 JoAnn Trial1,2*
JoAnn Trial1,2* Katarzyna A. Cieslik1,2 Sandra B. Haudek1,2
Katarzyna A. Cieslik1,2 Sandra B. Haudek1,2 Clemens Duerrschmid1,2 Mark L. Entman1,2
Clemens Duerrschmid1,2 Mark L. Entman1,2
- 1Division of Cardiovascular Sciences, Department of Medicine, DeBakey Heart Center, Baylor College of Medicine, Houston, TX, USA
- 2Houston Methodist, Houston, TX, USA
We have demonstrated that cardiac fibrosis arises from the differentiation of monocyte-derived fibroblasts. We present here evidence that this process requires sequential Th1 and Th2 induction promoting analogous M1 (classically activated) and M2 (alternatively activated) macrophage polarity. Our models are: (1) mice subjected to daily repetitive ischemia and reperfusion (I/R) without infarction and (2) the in vitro transmigration of human mononuclear leukocytes through human cardiac microvascular endothelium. In the mouse heart, leukocytes entered after I/R in response to monocyte chemoattractant protein-1 (MCP-1), which is the major cytokine induced by this protocol. Monocytes within the heart then differentiated into fibroblasts making collagen while bearing the markers of M2 macrophages. T cells were seen in these hearts as well as in the human heart with cardiomyopathy. In the in vitro model, transmigration of the leukocytes was likewise induced by MCP-1 and some monocytes matured into fibroblasts bearing M2 markers. In this model, the MCP-1 stimulus induced a transient Th1 and M1 response that developed into a predominantly Th2 and M2 response. An increase in the Th2 product IL-13 was present in both the human and the mouse models, consistent with its known role in fibrosis. In these simplified models, in which there is no cell death to stimulate an anti-inflammatory response, there is nonetheless a resolution of inflammation enabling a profibrotic environment. This induces the maturation of monocyte precursors into fibroblasts.
Introduction
The immune system uses similar tools whether responding to infections or sterile tissue injury. The same steps of an inflammatory response followed by resolution and healing take place under both circumstances (1). For example, the liberation of heat shock proteins (2), mitochondrial DNA (3), HMGB1 (high-mobility group box 1) (4, 5), and a host of other internal components from dead cells [alarmins (6)] can trigger inflammation in the absence of infection. Others, such as adenosine (7), apoptotic cell membranes (8, 9), or self DNA (10), can at the same time initiate the anti-inflammatory response that will bring the organism back to homeostatic balance. Both types of influences may act via chemokines that attract different subsets of immunocytes or may skew the subsequent maturation of the cells (11). In contrast to that schema, this report describes and defines similar responses, both proinflammatory and anti-inflammatory, in the absence of cell death and its associated signaling.
Our murine model of intermittent cardiac ischemia and reperfusion (I/R) in vivo, despite the absence of cell death, still results in an immune response. This treatment, which leads to cardiomyopathy, is the daily repetitive brief (15 min) I/R of a coronary artery, in which the major chemokine induced is monocyte chemoattractant protein-1 (MCP-1) and histology reveals interstitial fibrosis but no necrosis (12). We have shown that the fibrosis results from an influx of CD34+CD45+ monocytes that become fibroblasts making collagen (13). As in many models of fibrosis, these cells bear the markers of M2 macrophages while also assuming the shape and markers of fibroblasts (14). Because this model does not result in the death of cardiomyocytes, the mediators inducing the monocyte influx and transition into fibroblasts are limited in comparison to those accompanying a myocardial infarct, including both proinflammatory and anti-inflammatory or profibrotic influences (12). Neither the influx nor the fibrosis occurs in animals when MCP-1 is genetically deleted, indicating the necessity of this chemokine for initiation of the immune response leading to fibrosis (15, 16).
To simulate these events using human cells in vitro, we designed a transendothelial migration (TEM) assay using human cardiac microvascular endothelial cells (HCMEC) growing on a model basement membrane over an insert with 8 μm pores. Human mononuclear cells are attracted to migrate through the endothelium and the pores in response to MCP-1 below the insert (17). Some of the transmigrating monocytes become M1 macrophages, whereas others become M2 macrophages that mature into fibroblasts, and the numbers of the latter vary with different stimuli. We have used this TEM model to investigate the response of monocytes to Fc receptor engagement (17), angiotensin-II (18), and ROCK-1 knockdown (19), with parallel results to the corresponding in vivo mouse models. As with the in vivo models, MCP-1 is necessary for the migration of the monocytes through the endothelium (17). Classically, MCP-1 is regarded as a stimulus for M1 development, but MCP-1 may be accompanied by other chemokines to which a subset of monocytes capable of maturation into M2 macrophages may migrate (20). A major inducer of the M2 phenotype is the Th2 cell product, IL-13 (11), and addition or production of IL-13 is required for the formation of monocyte-derived fibroblasts in our in vitro assay (21). This suggests that it may be pertinent to human disease states.
Since IL-13 has been associated with fibrosis in the aging heart and in other organs (21–23), we elected to investigate its presence and kinetics in our cardiomyopathy model as well as in our in vitro model. Although IL-13 can be made by many kinds of cells, the source of it in an inflammatory infiltrate is likely to be predominantly from T cells, and so we verified the presence of T lymphocytes. Our data suggest that the presence of a Th2 response is integral to the fibrosis in these models of immune activation, in which there is an initial induction of the Th1 and M1 responses that then evolve into a Th2/M2 response in the absence of cell death. The Th2 response is obligate for the formation of myeloid fibroblasts and the development of a fibrotic cardiomyopathy.
Results
Murine In vivo Model
To quantify the number of M2 macrophages and T lymphocytes in heart tissue from shams or animals treated with the I/R protocol, we digested the heart to isolate cells and performed flow cytometry to measure the expression of the markers CD45 (cells of hematopoietic origin), CD301 (M2 macrophages), and CD3 (T cells). These markers were combined with fluorescence from calcein, which identifies viable cells. Mechanical disruption and digestion of the heart tissue as optimized in our protocol breaks cardiomyocytes apart, so the live cells are all non-myocytes, representing endothelial cells, smooth muscle cells, fibroblasts, and any hematopoietic cells that may have entered the heart. In Figure 1, it can be seen that the number of CD45+CD301+ M2 macrophages increased after I/R, as did the number that were CD45+CD301+collagen type I+ (and 38 ± 7% of the CD45+CD301+ were positive for collagen type I), consistent with our previous histological results that these cells are in the heart and are making collagen (13, 14). In addition, it can be seen that there were also T lymphocytes, identified by their CD45 and CD3 markers. Of note, in human heart tissue from cases of cardiomyopathy, we also found CD45+ cells in a perivascular location expressing either a myofibroblast marker (α-smooth muscle actin) or the T cell marker CD3, as well as CD301+ α-smooth muscle actin+ cells (Figure 2).
