



- Department of Microbiology and Immunology, Indiana University School of Medicine, Indianapolis, IN, USA
The nicotinamide adenine dinucleotide phosphate (NADPH) oxidase expressed in phagocytes is a multi-subunit enzyme complex that generates superoxide (O2.−). This radical is an important precursor of hydrogen peroxide (H2O2) and other reactive oxygen species needed for microbicidal activity during innate immune responses. Inherited defects in NADPH oxidase give rise to chronic granulomatous disease (CGD), a primary immunodeficiency characterized by recurrent infections and granulomatous inflammation. Interestingly, CGD, CGD carrier status, and oxidase gene polymorphisms have all been associated with autoinflammatory and autoimmune disorders, suggesting a potential role for NADPH oxidase in regulating adaptive immune responses. Here, NADPH oxidase function in antigen processing and presentation is reviewed. NADPH oxidase influences dendritic cell (DC) crosspresentation by major histocompatibility complex class I molecules through regulation of the phagosomal microenvironment, while in B lymphocytes, NADPH oxidase alters epitope selection by major histocompatibility complex class II molecules.
NADPH Oxidase
The phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex catalyzes electron transfer from NADPH to molecular oxygen and thus generates superoxide (O2.−), the precursor of H2O2 and other reactive oxygen species (ROS) necessary for the microbicidal activity of neutrophils and other phagocytes during innate immune responses. The active phagocyte oxidase (phox) complex consists of five subunits and the small GTPase Rac. The catalytic core of the complex, which displays little to no activity in resting phagocytic cells, is comprised of two transmembrane subunits, gp91phox (often referred to as NOX2) and p22phox. These proteins form a heterodimer known as flavocytochrome b558 in vesicular and plasma membranes of neutrophils and other leukocytes. In response to stimuli, the regulatory subunits p47phox, p67phox, and p40phox, which together exist as a complex in the cytosol, translocate along with Rac-GTP to the membrane-bound catalytic core to form the active enzyme complex (Figure 1) (1–3).
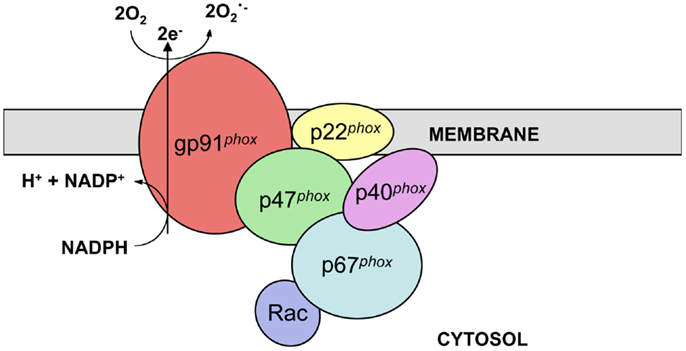
Figure 1. NADPH oxidase structure. NADPH oxidase is a multi-subunit enzyme complex present in the vesicular and plasma membranes of leukocytes. Two transmembrane subunits, gp91phox and p22phox, make up flavocytochrome b558, the catalytic core of the complex. This heterodimer catalyzes the transfer of electrons from cytosolic NADPH to molecular oxygen, thus generating superoxide. Flavocytochrome b558 is regulated by association with the subunits p47phox, p67phox, and p40phox along with the small GTPase Rac. These regulatory subunits are present in the cytosol of resting cells and translocate to the catalytic core following stimulation.
Chronic Granulomatous Disease
Inherited mutations in the subunits of NADPH oxidase result in chronic granulomatous disease (CGD), a primary immunodeficiency disorder affecting at least 1 in 250,000 individuals in the US (4). X-linked CGD, the most common genetic subgroup, is caused by mutations in gp91phox. X-linked CGD accounts for approximately 70% of CGD cases and affects mostly males, although female carriers with skewed X-chromosome inactivation can also display a CGD phenotype (4–7). Mutations in p22phox, p47phox, and p67phox can also give rise to CGD through autosomal recessive inheritance. Patients with autosomal recessive forms of CGD are often diagnosed later in life, exhibit a milder clinical phenotype, and live longer than patients with X-linked CGD (4, 7). Isolated cases of children with a CGD phenotype having mutations in Rac2 (8) and p40phox (9) have also been described.
