ECM (63, 64). Although both CD4+ αβ T and CD8+ αβ T cells have a similar potential to produce IFN-γ, CD8+ αβ T cells are not an important IFN-γ producer for the development of ECM (61). IFN-γ-producing CD4+ αβ T cells promote recruitment of cytotoxic CD8+ αβ T cells to the brain; in contrast, CD4+ αβ T cells dot not accumulate markedly in the brain (60). Because depletion of NK cells can prevent ECM after infection with P. berghei ANKA, NK cells are also associated with IFN-γ responses in the development of ECM. NK cells activate CD8α+ DCs that prime CD8+ αβ T cells after infection with P. berghei ANKA (65). Furthermore, depletion of γδ T cells can also prevent ECM after infection with P. berghei ANKA. Therefore γδ T cells would be also associated with IFN-γ responses in the development of ECM (66). IFN-γ has both protective and pathologic effects on the immune response to Plasmodium infections. Therefore, simply inducing a reduction in blood IFN-γ levels by treatment with an antibody or drug may not be an effective way to treat cerebral malaria. Further studies focused on the regulation of CD8+ αβ T-cell activation by DCs are needed to develop a preventative therapy for cerebral malaria.
Does IFN-γ Induce Effective Hematopoiesis in the Host?
IFN-γ is an important mediator of hematopoietic stem cell and progenitor cell activation during bacterial infections (67–69). Furthermore, rodent Plasmodium infection induces the generation of uncommon myelolymphoid progenitor cells, which express IL7-R+ and c-Kithivia IFN-γ signaling. These myelolymphoid progenitor cells can differentiate into both myeloid cells and lymphoid cells (70). Host protection from malaria depends on hematopoietic differentiation (i.e., hematopoiesis) to supply many types of differentiated cells. First, differentiation of erythroid progenitor cells is necessary to avoid the development of severe anemia. Second, differentiation of myeloid progenitor cells is necessary to supply a high number of phagocytic cells for the clearance of Plasmodium parasites. Hematopoiesis is a fundamental process for “curing” malaria and other infections. Moreover, several studies have shown IFN-γ-induced inhibition of some steps of hematopoiesis (71–73). Considering important roles of IFN-γ in protective immunity against Plasmodium parasites, IFN-γ would be a key factor for regulation of effective hematopoiesis in malaria. There are many cell types of IFN-γ producers in Plasmodium infection as mentioned above. Therefore, some of the IFN-γ producers in Plasmodium infection may play crucial role for regulation of hematopoiesis by the effect of IFN-γ or a combination of IFN-γ and other factors, which are produced from the IFN-γ-producing cells.
Host Immune Responses of γδ T Cells in Malaria
Varieties of γδ T-Cell Function
γδ T cells play various roles, including in protective immunity against pathogens, in the curing of injured tissue, tumor surveillance, and as a bridge between innate and adaptive immunity. These multiple functions of γδ T cells are thought to be due to their abilities to produce various cytokines and chemokines. Such abilities are broadly restricted by the Vγ and Vδ repertoires of γδ T cells. The distribution of γδ T-cell subsets differs depending on their resident tissue (74). To examine the physiologic role(s) in the immune response against Plasmodium parasites, γδ T-cell subsets should be compared carefully between mice and humans based on their abilities, such as cytokine production or ligands for activation.
γδ T-Cell-Related Protective Immunity Against Blood-Stage Plasmodium Infection
Although previous reports of in vitro and in vivo experiments have suggested that γδ T cells are associated with protective immune responses during malaria infection, the functions of γδ T cells in the spleen remain largely unknown (75–79). We recently reported on the mechanism of γδ T-cell-related protective immunity against Plasmodium parasites using the rodent malaria parasite P. berghei XAT (32, 80) (Figure 3). P. berghei XAT is an attenuated strain derived from the lethal P. berghei NK65 strain (32, 81). P. berghei XAT was developed for investigations of vaccines and protective immune responses against Plasmodium parasites owing to its potent ability to induce immune memory, even against lethal P. berghei NK65 strains. Previous studies have reported the essential cytokines and immune cells required for the clearance of P. berghei XAT. Since γδ T-cell-deficient mice on a C57BL/6 background are unable to control P. berghei XAT infection, γδ T cells are essential for protective immunity against the parasites (32). Other studies using P. chabaudi parasites have also suggested that γδ T cells are related to, but not essential for, protective immunity against the parasites (76, 78). Thus, this P. berghei XAT strain is useful for investigating the mechanism(s) of γδ T-cell-related protective immunity against Plasmodium parasites. In general, the immunologic functions of γδ T cells are largely similar to those of NK cells. The difference in the need for γδ T cells between protective immunity against the two parasite strains may be influenced by the contribution of NK cells to protective immunity.