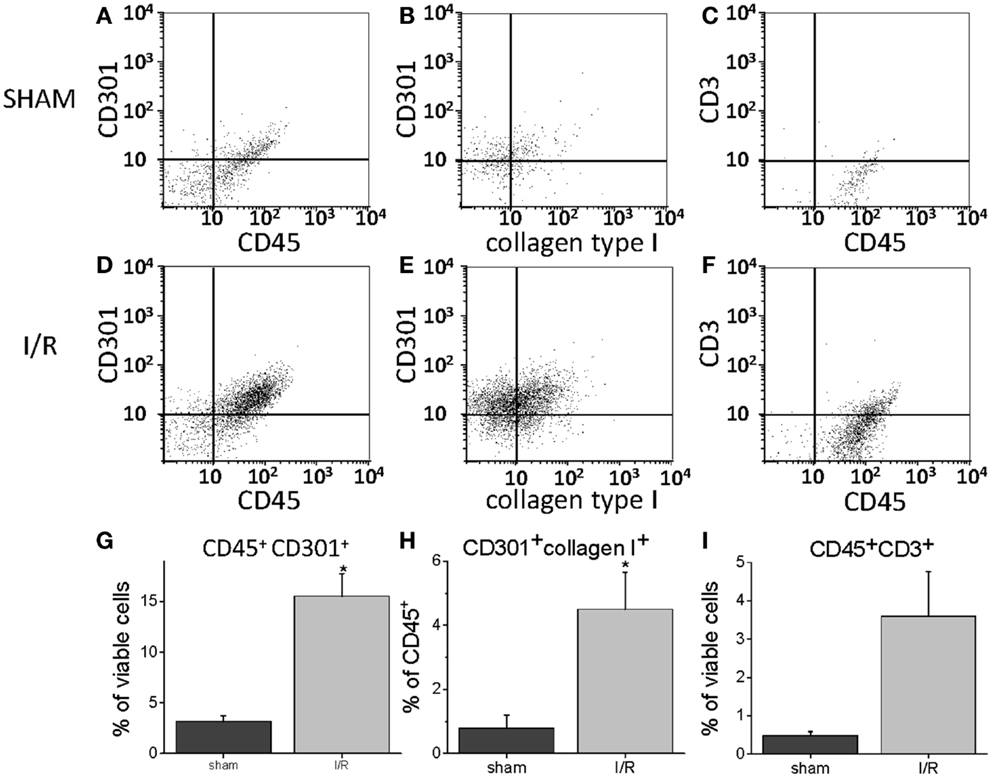
Figure 1. Flow cytometry for the indicated markers on cells harvested from mouse hearts after 5 days of I/R or from sham-treated animals. (A) Through (C) are representative histograms for sham-treated animals. (D) Through (F) are representative histograms for I/R animals. (G) Through (I) are summary data graphs. (A,D,G) Represent the percent of all viable cells (calcein positive) that were positive for both CD45 and CD301; (B,E,H) are the percent of all cells positive for CD45 that were also positive for both CD301 and collagen type I; (C,F,I) are the percent of all viable cells that were positive for both CD45 and CD3 (n = 3, *p < 0.05).
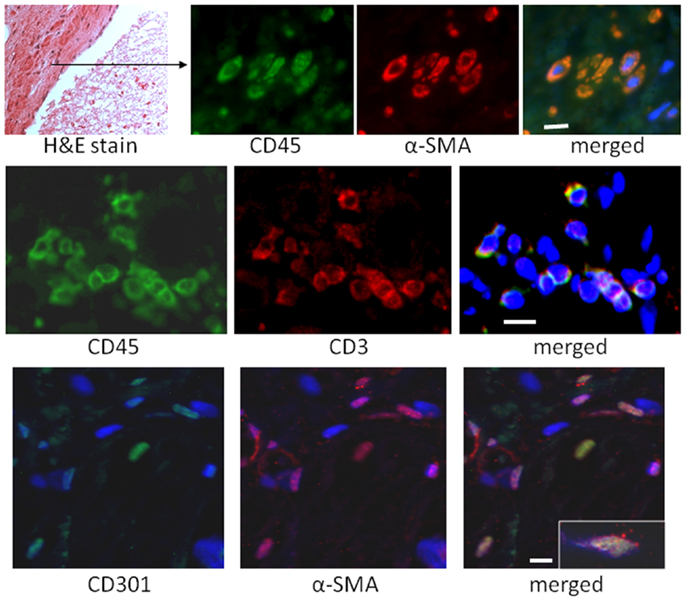
Figure 2. Sections of human hearts with cardiomyopathy were stained with hematoxylin and eosin (H&E) or by immunofluorescence for the indicated markers. The white bars are 10 μm.
Murine Cytokines
In our mouse model, we were interested in the temporal gene expression of MCP-1 and IL-13. Our results showed high levels of MCP-1 mRNA early during the I/R response that waned thereafter (Figure 3). The Th2 product, IL-13, showed a later peak, but was present throughout the 2-week period of I/R. Neither of these responses took place in mice deficient in MCP-1 expression (genetic knockout, KO). IL-33, another mediator of interest because of its ability to induce IL-13, was increased with a similar pattern to that of IL-13. Unlike IL-13, IL-33 was induced in both the wild-type (WT) and the KO mice, and its expression was maintained at 14 days in the KO but not the WT mice.
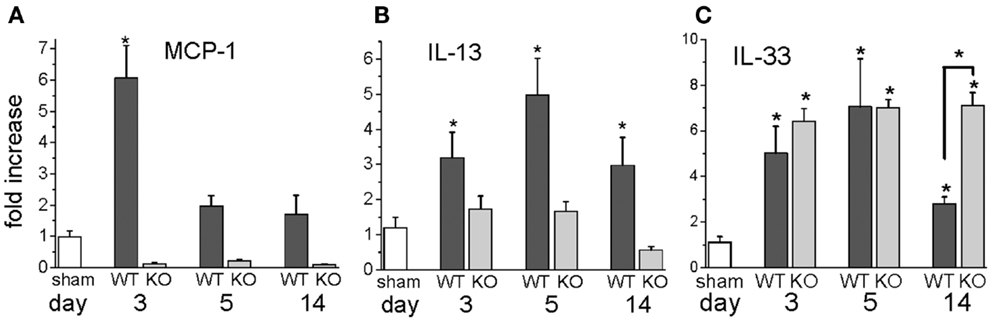
Figure 3. Gene expression levels for MCP-1, IL-13, and IL-33 in wild-type (WT) or MCP-1 knockout (KO) after the indicated number of days of I/R, expressed as the fold increase over sham levels. (A) the fold increase of MCP-1 in WT and KO animals over sham levels; (B) the fold increase of IL-13 in WT and KO animals over sham levels; (C) the fold increase of IL-33 in WT and KO over sham levels (n = 3 for WT and for KO shams; there was no difference between these groups, so the numbers were combined for the sham levels shown in the graph; *p < 0.05). The graph for IL-13 is reprinted with permission from (27).
