

- Department of Immunology, St. Jude Children’s Research Hospital, Memphis, TN, USA
NOD-like receptors (NLRs) are a class of cytoplasmic pattern-recognition receptors. Although most NLRs play some role in immunity, their functions range from regulating antigen presentation (NLRC5, CIITA) to pathogen/damage sensing (NLRP1, NLRP3, NLRC1/2, NLRC4) to suppression or modulation of inflammation (NLRC3, NLRP6, NLRP12, NLRX1). However, NLRP2, NLRP5, and NLRP7 are also involved in non-immune pathways such as embryonic development. In this review, we highlight some of the least well-understood aspects of NLRs, including the mechanisms by which they sense pathogens or damage. NLRP3 recognizes a diverse range of stimuli and numerous publications have presented potential unifying models for NLRP3 activation, but no single mechanism proposed thus far appears to account for all possible NLRP3 activators. Additionally, NLRC3, NLRP6, and NLRP12 inhibit NF-κB activation, but whether direct ligand sensing is a requirement for this function is not known. Herein, we review the various mechanisms of sensing and activation proposed for NLRP3 and other inflammasome activators. We also discuss the role of NLRC3, NLRP6, NLRP12, and NLRX1 as inhibitors and how they are activated and function in their roles to limit inflammation. Finally, we present an overview of the emerging roles that NLRP2, NLRP5, and NLRP7 play during embryonic development and postulate on the potential pathways involved.
Introduction
Innate immunity is initiated by germline-encoded pattern-recognition receptors (PRRs). Among these, the nucleotide oligomerization and binding domain (NOD)-like receptors (NLRs) comprise a large receptor family of more than 20 members (1–4). Only about half of the NLRs have been characterized in any detail. However, it is well documented that NLRs play a critical role in protection against infectious diseases, including bacteria (5, 6), viruses (7, 8), fungi (9, 10), protists (11, 12), and helminthes (13). Of the NLRs which have been studied, most of them fall into one of four categories: (1) Inflammasome activators, (2) Activators of Nuclear Factor-κB (NF-κB) and mitogen activated protein kinase (MAPK), (3) Inhibitors of inflammatory signaling, (4) and trans-activators of MHC expression. However, several NLRs have definite roles in embryogenesis, uterine implantation, and fetal development (14). Intriguingly, some NLRs appear to play multiple roles within inflammation or development. This suggests alternative functions for some NLRs in different cell types or multiple activation mechanisms with separate downstream effects for other NLRs.
One set of NLRs that regulates NF-κB and MAPK are NLRC1 (NOD1 or CARD4) and NLRC2 (NOD2 or CARD15). NLRC1 recognizes iE-DAP, a subunit of peptidoglycan found in some bacterial cell walls (5, 15). NLRC2 recognizes MDP, another peptidoglycan fragment (16–19). NLRC1 and NLRC2 then act through the adaptor RIPK2 to activate NF-κB and MAPK signaling (20–22). However, there are now multiple reports that demonstrate NLRC2 can respond to cytosolic RNA during viral infection (23–25). Although viral RNA also induces an interaction between NLRC2 and RIPK2, this appears to regulate autophagy mechanisms, instead of NF-κB, and subsequently represses inflammasome activation and prevents immunopathology (25). Furthermore, viral RNA mediated activation causes NLRC2 to interact with the antiviral adaptor protein MAVS. This interaction was shown to be essential for the production of IFN-β during viral infection and for suppressing virus replication (24). Additionally, NLRC2 regulates other antiviral pathways like 2′-5′ oligoadenylate synthease (OAS2), which activates RNAse L and degrades viral RNA, thus potentiating antiviral signaling (23). It is not clear how NLRC2 would bind to both MDP and RNA, but one possibility is that additional upstream adaptor proteins, which have yet to be discovered, actually provide specificity.
In the case of NLRC4, it is activated by bacterial flagellin (26, 27) or the rod complex of bacterial type III secretion systems (T3SS) (28). Once activated, NLRC4 forms a multimeric complex, known as the inflammasome, with the adaptor ASC and caspase-1 (26, 27). Inflammasome formation results in a proinflammatory cell death termed pyroptosis (29) and the release of IL-1β and IL-18 (30–34). Recently, the ability of NLRC4 to recognize flagellin and T3SS components was tagged to an association between NLRC4 and another class of NOD proteins known as NAIPs. NAIP5 and NAIP6 in the mouse recognize flagellin and NAIP2 recognizes T3SS rod complexes respectively and then activate NLRC4 (35–37). Furthermore, NAIP1 in mice activates NLRC4 in response to the needle protein of some T3SS (38). In human cells, only one NAIP exists, and this recognizes the needle protein of T3SS similar to mouse NAIP1 (38). These results demonstrate that one mechanism for the recognition of multiple ligands by NLRs is the presence of upstream adaptor proteins like NAIPs.
Distinct NLRs recognize microbial or viral components such as peptidoglycan, flagellin, or viral RNA. These pathogen specific molecules are known as pathogen-associated molecular patterns (PAMPs). Alternatively, some NLRs, like NLRP3, detect damage associated molecular patterns (DAMPs). DAMPs consist of byproducts of pathogen invasion or sterile cellular damage such as uric acid crystals, reactive oxygen species (ROS), or extracellular ATP release (39–42). Sensing of DAMPs by NLRP3 is not only critical for detection and clearance of pathogens but also for protection and repair of tissues during inflammation (43, 44). NLRP3 is one of the most ubiquitously important NLRs. Once activated, NLRP3 also forms an inflammasome with the adaptor ASC and caspase-1 (45, 46). NLRP3 responds to an incredibly broad range of pathogens making it unlikely that it senses PAMPs directly. Many lines of evidence support a role for NLRP3 in DAMP sensing, where damage to the host results in the release of certain danger signals not present under homeostatic conditions. ROS, potassium efflux, and release of proteases from endosomes have all been reported to activate NLRP3 (41, 42, 47–51). Although much is know about the range of stimuli that can activate NLRP3, much research remains to be done to understand how NLRP3 becomes activated.