Phagocytes lacking functional NADPH oxidase are unable to produce ROS necessary to kill certain pathogenic bacteria and fungi. Thus, patients with CGD experience recurrent infections, most often with catalase positive organisms. Patients are also prone to develop severe granulomas resulting in obstructive lesions in the esophagus, stomach, and urinary tract (4, 7, 10, 11). In a cohort of 368 CGD patients from the US, pneumonia was the most common form of infection (79% of patients with at least one episode), and 41% of these infections were caused by Aspergillus spp. Abscesses were also common (68% of patients with at least one episode). Additionally, suppurative adenitis, osteomyelitis, septicemia, cellulitis, and meningitis were reported (4). Similar data were described in a European cohort of 429 CGD patients (7). In both cohorts, the most common causes of death were infections involving Aspergillus spp. and Burkholderia cepacia (4, 7).
CGD and Autoimmunity
Remarkably, CGD is also associated with a number of autoinflammatory and autoimmune disorders. Several patients with systemic lupus erythematosus (SLE) and discoid lupus erythematosus (DLE) have been described (4, 7, 12, 13). In addition, lupus has also been widely reported in female carriers of X-CGD (4, 7, 12–15). A number of other studies have pointed to an association of CGD with autoimmune arthritis (7, 15, 16). CGD patients with colitis-like symptoms (4, 9) similar to inflammatory bowel disease, myasthenia gravis (4), immune thrombocytopenia (4, 7), sarcoidosis (17), pericardial effusion, IgA nephropathy, and aphthous stomatitis (15) have also been described.
In addition to this well-established connection between CGD and autoimmunity, genes encoding oxidase subunits have been associated with autoinflammatory and autoimmune disorders in genome-wide association studies (GWAS). A variation in NCF2, the gene encoding p67phox, has been identified as an important risk factor for SLE (18). NCF4, encoding p40phox, has been associated with rheumatoid arthritis (19) and Crohn’s disease (20, 21). In the absence of NADPH oxidase activity, murine dendritic cells (DC) produce elevated levels of inflammatory cytokines that alter helper T cell differentiation and promote development of collagen-induced arthritis (22, 23). These findings suggest NADPH oxidase may be playing an important role not only in innate immune responses, but also in adaptive immune responses.
NADPH Oxidase and Crosspresentation
Several studies have looked into the relationship between NADPH oxidase activity and antigen crosspresentation. DC, and to a lesser extent, macrophages, are capable of crosspresentation, a process by which peptides derived from exogenous antigens are loaded onto and presented by major histocompatibility complex class I (MHC-I) molecules. During crosspresentation, exogenous antigens, either particulate or soluble, are phagocytosed or endocytosed. These antigens are proteolytically processed to short peptides, 8–10 amino acids long, and subsequently loaded onto MHC-I. Crosspresentation has been implicated in infection responses where uninfected DC play a critical role in CD8+ T cell activation (24–26). Crosspresentation of self and environmental antigens may also play a role in the maintenance of central and peripheral tolerance, implicating crosspresentation in the development of autoimmune disorders and as a potential target for immunotherapy (27–30).
The pathways by which exogenous antigens encounter MHC-I are incompletely understood. Some studies have shown crosspresentation is dependent on proteasome activity, suggesting exogenous antigens are transported into the cytosol for processing (31, 32). In DC, studies have shown phagosomes fuse with the endoplasmic reticulum (ER), and antigenic peptides processed in the cytosol are transported by TAP back into the phagosome lumen before being loaded onto MHC-I (33). Other studies indicate exogenous antigens are processed within phagosomes and endosomes prior to intersecting MHC-I, independent of the proteasome and TAP (Figure 2) (34, 35). In DC, Amigorena and colleagues identified a central role for NADPH oxidase in modulating the efficiency of antigen degradation and MHC-I crosspresentation to CD8+ T cells.