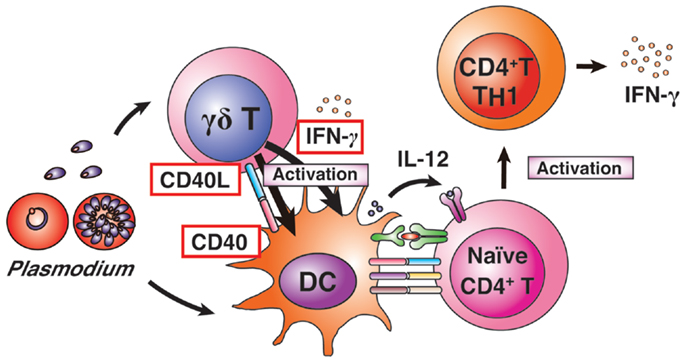
Figure 3. γδ T-cell-related protective immunity. γδ T cells and DCs are activated by Plasmodium infection. Activated γδ T cells begin to express CD40L and produce IFN-γ. The activated γδ T cells enhance MHC II and co-stimulatory factors on DCs, and induce high levels of IL-12 production from DCs. The CD40L-expressing γδ T cells enhance DC activation. Activated DCs induce differentiation of naïve CD4+ T cells into IFN-γ-producing TH1 cells.
Although malaria infections in humans, monkeys, and mice lead to increased numbers of γδ T cells in the blood and spleen, the γδ T-cell population is still minor in relation to the total number of lymphocytes (82). It may be possible to explain why such a minor population of lymphocytes plays such key roles in protective immunity against malaria parasites. First, γδ T cells can directly recognize malarial parasites, making them major producers of IFN-γ in response to malaria antigens during the early phase of infection. In contrast, NK cells are reciprocally regulated by DCs. Thus, DC activation is needed first to activate NK cells in malaria infection (3, 83). NK cells are unexpectedly minor producers of IFN-γ in response to malarial antigens in human blood in experimental infections with P. falciparum parasites (59). Nevertheless, NK cells also become key players in protective immunity against malarial parasites, as has been shown in many studies (2, 3, 83), although NK cells are not required for the control of P. berghei XAT parasites (35). Second, γδ T cells have the ability to interact readily with other central immune players, such as DCs. Our recent report showed that about 30% of splenic γδ T cells were localized in the vicinity of DCs even under naïve conditions. A more than twofold increase in the percentage of splenic γδ T cells that adhered to DCs was observed in the early phase of infection (32). The molecular mechanism of this adhesive ability of γδ T cells to DCs remains to be determined. As γδ T cells can produce chemokines, some γδ T-cell-produced chemokines may attract DCs and also αβ T cells to help antigen presentation by DCs (84, 85). Furthermore, our study provided evidence that γδ T cells boost DC activation for protective immunity against P. berghei XAT parasites via CD40 ligand expression on γδ T cells. CD40L-CD40 signaling induces the expression of MHC II and co-stimulatory factors, such as CD40, CD80, and CD86 (32). Such signaling to DCs would be synergistically activated by the uptake of Plasmodium antigens (86).
Memory Phenotype of γδ T Cells in Malaria
For long-standing protective immunity against pathogens, memory lymphocytes maintain phenotypes to respond against pathogens and live for a long period after infections or vaccination with antigens (87, 88). In general, memory T cells are converted from naïve T cells through three phases (expansion, contraction, and memory phases; Figure 4). In the expansion phase, effector or killer T cells activate and proliferate after pathologic infection. Soon after pathogen clearance, the numbers of expanded antigen-specific T cells are reduced by induction of apoptosis in the contraction phase. Then, a fraction of the remaining antigen-specific T cells become memory T cells, resulting in long-lasting and rapid protective immunity against re-infection with pathogens (memory phase). Memory plasma cells and B cells maintain the potential to generate antigen-specific antibodies, and memory CD4+ T cells and CD8+ T cells maintain rapid helper and killer responses to antigens, resulting in rapid elimination of re-infecting pathogens. In malaria, those memory lymphocytes would also exist in human subjects who have been infected with Plasmodium parasites previously (4, 5). However, there is still controversy over the duration of memory lymphocyte maintenance (89, 90). Previous studies have shown that human γδ T cells generate rapid and amplified responses to a secondary challenge of bacteria and viruses, such as mycobacteria and cytomegalovirus (91, 92). These data suggest that human γδ T cells have the ability to develop memory cells for protection against re-infection. On the other hand, a recent report suggested that human γδ T cells develop effector memory cells after infection with P. falciparum parasites (5).
