
- Institut National de la Santé et de le Recherche Médicale, Unité 1013, Université Paris Descartes, Sorbonne Paris Cité, Faculté de médecine, Paris, France
MHC class II molecules expressed by professional antigen presenting cells (pAPCs) such as macrophages, B cells, and dendritic cells (DCs) play a fundamental role in presenting peptides to CD4+ T cells. However, to elicit CD4+-T cells immunity, pAPCs need an additional signal, which can be delivered by toll-like receptors (TLRs) molecules. TLRs recognize microbial patterns and are critical in initiating immune responses. Proteases, which provide peptide ligands for the MHC class II antigenic presentation pathway, were recently shown to cleave and activate intracellular TLRs in endosomal compartments. Here, I give an overview on the individual roles of the most well studied proteases in both antigen and TLRs processing.
MHC Class II Pathway
A complex series of biosynthetic and proteolytic events must ensure correct assembly, trafficking, and peptide loading in order for the MHC class II molecules to efficiently interact with their specific T-cell Receptor (TCR) expressed on CD4+ helper T cells (1, 2). MHC class II molecules consist of an α and β chain which dimerize shortly after synthesis in the endoplasmic reticulum (ER) to create the groove in which antigenic peptides are accommodated. Newly synthesized αβ dimers assemble with a non-polymorphic glycoprotein, the invariant chain (Ii), which fills the peptide-binding groove of αβ dimers and prevent loading of premature peptides in the ER. In addition, the cytoplasmic tail of Ii contains targeting signals that deliver the MHC class II-Ii complexes from the trans-Golgi network directly into the endocytic pathway (3) where exogenous antigen are internalized. Ii is then sequentially cleaved leaving a C-terminal (C-ter) portion: the class II-associated invariant chain peptide or CLIP in the peptide-binding groove. Within the lysosomes, the chaperone molecule HLA-DM interacts with MHCII-CLIP and catalyzes the exchange of CLIP for exogenous peptides. Newly formed MHCII-peptides complexes are then targeted to the plasma membrane where they interact with CD4+ T cells.
Antigen Processing
The presentation of antigens by MHC class II molecules is strictly dependent on the action of endocytic proteases. Indeed, these enzymes not only degrade proteins in order to produce the antigenic peptides but they also process the Ii chain. Limited antigen proteolysis is required for MHC class II-peptide loading and peptides of 9–16 residues are presented to CD4+ T cells. A balanced proteolytic environment is therefore required to ensure adequate antigen processing while preventing complete destruction. The three main classes of intracellular proteases residing in the lysosomal/endosomal compartments and participating in antigen degradation are cysteine (cathepsin B, F, H, L, S, Z, and AEP, for asparaginylendopeptidase), aspartate (cathepsin D, E), and serine (cathepsin A, G) proteases. The protease nomenclature designates the amino acid of the protease active site that catalyzes hydrolysis of the substrate peptide bond. With the exception of AEP, most endocytic proteases display broad cleavage specificity. Indeed, AEP cleaves on the carboxyl terminal sides of asparagines residues whereas other cysteine proteases recognize hydrophobic motifs.
The use of specific protease inhibitors and mice deficient for murine proteases has helped to identify the key enzymes in Ii processing. Indeed, the final steps of Ii degradation are dependent on cathepsin L or S and in their absence an Ii N-terminal fragment of approximately of 10 kDa (p10) accumulates on MHC class II molecules (4–6). However, it is less obvious which enzymes are involved in antigen processing but a critical role of cathepsin S and L has been confirmed in the generation of MHC II-peptides complexes albeit in different cell types (4–8). In addition, cathepsin F was reported to compensate for the loss of cathepsins S and L in macrophages (9). Furthermore, in a recent human study, cathepsin S expressed in thymic dendritic cells (DCs) was shown to be responsible for the destruction of a certain number of epitopes from two auto-antigens involved in experimental allergic encephalitis and in diabetes (10).
Concerning serine proteases, a latest study has described the importance of cathepsin G in generating several proinsulin peptides in vitro and in human cells. Indeed, cathepsin G activity was found to be elevated in PBMC from diabetic patients and blocking its activity resulted in the abrogation of the proliferation of specific proinsulin T cells (11).