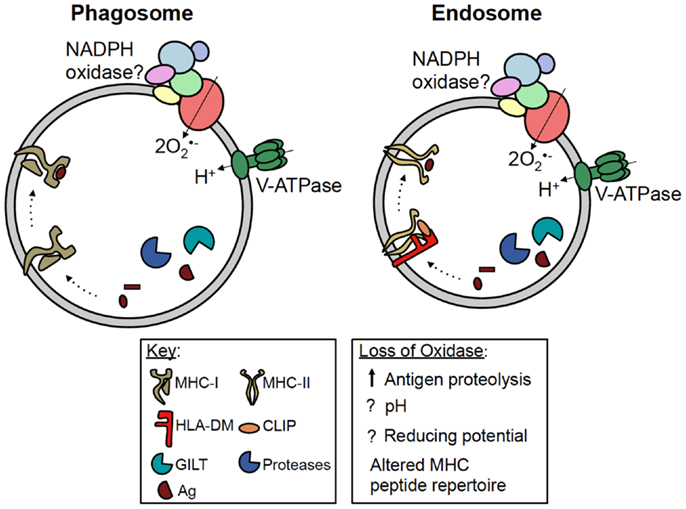
Figure 2. Potential roles of NADPH oxidase in antigen presentation. MHC-I and MHC-II are both present in phagosomes and endosomes of antigen presenting cells. For simplicity, MHC-I has been shown in a phagosome, while MHC-II has been shown in an endosome. During crosspresentation, exogenous antigens can be processed within these compartments by proteases, prior to loading onto MHC-I. NADPH oxidase has been shown to regulate antigen processing and MHC-I crosspresentation in DC; however, whether NADPH oxidase regulates this process by modulating the phagosomal pH or redox microenvironment is currently debated. During classical MHC-II presentation, exogenous antigens are processed within vesicular compartments by proteases and GILT. Following the removal of CLIP by HLA-DM, processed peptides are loaded onto MHC-II. NADPH oxidase can also regulate MHC-II presentation in B cells by altering the peptide repertoire displayed by MHC-II, possibly in favor of self antigens. However, the mechanism underlying this phenomenon is still unclear. Whether or not NADPH oxidase, MHC-I and -II, and the antigen processing machinery co-localize within the same vesicular compartment also remains to be elucidated.
To investigate in vivo oxidase function in crosspresentation, wild type and gp91phox-deficient mice were immunized with ovalbumin coupled to an antibody specific for the receptor DEC205, targeting this antigen-antibody complex to murine DC. CD8+ T cells that recognize epitopes from ovalbumin in the context of MHC-I were adoptively transferred into recipient wild type or oxidase-deficient mice, followed by an analysis of T cell activation (36). Ovalbumin-specific T cell activation was significantly less efficient in gp91phox-deficient mice compared to wild type mice. Results in vitro also showed gp91phox-deficient DC did not efficiently crosspresent ovalbumin to antigen-specific CD8+ T cells compared to wild type DC. However, presentation of the pre-processed ovalbumin SIINFEKL peptide was similar in both DC populations. Together, these data suggest gp91phox is important for antigen processing and crosspresentation in murine DC (36).
Similarly, in vitro human CD8+ T cell recognition of an HLA-A2-restricted extended MelanA/MART-1 peptide was reduced in DC from CGD patients and in DC treated with an oxidase inhibitor compared to wild type DC, whereas presentation of a shorter form of this peptide was not affected (37). Chemical inhibition of the oxidase did not alter DC phagocytosis. Given that crosspresentation of the long, but not short, MelanA/MART-1 peptide requires processing by DC, these results indicate an important role for NADPH oxidase in antigen processing and crosspresentation by human DC (37).
Amigorena and colleagues found this effect of NADPH oxidase on crosspresentation occurs in murine CD8+ but not CD8− DC. Rac2, the small GTPase which facilitates oxidase subunit assembly on phagosomal membranes, was also required for efficient crosspresentation (38).
Precisely how loss of oxidase function disrupts MHC-I crosspresentation remains controversial. Amigorena and colleagues have proposed in DC that NADPH oxidase promotes an alkalinization of the phagosome lumen thus limiting antigen proteolysis and preserving epitopes for crosspresentation by MHC-I (36–39). FACS-based studies using pH sensitive fluorophores coupled to latex beads revealed pH values of 7.5 or higher after phagocytosis in phagosomes of DC with functional oxidase, while rapid acidification of phagosomes was observed in DC from gp91phox-deficient mice (36). Loss of ovalbumin on coated beads after phagocytosis suggested enhanced degradation of this antigen in gp91phox-deficient cells compared with wild type DC, consistent with a role for the oxidase in limiting antigen processing for crosspresentation (36).
Yates and colleagues also found in the absence of functional NADPH oxidase, the rate of phagosomal proteolysis in DC was significantly increased compared to cells with normal oxidase activity (40). However, results of excitation ratio fluorometry and real-time fluorometric confocal microscopy experiments suggested NADPH oxidase activity does not prevent acidification of DC phagosomes (40).