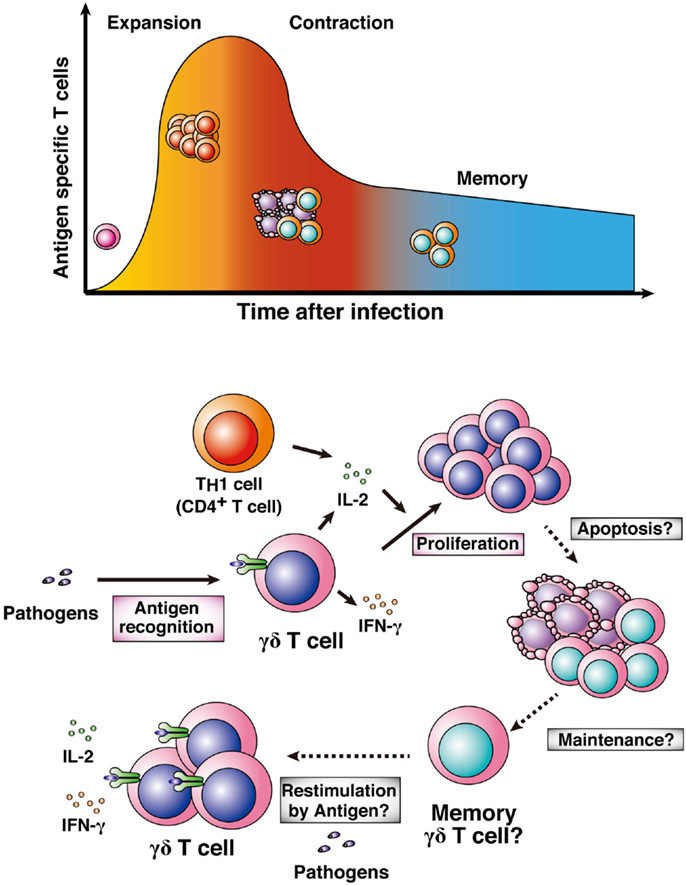
Figure 4. Memory γδ T cells. Naïve T cells are activated by pathologic infections and differentiate into effector or killer T cells. Activated T cells clonally expand in response to particular antigens (expansion phase). After pathogen clearance, high proportions of the expanded antigen-specific T cells undergo apoptosis (contraction phase). Some population of the surviving antigen-specific T cells is maintained as memory T cells (memory phase). γδ T cells also expand after infection with pathogens, including Plasmodium parasites. High-level proliferation of γδ T cells is involved in generating IL-2 from TH1 cells or the γδ T cells themselves. It remains to be determined whether antigen-specific γδ T cells shift into a contraction phase and then a memory phase.
Some details about memory γδ T cells in malaria remain to be uncovered (Figure 4). The first point is the importance of memory γδ T cells in malaria. Although the numbers of malaria antigen-specific γδ T cells certainly increase in the year after primary infection, whether the memory γδ T cells are important for effective protection against re-infection with Plasmodium parasites remains to be determined. The second point is the developmental process of memory γδ T cells. γδ T cells function not only in adaptive immunity but also in innate immunity. Thus, the process for the development of memory γδ T cells may differ from that of general memory T cells in malaria or in other infections. The third point is the localization of memory γδ T cells. A recent report showed that memory T cells in lymph nodes and peripheral tissues translocate to bone marrow for the long term (93). It would be difficult to examine whether this hypothesis applies to memory γδ T cells, and other memory lymphocytes, in human subjects. Thus, rodent malaria models are needed for examining the location of memory γδ T cells.
Concluding Remarks
Many studies have demonstrated that IFN-γ is responsible for protective immunity against Plasmodium parasites. The critical cellular source(s) of IFN-γ in malaria patients remains to be determined. γδ T cells are not only a major candidate as the key IFN-γ producer, but are also critical modulators of protective immunity against Plasmodium parasites. We should continue to investigate the roles of γδ T cells in host defenses against Plasmodium parasites. Thus, it is important to investigate whether the abilities of γδ T cells to produce IFN-γ and mediate immune responses against malarial antigens persist as memory cells. Mechanisms of immune responses in lymphoid tissues, especially in the spleen, to blood-stage malaria are gradually being uncovered by studies using rodent malaria models. To investigate further the relationship between IFN-γ and γδ T cells in immune responses against malaria, we should make more effort to study the issues using not only rodent malaria models, but also humanized mice that can be infected with P. falciparum parasites. To make this a reality, we first need to refine the humanized mice. Based on studies of γδ T-cell-related protective immunity, we may be able to develop novel strategies for a malaria vaccine.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Moritani Scholarship Foundation, the Japan Prize Foundation to Shin-Ichi Inoue. This work was also supported by Grants-in-Aid for Young Scientists (B) to Shin-Ichi Inoue (No. 23790462) and Mamoru Niikura (No. 24790405) and a Grant-in-Aid for Scientific Research (C) to Fumie Kobayashi (No. 23590493) from the Japan Society for the Promotion of Science (JSPS). We thank Mrs. Natsuki Yokota for her support of our work.