Together the groups of Alan Barrett and Colin Watts described few years ago a novel lysosomal cysteine protease, AEP. This asparagine endopeptidase was shown to initiate processing of the tetanus toxin antigen in human B cells (12, 13), to be capable of destroying an immunodominant peptide of myelin basic protein (MBP, 85–99) an auto antigen implicated in the autoimmune disease multiple sclerosis (14) and to perform the early step of Ii chain degradation in human B-EBV cells (15). AEP participation was clearly demonstrated in processing antigens in human APCs. However, its role was less clear in mice. Indeed, it was reported that in AEP-deficient cells, antigen processing of Ii chain and two other antigens were weakly altered (16, 17).
An additional lysosomal processing enzyme, named GILT or gamma interferon inducible lysosomal thiol reductase, was described to generate T cell epitopes by reducing proteins disulfide bonds. Indeed, by catalyzing the reduction of disulfide bonds, GILT generates proteins partially denatured more susceptible to proteolysis. GILT is now reported to be required for the presentation of many peptide ligands of hen egg lysozyme, ribonuclease A, human IgG, the melanoma differentiation antigens tyrosine and TRP1, the human immunodeficiency virus-1 envelope proteins and the allergens Derp1 and Blag2 which all contain disulfide bonds (18–22).
Another important role of AEP and GILT is to regulate the expression or the maturation of other cysteine proteases. Lysosomal proteases reach the endocytic compartments as proforms or zymogens where their propeptide is removed by proteolysis. The resulting single-chain form is then cleaved into a mature form or a two-chain form (heavy and light chains). For example, GILT has been described to regulate the expression of cathepsin B specifically in B cells (23) and AEP-deficient mice exhibit a defect in the maturation of Cat B, Cat D, Cat H, and Cat L in kidney and bone marrow derived dendritic cells (BMDCs) (16, 24, 25) and an increase in Cat K expression. How exactly AEP mediates these events are still unclear.
Acidic pH is a prerequisite for maturation and activity of most of these intracellular proteases and so their maximal activity is found in lysosomal compartments. Indeed, chloroquine, a lysomotropic agent, which was shown to abrogate MHC class II antigen presentation because of inhibiting enzymatic activities in lysosomes, also altered intracellular toll-like receptors (TLRs) signaling (26). Accordingly, a new role for the endocytic proteases was discovered.
TLRs Processing
Toll-like receptors are proteins, which recognize conserved molecules from microorganisms and in DCs, they are crucial in linking innate to adaptive immunity. TLRs contain several leucine rich repeats (LRR) in an extracellular loop, a trans-membrane domain, and a cytosolic domain and are expressed either at the plasma membrane or in the endosomal/lysosomal organelles. TLR stimulation is linked to MyD88 or TRIF-dependent signaling pathways that regulate the activation of different transcription factors, such as NF-κB and IRF (27). Specific interaction between TLRs and their ligands activates NF-κB resulting in enhanced inflammatory cytokine responses, induction of DCs maturation and expression of chemokine receptors. TLRs expressed at the plasma membrane recognized Gram-negative bacteria and endosomal TLRs sense viral and bacterial nucleic acids such as double/single-stranded RNA or double stranded DNA. Endogenous ligands called DAMPs (for damage associated molecular patterns) may also activate TLRs during self-tissues or cell damage (28). Several published results demonstrated that intracellular TLRs require partial proteolysis in endosomes for full-activation. Indeed, many groups have now reported that murine TLR9 is non-functional until it is subjected to proteolytic cleavage in the endosomes (24, 29–31). Upon stimulation, full-length (FL) TLR9 is cleaved into a C-ter fragment sufficient for signaling. This cleavage is realized by several proteases in different cells. Addition of Z-FA-FMK, a broad inhibitor of cathepsins, or specific inhibitors of cathepsins B, L, and S partially or completely impaired TLR9 signaling in macrophages and B cell lines. In primary cells such as BMDCs, deficiency of cathepsin L led to partial reduction (about 50%) of TLR9 function. In cathepsin K deficient DCs, TLR9 signaling was totally abrogated (32). Moreover, inhibition of cathepsin K activity exerted beneficial effects on collagen-induced arthritis in mice, an autoimmune disease induced after injection of type II collagen and complete freund adjuvant containing TLR9 agonist such as CpG-ODN (32). In addition, in DCs and mice lacking AEP, even though TLR9 cleavage in phagosomal compartments was still occurring, CD4+ antigen specific T cell proliferation was greatly reduced upon CpG and ovalbumin stimulation (24). These results correlated with some in vitro digestion assays where FL TLR9 was shown to be proteolyzed into a C-ter fragment by cathepsins K, L, S, or AEP (24, 29–31). Altogether, these results described clearly the involvement of several distinct lysosomal proteases for TLR9 function.