To determine an alternative mechanism by which NADPH oxidase activity limits antigen proteolysis, the effect of oxidase activity on different classes of proteases was examined. Phagosomal hydrolysis of fluorogenic substrates for cysteine cathepsins B/S/L and aspartic cathepsins D/E was monitored in oxidase-proficient and -deficient DC in real time. NADPH oxidase ROS production inhibited phagosomal cysteine, but not aspartic, cathepsin activity. This inhibition also occurred in the presence of H2O2 in a reconstituted system of lysosomal extracts and could be reversed upon addition of reduced glutathione, suggesting oxidase activity may limit antigen proteolysis through redox modulation of cysteine cathepsin activity. Using an assay with bead coupled cysteine-linked fluorochromes, disulfide bond reducing capacity was also found to be markedly inhibited within phagosomes of wild type DC compared to gp91phox-deficient DC, suggesting NADPH oxidase activity may also be important for regulating disulfide bond reduction in antigen processing events (40).
Discrepancies regarding the effect of NADPH oxidase activity on phagosomal pH in DC remain. However, all studies highlight a role for NADPH oxidase in limiting antigen proteolysis in phagosomes (36, 40). Whether the reduced antigen crosspresentation by DC outlined in these reports perturbs the development of self tolerance in CGD patients remains unclear.
NADPH Oxidase and MHC-II Antigen Presentation
A new genetic subgroup of CGD was recently defined upon identification of a patient with autosomal recessive mutations in both copies of NCF4, the gene encoding p40phox (9). Familial studies to track p40phox mutations in this patient revealed the paternal allele encodes a truncated protein due to the presence of a premature stop codon. The maternal allele contains an R105Q missense mutation in the phospholipid binding PX (phox homology) domain. In neutrophils, this mutation results in a loss of p40phox phosphatidylinositol (3,4,5)-triphosphate binding followed by premature dissociation from the neutrophil phagosome and ultimately impaired production of intracellular ROS by the oxidase complex (9).
This patient did not initially present with recurrent pathogenic infections characteristic of CGD, but rather with severe granulomatous colitis (9). NCF4 has been associated with ileal Crohn’s disease (20, 21) as well as rheumatoid arthritis (19) in GWAS. These linkages to autoinflammatory and autoimmune disorders suggest a potential role for p40phox in regulating adaptive immune responses. Supporting this hypothesis, human B lymphocytes lacking functional p40phox exhibit altered antigen presentation by major histocompatibility complex class II (MHC-II) molecules (41).
MHC-II molecules are heterodimeric transmembrane proteins composed of α and β subunits. Within professional antigen presenting cells, these αβ heterodimers are loaded with peptides derived from processed protein antigens prior to cell surface presentation to CD4+ T cells. MHC-II assemble in the ER where they bind invariant chain (Ii) (42). Ii stabilizes the MHC-II heterodimer and directs the trafficking of MHC-II through the Golgi and into the endosomal network (43). Within this network, Ii is systematically cleaved by acid proteases leaving class II-associated invariant chain peptide (CLIP) bound to the MHC-II binding groove (44–46). In endosomes, HLA-DM, a non-classical MHC-II protein, catalyzes the removal of CLIP, and further edits peptides that bind MHC-II to favor the presentation of stable antigenic epitopes (Figure 2) (47, 48).
Antigenic peptides capable of binding MHC-II are generated within the endosomal network by proteases active at acidic pH, including cathepsins and asparaginyl endopeptidase (AEP) (49). Antigen proteolysis can be influenced by gamma-interferon-induced lysosomal thiol reductase (GILT). This enzyme reduces antigen disulfides to favor protein unfolding at acidic pH (50, 51). Whether or not NADPH oxidase co-localizes with MHC-II, GILT, and acidic proteases in endosomes remains unclear (Figure 2).
Interestingly, evidence also suggests a role for ROS in regulating antigen presentation by MHC-II. In phagocytes, nitric oxide production is required for processing bacterial polysaccharides into fragments capable of binding MHC-II for presentation to T cells (52). Conflicting reports with B lymphocytes also suggest roles for superoxide and NADPH oxidase in MHC-II presentation, yet these early studies employed samples from reported CGD patients without genetic analysis to confirm defective oxidase subunits (53, 54). Although B lymphocytes express functional NADPH oxidase subunits (55), the precise function of this enzyme complex in these cells remains unclear. Analysis of CGD patient B cells (56, 57) revealed alterations in circulating memory B cells; however, inactivation of the oxidase also impacts T lymphocyte development and function (58–60).