We also measured specific cytokines that would characterize the T cells in the mouse heart as Th1 or Th2 by performing protein arrays on sham and I/R hearts after 3 days of treatment (Table 1). The Th2-related IL-13 protein was the most elevated protein in I/R over sham levels. The Th2 cytokine IL-4, by contrast, was not as increased, and neither was the major Th1 cytokine, IFN-γ.
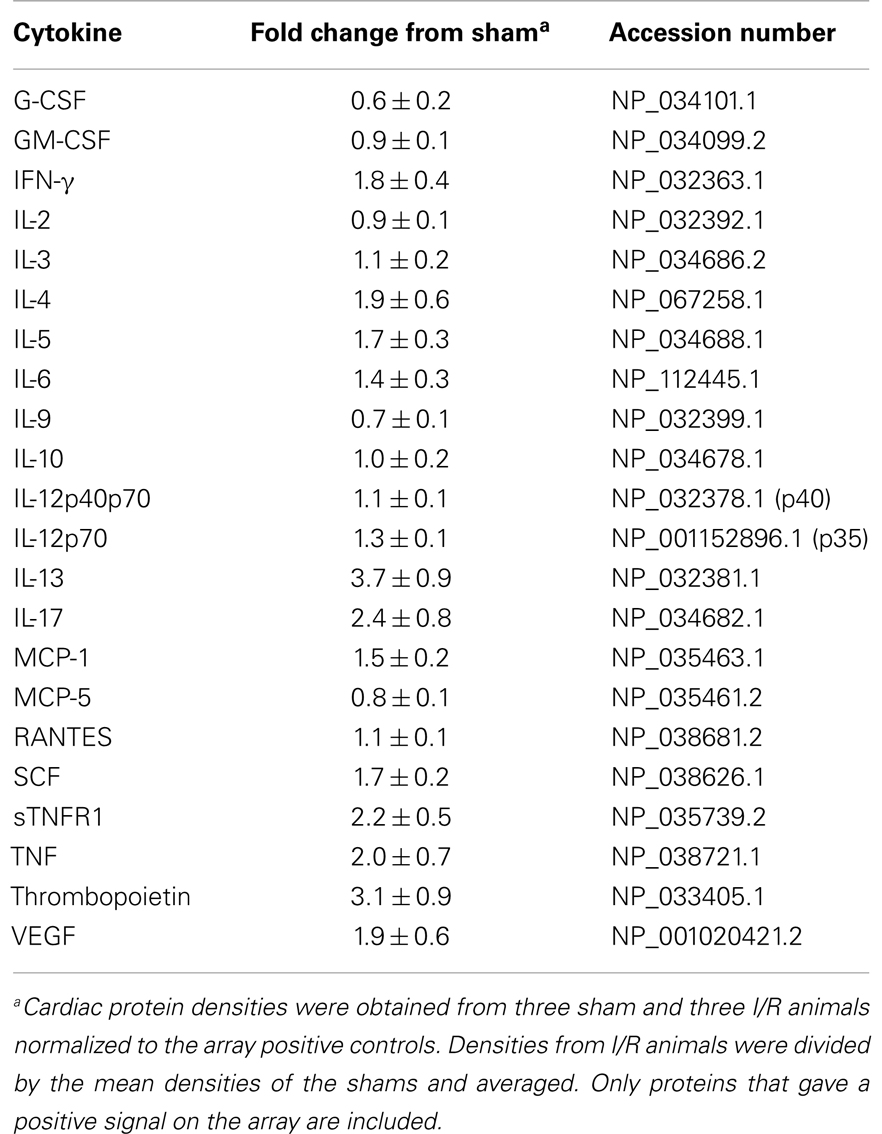
Table 1. Protein array data from hearts of animals subjected to 3 days of I/R.
Human In vitro Model
In our human in vitro TEM assay, we stained macrophages for M1 versus M2 markers after two different times of migration, early (migrating from 0 to 16 h) and late (cells migrating during the interval of 16–96 h), plus a group with an additional 6 days of culture post-migration. Macrophages developing from monocytes that had migrated early had a round morphology with markers of M1 macrophages, CD86 and NOS2 (nitric oxide synthase 2 or inducible NOS) (Figure 4). Cells bearing CD86 were 81 ± 2% of the total cells counted (out of 1038 counted from three donors). No cells bearing CD150, CD206, or CD301 were found at the early time interval. Many macrophages from monocytes that migrated later were elongated and had an M2 marker, CD206, as well as making prolylhydroxylase (the last enzyme in the pathway of collagen synthesis). Cells positive for CD206 were 64 ± 5%, for CD86 were 8 ± 2%, and double positive for CD206 and CD86 were 5 ± 1% of the total cells (1019), indicating the possibility of M1 to M2 conversion. No CD150 or CD301 positive cells were found in this time frame. With further culture (6 days), some of the elongated cells became positive for CD301, another marker for M2 macrophages (24). Cells positive for CD301 were 23 ± 5% of the total (471). No CD150+ cells were seen. Cells positive for CD206 were 61 ± 4, for CD86 were 3.4 ± 0.7, and double positive were 1.9 ± 0.5 out of a total of 924.