Concerning TLR7, the literature is scarce but it was firstly reported the generation of a TLR7 proteolytic fragment (33) on a SDS gel, which by analogy with TLR9 was identified as the TLR7 C-ter fragment. Then, using different wild type or AEP-deficient primary cells, such as DCs, plasmacytoid dendritic cells (pDCs) or epithelial cells, it was shown that TLR7 was also subjected to similar proteolytic maturation than TLR9 and required AEP for proper signaling. In addition, infected mice lacking AEP with influenza virus, a single-stranded RNA sensed by TLR7, developed much less inflammation and exhibited significant reduced CD8+ T cells priming (34). Recently, it was shown that, in contrast to TLR9, TLR7 N-terminal fragment (N-Ter) remained linked by a disulfide bond with TLR7 C-ter. Cysteine 98 in TLR7 N-ter and Cysteine 445 in TLR7 C-ter were required for this disulfide bond and mutating one of them abrogated TLR proteolytic cleavage and RNA sensing (35). It will be interesting to investigate whether this disulfide bond in TLR7 is also required for TLR7 signaling in pAPCs such as DCs and the eventual role of GILT in maintaining or reducing this disulfide bond. Nevertheless, it still uncertain whether or not human TLR7 and TLR9 require proteolysis for their function. Only one report to date has shown that blocking AEP activity in human pDCs totally abrogate TLR9 signaling (24).
The results concerning TLR3 processing are less obvious. In the mouse and human system, it was described that Z-FA-FMK did alter TLR3 processing but not its signaling in a macrophage cell line and in HEK 293 cells overexpressing TLR3 and UNC93B1 (36). In contrast, inhibition of TLR3 processing into a C-ter fragment and subsequently its signaling was reported by the group of S. Lebecque in human monocyte derived DCs incubated with Z-FA-FMK (37). Moreover, the identity of the proteases involved in TLR3 proteolysis is still a matter of debate. Yet, cathepsins B and H seem to be good candidates (38) to generate the TLR3 C-terminal fragment as it was observed that in cells silenced for these two genes, the generation of the TLR3 C-ter fragment was reduced.
Intracellular TLRs traffic from the ER to lysosomal compartments where they respond to their ligands. UNC93B1, an ER resident protein, facilitates their trafficking (39, 40). In mice and cells defective for UNC93B1, harboring a mutation in the trans-membrane domain, TLR7 and TLR9 remain in the ER and fail to respond to TLRs stimulation. As a consequence, mice expressing mutated UNC93B1 (3d mice) are more susceptible to bacterial and viral infection (41, 42). Beyond the role of UNC93B1 in intracellular TLRs trafficking, UNC93B1 is also important in the MHC class I cross presentation and MHC class II pathways (42). Indeed, mice and DCs mutated for UNC93B1 are unable to present exogenous antigens to CD4+ and CD8+ T cells. However, despite abundant work on UNC93B1, little is known about the molecular mechanism leading to MHC II antigen presentation defect in pAPCs expressing a faulty UNC93B1 protein. Interestingly, recently, MHCII has been shown to promote full-activation of TLR9. Following stimulation of TLR3 and 9, MHCII forms a complex with CD40 and the phosphorylated Bruton tyrosine kinase (Btk) in lysosomes (43). This prolonged interaction maintains Btk activated and increases proinflammatory cytokines and type I interferon secretion by DCs and macrophages following TLRs stimulation.
In addition to the role of TLRs in DCs activation via the up regulation of costimulatory molecules, TLR7 and 9 stimulation induce a drop of pH in the early endosomes of DCs (24, 34). This acidic pH, which boost protease activities, probably also favor processing of exogenous antigen and MHC class II presentation.
Conclusion
The endosomal/phagosomal pathway is a key meeting point between proteins regulating innate and adaptive immunity (Figure 1). MHCII, UNC93B1, and proteases have been shown to regulate both TLRs signaling and MHCII presentation. Understanding how proteases are regulated in specific APCs and identifying new components in TLRs activation, especially upon pathogen infection, will be no doubt important for controlling specific unwanted immune responses.