The ability of NLRP3 to respond to multiple PAMPs or DAMPs from such a broad range of pathogens strongly indicates the presence of upstream adaptors, as is the case for NLRC4, or common danger signals which funnel into one pathway. In the case of NLRC2, the different signaling pathways activated by MDP or viral RNA would suggest that different modes of NLRC2 activation lead to different protein conformations or other alterations in NLRC2 activity. This is subsequently responsible for activation of NF-κB, autophagy, or antiviral signaling. Indeed, these are some of the great-unsolved mysteries of NLR biology. In addition, other NLRs, like NLRC3, NLRP6, NLRP12, and NLRX1, play inhibitory roles during inflammation. Yet how these proteins are activated or perform their inhibitory functions is not well understood. Finally, there are numerous NLRs for which there are different reports indicating a multiplicity of potential functions. In this review, we discuss several of these unsolved mysteries and potential future directions in the field of NLR biology.
Activation Mechanisms of NLRP3
NLRP3 was initially described as an activator of caspase-1 in 2002 and was subsequently associated with autoinflammatory periodic fevers like Muckle–Wells syndrome or bacterial infection (45, 46). Since then, there have been many proposed mechanism for how NLRP3 is activated. There is no evidence that NLRP3 interacts directly with any PAMP. Although NLRP3 is activated in response to bacteria and viral RNA (7, 52), lipopolysaccharide, and MDP (53), most PAMPs appear to only be required for the transcriptional up-regulation of NLRP3 and pro-IL-1β (54). Once NLRP3 is upregulated, a second signal, generally a DAMP or a pore forming toxin like nigericin, is required for NLRP3 to interact with ASC and caspase-1 to form an active inflammasome. This second signal is frequently associated with the production of ROS or endosomal rupture (41, 42, 49, 50). Changes in intracellular and extracellular calcium (55–58) and potassium efflux (47, 48, 51) have also been proposed to activate NLRP3, as have changes in cytosolic or extracellular pH (59, 60). This dichotomy of signals for priming and activation is required for NLRP3 inflammasome formation. The big question that remains is how ROS, ion flux, or other DAMPs regulate NLRP3 (Figure 1). One possibility is that the structure of NLRP3 is sensitive to changes in ion concentrations, and exposure of the pyrin effector domain occurs when ion concentrations deviate from their homeostatic state (48, 51). Alternatively, protein sensors of cellular redox or ion sensors could regulate NLRP3 activation following their own activation. Studies into the structure of NLRP3 and the effects of different ions on the ATPase activity, pyrin effector domain exposure, and ASC binding affinity of NLRP3 would greatly increase our understanding of how NLRP3 is activated.
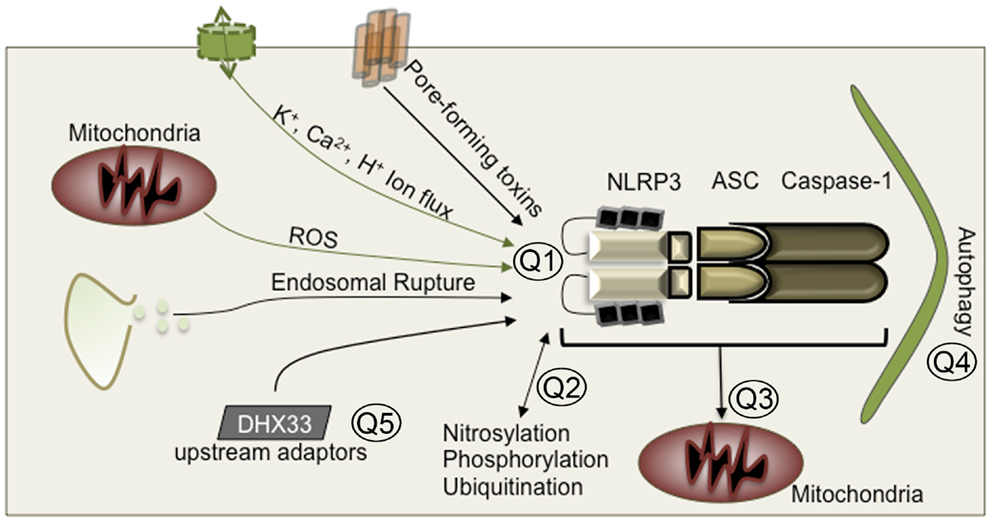
Figure 1. Unsolved mysteries in NLRP3 biology. Q1: Is there a common DAMP that activates NLRP3? Do DAMPs directly activate NLRP3? Do DAMPs induce structural rearrangement of NLRP3? Q2: How do post-translational modifications regulate activation on a structural level? Q3: Is mitochondrial localization essential for NLRP3 inflammasome formation? Q4: How does autophagy inhibit NLRP3? Does autophagy directly engulf NLRP3 inflammasomes? Does it engulf damaged mitochondria where NLRP3 is localized? Does autophagy merely remove the source of DAMPs? Q5: Are there additional upstream sensors or adaptors that facilitate NLRP3 activation?
Proteomic studies directed at understanding the NLRP3 interactome using different activators of NLRP3 may also provide further insight into potential upstream regulators such as NAIPs or ion/ROS sensors. A recent paper by Mitoma et al. (61) found in human macrophages that NLRP3 is activated in responses to double stranded RNA through an interaction with the RNA helicase DHX33 (61). Protein kinase R (PKR) is an RNA dependent kinase involved in antiviral defenses. Activation of NLRP3 was also proposed to be dependent on PKR, although phosphorylation of NLRP3 was not required (62). However, another group attempted to examine the role of PKR mediated activation of NLRP3 but found no role for PKR (63). Finally, the adaptor protein MAVS, which is required for antiviral signaling downstream of the RNA helicases RIG-I and MDA5, has been shown to interact with NLRP3 and regulate its activation and localization to the mitochondria (64). Although this was shown in the context of RNA transfection, LPS + ATP treatment or nigericin, how LPS or nigericin could activate MAVS remains to be investigated further. In all, there is a significant body of research that would indicate the presence of upstream PRR that tie into the NLRP3 pathway (Figure 1).