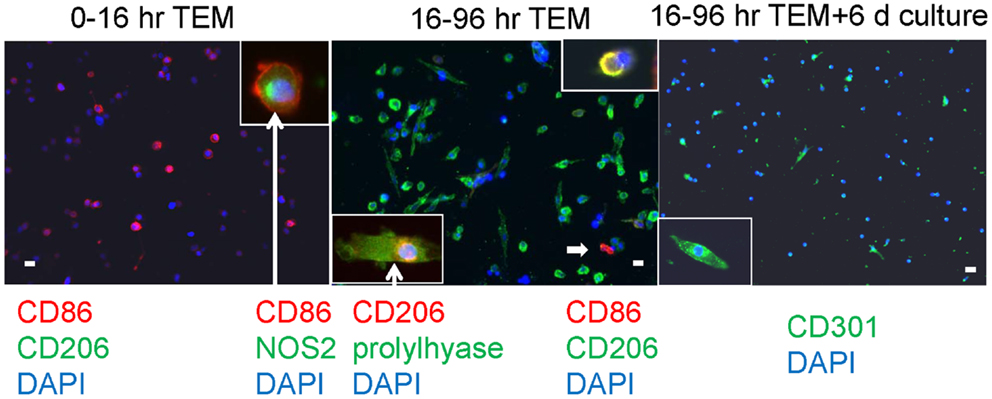
Figure 4. Human PBMC were allowed to migrate for the indicated times through human cardiac microvascular endothelium in response to MCP-1 and thereafter adhered to poly d-lysine coated coverslips. The cells were stained for the external markers CD86, CD206, and CD301, and after fixation and permeabilization, for the internal markers NOS2 and prolylhydroxylase (labeled prolylhyase in the figure). DAPI was used to stain cell nuclei blue. The insets are cells at either higher magnification (CD301) or cells with external staining and an internal marker (CD86 plus NOS2 and CD206 plus prolylhydroxylase) with white arrows extending from the label. The white arrow within the center panel indicates a CD86 positive cell in the low magnification field, and the inset at top right shows a cell that is double positive for CD86 and CD206. White bars represent 20 μm.
Human Cell Identification
To characterize further the kinetics of macrophage M2 development, we performed quantitative PCR on in vitro human transmigrated cells from two different time frames (0–16 and 16–96 h). The cells were expanded with the mitogens phorbol myristate acetate (PMA) and calcium ionomycin. To relate gene expression to a negative control (either unmigrated cultured total leukocytes or freshly isolated uncultured mononuclear cells) would not have been productive because many genes are not expressed in these cells. Therefore, we calculated the gene expression as a percentage of the theoretical maximum expression from the same donor’s total unmigrated mononuclear leukocytes cultured with mitogens. The mitogen-induced signals from the total cells should not be biased toward one lineage because they contain all of the starting cell populations. It can be seen in Figure 5 that two transcription factors involved in M2 development, KLF4 and SOCS1 (25, 26), were present early but also increased with later transmigration. Flow cytometry indicated that CD3+ T cells had also transmigrated, and their distribution of CD4 and CD8 subsets after 4 days of migration was similar to that of the original T cells (fresh CD3+ MNLs were 73 ± 4% CD4+ and 24 ± 7% CD8+ while transmigrated CD3+ cells were 69 ± 3% CD4+ and 22 ± 6% CD8+, n = 3). An analysis of their gene expression showed increased levels of IL-13 mRNA with time [Figure 6 and as we have found in the in vivo model (27)] in conjunction with dramatically decreased levels of IFN-γ mRNA (Figure 6). These data indicated that the lymphokine kinetics were compatible with an early Th1 response (IFN-γ) followed by a Th2 response (IL-13) and downregulation of Th1 gene expression.
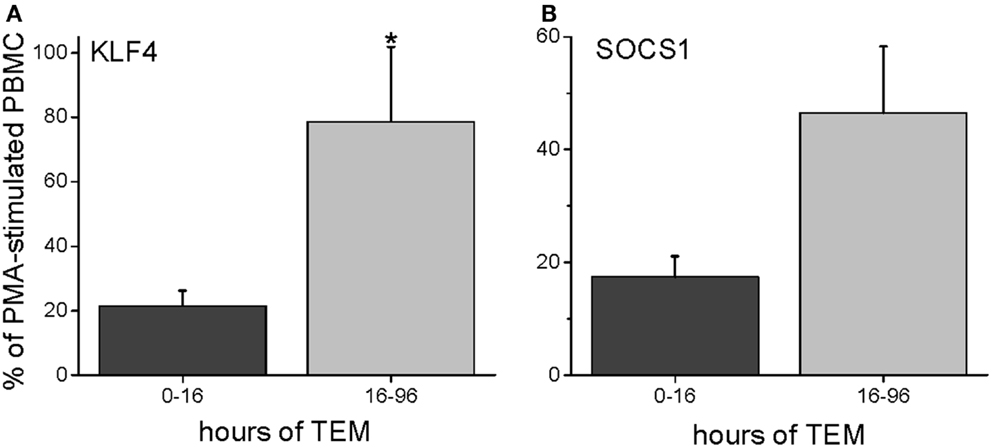
Figure 5. Human PBMC transmigrated for 0–16 or 16–96 h were expanded with mitogens (PMA and calcium ionomycin) and harvested for RNA. The gene expression levels were expressed as a percentage of mRNA levels in unmigrated mitogen-expanded total PBMCs (PMA = 100%), giving the percent levels of KLF4 (A) or SOCS1 (B) (n = 3 donors in duplicate).
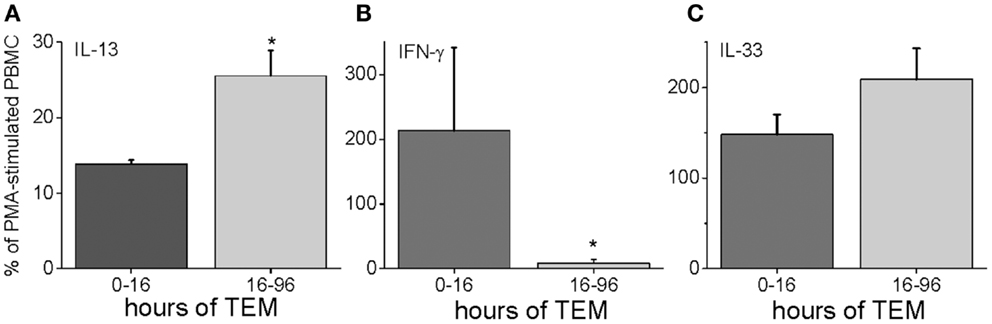
Figure 6. Human PBMC transmigrated for 0–16 or 16–96 h were expanded with mitogens and harvested for RNA. Their gene expression levels were expressed as a percentage of mRNA levels in unmigrated mitogen-expanded total PBMCs for IL-13 (A), IFN-γ (B), and IL-33 (C) (n = 3 donors in duplicate, *p < 0.05 for the comparison of the indicated group from the total PBMC level).
IL-13 can also be upregulated by IL-33, which is a member of the IL-1 family (28, 29). We measured the gene expression of IL-33 in our human model, and found that it was highly expressed both early and late during TEM (Figure 6).