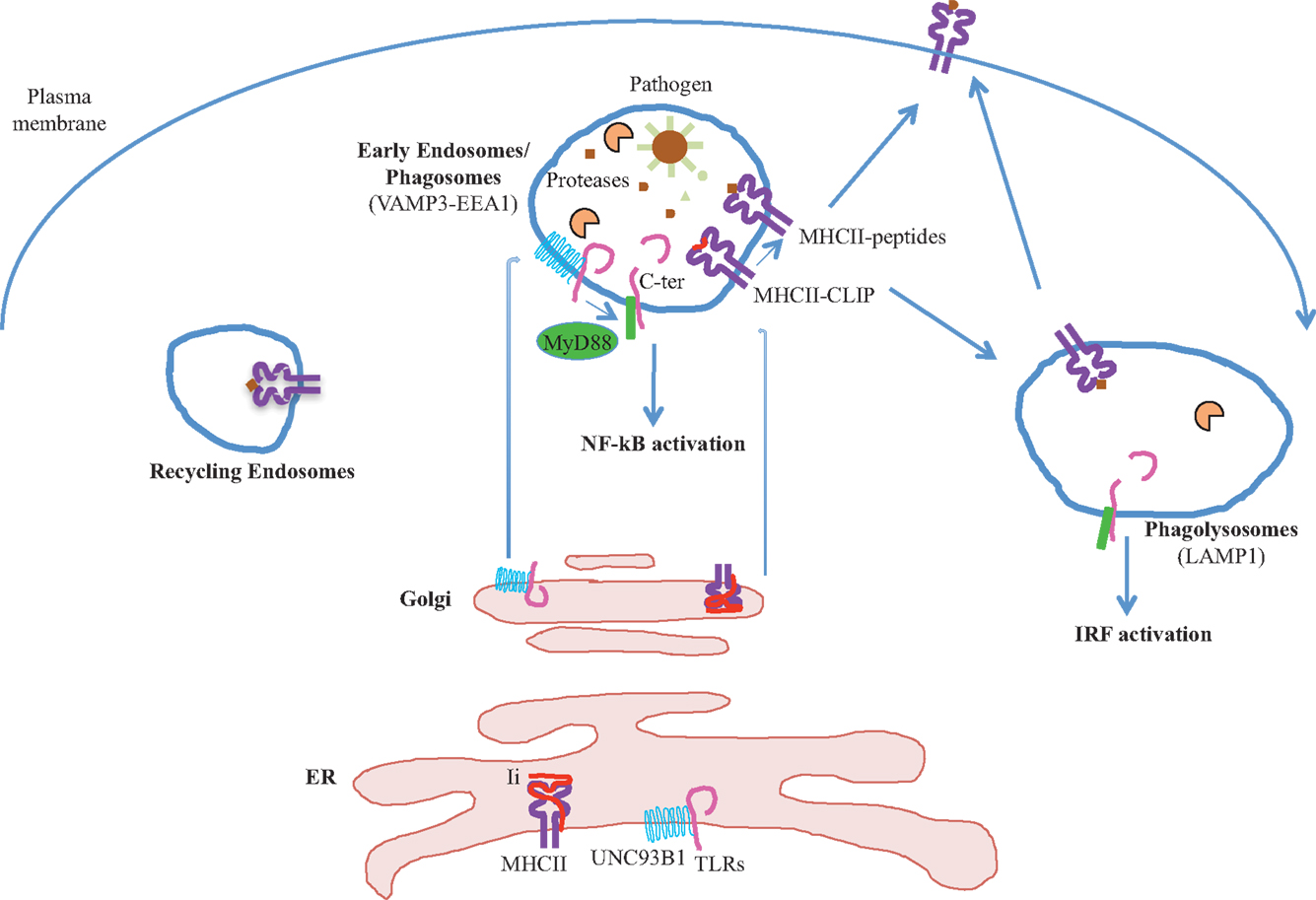
Figure 1. Endosomal proteases process internalized antigens and intracellular TLRs. Both intracellular TLRs and MHCII molecules associate with their specific chaperones proteins in the ER, UNC93B1, and Ii chains respectively, and traffic toward the endosomes. In the endosomes, TLRs, Ii chain, and exogenous antigens are cleaved by proteases. Peptides are loaded on MHCII molecules and the complexes MHCII-peptides are then target to the plasma membrane to interact with their specific TCR expressed by CD4+ T cells. Cleaved TLRs associate with the adaptor molecule MyD88 that trigger either NF-κB or IRF activation. These two events, formation of MHC II-peptide complexes and TLRs activation in APCs, are critical for inducing CD4 T cell responses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by an INSERM Grant (ANR 2010 MIDI 008 01).