References
1. Amino R, Thiberge S, Martin B, Celli S, Shorte S, Frischknecht F, et al. Quantitative imaging of Plasmodium transmission from mosquito to mammal. Nat Med (2006) 12:220–4. doi:10.1038/nm1350
2. Artavanis-Tsakonas K, Riley EM. Innate immune response to malaria: rapid induction of IFN-γ from human NK cells by live Plasmodium falciparum-infected erythrocytes. J Immunol (2002) 169:2956–63.
3. Ing R, Stevenson MM. Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect Immun (2009) 77:770–82. doi:10.1128/IAI.00994-08
4. Roestenberg M, McCall M, Hopman J, Wiersma J, Luty AJ, van Gemert GJ, et al. Protection against a malaria challenge by sporozoite inoculation. N Engl J Med (2009) 361:468–77. doi:10.1056/NEJMoa0805832
5. Teirlinck AC, McCall MB, Roestenberg M, Scholzen A, Woestenenk R, de Mast Q, et al. Longevity and composition of cellular immune responses following experimental Plasmodium falciparum malaria infection in humans. PLoS Pathog (2011) 7:e1002389. doi:10.1371/journal.ppat.1002389
6. Flesch IE, Hess JH, Huang S, Aguet M, Rothe J, Bluethmann H, et al. Early interleukin 12 production by macrophages in response to mycobacterial infection depends on interferon γ and tumor necrosis factor α. J Exp Med (1995) 181:1615–21. doi:10.1084/jem.181.5.1615
7. Hilkens CM, Kalinski P, de Boer M, Kapsenberg ML. Human dendritic cells require exogenous interleukin-12-inducing factors to direct the development of naive T-helper cells toward the Th1 phenotype. Blood (1997) 90:1920–6.
8. Ohteki T, Fukao T, Suzue K, Maki C, Ito M, Nakamura M, et al. Interleukin 12-dependent interferon γ production by CD8α+ lymphoid dendritic cells. J Exp Med (1999) 189:1981–6. doi:10.1084/jem.189.12.1981
9. Suzue K, Asai T, Takeuchi T, Koyasu S. In vivo role of IFN-γ produced by antigen-presenting cells in early host defense against intracellular pathogens. Eur J Immunol (2003) 33:2666–75. doi:10.1002/eji.200323292
10. Moretto MM, Weiss LM, Combe CL, Khan IA. IFN-γ-producing dendritic cells are important for priming of gut intraepithelial lymphocyte response against intracellular parasitic infection. J Immunol (2007) 179:2485–92.
11. Buchmeier NA, Schreiber RD. Requirement of endogenous interferon-γ production for resolution of Listeria monocytogenes infection. Proc Natl Acad Sci U S A (1985) 82:7404–8. doi:10.1073/pnas.82.21.7404
12. Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, et al. Immune response in mice that lack the interferon-γ receptor. Science (1993) 259:1742–5. doi:10.1126/science.8456301
13. Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science (2011) 332:717–21. doi:10.1126/science.1201711
14. Green AM, Difazio R, Flynn JL. IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol (2013) 190:270–7. doi:10.4049/jimmunol.1200061
15. Murray HW, Rubin BY, Rothermel CD. Killing of intracellular Leishmania donovani by lymphokine-stimulated human mononuclear phagocytes. Evidence that interferon-γ is the activating lymphokine. J Clin Invest (1983) 72:1506–10. doi:10.1172/JCI110972
16. Torrico F, Heremans H, Rivera MT, Van ME, Billiau A, Carlier Y. Endogenous IFN-γ is required for resistance to acute Trypanosoma cruzi infection in mice. J Immunol (1991) 146:3626–32.
17. Scharton-Kersten TM, Wynn TA, Denkers EY, Bala S, Grunvald E, Hieny S, et al. In the absence of endogenous IFN-γ, mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J Immunol (1996) 157:4045–54.
18. Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, et al. A cluster of interferon-γ-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity (2012) 37:302–13. doi:10.1016/j.immuni.2012.06.009
19. Micallef MJ, Ohtsuki T, Kohno K, Tanabe F, Ushio S, Namba M, et al. Interferon-γ-inducing factor enhances T helper 1 cytokine production. Eur J Immunol (1996) 26:1647–51. doi:10.1002/eji.1830260736
20. Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity (1998) 8:383–90. doi:10.1016/S1074-7613(00)80543-9
21. Okamoto I, Kohno K, Tanimoto T, Ikegami H, Kurimoto M. Development of CD8+ effector T cells is differentially regulated by IL-18 and IL-12. J Immunol (1999) 162:3202–11.