Although multiple upstream sensors may regulate NLRP3, it is possible that NLRP3 interacting partners regulate its activation through the addition or removal of post-translational modifications. Post-translational modification of NLRs has been reported to regulate their activation. For example, phosphorylation of NLRC4 by PKCδ regulates its activation during Salmonella typhimurium infection in macrophages (65). In the case of NLRP3, nitric oxide produced during chronic inflammation in vivo during Mycobacterium tuberculosis infection results in nitrosylation of NLRP3 and inhibition of inflammasome activation (66). Similarly, the addition of NO donor compounds to macrophages or induction of NO by IFN-γ treatment inhibited NLRP3 activation (66, 67). The role of NO for NLRP3 inhibition during LPS-induced sepsis in mice has also been reported (68). Therefore, proteins that can regulate the nitrosylation status of NLRP3 may be able to regulate its activation. Ubiquitination and deubiquitination were also found to regulate NLRP3 activation (69, 70). Thus far, deubiquitination by the BRCC3 deubiquitinase is the only post-translational modification that is reported to activate NLRP3 (70). It is clear that post-translational modifications can affect NLRP3 activation, although how ubiquitination, or nitrosylation affect the function of NLRP3 needs further biochemical examination (Figure 1).
The cellular autophagy pathway, which is required for recycling damaged organelles and proteins, has been reported to inhibit NLRP3 activation. Ubiquitinated inflammasomes are degraded through the autophagy pathway (71). This report, in combination with those above, may indicate that deubiquitination of NLRP3 prevents autophagic degradation and allows for inflammasome formation. Alternatively, the removal of damaged mitochondria, which produce NLRP3 activators like ROS or release of mitochondrial DNA into the cytosol, constitutes another mechanism by which autophagy regulates NLRP3 activation (25, 72, 73). It is also possible that autophagosomal degradation of damaged mitochondria simultaneously removes inflammasomes. Several recent publications demonstrate that NLRP3 inflammasome formation is dependent on localization to the mitochondria (64, 74). However, another report demonstrated that inflammasome activation was not associated with any organelle but occurred in the cytosol (75). Why there are conflicting reports regarding the mechanisms that activate NLRP3 are unclear. However, in the case of cellular localization, differences in fixation or staining methodologies may result in aggregation of inflammasomes with mitochondria or their disassociation, respectively. In all, mitochondria appear to play a role in the regulation of NLRP3 inflammasome activation, but whether they serve as an activation platform, a source of stimuli, or both requires further investigation (Figure 1).
To more fully understand NLRP3 regulation, the interactome of NLRP3 including kinases and ubiquitin ligases still need to be discovered and the regulation of post-translational pathways examined. Clearly there is need for a concerted effort from biochemists, molecular and structural biologists, and immunologists to collaborate on these issues. As NLRP3 is associated with numerous autoinflammatory and autoimmune diseases, understanding how NLRP3 is regulated will be necessary for understanding and potentially preventing disease development, as well as for the design of inhibitors which are useful under specific inflammatory conditions.
Regulation of Inhibitory NLRs
Intriguingly, all inhibitory NLRs studied thus far have been found to inhibit NF-κB activation. NLRP12 was examined during colon inflammation and colon tumorigenesis and found to negatively regulate NF-κB down stream of Toll-like receptors (TLRs) (76, 77) or to regulate the alternative NF-κB pathway downstream of TNF family receptors (76, 78). NLRP12 appears to interact with NF-κB–inducing kinase (NIK), interleukin-1 receptor-associated kinase 1 (IRAK1), and TNF receptor-associated factor 3 (TRAF3), which are known mediators of NF-κB signaling (78, 79). These interactions appear to regulate the phosphorylation of IRAK1 and the degradation of NIK, thus resulting in inhibition of the alternative NF-κB pathway. However, the mechanism by which NLRP12 inhibits TLR mediated activation of the classical NF-κB pathway is not known (Figure 2).
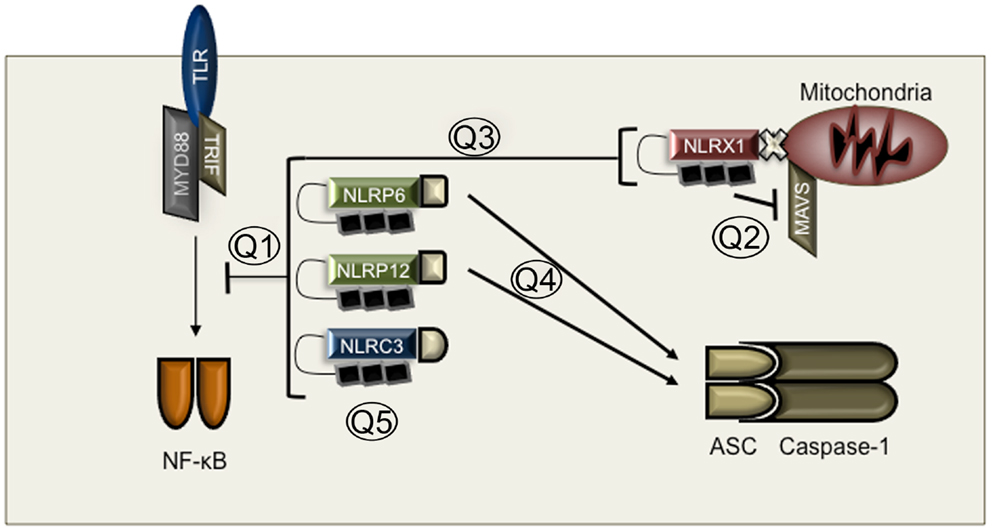
Figure 2. Mechanisms of inhibitory NLRs. Q1: How do inhibitory NLRs function? Is PAMP recognition required for inhibitory NLR function? Is NLR expression sufficient for inhibitory function? Q2: Is NLRX1 an inhibitor of MAVS or a modulator of mitochondrial ROS? Q3: How does NLRX1 inhibit NF-κB if it is localized to the mitochondria? Q4: Do NLRP6 and NLRP12 regulate inflammasome activation and how? Does gut flora play a role in inflammasome activation in the absence of NLRP6 and NLRP12. Q5: Why are there so many inhibitory NLRs? Do inhibitory NLRs play redundant or context specific roles?