To examine the role of the oxidase in cytoplasmic antigen presentation by MHC-II, p40phox expression in a human B cell line was disrupted using lentiviral-driven shRNA (41). In B lymphocytes, p40phox deficiency altered the profile of antigenic epitopes displayed by MHC-II, with a shift toward preferential presentation of self membrane-associated antigens. Studies using a panel of antigen-specific T cells revealed MHC-II presentation of epitopes from cytoplasmic and exogenous antigens was reduced with knockdown of p40phox in B cells. Yet, MHC-II presentation of epitopes from membrane antigens was enhanced for B cells deficient in p40phox when compared to parental or control cells with normal levels of this subunit. MHC-II presentation of synthetic short peptides by p40phox-deficient B cells was comparable to control cells, revealing that loss of this subunit did not influence peptide binding to surface MHC-II or T cell recognition of these complexes. Consistent with these results, surface levels of HLA-DR in parental and control shRNA-treated cells were equivalent (41). These results suggest that diminished p40phox expression may modulate the balance of foreign versus self antigen presented in the context of MHC-II in human B cells.
To determine whether spontaneously arising mutations in human p40phox influence MHC-II presentation, B lymphoblastoid cells were generated from the CGD patient with mutations in NCF4 (9) and transduced to express the MHC-II allele DRβ1*0401 (61). These patient-derived cells were also transduced to express functional wild type p40phox or the mutant R105Q allele (41). In agreement with previous results, reduced CD4+ T cell responses were detected in response to exogenous antigens in patient B cells with mutated p40phox compared to patient cells reconstituted with wild type p40phox. MHC-II presentation of epitopes from endogenous, self membrane antigens was similar regardless of the form of p40phox expressed by these B cells. T cell responses to synthetic peptides presented in the context of MHC-II on patient-derived B cells were comparable for cells expressing mutant or wild type forms of p40phox (41).
Notably, presentation of exogenous tetanus toxoid was also reduced in B cells from CGD patients with mutations in gp91phox as well as B cells from the patient with mutations in p40phox. No differences were observed in the expression levels of key components of the MHC-II pathway including HLA-DR, -DM, or -DO in B cells from the patient with NCF4 mutations. While reduced presentation of exogenous antigens was consistently observed with reduced oxidase p40phox, the ability of these B cells to endocytose antigen was similar to cells from healthy individuals. Flow cytometric analysis revealed patient-derived B cells deficient in p40phox spontaneously produced less intracellular superoxide compared to these CGD cells with restored wild type p40phox expression. Yet as observed in patient neutrophils (9), extracellular ROS production in response to PMA was similar for the patient and wild type p40phox-reconstituted B cells (41). Taken together, these data suggest the altered profile of epitopes selected for presentation in p40phox-deficient B cells may be linked to diminished intracellular superoxide production.
While p40phox is known to regulate oxidase activation in response to specific stimuli in neutrophils, this marks the first demonstration of a role for p40phox in antigen presentation and B cell functions. These results suggest functional p40phox expression can influence the peptide repertoire presented by MHC-II on B cells and the subsequent activation of CD4+ T cells. Thus, p40phox deficiency may contribute to autoimmune and autoinflammatory disease predisposition in patients with CGD or oxidase gene polymorphisms.
Conclusion
Inherited defects in NADPH oxidase give rise to CGD, a primary immunodeficiency that has been linked with a number of autoinflammatory and autoimmune disorders. Genes encoding the subunits of NADPH oxidase have also been associated with these disorders in GWAS. Together, these observations suggest a potential role for NADPH oxidase in regulating adaptive immune responses. Currently, studies have shown NADPH oxidase activity limits antigen proteolysis in DC phagosomes to promote MHC-I crosspresentation; however, the mechanism underlying this effect remains unclear. Some studies suggest NADPH oxidase activity limits antigen proteolysis by preventing phagosome acidification, while others suggest it alters the local redox microenvironment within the phagosome. Studies from our laboratory have shown NADPH oxidase also modulates MHC-II presentation in B cells and that defects in oxidase activity may alter the peptide repertoire displayed by MHC-II. This phenomenon could contribute to the development of autoimmunity in patients with CGD or polymorphisms in oxidase genes. Future studies are needed to further establish this link as well as to elucidate the mechanism by which NADPH oxidase regulates antigen presentation.
Author Contributions
Gail J. Gardiner performed the literature review of NADPH oxidase, CGD, connections to autoimmunity, NADPH oxidase and crosspresentation, and NADPH oxidase and MHC-II presentation and also wrote the manuscript. Sarah N. Deffit reviewed crosspresentation and MHC-II presentation and wrote these sections of the manuscript. Shawna McLetchie and Liliana Pérez designed the figures and wrote the figure legends. Crystal C. Walline reviewed CGD connections to autoimmunity and edited the manuscript. Janice S. Blum edited the manuscript and oversaw its preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health Grants R01 AI079065 and an Indiana CTSI Core Grant. Gail J. Gardiner is supported by the Merilyn Hester Scholarship Fund, Sarah N. Deffit by NIH T32 DK007519, and Liliana Pérez by NIH T32 AI060519.