Discussion
Under homeostatic regulation, inflammation is followed by its resolution (1). In infections, removal of the responsible agent leads to the quieting of the immune response. In injury, cell death by various modes leads to active anti-inflammatory signaling. In our in vivo and in vitro models, in which cell death is avoided, it is clear that the same transition from proinflammatory to anti-inflammatory responses can take place, as well as a subsequent profibrotic environment. In both simplified in vivo and in vitro models, the initiating factor is MCP-1, considered a proinflammatory signal, but the end stage is fibrosis. In the in vivo model, the shift from inflammation to fibrosis may be mediated at least partially by IL-13, which is well known for this effect (30, 31). Indeed, the targets of IL-13, M2 macrophages, were present in the heart at that time (3 days). Some of these cells were making collagen, indicating that they had already responded to profibrotic mediators. The source of the IL-13 may be the CD3+ T cells that transmigrated in response to MCP-1, because IL-13 was lacking in the MCP-1 knockout. We also demonstrate that T cells were found in the human heart with cardiomyopathy; this indicates that our in vivo model may be relevant to human disease. Another stimulus for the upregulation of IL-13 in WT animals may be increased IL-33, expected to be made by infiltrating immune cells. However, IL-33 gene expression occurred even in the absence of MCP-1. IL-33 can be secreted from cells under mechanical strain (32), and is constitutively expressed in endothelial cells (33), so these may be sources of IL-33 that are independent of an inflammatory infiltrate. In the MCP-1 KO animals, IL-33 gene expression did not decline at the 14-day time point, indicating the possibility that MCP-1 could be responsible for the eventual downregulation of IL-33 mRNA.
As in the murine in vivo model, the presence of MCP-1 in the human in vitro model induced first a Th1 (IFN-γ) and M1 (CD86, NOS2) response that within a day shifted to a Th2 (IL-13) and M2 (CD206, KLF4, and SOCS1) plus fibrotic (prolylhydroxylase) response without the addition of any further mediators. Therefore, the proinflammatory response uncomplicated by known external stimuli for its resolution was down regulated and evolved into a fibrotic response. A resolution to this conundrum may be found in further investigation of the action of MCP-1.
The view of MCP-1 as being responsible only for inflammation has been challenged by findings in other models; effects were noted either directly on myeloid cells or indirectly via T cell production of cytokines. Human monocytes in vitro and in tumor environments can be induced by MCP-1 to undergo M2 polarization (34). T cells treated with MCP-1 elaborate IL-4 and IL-13 (Th2 cytokines) (35, 36) and MCP-1-deficient mice cannot mount Th2 responses (37). Some of the influence of MCP-1 on the promotion of Th2 responses may be mediated through MCP-1-induced protein-1 (MCPIP1), a down regulator of inflammatory responses that degrades cytokine mRNAs and deubiquitinates TNF receptor-associated factor family proteins (38, 39), thus allowing an unfettered Th2 response. MCP-1 may induce both inflammation and anti-inflammation, perhaps at different times or in different environments, and the cessation of its proinflammatory effects may operate through the induction of anti-inflammatory gene products.
In previous studies of myocardial infarction injury (with extensive cell death), the presence of two monocyte subsets, one inflammatory and the other reparative, was noted in mouse models (40) and in human patients (41). In myocardial infarction, the proinflammatory macrophages are thought to be required for terminal digestion of tissue and removal of necrotic debris, which would not be necessary in our models. Later after infarction, reparative macrophages (M2) dominate. Thus, there is the generation of a temporal pattern such that the influx of inflammatory macrophages (phenotypically and functionally identical to M1 macrophages) early after injury is followed by reparative macrophages (identified as M2) (11, 42). In our models, despite the absence of cardiac injury sufficient to cause cell death, there was a reprise of this temporal pattern, including a transient M1 response followed by a robust M2 response, despite there being nothing to “repair.” In fact, our previous studies demonstrated a transition to a state of unrestrained fibrosis resulting from dysregulation of immune function in M2 macrophages (14, 21). One cause may be that IL-13 can drive further production of MCP-1, which may set up a profibrotic feed forward loop (43). Thus the fibrotic response may represent dysregulation of a potentially protective immunological evolution (21).
Members of the adaptive immune system recognize foreign or altered self molecules via their multichain immunoreceptors, and innate immune cells recognize conserved pathogen-associated molecules through their pattern recognition receptors. In the absence of infection or cognate antigen presentation, however, there can be cytokine-induced cytokine production by both conventional and innate lymphoid cells (44). For example, the IL-1 family member IL-33 (an alarmin) causes antigen-independent IL-13 production (28, 29). IL-33 gene expression was present in both our models, although we do not know if this was an ancillary or primary stimulus for IL-13 production. The lack of IL-13 and presence of IL-33 in the MCP-1 knockout animals may be interpreted as the production of IL-33 in resident cells, which thereafter cannot induce the production of IL-13 from an absent inflammatory infiltrate. In either case (antigen recognition or cytokine-induced), we have demonstrated that MCP-1 alone can initiate an immune response that moves rapidly through a Th1/M1 inflammatory stage and into a Th2/M2 anti-inflammatory and profibrotic environment. Thereafter, the establishment of a chronically fibrotic state may depend on dysregulation of the immune response, as in diseases such as systemic sclerosis (45). Although MCP-1 and its receptor CCR2 historically have been targeted to reduce inflammation [reviewed in (46)], the possibility that it may initiate chronic fibrosis without chronic inflammation has not been considered. Our simplified models may allow an examination of this issue, as we propose that MCP-1 may initiate both an M1 and an M2 response leading to fibrosis, or promote M1 to M2 conversion.
The data from a murine model with daily episodes of brief ischemia not inducing infarction and an in vitro model of mononuclear cell TEM driven by MCP-1 both demonstrate the presence of a sequential inflammatory reaction occurring without evidence of tissue injury or infection and leading to interstitial fibrosis. The reaction is initiated by MCP-1-induced TEM of mononuclear cells (monocytes and T lymphocytes) and does not occur in the absence of MCP-1. Initially, the mononuclear cells are predominantly in an inflammatory phenotype (M1, Th1) and secrete cytokines and lymphokines appropriate to that phenotype (Figure 7).