Currently, it is unclear how the inhibitory function of NLRP12 is regulated (Figure 2). ATP binding appears to be a requirement for activation (79) but the mechanism by which NLRP12 structural rearrangement occurs to permit ATP binding has not been examined. NLRP12 expression increases following NF-κB activation (80). It is also apparent that NLRP12 interacts with other proteins which regulate its function, including HSP90, which stabilizes NLRP12 and prevents its proteasomal degradation (81). Whether expression alone is sufficient for its inhibitory function, or if NLRP12 is regulated by post-translational modifications of some kind is unclear.
Recently, Nlrc3-deficient mice were generated and inflammation examined in response to LPS treatment (82). Sub-lethal LPS administration resulted in increased IL-6, increased macrophage numbers and increased hypothermia in Nlrc3−/− mice. Examination of Nlrc3−/− macrophages showed that there was enhanced NF-κB activation down stream of TLR signaling (82). Mechanistically, NLRC3 appears to regulate TRAF6 activation by modulating its K63-linked ubiquitination and stability. Once again, the mechanisms that regulate NLRC3 activation remain to be examined (Figure 2).
Similar to NLRC3 and NLRP12, NLRP6 also inhibits NF-κB activation down stream of TLR signaling. NLRP6 was shown to suppress NF-κB activation during Listeria monocytogenes and Salmonella typhimurium infection, and in the absence of NLRP6, bacteria were cleared more rapidly (83). In other studies, Nlrp6 deficiency predisposes mice to increased inflammation in models of colitis and to increased tumorigenesis in colon cancer models (84, 85). However, the mechanisms proposed for susceptibility to colitis and tumorigenesis are reportedly due to NLRP6 mediated inflammasome activation. Nlrp6−/− mice have reduced IL-18 in the colon in these models (84, 85). It should be noted, though, that no biochemical or molecular evidence for an NLRP6 inflammasome has been presented to date. It is therefore possible that NLRP6 regulates inflammasome activation indirectly. In fact, there are significant differences in the gut microbiota in Nlrp6−/− mice in the above models, which could result in altered inflammasome activation (Figure 2).
NLRP12 has also been proposed to form an inflammasome. During Yersinia pestis infection, NLRP12 is reported to recognize acylated lipid A and Nlrp12−/− mice were more susceptible to infection and had reduced IL-18 levels (86). In humans, NLRP12 polymorphisms are associated with inflammasome activation during periodic fever syndromes (87, 88). It is possible that both NLRP12 and NLRP6 have regulatory roles during NF-κB activation as well as in inflammasome formation. To verify these functions, however, much needs to be done on the molecular and biochemical level to determine the mechanisms by which these proteins activate the inflammasome and what stimuli activate them to form an inflammasome verses inhibit NF-κB activation. Finally, as discussed above, several NLR deficient mouse strains have been found to harbor altered gut microbiota compared to WT controls. The ability of the microbiome to regulate immunity is clear, but the exact effects of these changes are still not well-understood. Especially during models of colon inflammation, differences in gut flora between mice may be an essential factor in the phenotypes observed. The use of germ free or gnotobiotic mice for studying the roles of NLRs in general, but NLRP12 and NLRP6 in particular, may help resolve their functions as immune activators or repressors and the mechanisms by which they perform these functions.
The last NLR with a proposed inhibitory function is NLRX1. NLRX1 was originally reported to inhibit antiviral signaling through inhibition of the adaptor MAVS (8, 89). Subsequently, NLRX1 was shown to inhibit TLR mediated activation of NF-κB (90). How NLRX1 inhibits NF-κB is not clear though, as NLRX1 is localized to the mitochondria. Furthermore, the role of NLRX1 as an inhibitor is debated. Several groups have found no role for NLRX1 in regulating MAVS but have instead reported NLRX1 as a modulator of mitochondrial ROS (91–93). Recently, the crystal structure of NLRX1 was solved along with biochemical evidence for the binding of NLRX1 to the viral RNA mimic poly(I:C) (94). Although this finding would support a role for NLRX1 in antiviral signaling, the exact function of NLRX1 will require further examination (Figure 2).
As discussed above, the in vivo importance of inhibitory NLRs has been demonstrated in various models of inflammation. However, why there are so many NLRs that inhibit NF-κB signaling is a conundrum. If expression of these inhibitory NLRs alone were sufficient to suppress NF-κB activation, then why would there need to be four. One possibility is that they function as a whole to modulate NF-κB activation appropriately. Another possibility is that they are activated only in response to certain infections or stimuli. However, treatment with LPS or poly(I:C) both resulted in increased NF-κB activation in Nlrp12−/− macrophages (77), suggesting that ligand recognition is not required for its function. Understanding the individual and combined roles of NLRC3, NLRP6, NLRP12, and NLRX1 during specific infections or models of inflammation will be important as this field moves forward (Figure 2).