22. Kamath AT, Sheasby CE, Tough DF. Dendritic cells and NK cells stimulate bystander T cell activation in response to TLR agonists through secretion of IFN-αβ and IFN-γ. J Immunol (2005) 174:767–76.
23. Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon-γ upon combined stimulation with interleukin (IL)-12 and IL-18: a novel pathway of autocrine macrophage activation. J Exp Med (1998) 187:2103–8. doi:10.1084/jem.187.12.2103
24. Otani T, Nakamura S, Toki M, Motoda R, Kurimoto M, Orita K. Identification of IFN-γ-producing cells in IL-12/IL-18-treated mice. Cell Immunol (1999) 198:111–9. doi:10.1006/cimm.1999.1589
25. Fricke I, Mitchell D, Mittelstädt J, Lehan N, Heine H, Goldmann T, et al. Mycobacteria induce IFN-γ production in human dendritic cells via triggering of TLR2. J Immunol (2006) 176:5173–82.
26. Hiromatsu K, Yoshikai Y, Matsuzaki G, Ohga S, Muramori K, Matsumoto K, et al. A protective role of γδ T cells in primary infection with Listeria monocytogenes in mice. J Exp Med (1992) 175:49–56. doi:10.1084/jem.175.1.49
27. Rosat JP, MacDonald HR, Louis JA. A role for γδ T cells during experimental infection of mice with Leishmania major. J Immunol (1993) 150:550–5.
28. Hisaeda H, Nagasawa H, Maeda K, Maekawa Y, Ishikawa H, Ito Y, et al. γδ T cells play an important role in hsp65 expression and in acquiring protective immune responses against infection with Toxoplasma gondii. J Immunol (1995) 155:244–51.
29. Wang T, Gao Y, Scully E, Davis CT, Anderson JF, Welte T, et al. γδ T cells facilitate adaptive immunity against West Nile virus infection in mice. J Immunol (2006) 177:1825–32.
30. Déchanet J, Merville P, Lim A, Retière C, Pitard V, Lafarge X, et al. Implication of γδ T cells in the human immune response to cytomegalovirus. J Clin Invest (1999) 103:1437–49. doi:10.1172/JCI5409
31. Egan CE, Dalton JE, Andrew EM, Smith JE, Gubbels MJ, Striepen B, et al. A requirement for the Vγ1+ subset of peripheral γδ T cells in the control of the systemic growth of Toxoplasma gondii and infection-induced pathology. J Immunol (2005) 175:8191–9.
32. Inoue S, Niikura M, Takeo S, Mineo S, Kawakami Y, Uchida A, et al. Enhancement of dendritic cell activation via CD40 ligand-expressing γδ T cells is responsible for protective immunity to Plasmodium parasites. Proc Natl Acad Sci U S A (2012) 109:12129–34. doi:10.1073/pnas.1204480109
33. Waki S, Uehara S, Kanbe K, Ono K, Suzuki M, Nariuchi H. The role of T cells in pathogenesis and protective immunity to murine malaria. Immunology (1992) 75:646–51.
34. Yoshimoto T, Takahama Y, Wang CR, Yoneto T, Waki S, Nariuchi H. A pathogenic role of IL-12 in blood-stage murine malaria lethal strain Plasmodium berghei NK65 infection. J Immunol (1998) 160:5500–5.
35. Yoneto T, Yoshimoto T, Wang CR, Takahama Y, Tsuji M, Waki S, et al. Gamma interferon production is critical for protective immunity to infection with blood-stage Plasmodium berghei XAT but neither NO production nor NK cell activation is critical. Infect Immun (1999) 67:2349–56.
36. Favre N, Ryffel B, Bordmann G, Rudin W. The course of Plasmodium chabaudi chabaudi infections in interferon-γ receptor deficient mice. Parasite Immunol (1997) 19:375–83. doi:10.1046/j.1365-3024.1997.d01-227.x
37. van der Heyde HC, Pepper B, Batchelder J, Cigel F, Weidanz WP. The time course of selected malarial infections in cytokine-deficient mice. Exp Parasitol (1997) 85:206–13. doi:10.1006/expr.1996.4132
38. Amani V, Vigário AM, Belnoue E, Marussig M, Fonseca L, Mazier D, et al. Involvement of IFN-γ receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur J Immunol (2000) 30:1646–55. doi:10.1002/1521-4141(200006)30:6<1646::AID-IMMU1646>3.0.CO;2-0
39. Su Z, Stevenson MM. Central role of endogenous γ interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect Immun (2000) 68:4399–406. doi:10.1128/IAI.68.8.4399-4406.2000
40. Cigel F, Batchelder J, Burns JM Jr, Yañez D, van der Heyde H, Manning DD, et al. Immunity to blood-stage murine malarial parasites is MHC class II dependent. Immunol Lett (2003) 89:243–9. doi:10.1016/S0165-2478(03)00152-4
41. Süss G, Eichmann K, Kury E, Linke A, Langhorne J. Roles of CD4- and CD8-bearing T lymphocytes in the immune response to the erythrocytic stages of Plasmodium chabaudi. Infect Immunity (1988) 56:3081–8.