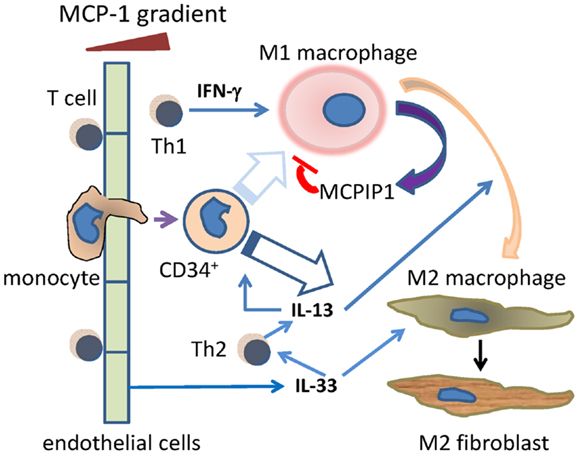
Figure 7. Schema of the Th1/M1 and Th2/M2 responses after cells move through blood vessel endothelium. Monocytes and T cells migrate through endothelium, initially in response to MCP-1, and including a primitive (CD34+) monocyte. The Th1 product IFN-γ drives the migrated monocytes toward the M1 phenotype, although this may be less likely for the CD34+ subset (faded arrow). MCP-1 itself induces migrated activated macrophages to secrete MCPIP1 (MCP-1-induced protein-1), which limits the activated M1 phenotype. During that initial Th1/M1 phase, Th2 activities such as the secretion of IL-13 begin, and become dominant at later time points. This may be aided by endothelial cell secretion of IL-33. IL-13 may promote some M1–M2 transition as well as direct monocyte to M2 maturation. The resulting M2 macrophages can become collagen-secreting myeloid fibroblasts.
As the reaction progresses, mononuclear cells of an “anti-inflammatory” phenotype begin to appear and subsequently dominate the cell population. This is followed by the appearance of a population of fibroblasts expressing collagen while still bearing the markers of their myeloid origin (CD45) and alternative polarization (CD206, CD301). The data suggest that the signaling cascade also contains intrinsic “brake mechanisms” that limit the intensity and duration of the acute inflammatory response. From the data presented, we suggest potential signaling molecules that may orchestrate this “braking” response as well as those initiating the transition from M1 to M2 phenotypes (Figure 7). We also suggest that our previous findings of CD34+CD45+ cells infiltrating the mouse heart during injuries may provide evidence for a relatively primitive monocyte precursor for either or both the M1 and M2 macrophages, although the finding of collagen in these cells suggests that they may be destined preferentially for the M2 phenotype (13, 16).
As described above, IL-33 is a powerful inducer of IL-13 and the Th2 phenotype without requiring an antigen response. Our data suggest that it also may be an important factor in the initiation of a Th2 response in an area of inflammation without tissue necrosis. In addition, MCPIP1 is secreted from activated M1 macrophages and has been observed to block M1 signaling (47), so this modulator may also aid in the transition from a Th1/M1 to a Th2/M2 response. Our previous and current data suggest that the chronic presence of M2 macrophages is associated with some of them undergoing maturation into secretory myofibroblasts. The mechanism and signaling associated with this maturation has not been well characterized although the phenomenon has been described in vivo and in vitro (13, 17, 48, 49). When the stimulus continues, as it does in our in vivo model, even the basic response may change its nature to that of a chronic pathology (fibrosis) very different from the initial inflammation. Having thus identified the sequential immune responses and the initial signals involved, we will investigate further pathways and additional responses involved in chronic fibrosis in future studies.
Materials and Methods
Animals and Ischemia Reperfusion Protocol
Ten to twelve week old B6.129S4-Ccl2tm1Rol/J (MCP-1-KO) and C57BL/6J WT mice (both from Jackson Laboratory) were subjected to closed chest surgery as previously described (12). Briefly, 1 week after suture implantation, 15-min occlusions of the left anterior descending artery were performed for the number of indicated days, allowing a 24-h reperfusion period in between. Mice were euthanized 5 h after the last ischemic episode. All mice were fed standard mouse chow and water ad libitum. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. All animals were treated in accordance with the guidelines of the Baylor College of Medicine Animal Care and Research Advisory Committee.
Human Heart Tissue
Tissue pieces from human hearts with cardiomyopathy were obtained under an Institutional Review Board-approved protocol (50). The formalin-fixed tissue was paraffin embedded and sectioned.
Mouse Cell Identification
After 5 days of I/R, mice were anesthetized with 2% isoflurane followed by cervical dislocation. Hearts were isolated and non-myocytes were isolated by enzymatic digestion using 0.1 mg/ml Liberase TH Research Grade (Roche Applied Science), as described previously (13, 18). For flow cytometric analysis, 1–3 × 105 freshly isolated cells were incubated with 0.5 μg biotin-conjugated anti-CD45 antibody (BD Biosciences) followed by PE/Cy-5-conjugated streptavidin (BD Biosciences), together with either 0.5 μg anti-CD301 (Abcam Inc) + PE-conjugated anti-rat secondary antibody (Jackson ImmunoResearch), or PE-conjugated anti-CD3ϵ (Beckman Coulter). Fifty nanomolars calceinAM (Invitrogen Molecular Probes) was added to measure cell viability (only live cells metabolize calcein to an intracellular, green-fluorescent permanent cell dye). A subset of cells were incubated with antibodies against CD45 and CD301 as described above, then fixed and permeabilized (kit from BD Pharmingen), and incubated over night with 1 μg anti-collagen type I antibody (Rockland), followed by FITC-conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch). Fluorescence intensities were measured on a Beckman Coulter Epics XL.MCL using EXPO32 software.
Human Cell Identification
Human heart sections were stained with H&E according to standard procedures (12). Before immunofluorescence staining, tissue sections were autoclaved in citrate buffer pH 6 for antigen retrieval and then permeabilized in 1% Triton X-100; after staining, sections were exposed to 0.3% Sudan Black in 70% alcohol to quench autofluorescence. Tissue sections were stained as described previously (17, 18) using anti-CD45 (BD Biosciences), -α-SMA (Sigma), -CD3ϵ (Beckman Coulter), and -CD301 (Dendritics).
Cells that transmigrated through an endothelial barrier were allowed to attach to poly d-lysine-coated coverslips (Neuvitro) at the bottom of the lower well. The cells were fixed and stained with anti-CD86 (BioLegend), -CD301 (Dendritics), or appropriate isotype controls. For the internal antigens, the cells were also permeabilized (BD Pharmingen) and stained with anti-prolylhydroxylase (Millipore), -CD206 (Epitomics), or -NOS2 (Abcam Inc). These were followed by affinity-purified F(ab′)2 IgG specific fluorochrome-conjugated secondary antibodies (Jackson ImmunoResearch) and the coverslips were mounted with the cell nucleus stain DAPI. Microscopy was performed on an Olympus AX70 using a QImaging Retiga 2000R camera. Colors were assigned and merged using ImageJ software (version 1.46r, NIH).
For cytometry, mononuclear leukocytes were stained fresh or after a 4-day transmigration with anti-CD3 plus either anti-CD4 or -CD8 (all from R&D Systems). Cells were analyzed on a Cell Lab Quanta SC flow cytometer (Beckman Coulter) using the Quanta Analysis software.