NLRs as Double Agents
As discussed in the last section, NLRP12 and NLRP6 have roles in inhibiting inflammation by modulating NF-κB activation (77–79, 83). In addition, both of these NLRs are reported to regulate inflammasome activation (84–86). As discussed in the introduction, NLRC2 is able to respond to both MDP and viral RNA and activates distinct pathways including NF-κB, autophagy, or antiviral signaling (Table 1). All of these pathways are important for inflammation and immunity. However, NLRs are also implicated in numerous non-inflammatory roles. NLRC1 and NLRC2 have been shown to regulate the differentiation of human umbilical cord blood-derived mesenchymal stem cells (MSC). Although NLRC1 and NLRC2 had no effect on MSC proliferation, they enhanced their differentiation into chondrocytes and osteocytes and inhibited adipocyte formation in vitro (95). The ability of NLRC1 and NLRC2 to regulate MSC differentiation was associated with increased ERK1/2 MAPK signaling; a known function of these NLRs (Table 1). The ability of NLRs to affect MSC may play an important part of wound healing and the resolution of inflammation. In fact, NLRP3 was found to play an important function in tissue repair in the lung during influenza A virus infection, although this was likely due to impaired recruitment of macrophages or other cells necessary for wound repair and healing (43).

Table 1. Functionally distinct roles of NLRs in biology.
The role of NLRs in tissue repair or MSC differentiation may be a logical progression following inflammation but several additional NLRs have been reported to regulate seemingly disparate functions. NLRP2 is reported to inhibit NF-κB activation (96, 97) and to enhance caspase-1 activation (96). In addition, siRNA mediated knockdown of NLRP2 in primary human astrocytes was recently reported to impair inflammasome activation (98). How NLRP2 affects inflammasome activation is not entirely clear, as knockdown of NLRP2 resulted in decreased caspase-1 expression as well. Furthermore, the stimulus used for NLRP2 activation was the NLRP3 activator extracellular ATP (98). These findings might indicate that NLRP2 regulates the expression of key NLRP3 inflammasome components as opposed to a novel NLRP2 specific inflammasome. In addition to the role for NLRP2 in inflammasome activation and inhibition of NF-κB signaling, NLRP2 has a definite role in embryonic development (Table 1). A truncation mutation of NLRP2 was found in association with Beckwith–Wiedemann Syndrome (BWS) (99). The NLRP2 mutation resulted in developmental defects that stemmed from altered DNA methylation and gene expression initially present in the maternal oocyte (maternal imprinting) and perpetuated in the fertilized embryo and developing fetus (99). Another study found some association between NLRP2 and recurrent miscarriages (100). Finally, siRNA knockdown of NLRP2 in murine oocytes or embryos leads to nearly complete developmental arrest (101).
Other NLRs have also been proposed to regulate inflammasome activation and development. NLRP7 regulates inflammasome activation in response to acylated lipopeptides like FSL-1 or triacylated Pam3CSK4 (102). In addition, NLRP7 is associated with recurrent miscarriages and recurrent hydatidiform molar pregnancies (100, 103–105). The above findings definitely support roles for NLRP2 and NLRP7 in inflammation and development. Interestingly, NLRP7 is not present in the mouse genome and appears to have arisen from a gene duplication event from NLRP2 (103). Therefore, it is not surprising that these two NLRs possess similar functions, but how they regulate both inflammasome activation and development is currently unknown (Table 1). Indeed, the role of NLRs in development is severely understudied, and many biochemical and cell specific studies on the function of these NLRs are needed to understand their differential roles. One possibility is that inflammasome activation is the mechanism by which NLRP2 and NLRP7 regulate embryonic development. The role of IL-1β in oocyte maturation and development has been appreciated for over a decade and has been reviewed previously (106, 107). Intrafollicular injection of IL-1β in horses induces ovulation but also inhibits embryo development (108), which is similar to the developmental arrest seen with NLRP2 and NLRP7 mutations. Furthermore, treatment of rabbit ovaries in vitro with IL-1β also arrests developing embryos (109). However, a lack of IL-1β signaling does not significantly affect fertility and embryo viability as IL-1 receptor deficient mice reproduce normally (110). Therefore, increased levels of IL-1β in patients with NLRP2 and NLRP7 mutations may be the cause of developmental arrest. However, much additional research on the roles of NLRP2 and NLRP7 needs to be performed before any conclusions can be reached regarding their functions in development.
Conclusion
The role of NLRs in immune function is unequivocal. However, there is much molecular, biochemical and structural research which remains to be done to better understand how NLRs are activated and regulated. Due to the diversity of functions among NLRs, understanding their activation and regulation should provide a cornucopia of new opportunities to modulate the immune system. The activation of proinflammatory NLRs has already been demonstrated to be important for the function of many adjuvants used in research or in the clinic (111, 112). Targeting NLRs specifically for the generation of novel adjuvants may provide for more effective vaccines. On the other hand, targeting NLRs may provide for new treatments against numerous diseases such as arthritis (40, 113), diabetes (114, 115), colitis (44, 85, 116), multiple sclerosis (117–119), Alzheimer’s (49, 120), and many other diseases associated with mutations or disregulation of NLRs.
Several unstudied NLRs have recently been assigned some putative functions. NLRP10 has been reported to play a critical role in the induction of Th1 and Th17 mediated T cell responses through a defect in dendritic cells migration during Candida albicans infection (121, 122). As discussed above, NLRP7 was recently reported to assemble an inflammasome in response to bacterial diacylated lipopeptides (102). The fact that after a decade of research, new inflammasome activators are still being discovered may indicate that more NLRs fill this function than those previously described. Furthermore, recent studies have also validated roles for NLRP5 in embryonic development, although the exact mechanisms underlying these observations have not been elucidated (123–125). With more than 10 NLRs unstudied, it will be of interest to determine the function of these remaining NLRs in inflammation and development.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Paras Anand and Si Ming Man for help in preparing this manuscript. This work was supported by grants to Thirumala-Devi Kanneganti from the National Institutes of Health through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Award Number AR056296), the National Institute of Allergy and Infectious Disease (Award Number AI101935) and the National Cancer Institute (Award Number CA163507). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also funded by the American Lebanese Syrian Associated Charities to Thirumala-Devi Kanneganti.
References
1. Inohara N, Nunez G. The NOD: a signaling module that regulates apoptosis and host defense against pathogens. Oncogene (2001) 20:6473–81. doi:10.1038/sj.onc.1204787
2. Harton JA, Linhoff MW, Zhang J, Ting JP. Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J Immunol (2002) 169:4088–93.
3. Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem (2002) 277:29874–80. doi:10.1074/jbc.M203915200
4. Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity (2007) 27:549–59. doi:10.1016/j.immuni.2007.10.002
5. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol (2003) 4:702–7. doi:10.1038/ni945
6. Anand PK, Kanneganti TD. Targeting NLRP6 to enhance immunity against bacterial infections. Future Microbiol (2012) 7:1239–42. doi:10.2217/fmb.12.94
7. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Critical role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem (2006) 281:36560–8. doi:10.1074/jbc.M607594200
8. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature (2008) 451:573–7. doi:10.1038/nature06501
9. Tomalka J, Ganesan S, Azodi E, Patel K, Majmudar P, Hall BA, et al. A novel role for the NLRC4 inflammasome in mucosal defenses against the fungal pathogen Candida albicans. PLoS Pathog (2011) 7:e1002379. doi:10.1371/journal.ppat.1002379
10. van de Veerdonk FL, Joosten LA, Shaw PJ, Smeekens SP, Malireddi RK, Van Der Meer JW, et al. The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Eur J Immunol (2011) 41:2260–8. doi:10.1002/eji.201041226
11. Shaw MH, Reimer T, Sanchez-Valdepenas C, Warner N, Kim YG, Fresno M, et al. T cell-intrinsic role of Nod2 in promoting type 1 immunity to Toxoplasma gondii. Nat Immunol (2009) 10:1267–74. doi:10.1038/ni.1816
12. Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog (2009) 5:e1000559. doi:10.1371/journal.ppat.1000559
13. Ritter M, Gross O, Kays S, Ruland J, Nimmerjahn F, Saijo S, et al. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc Natl Acad Sci U S A (2010) 107:20459–64. doi:10.1073/pnas.1010337107
14. Kufer TA, Sansonetti PJ. NLR functions beyond pathogen recognition. Nat Immunol (2011) 12:121–8. doi:10.1038/ni.1985
15. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science (2003) 300:1584–7. doi:10.1126/science.1084677
16. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278:8869–72. doi:10.1074/jbc.C200651200
17. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem (2003) 278:5509–12. doi:10.1074/jbc.C200673200
18. Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science (2005) 307:731–4. doi:10.1126/science.1104911
19. Grimes CL, Ariyananda Lde Z, Melnyk JE, O’Shea EK. The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J Am Chem Soc (2012) 134:13535–7. doi:10.1021/ja303883c
20. Bertin J, Nir WJ, Fischer CM, Tayber OV, Errada PR, Grant JR, et al. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-kappaB. J Biol Chem (1999) 274:12955–8. doi:10.1074/jbc.274.19.12955
21. Girardin SE, Tournebize R, Mavris M, Page AL, Li X, Stark GR, et al. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep (2001) 2:736–42. doi:10.1093/embo-reports/kve155
22. Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol (2007) 178:2380–6.
23. Dugan JW, Albor A, David L, Fowlkes J, Blackledge MT, Martin TM, et al. Nucleotide oligomerization domain-2 interacts with 2′-5′-oligoadenylate synthetase type 2 and enhances RNase-L function in THP-1 cells. Mol Immunol (2009) 47:560–6. doi:10.1016/j.molimm.2009.09.025
24. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol (2009) 10:1073–80. doi:10.1038/ni.1782
25. Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol (2013) 14:480–8. doi:10.1038/ni.2563
26. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol (2006) 7:576–82. doi:10.1038/ni1346
27. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol (2006) 7:569–75. doi:10.1038/ni1344
28. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A (2010) 107:3076–80. doi:10.1073/pnas.0913087107
29. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun (2005) 73:1907–16. doi:10.1128/IAI.73.4.1907-1916.2005
30. Black RA, Kronheim SR, Sleath PR. Activation of interleukin-1 beta by a co-induced protease. FEBS Lett (1989) 247:386–90. doi:10.1016/0014-5793(89)81376-6
31. Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science (1992) 256:97–100. doi:10.1126/science.1373520
32. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature (1992) 356:768–74. doi:10.1038/356768a0
33. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature (1997) 386:619–23. doi:10.1038/386619a0
34. Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science (1997) 275:206–9. doi:10.1126/science.275.5297.206
35. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477:592–5. doi:10.1038/nature10394
36. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi:10.1038/nature10510
37. Halff EF, Diebolder CA, Versteeg M, Schouten A, Brondijk TH, Huizinga EG. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J Biol Chem (2012) 287:38460–72. doi:10.1074/jbc.M112.393512
38. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A (2013) 110:14408–13. doi:10.1073/pnas.1306376110
39. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi:10.1038/nature04515
40. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440:237–41. doi:10.1038/nature04516
41. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (2008) 320:674–7. doi:10.1126/science.1156995
42. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-Tekippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity (2009) 30:556–65. doi:10.1016/j.immuni.2009.02.005
43. Thomas PG, Dash P, Aldridge JR Jr., Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity (2009) 30:566–75. doi:10.1016/j.immuni.2009.02.006
44. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32:379–91. doi:10.1016/j.immuni.2010.03.003
45. Manji GA, Wang L, Geddes BJ, Brown M, Merriam S, Al-Garawi A, et al. PYPAF1, a PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NF-kappa B. J Biol Chem (2002) 277:11570–5. doi:10.1074/jbc.M112208200
46. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity (2004) 20:319–25. doi:10.1016/S1074-7613(04)00046-9
47. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem (1994) 269:15195–203.
48. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ (2007) 14:1583–9. doi:10.1038/sj.cdd.4402195
49. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9:857–65. doi:10.1038/ni.1636
50. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9:847–56. doi:10.1038/ni.1631
51. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity (2013) 38:1142–53. doi:10.1016/j.immuni.2013.05.016
52. Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature (2006) 440:233–6. doi:10.1038/nature04517
53. Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol (2004) 14:1929–34. doi:10.1016/j.cub.2004.10.027
54. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183:787–91. doi:10.4049/jimmunol.0901363
55. Ito M, Yanagi Y, Ichinohe T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog (2012) 8:e1002857. doi:10.1371/journal.ppat.1002857
56. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature (2012) 492:123–7. doi:10.1038/nature11588
57. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A (2012) 109:11282–7. doi:10.1073/pnas.1117765109
58. Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun (2012) 3:1329. doi:10.1038/ncomms2339
59. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol (2010) 11:404–10. doi:10.1038/ni.1861
60. Rajamaki K, Nordstrom T, Nurmi K, Akerman KE, Kovanen PT, Oorni K, et al. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J Biol Chem (2013) 288:13410–9. doi:10.1074/jbc.M112.426254
61. Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, et al. The DHX33 RNA Helicase Senses Cytosolic RNA and Activates the NLRP3 Inflammasome. Immunity (2013) 39:123–35. doi:10.1016/j.immuni.2013.07.001
62. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature (2012) 488:670–4. doi:10.1038/nature11290
63. He Y, Franchi L, Nunez G. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur J Immunol (2013) 43:1147–52. doi:10.1002/eji.201243187
64. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153:348–61. doi:10.1016/j.cell.2013.02.054
65. Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature (2012) 490:539–42. doi:10.1038/nature11429
66. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol (2013) 14:52–60. doi:10.1038/ni.2474
67. Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, et al. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol (2012) 189:5113–7. doi:10.4049/jimmunol.1202479
68. Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res (2013) 23:201–12. doi:10.1038/cr.2013.6
69. Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem (2012) 287:36617–22. doi:10.1074/jbc.M112.407130
70. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell (2013) 49:331–8. doi:10.1016/j.molcel.2012.11.009
71. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13:255–63. doi:10.1038/ni.2215
72. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi:10.1038/ni.1980
73. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi:10.1038/nature09663
74. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol (2013) 14:454–60. doi:10.1038/ni.2550
75. Wang Y, Yang C, Mao K, Chen S, Meng G, Sun B. Cellular localization of NLRP3 inflammasome. Protein Cell (2013) 4:425–31. doi:10.1007/s13238-013-2113-2
76. Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem (2005) 280:39914–24. doi:10.1074/jbc.M502820200
77. Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell (2011) 20:649–60. doi:10.1016/j.ccr.2011.10.022
78. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity (2012) 36:742–54. doi:10.1016/j.immuni.2012.03.012
79. Ye Z, Lich JD, Moore CB, Duncan JA, Williams KL, Ting JP. ATP binding by monarch-1/NLRP12 is critical for its inhibitory function. Mol Cell Biol (2008) 28:1841–50. doi:10.1128/MCB.01468-07
80. Chattoraj P, Yang Q, Khandai A, Al-Hendy O, Ismail N. TLR2 and Nod2 mediate resistance or susceptibility to fatal intracellular Ehrlichia infection in murine models of ehrlichiosis. PLoS ONE (2013) 8:e58514. doi:10.1371/journal.pone.0058514
81. Arthur JC, Lich JD, Aziz RK, Kotb M, Ting JP. Heat shock protein 90 associates with monarch-1 and regulates its ability to promote degradation of NF-kappaB-inducing kinase. J Immunol (2007) 179:6291–6.
82. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol (2012) 13:823–31. doi:10.1038/ni.2378
83. Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature (2012) 488:389–93. doi:10.1038/nature11250
84. Chen G, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol (2011) 186:7187–94. doi:10.4049/jimmunol.1100412
85. Elinav E, Strowig T, Kau A, Henao-Mejia J, Thaiss C, Booth C, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi:10.1016/j.cell.2011.04.022
86. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity (2012) 37:96–107. doi:10.1016/j.immuni.2012.09.001
87. Jeru I, Hentgen V, Normand S, Duquesnoy P, Cochet E, Delwail A, et al. Role of interleukin-1beta in NLRP12-associated autoinflammatory disorders and resistance to anti-interleukin-1 therapy. Arthritis Rheum (2011) 63:2142–8. doi:10.1002/art.30378
88. Jeru I, Le Borgne G, Cochet E, Hayrapetyan H, Duquesnoy P, Grateau G, et al. Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum (2011) 63:1459–64. doi:10.1002/art.30241
89. Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity (2011) 34:854–65. doi:10.1016/j.immuni.2011.03.026
90. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity (2011) 34:843–53. doi:10.1016/j.immuni.2011.02.022
91. Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep (2008) 9:293–300. doi:10.1038/sj.embor.7401161
92. Rebsamen M, Vazquez J, Tardivel A, Guarda G, Curran J, Tschopp J. NLRX1/NOD5 deficiency does not affect MAVS signalling. Cell Death Differ (2011) 18:1387. doi:10.1038/cdd.2011.64
93. Soares F, Tattoli I, Wortzman ME, Arnoult D, Philpott DJ, Girardin SE. NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate Immun (2013) 19:438–48. doi:10.1177/1753425912467383
94. Hong M, Yoon SI, Wilson IA. Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity (2012) 36:337–47. doi:10.1016/j.immuni.2011.12.018
95. Kim HS, Shin TH, Yang SR, Seo MS, Kim DJ, Kang SK, et al. Implication of NOD1 and NOD2 for the differentiation of multipotent mesenchymal stem cells derived from human umbilical cord blood. PLoS ONE (2010) 5:e15369. doi:10.1371/journal.pone.0015369
96. Bruey JM, Bruey-Sedano N, Newman R, Chandler S, Stehlik C, Reed JC. PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages. J Biol Chem (2004) 279:51897–907. doi:10.1074/jbc.M406741200
97. Fontalba A, Gutierrez O, Fernandez-Luna JL. NLRP2, an inhibitor of the NF-kappaB pathway, is transcriptionally activated by NF-kappaB and exhibits a nonfunctional allelic variant. J Immunol (2007) 179:8519–24.