References
1. Bokoch GM, Knaus UG. NADPH oxidases: not just for leukocytes anymore! Trends Biochem Sci (2003) 28:502–8. doi: 10.1016/S0968-0004(03)00194-4
2. Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol (2004) 122:277–91. doi:10.1007/s00418-004-0679-8
3. Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J (2005) 386:401–16. doi:10.1042/BJ20041835
4. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) (2000) 79:155–69. doi:10.1097/00005792-200005000-00003
5. Romera Modamio G, Martin Mateos MA, Gonzalez Ensenat MA, Pastor Gomez MA. Chronic granulomatous disease: a case study of a symptomatic carrier. J Investig Allergol Clin Immunol (1997) 7:57–61.
6. Lewis EM, Singla M, Sergeant S, Koty PP, Mcphail LC. X-linked chronic granulomatous disease secondary to skewed X chromosome inactivation in a female with a novel CYBB mutation and late presentation. Clin Immunol (2008) 129:372–80. doi:10.1016/j.clim.2008.07.022
7. van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS One (2009) 4:e5234. doi:10.1371/journal.pone.0005234
8. Williams DA, Tao W, Yang F, Kim C, Gu Y, Mansfield P, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood (2000) 96:1646–54.
9. Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood (2009) 114:3309–15. doi:10.1182/blood-2009-07-231498
10. Segal BH, Veys P, Malech H, Cowan MJ. Chronic granulomatous disease: lessons from a rare disorder. Biol Blood Marrow Transplant (2011) 17:S123–31. doi:10.1016/j.bbmt.2010.09.008
11. Segal BH, Grimm MJ, Khan AN, Han W, Blackwell TS. Regulation of innate immunity by NADPH oxidase. Free Radic Biol Med (2012) 53:72–80. doi:10.1016/j.freeradbiomed.2012.04.022
12. Manzi S, Urbach AH, Mccune AB, Altman HA, Kaplan SS, Medsger TA Jr, et al. Systemic lupus erythematosus in a boy with chronic granulomatous disease: case report and review of the literature. Arthritis Rheum (1991) 34:101–5. doi:10.1002/art.1780340116
13. Martin-Villa JM, Corell A, Ramos-Amador JT, Ruiz-Contreras J, Arnaiz-Villena A. Higher incidence of autoantibodies in X-linked chronic granulomatous disease carriers: random X-chromosome inactivation may be related to autoimmunity. Autoimmunity (1999) 31:261–4. doi:10.3109/08916939908994071
14. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol (2007) 148:79–84. doi:10.1111/j.1365-2249.2007.03321.x
15. De Ravin SS, Naumann N, Cowen EW, Friend J, Hilligoss D, Marquesen M, et al. Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol (2008) 122:1097–103. doi:10.1016/j.jaci.2008.07.050
16. Lee BW, Yap HK. Polyarthritis resembling juvenile rheumatoid arthritis in a girl with chronic granulomatous disease. Arthritis Rheum (1994) 37:773–6. doi:10.1002/art.1780370524
17. De Ravin SS, Naumann N, Robinson MR, Barron KS, Kleiner DE, Ulrick J, et al. Sarcoidosis in chronic granulomatous disease. Pediatrics (2006) 117:e590–5. doi:10.1542/peds.2005-1349
18. Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A (2012) 109:E59–67. doi:10.1073/pnas.1113251108
19. Olsson LM, Lindqvist AK, Kallberg H, Padyukov L, Burkhardt H, Alfredsson L, et al. A case-control study of rheumatoid arthritis identifies an associated single nucleotide polymorphism in the NCF4 gene, supporting a role for the NADPH-oxidase complex in autoimmunity. Arthritis Res Ther (2007) 9:R98. doi:10.1186/ar2299
20. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39:596–604. doi:10.1038/ng2032
21. Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR. Confirmation of association of IRGM and NCF4 with ileal Crohn’s disease in a population-based cohort. Genes Immun (2008) 9:561–5. doi:10.1038/gene.2008.49
22. George-Chandy A, Nordstrom I, Nygren E, Jonsson IM, Postigo J, Collins LV, et al. Th17 development and autoimmune arthritis in the absence of reactive oxygen species. Eur J Immunol (2008) 38:1118–26. doi:10.1002/eji.200737348
23. Jendrysik MA, Vasilevsky S, Yi L, Wood A, Zhu N, Zhao Y, et al. NADPH oxidase-2 derived ROS dictates murine DC cytokine-mediated cell fate decisions during CD4 T helper-cell commitment. PLoS One (2011) 6:e28198. doi:10.1371/journal.pone.0028198
24. Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity (2006) 25:153–62. doi:10.1016/j.immuni.2006.04.017