Protein Array
After 3 days of I/R, hearts were harvested and protein was isolated using Cell Lysis Buffer (RayBiotech) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Protein (250 μg) was loaded onto standard mouse cytokine antibody array membranes (cytokine array C1, RayBiotech). Membranes were processed according to the manufacturer’s instructions, images on film were scanned, and densitometry was assessed by ImageJ software.
Transendothelial Migration
As detailed earlier (17, 18), normal blood was obtained from volunteers under a protocol approved by the Institutional Review Board of Baylor College of Medicine. Anticoagulated blood was fractionated by Ficoll-Hypaque gradient centrifugation (Histopaque-1077; Sigma) to collect mononuclear cells (PBMC). For a TEM assay, HCMEC (Lonza, passages 4–9) were seeded on insert membranes with 8 μm pores. Human PBMC [25 × 104 in RPMI 1640 with no serum but with ITS-X (Sigma) and antibiotic-antimycotic (Invitrogen)] were then added to each insert and the same medium including 650 ng/ml MCP-1 (R&D Systems) for chemoattraction was added to the well below. PBMC were allowed to migrate for 16 h, and then the inserts were moved into new medium-containing wells (without additional MCP-1) for another 80 h (96 h total migration time, but separate from the 0–16 h time frame for migration). At the end of each migration period, the migrated cells were expanded in medium plus 5% FBS (HyClone) plus PMA plus calcium ionomycin (both from Sigma) for 3 days and then harvested for RNA.
RNA Isolation and Transcriptional Expression
Cells were lysed and total RNA was isolated with TRIzol reagent (Life Technologies) and treated with RNAse-free DNaseI (Qiagen). Complimentary DNA was synthesized from 1 μg of RNA with a Verso cDNA synthesis kit (Thermo Scientific) using random hexamer and oligo-dT primers (3:1). Real-time PCR amplification reactions were performed with SsoAdvancedTM SYBR Green Supermix (Bio-Rad) in triplicate using a CFX96 thermal cycler (Bio-Rad). Cycling conditions (annealing temperature/time) were optimized for each primer pair. Gene expression was measured by the ΔΔCT method and was normalized to HPRT or 18S ribosomal RNA levels. Primers were designed using Primer 3 1, checked for homologies using BLAST 2 and for secondary structures using the mfold server 3. All primer pairs were verified to adhere to the MIQE guidelines using a temperature gradient and serial dilutions (51). Primer sequences (all in 5′–3′):
Human:
KLF4: ACTCGCCTTGCTGATTGTCT and CCCCAAAGTCAACGAAGAGA
SOCS1: CGAGCATCCGCGTGCACTTT and GCAGCTCGAAGAGGCAGTCGAA
IL-13: CAATGGCAGCATGGTATGG and AGAATCCGCTCAGCATCC
IFN-γ: TCCCATGGGTTGTGTGTTTA and GGGTCACCTGACACATTCAA
IL-33: ACCCAAATGCTACCACTGGA and TCCACACATGCTAAGCACTCTT
HPRT-1: GACCAGTCAACAGGGGACAT and CTTGCGACCTTGACCATCTT
Mouse:
IL-13: TCCAATTGCAATGCCATCTA and TGAGTCCACAGCTGAGATGC
MCP-1: TCCACAACCACCTCAAGCACTTC and GGCATCACAGTCCGAGTCACAC
IL-33: GGCTGCTTGCTTTCCTTATG and CCGTTACGGATATGGTGGTC
18sRNA: ACCGCAGCTAGGAATAATGGA and GCCTCAGTTCCGAAAACCA
HPRT-1: GCCCCAAAATGGTTAAGGTT and TTGCGCTCATCTTAGGCTTT
Statistical Analysis
All data are expressed as mean ± SE. A two-tailed, unpaired Student’s t-test or Mann–Whitney test was used to determine a significant difference between two groups. The Kruskal–Wallis ANOVA test with Dunn’s post-test was used for three or more groups. A p-value <0.05 was considered statistically significant.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health RO1 HL-089792, The American Heart Association, the Hankamer Foundation, and the Medallion Foundation. The authors wish to thank Dorellyn B. Lee and Hao Zhang for excellent technical assistance and Sharon Malinowski for editorial assistance with the manuscript. This research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
1. Stoecklein VM, Osuka A, Lederer JA. Trauma equals danger – damage control by the immune system. J Leukoc Biol (2012) 92(3):539–51. doi:10.1189/jlb.0212072
2. Mathur S, Walley KR, Wang Y, Indrambarya T, Boyd JH. Extracellular heat shock protein 70 induces cardiomyocyte inflammation and contractile dysfunction via TLR2. Circ J (2011) 75(10):2445–52. doi:10.1253/circj.CJ-11-0194
3. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
4. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol (2011) 29:139–62. doi:10.1146/annurev-immunol-030409-101323
5. Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol (2007) 81(1):59–66. doi:10.1189/jlb.0306180
6. Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol (2005) 17(4):359–65. doi:10.1016/j.coi.2005.06.002
7. Zhang N, Yang D, Dong H, Chen Q, Dimitrova DI, Rogers TJ, et al. Adenosine A2a receptors induce heterologous desensitization of chemokine receptors. Blood (2006) 108(1):38–44. doi:10.1182/blood-2005-06-2599
8. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101(4):890–8. doi:10.1172/JCI1112
9. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390(6658):350–1. doi:10.1038/37022
10. Seledtsov VI, Seledtsova GV. A balance between tissue-destructive and tissue-protective immunities: a role of toll-like receptors in regulation of adaptive immunity. Immunobiology (2012) 217(4):430–5. doi:10.1016/j.imbio.2011.10.011
11. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi:10.1016/j.it.2004.09.015
12. Dewald O, Frangogiannis NG, Zoerlein M, Duerr GD, Klemm C, Knuefermann P, et al. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc Natl Acad Sci U S A (2003) 100:2700–5. doi:10.1073/pnas.0438035100
13. Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci U S A (2006) 103(48):18284–9. doi:10.1073/pnas.0608799103
14. Crawford JR, Haudek SB, Cieslik KA, Trial J, Entman ML. Origin of developmental precursors dictates the pathophysiologic role of cardiac fibroblasts. J Cardiovasc Transl Res (2012) 5(6):749–59. doi:10.1007/s12265-012-9402-7
15. Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation (2007) 115(5):584–92. doi:10.1161/CIRCULATIONAHA.106.646091
16. Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, et al. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol (2010) 49(3):499–507. doi:10.1016/j.yjmcc.2010.05.005
17. Haudek SB, Trial J, Xia Y, Gupta D, Pilling D, Entman ML. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci U S A (2008) 105(29):10179–84. doi:10.1073/pnas.0804910105
18. Duerrschmid C, Crawford JR, Reineke E, Taffet GE, Trial J, Entman ML, et al. TNF receptor 1 signaling is critically involved in mediating angiotensin-II-induced cardiac fibrosis. J Mol Cell Cardiol (2013) 57:59–67. doi:10.1016/j.yjmcc.2013.01.006
19. Haudek SB, Gupta D, Dewald O, Schwartz RJ, Wei L, Trial J, et al. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc Res (2009) 83(3):511–8. doi:10.1093/cvr/cvp135
20. Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, et al. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med (2003) 197(12):1701–7. doi:10.1084/jem.20022156
21. Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol (2011) 50(1):248–56. doi:10.1016/j.yjmcc.2010.10.019
22. Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol (2004) 173(6):4020–9.
23. Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med (2001) 194(6):809–21. doi:10.1084/jem.194.6.809
24. Westcott DJ, Delproposto JB, Geletka LM, Wang T, Singer K, Saltiel AR, et al. MGL1 promotes adipose tissue inflammation and insulin resistance by regulating 7/4hi monocytes in obesity. J Exp Med (2009) 206(13):3143–56. doi:10.1084/jem.20091333
25. Feinberg MW, Cao Z, Wara AK, Lebedeva MA, Senbanerjee S, Jain MK. Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J Biol Chem (2005) 280(46):38247–58. doi:10.1074/jbc.M509378200
26. Whyte CS, Bishop ET, Ruckerl D, Gaspar-Pereira S, Barker RN, Allen JE, et al. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol (2011) 90(5):845–54. doi:10.1189/jlb.1110644
27. Entman ML, Cieslik KA, Carlson S, Haudek SB, Trial J. Myeloid fibroblast precursors in cardiac interstitial fibrosis – the origin of fibroblast precursors dictate the pathophysiologic role. In: Bucala R, editor. Fibrocytes in Health and Disease. Hackensack, NJ: World Scientific Press (2011). p. 197–228.
28. Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A (2009) 106(32):13463–8. doi:10.1073/pnas.0906988106
29. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol (2008) 20(6):791–800. doi:10.1093/intimm/dxn037
30. Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med (2006) 12(1):99–106. doi:10.1038/nm1332
31. Fuschiotti P, Medsger TA Jr, Morel PA. Effector CD8+ T cells in systemic sclerosis patients produce abnormally high levels of interleukin-13 associated with increased skin fibrosis. Arthritis Rheum (2009) 60(4):1119–28. doi:10.1002/art.24432
32. Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem (2012) 287(9):6941–8. doi:10.1074/jbc.M111.298703
33. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One (2008) 3(10):e3331. doi:10.1371/journal.pone.0003331
34. Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem (2009) 284(49):34342–54. doi:10.1074/jbc.M109.042671
35. Karpus WJ, Lukacs NW, Kennedy KJ, Smith WS, Hurst SD, Barrett TA. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J Immunol (1997) 158(9):4129–36.
36. Lehmann I, Roder-Stolinski C, Nieber K, Fischader G. In vitro models for the assessment of inflammatory and immuno-modulatory effects of the volatile organic compound chlorobenzene. Exp Toxicol Pathol (2008) 60(2-3):185–93. doi:10.1016/j.etp.2008.01.009
37. Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature (2000) 404(6776):407–11. doi:10.1038/35006097
38. Jura J, Skalniak L, Koj A. Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochim Biophys Acta (2012) 1823(10):1905–13. doi:10.1016/j.bbamcr.2012.06.029
39. Xu J, Fu S, Peng W, Rao Z. MCP-1-induced protein-1, an immune regulator. Protein Cell (2012) 3(12):903–10. doi:10.1007/s13238-012-2075-9
40. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204(12):3037–47. doi:10.1084/jem.20070885
41. Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol (2009) 54(2):130–8. doi:10.1016/j.jacc.2009.04.021
42. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11(11):723–37. doi:10.1038/nri3073
43. Murray LA, Argentieri RL, Farrell FX, Bracht M, Sheng H, Whitaker B, et al. Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2. Int J Biochem Cell Biol (2008) 40(10):2174–82. doi:10.1016/j.biocel.2008.02.016
44. Guo L, Junttila IS, Paul WE. Cytokine-induced cytokine production by conventional and innate lymphoid cells. Trends Immunol (2012) 33(12):598–606. doi:10.1016/j.it.2012.07.006
45. Chizzolini C, Brembilla NC, Montanari E, Truchetet ME. Fibrosis and immune dysregulation in systemic sclerosis. Autoimmun Rev (2011) 10(5):276–81. doi:10.1016/j.autrev.2010.09.016
46. Kanzler I, Liehn EA, Koenen RR, Weber C. Anti-inflammatory therapeutic approaches to reduce acute atherosclerotic complications. Curr Pharm Biotechnol (2012) 13(1):37–45. doi:10.2174/138920112798868557
47. Huang S, Miao R, Zhou Z, Wang T, Liu J, Liu G, et al. MCPIP1 negatively regulates toll-like receptor 4 signaling and protects mice from LPS-induced septic shock. Cell Signal (2013) 25(5):1228–34. doi:10.1016/j.cellsig.2013.02.009
48. Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med (1994) 1(1):71–81.
49. Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol (2003) 171(10):5537–46.
50. Bruckner BA, Stetson SJ, Farmer JA, Radovancevic B, Frazier OH, Noon GP, et al. The implications for cardiac recovery of left ventricular assist device support on myocardial collagen content. Am J Surg (2000) 180(6):498–501. doi:10.1016/S0002-9610(00)00553-5
Keywords: IL-13, monocytes, fibrosis, IL-33, MCP-1, M1, M2
Citation: Trial J, Cieslik KA, Haudek SB, Duerrschmid C and Entman ML (2013) Th1/M1 conversion to Th2/M2 responses in models of inflammation lacking cell death stimulates maturation of monocyte precursors to fibroblasts. Front. Immunol. 4:287. doi: 10.3389/fimmu.2013.00287
Received: 23 April 2013; Paper pending published: 24 July 2013;
Accepted: 03 September 2013; Published online: 18 September 2013.
Edited by:
Zoulfia Allakhverdi, University of Montreal, CanadaReviewed by:
Pablo Pelegrin, Hospital Universitario Virgen Arrixaca, SpainZoulfia Allakhverdi, University of Montreal, Canada
Copyright: © 2013 Trial, Cieslik, Haudek, Duerrschmid and Entman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: JoAnn Trial, Division of Cardiovascular Sciences, Department of Medicine, Baylor College of Medicine, BCM620, One Baylor Plaza, Houston, TX 77030, USA e-mail:anRyaWFsQGJjbS5lZHU=