42. Podoba JE, Stevenson MM. CD4+ and CD8+ T lymphocytes both contribute to acquired immunity to blood-stage Plasmodium chabaudi AS. Infect Immun (1991) 59:51–8.
43. Imai T, Shen J, Chou B, Duan X, Tu L, Tetsutani K, et al. Involvement of CD8+ T cells in protective immunity against murine blood-stage infection with Plasmodium yoelii 17XL strain. Eur J Immunol (2010) 40:1053–61. doi:10.1002/eji.200939525
44. Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol (2011) 13(2):188–95. doi:10.1038/ni.2180
45. Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med (2005) 201:19–25. doi:10.1084/jem.20041836
46. Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A (2007) 104:1919–24. doi:10.1073/pnas.0608745104
47. Seixas E, Moura Nunes JF, Matos I, Coutinho A. The interaction between DC and Plasmodium berghei/chabaudi-infected erythrocytes in mice involves direct cell-to-cell contact, internalization and TLR. Eur J Immunol (2009) 39:1850–63. doi:10.1002/eji.200838403
48. Gowda NM, Wu X, Gowda DC. TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J Immunol (2012) 188:5073–85. doi:10.4049/jimmunol.1102143
49. Morita CT, Beckman EM, Bukowski JF, Tanaka Y, Band H, Bloom BR, et al. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human γδ T cells. Immunity (1995) 3:495–507. doi:10.1016/1074-7613(95)90178-7
50. Behr C, Poupot R, Peyrat MA, Poquet Y, Constant P, Dubois P, et al. Plasmodium falciparum stimuli for human γδ T cells are related to phosphorylated antigens of mycobacteria. Infect Immun (1996) 64:2892–6.
51. Pichyangkul S, Saengkrai P, Yongvanitchit K, Stewart A, Heppner DG. Activation of γδ T cells in malaria: interaction of cytokines and a schizont-associated Plasmodium falciparum antigen. J Infect Dis (1997) 176:233–41. doi:10.1086/514029
52. Elloso MM, van der Heyde HC, Troutt A, Manning DD, Weidanz WP. Human γδ T cell subset-proliferative response to malarial antigen in vitro depends on CD4+ T cells or cytokines that signal through components of the IL-2R. J Immunol (1996) 157:2096–102.
53. Ribot JC, Debarros A, Mancio-Silva L, Pamplona A, Silva-Santos B. B7-CD28 costimulatory signals control the survival and proliferation of murine and human γδ T cells via IL-2 production. J Immunol (2012) 189:1202–8. doi:10.4049/jimmunol.1200268
54. Mokuno Y, Matsuguchi T, Takano M, Nishimura H, Washizu J, Ogawa T, et al. Expression of toll-like receptor 2 on γδ T cells bearing invariant Vγ6/Vδ1 induced by Escherichia coli infection in mice. J Immunol (2000) 165:931–40.
55. Deetz CO, Hebbeler AM, Propp NA, Cairo C, Tikhonov I, Pauza CD. Gamma interferon secretion by human Vγ2Vδ2 T cells after stimulation with antibody against the T-cell receptor plus the toll-like receptor 2 agonist Pam3Cys. Infect Immun (2006) 74:4505–11. doi:10.1128/IAI.00088-06
56. Pietschmann K, Beetz S, Welte S, Martens I, Gruen J, Oberg HH, et al. Toll-like receptor expression and function in subsets of human γδ T lymphocytes. Scand J Immunol (2009) 70:245–55. doi:10.1111/j.1365-3083.2009.02290.x
57. Naik RS, Branch OH, Woods AS, Vijaykumar M, Perkins DJ, Nahlen BL, et al. Glycosylphosphatidylinositol anchors of Plasmodium falciparum: molecular characterization and naturally elicited antibody response that may provide immunity to malaria pathogenesis. J Exp Med (2000) 192:1563–76. doi:10.1084/jem.192.11.1563
58. Devilder MC, Allain S, Dousset C, Bonneville M, Scotet E. Early triggering of exclusive IFN-γ responses of human Vγ9Vδ2 T cells by TLR-activated myeloid and plasmacytoid dendritic cells. J Immunol (2009) 183:3625–33. doi:10.4049/jimmunol.0901571
59. D’Ombrain MC, Robinson LJ, Stanisic DI, Taraika J, Bernard N, Michon P, et al. Association of early interferon-gamma production with immunity to clinical malaria: a longitudinal study among Papua New Guinean children. Clin Infect Dis (2008) 47:1380–7. doi:10.1086/592971
60. Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, Viguier M, et al. On the pathogenic role of brain-sequestered αβ CD8+ T cells in experimental cerebral malaria. J Immunol (2002) 169:6369–75.