25. John B, Harris TH, Tait ED, Wilson EH, Gregg B, Ng LG, et al. Dynamic Imaging of CD8(+) T cells and dendritic cells during infection with Toxoplasma gondii. PLoS Pathog (2009) 5:e1000505. doi:10.1371/journal.ppat.1000505
26. Ballesteros-Tato A, Leon B, Lund FE, Randall TD. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat Immunol (2010) 11:216–24. doi:10.1038/ni.1838
27. Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med (1996) 184:923–30. doi:10.1084/jem.184.3.923
28. Hernandez J, Aung S, Redmond WL, Sherman LA. Phenotypic and functional analysis of CD8(+) T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J Exp Med (2001) 194:707–17. doi:10.1084/jem.194.6.707
29. Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med (2002) 196:1627–38. doi:10.1084/jem.20021598
30. Luckashenak N, Schroeder S, Endt K, Schmidt D, Mahnke K, Bachmann MF, et al. Constitutive crosspresentation of tissue antigens by dendritic cells controls CD8+ T cell tolerance in vivo. Immunity (2008) 28:521–32. doi:10.1016/j.immuni.2008.02.018
31. Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity (2006) 25:607–17. doi:10.1016/j.immuni.2006.08.017
32. Imai T, Kato Y, Kajiwara C, Mizukami S, Ishige I, Ichiyanagi T, et al. Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc Natl Acad Sci U S A (2011) 108:16363–8. doi:10.1073/pnas.1108372108
33. Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature (2003) 425:397–402. doi:10.1038/nature01911
34. Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity (2004) 21:155–65. doi:10.1016/j.immuni.2004.07.004
35. Bertholet S, Goldszmid R, Morrot A, Debrabant A, Afrin F, Collazo-Custodio C, et al. Leishmania antigens are presented to CD8+ T cells by a transporter associated with antigen processing-independent pathway in vitro and in vivo. J Immunol (2006) 177:3525–33.
36. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell (2006) 126:205–18. doi:10.1016/j.cell.2006.05.035
37. Mantegazza AR, Savina A, Vermeulen M, Perez L, Geffner J, Hermine O, et al. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood (2008) 112:4712–22. doi:10.1182/blood-2008-01-134791
38. Savina A, Peres A, Cebrian I, Carmo N, Moita C, Hacohen N, et al. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity (2009) 30:544–55. doi:10.1016/j.immuni.2009.01.013
39. Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, Dang PM, et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol (2007) 9:367–78. doi:10.1038/ncb1552
40. Rybicka JM, Balce DR, Chaudhuri S, Allan ER, Yates RM. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J (2012) 31:932–44. doi:10.1038/emboj.2011.440
41. Crotzer VL, Matute JD, Arias AA, Zhao H, Quilliam LA, Dinauer MC, et al. Cutting edge: NADPH oxidase modulates MHC class II antigen presentation by B cells. J Immunol (2012) 189:3800–4. doi:10.4049/jimmunol.1103080
42. Roche PA, Marks MS, Cresswell P. Formation of a nine-subunit complex by HLA class II glycoproteins and the invariant chain. Nature (1991) 354:392–4. doi:10.1038/354392a0
43. Bakke O, Dobberstein B. MHC class II-associated invariant chain contains a sorting signal for endosomal compartments. Cell (1990) 63:707–16. doi:10.1016/0092-8674(90)90137-4
44. Blum JS, Cresswell P. Role for intracellular proteases in the processing and transport of class II HLA antigens. Proc Natl Acad Sci U S A (1988) 85:3975–9. doi:10.1073/pnas.85.11.3975
45. Thomas LJ, Nguyen QV, Elliott WL, Humphreys RE. Proteolytic cleavage of Ii to p25. J Immunol (1988) 140:2670–4.
46. Morkowski S, Goldrath AW, Eastman S, Ramachandra L, Freed DC, Whiteley P, et al. T cell recognition of major histocompatibility complex class II complexes with invariant chain processing intermediates. J Exp Med (1995) 182:1403–13. doi:10.1084/jem.182.5.1403
47. Mellins E, Smith L, Arp B, Cotner T, Celis E, Pious D. Defective processing and presentation of exogenous antigens in mutants with normal HLA class II genes. Nature (1990) 343:71–4. doi:10.1038/343071a0
48. Denzin LK, Cresswell P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell (1995) 82:155–65. doi:10.1016/0092-8674(95)90061-6
49. Hsieh CS, Deroos P, Honey K, Beers C, Rudensky AY. A role for cathepsin L and cathepsin S in peptide generation for MHC class II presentation. J Immunol (2002) 168:2618–25.