98. Minkiewicz J, De Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia (2013) 61:1113–21. doi:10.1002/glia.22499
99. Meyer E, Lim D, Pasha S, Tee LJ, Rahman F, Yates JR, et al. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome). PLoS Genet (2009) 5:e1000423. doi:10.1371/journal.pgen.1000423
100. Huang JY, Su M, Lin SH, Kuo PL. A genetic association study of NLRP2 and NLRP7 genes in idiopathic recurrent miscarriage. Hum Reprod (2013) 28:1127–34. doi:10.1093/humrep/det001
101. Peng H, Chang B, Lu C, Su J, Wu Y, Lv P, et al. Nlrp2, a maternal effect gene required for early embryonic development in the mouse. PLoS ONE (2012) 7:e30344. doi:10.1371/journal.pone.0030344
102. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, De Almeida L, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity (2012) 36:464–76. doi:10.1016/j.immuni.2012.02.001
103. Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R, Kuick R, et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet (2006) 38:300–2. doi:10.1038/ng1740
104. Messaed C, Chebaro W, Di Roberto RB, Rittore C, Cheung A, Arseneau J, et al. NLRP7 in the spectrum of reproductive wastage: rare non-synonymous variants confer genetic susceptibility to recurrent reproductive wastage. J Med Genet (2011) 48:540–8. doi:10.1136/jmg.2011.089144
105. Ulker V, Gurkan H, Tozkir H, Karaman V, Ozgur H, Numanoglu C, et al. Novel NLRP7 mutations in familial recurrent hydatidiform mole: are NLRP7 mutations a risk for recurrent reproductive wastage? Eur J Obstet Gynecol Reprod Biol (2013) 170:188–92. doi:10.1016/j.ejogrb.2013.06.028
106. Gerard N, Caillaud M, Martoriati A, Goudet G, Lalmanach AC. The interleukin-1 system and female reproduction. J Endocrinol (2004) 180:203–12. doi:10.1677/joe.0.1800203
107. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi:10.1146/annurev.immunol.021908.132715
108. Caillaud M, Duchamp G, Gerard N. In vivo effect of interleukin-1beta and interleukin-1RA on oocyte cytoplasmic maturation, ovulation, and early embryonic development in the mare. Reprod Biol Endocrinol (2005) 3:26. doi:10.1186/1477-7827-3-26
109. Takehara Y, Dharmarajan AM, Kaufman G, Wallach EE. Effect of interleukin-1 beta on ovulation in the in vitro perfused rabbit ovary. Endocrinology (1994) 134:1788–93. doi:10.1210/en.134.4.1788
110. Abbondanzo SJ, Cullinan EB, McIntyre K, Labow MA, Stewart CL. Reproduction in mice lacking a functional type 1 IL-1 receptor. Endocrinology (1996) 137:3598–601. doi:10.1210/en.137.8.3598
111. Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature (2008) 453:1122–6. doi:10.1038/nature06939
112. Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A (2009) 106:870–5. doi:10.1073/pnas.0804897106
113. Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate, et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis (2013). doi:10.1136/annrheumdis-2013-203276
114. Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, et al. Glyburide inhibits the cryopyrin/Nalp3 inflammasome. J Cell Biol (2009) 187:61–70. doi:10.1083/jcb.200903124
115. Stienstra R, Van Diepen JA, Tack CJ, Zaki MH, Van De Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A (2011) 108:15324–9. doi:10.1073/pnas.1100255108
116. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411:599–603. doi:10.1038/35079107
117. Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol (2010) 185:974–81. doi:10.4049/jimmunol.0904145
118. Shaw PJ, Barr MJ, Lukens JR, McGargill MA, Chi H, Mak TW, et al. Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity (2011) 34:75–84. doi:10.1016/j.immuni.2010.12.015
119. Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, et al. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal (2012) 5:ra38. doi:10.1126/scisignal.2002767
120. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature (2013) 493:674–8. doi:10.1038/nature11729
121. Eisenbarth SC, Williams A, Colegio OR, Meng H, Strowig T, Rongvaux A, et al. NLRP10 is a NOD-like receptor essential to initiate adaptive immunity by dendritic cells. Nature (2012) 484:510–3. doi:10.1038/nature11012
122. Joly S, Eisenbarth SC, Olivier AK, Williams A, Kaplan DH, Cassel SL, et al. Cutting edge: Nlrp10 is essential for protective antifungal adaptive immunity against Candida albicans. J Immunol (2012) 189:4713–7. doi:10.4049/jimmunol.1201715
123. Tong ZB, Gold L, Pfeifer KE, Dorward H, Lee E, Bondy CA, et al. Mater, a maternal effect gene required for early embryonic development in mice. Nat Genet (2000) 26:267–8. doi:10.1038/81547
124. Wu X. Maternal depletion of NLRP5 blocks early embryogenesis in rhesus macaque monkeys (Macaca mulatta). Hum Reprod (2009) 24:415–24. doi:10.1093/humrep/den403
Keywords: inflammasomes, NOD-like receptors, DAMPs, PAMPs, innate immunity, caspase-1, embryonic development
Citation: Lupfer C and Kanneganti T-D (2013) Unsolved mysteries in NLR biology. Front. Immunol. 4:285. doi: 10.3389/fimmu.2013.00285
Received: 01 July 2013; Accepted: 02 September 2013;
Published online: 17 September 2013.
Edited by:
Thomas A. Kufer, University of Cologne, GermanyReviewed by:
Martin Sheldon, Swansea University, UKMohammed N. Al-Ahdal, King Faisal Specialist Hospital and Research Centre, Saudi Arabia
Copyright: © 2013 Lupfer and Kanneganti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thirumala-Devi Kanneganti, Department of Immunology, MS #351, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, TN 38105-3678, USA e-mail:dGhpcnVtYWxhLWRldmkua2FubmVnYW50aUBzdGp1ZGUub3Jn