61. Villegas-Mendez A, Greig R, Shaw TN, de Souza JB, Gwyer Findlay E, Stumhofer JS, et al. IFN-γ-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J Immunol (2012) 189:968–79. doi:10.4049/jimmunol.1200688
62. Yañez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J Immunol (1996) 157:1620–4.
63. Nitcheu J, Bonduelle O, Combadiere C, Tefit M, Seilhean D, Mazier D, et al. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J Immunol (2003) 170:2221–8.
64. Haque A, Best SE, Unosson K, Amante FH, de Labastida F, Anstey NM, et al. Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J Immunol (2011) 186:6148–56. doi:10.4049/jimmunol.1003955
65. Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS, et al. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8α+ dendritic cells. Proc Natl Acad Sci U S A (2008) 105:14509–14. doi:10.1073/pnas.0806727105
66. Yañez DM, Batchelder J, van der Heyde HC, Manning DD, Weidanz WP. γδ T-cell function in pathogenesis of cerebral malaria in mice infected with Plasmodium berghei ANKA. Infect Immun (1999) 67:446–8.
67. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature (2010) 465:793–7. doi:10.1038/nature09135
68. Katherine C, MacNamara KC, Oduro K, Martin O, Jones DD, McLaughlin M, et al. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN-γ signaling. J Immunol (2011) 186:1032–43. doi:10.4049/jimmunol.1001893
69. MacNamara KC, Jones M, Martin O, Winslow GM. Transient activation of hematopoietic stem and progenitor cells by IFNγ during acute bacterial infection. PLoS ONE (2011) 6:e28669. doi:10.1371/journal.pone.0028669
70. Belyaev NN, Brown DE, Diaz AI, Rae A, Jarra W, Thompson J, et al. Induction of an IL7-R+c-Kithi myelolymphoid progenitor critically dependent on IFN-γ signaling during acute malaria. Nat Immunol (2010) 11:477–85. doi:10.1038/ni.1869
71. Sato T, Selleri C, Young NS, MacIejewski JP. Hematopoietic inhibition by interferon-γ is partially mediated through interferon regulatory factor-1. Blood (1995) 86:3373–80.
72. de Bruin AM, Buitenhuis M, van der Sluijs KF, van Gisbergen KP, Boon L, Nolte MA. Eosinophil differentiation in the bone marrow is inhibited by T cell-derived IFN-γ. Blood (2010) 116:2559–69. doi:10.1182/blood-2009-12-261339
73. Libregts SF, Gutiérrez L, de Bruin AM, Wensveen FM, Papadopoulos P, van Ijcken W, et al. Chronic IFN-γ production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood (2011) 118:2578–88. doi:10.1182/blood-2010-10-315218
74. Falini B, Flenghi L, Pileri S, Pelicci P, Fagioli M, Martelli MF, et al. Distribution of T cells bearing different forms of the T cell receptor gamma/delta in normal and pathological human tissues. J Immunol (1989) 143:2480–8.
75. van der Heyde HC, Elloso MM, Chang WL, Kaplan M, Manning DD, Weidanz WP. γδ T cells function in cell-mediated immunity to acute blood-stage Plasmodium chabaudi adami malaria. J Immunol (1995) 154:3985–90.
76. Seixas EM, Langhorne J. γδ T cells contribute to control of chronic parasitemia in Plasmodium chabaudi infections in mice. J Immunol (1999) 162:2837–41.