50. Maric M, Arunachalam B, Phan UT, Dong C, Garrett WS, Cannon KS, et al. Defective antigen processing in GILT-free mice. Science (2001) 294:1361–5. doi:10.1126/science.1065500
51. Li P, Haque MA, Blum JS. Role of disulfide bonds in regulating antigen processing and epitope selection. J Immunol (2002) 169:2444–50.
52. Lewis CJ, Cobb BA. Carbohydrate oxidation acidifies endosomes, regulating antigen processing and TLR9 signaling. J Immunol (2010) 184:3789–800. doi:10.4049/jimmunol.0903168
53. Heijnen CJ, Van Der Meer JW, Zegers BJ. Altered antigen-presentation in the induction of the in-vitro antigen-specific T helper cell function in patients with chronic granulomatous disease. Clin Exp Immunol (1986) 66:111–7.
54. Barbey C, Tiercy JM, Fairweather N, Niemann H, Seger R, Corradin G. Processing and presentation of tetanus toxin by antigen-presenting cells from patients with chronic granulomatous disease (CGD) to human specific T cell clones are not impaired. Clin Exp Immunol (1994) 95:227–31. doi:10.1111/j.1365-2249.1994.tb06515.x
55. Kobayashi S, Imajoh-Ohmi S, Kuribayashi F, Nunoi H, Nakamura M, Kanegasaki S. Characterization of the superoxide-generating system in human peripheral lymphocytes and lymphoid cell lines. J Biochem (1995) 117:758–65.
56. Bleesing JJ, Souto-Carneiro MM, Savage WJ, Brown MR, Martinez C, Yavuz S, et al. Patients with chronic granulomatous disease have a reduced peripheral blood memory B cell compartment. J Immunol (2006) 176:7096–103.
57. Moir S, De Ravin SS, Santich BH, Kim JY, Posada JG, Ho J, et al. Humans with chronic granulomatous disease maintain humoral immunologic memory despite low frequencies of circulating memory B cells. Blood (2012) 120:4850–8. doi:10.1182/blood-2012-05-430959
58. Hasui M, Hattori K, Taniuchi S, Kohdera U, Nishikawa A, Kinoshita Y, et al. Decreased CD4+CD29+ (memory T) cells in patients with chronic granulomatous disease. J Infect Dis (1993) 167:983–5. doi:10.1093/infdis/167.4.983
59. Horvath R, Rozkova D, Lastovicka J, Polouckova A, Sedlacek P, Sediva A, et al. Expansion of T helper type 17 lymphocytes in patients with chronic granulomatous disease. Clin Exp Immunol (2011) 166:26–33. doi:10.1111/j.1365-2249.2011.04449.x
60. Montes-Berrueta D, Ramirez L, Salmen S, Berrueta L. Fas and FasL expression in leukocytes from chronic granulomatous disease patients. Invest Clin (2012) 53:157–67.
Keywords: NADPH oxidase, B lymphocytes, chronic granulomatous disease, autoimmunity, antigen presentation
Citation: Gardiner GJ, Deffit SN, McLetchie S, Pérez L, Walline CC and Blum JS (2013) A role for NADPH oxidase in antigen presentation. Front. Immunol. 4:295. doi: 10.3389/fimmu.2013.00295
Received: 26 July 2013; Accepted: 07 September 2013;
Published online: 23 September 2013.
Edited by:
Laura Santambrogio, Albert Einstein College of Medicine, USAReviewed by:
Stephanie Hugues, Université de Genève, SwitzerlandPeter Cresswell, Yale University Medical Center, USA
Copyright: © 2013 Gardiner, Deffit, McLetchie, Pérez, Walline and Blum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Janice S. Blum, Department of Microbiology and Immunology, Indiana University School of Medicine, 635 Barnhill Drive, MS 420, Indianapolis, IN 46202, USA e-mail:amJsdW1AaXVwdWkuZWR1