77. Bakir HY, Tomiyama-Miyaji C, Watanabe H, Nagura T, Kawamura T, Sekikawa H, et al. Reasons why DBA/2 mice are resistant to malaria infection; expansion of CD3int B220+ γδ T cells with double-negative CD4-CD8-phenotype in the liver. Immunology (2006) 117:127–35. doi:10.1111/j.1365-2567.2005.02273.x
78. Weidanz WP, LaFleur G, Brown A, Burns JM Jr, Gramaglia I, van der Heyde HC. γδ T cells but not NK cells are essential for cell-mediated immunity against Plasmodium chabaudi malaria. Infect Immun (2010) 78:4331–40. doi:10.1128/IAI.00539-10
79. Costa G, Loizon S, Guenot M, Mocan I, Halary F, de Saint-Basile G, et al. Control of Plasmodium falciparum erythrocytic cycle: γδ T cells target the red blood cell-invasive merozoites. Blood (2011) 118:6952–62. doi:10.1182/blood-2011-08-376111
80. Kobayashi F, Niikura M, Waki S, Matsui T, Fujino T, Tsuruhara T, et al. Plasmodium berghei XAT: contribution of γδ T cells to host defense against infection with blood-stage nonlethal malaria parasite. Exp Parasitol (2007) 117:368–75. doi:10.1016/j.exppara.2007.05.002
81. Waki S, Tamura J, Imanaka M, Ishikawa S, Suzuki M. Plasmodium berghei: isolation and maintenance of an irradiation attenuated strain in the nude mouse. Exp Parasitol (1982) 53:335–40. doi:10.1016/0014-4894(82)90076-5
82. Nakazawa S, Brown AE, Maeno Y, Smith CD, Aikawa M. Malaria-induced increase of splenic γδ T cells in humans, monkeys, and mice. Exp Parasitol (1994) 79:391–8. doi:10.1006/expr.1994.1101
83. Horowitz A, Newman KC, Evans JH, Korbel DS, Davis DM, Riley EM. Cross-talk between T cells and NK cells generates rapid effector responses to Plasmodium falciparum-infected erythrocytes. J Immunol (2010) 184:6043–52. doi:10.4049/jimmunol.1000106
84. Hudspeth K, Fogli M, Correia DV, Mikulak J, Roberto A, Della Bella S, et al. Engagement of NKp30 on Vδ1 T cells induces the production of CCL3, CCL4, and CCL5 and suppresses HIV-1 replication. Blood (2012) 119:4013–6. doi:10.1182/blood-2011-11-390153
85. Vermijlen D, Ellis P, Langford C, Klein A, Engel R, Willimann K, et al. Distinct cytokine-driven responses of activated blood γδ T cells: insights into unconventional T cell pleiotropy. J Immunol (2007) 178:4304–14.
86. Schulz O, Edwards AD, Schito M, Aliberti J, Manickasingham S, Sher A, et al. CD40 triggering of heterodimeric IL–12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity (2000) 13:453–62. doi:10.1016/S1074-7613(00)00045-5
87. Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol (2002) 2:251–62. doi:10.1038/nri778
88. Kalia V, Sarkar S, Gourley TS, Rouse BT, Ahmed R. Differentiation of memory B and T cells. Curr Opin Immunol (2006) 18:255–64. doi:10.1016/j.coi.2006.03.020
89. Freitas do Rosário AP, Muxel SM, Rodríguez-Málaga SM, Sardinha LR, Zago CA, Castillo-Méndez SI, et al. Gradual decline in malaria-specific memory T cell responses leads to failure to maintain long-term protective immunity to Plasmodium chabaudi AS despite persistence of B cell memory and circulating antibody. J Immunol (2008) 181:8344–55.
90. Stephens R, Langhorne J. Effector memory Th1 CD4 T cells are maintained in a mouse model of chronic malaria. PLoS Pathog (2010) 6:e1001208. doi:10.1371/journal.ppat.1001208
91. Shen Y, Zhou D, Qiu L, Lai X, Simon M, Shen L, et al. Adaptive immune response of Vγ2Vδ2 T cells during mycobacterial infections. Science (2002) 295:2255–8. doi:10.1126/science.1068819
92. Pitard V, Roumanes D, Lafarge X, Couzi L, Garrigue I, Lafon ME, et al. Long-term expansion of effector/memory Vδ2-γδ T cells is a specific blood signature of CMV infection. Blood (2008) 112:1317–24. doi:10.1182/blood-2008-01-136713
93. Tokoyoda K, Zehentmeier S, Hegazy AN, Albrecht I, Grün JR, Löhning M, et al. Professional memory CD4+ T lymphocytes preferentially reside and rest in the bone marrow. Immunity (2009) 30:721–30. doi:10.1016/j.immuni.2009.03.015
Keywords: IFN-γ, γδ T cells, malaria, dendritic cells, αβ T cells, memory cells, hematopoiesis
Citation: Inoue S-I, Niikura M, Mineo S and Kobayashi F (2013) Roles of IFN-γ and γδ T cells in protective immunity against blood-stage malaria. Front. Immunol. 4:258. doi: 10.3389/fimmu.2013.00258
Received: 22 March 2013; Accepted: 15 August 2013;
Published online: 29 August 2013.
Edited by:
Howard M. Johnson, University of Florida, USACopyright: © 2013 Inoue, Niikura, Mineo and Kobayashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fumie Kobayashi, Department of Infectious Diseases, Kyorin University School of Medicine, 20-2, Shinkawa 6, Mitaka, Tokyo 181-8611, Japan e-mail:ZnVtZnVtQGtzLmt5b3Jpbi11LmFjLmpw