References
1. Watts C. Antigen processing in the endocytic compartment. Curr Opin Immunol (2001) 13:26–31. doi: 10.1016/S0952-7915(00)00177-1
2. Wolf PR, Ploegh HL. How MHC class II molecules acquire peptide cargo: biosynthesis and trafficking through the endocytic pathway. Annu Rev Cell Dev Biol (1995) 11:267–306. doi:10.1146/annurev.cb.11.110195.001411
3. Stumptner-Cuvelette P, Benaroch P. Multiple roles of the invariant chain in MHC class II function. Biochim Biophys Acta (2002) 1542:1–13. doi:10.1016/S0167-4889(01)00166-5
4. Nakagawa TY, Brissette WH, Lira PD, Griffiths RJ, Petrushova N, Stock J, et al. Impaired invariant chain degradation and antigen presentation and diminished collagen-induced arthritis in cathepsin S null mice. Immunity (1999) 10:207–17. doi:10.1016/S1074-7613(00)80021-7
5. Riese RJ, Wolf PR, Bromme D, Natkin LR, Villadangos JA, Ploegh HL, et al. Essential role for cathepsin S in MHC class II-associated invariant chain processing and peptide loading. Immunity (1996) 4:357–66. doi:10.1016/S1074-7613(00)80249-6
6. Nakagawa T, Roth W, Wong P, Nelson A, Farr A, Deussing J, et al. Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science (1998) 280:450–3. doi:10.1126/science.280.5362.450
7. Hsieh CS, deRoos P, Honey K, Beers C, Rudensky AY. A role for cathepsin L and cathepsin S in peptide generation for MHC class II presentation. J Immunol (2002) 168:2618–25.
8. Pluger EB, Boes M, Alfonso C, Schroter CJ, Kalbacher H, Ploegh HL, et al. Specific role for cathepsin S in the generation of antigenic peptides in vivo. Eur J Immunol (2002) 32:467–76. doi:10.1002/1521-4141(200202)32:2<467::AID-IMMU467>3.0.CO;2-Y
9. Shi GP, Bryant RA, Riese R, Verhelst S, Driessen C, Li Z, et al. Role for cathepsin F in invariant chain processing and major histocompatibility complex class II peptide loading by macrophages. J Exp Med (2000) 191:1177–86. doi:10.1084/jem.191.7.1177
10. Stoeckle C, Quecke P, Ruckrich T, Burster T, Reich M, Weber E, et al. Cathepsin S dominates autoantigen processing in human thymic dendritic cells. J Autoimmun (2012) 38:332–43. doi:10.1016/j.jaut.2012.02.003
11. Zou F, Schafer N, Palesch D, Brucken R, Beck A, Sienczyk M, et al. Regulation of cathepsin G reduces the activation of proinsu1lin-reactive T cells from type 1 diabetes patients. PLoS ONE (2011) 6:e22815. doi:10.1371/journal.pone.0022815
12. Antoniou AN, Blackwood SL, Mazzeo D, Watts C. Control of antigen presentation by a single protease cleavage site. Immunity (2000) 12:391–8. doi:10.1016/S1074-7613(00)80191-0
13. Manoury B, Hewitt EW, Morrice N, Dando PM, Barrett AJ, Watts C. An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation. Nature (1998) 396:695–9. doi:10.1038/25379
14. Manoury B, Mazzeo D, Fugger L, Viner N, Ponsford M, Streeter H, et al. Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat Immunol (2002) 3:169–74. doi:10.1038/ni754
15. Manoury B, Mazzeo D, Li DN, Billson J, Loak K, Benaroch P, et al. Asparagine endopeptidase can initiate the removal of the MHC class II invariant chain chaperone. Immunity (2003) 18:489–98. doi:10.1016/S1074-7613(03)00085-2
16. Maehr R, Hang HC, Mintern JD, Kim YM, Cuvillier A, Nishimura M, et al. Asparagine endopeptidase is not essential for class II MHC antigen presentation but is required for processing of cathepsin L in mice. J Immunol (2005) 174:7066–74.
17. Matthews SP, Werber I, Deussing J, Peters C, Reinheckel T, Watts C. Distinct protease requirements for antigen presentation in vitro and in vivo. J Immunol (2010) 184:2423–31. doi:10.4049/jimmunol.0901486
18. Maric M, Arunachalam B, Phan UT, Dong C, Garrett WS, Cannon KS, et al. Defective antigen processing in GILT-free mice. Science (2001) 294:1361–5. doi:10.1126/science.1065500
19. Rausch MP, Irvine KR, Antony PA, Restifo NP, Cresswell P, Hastings KT. GILT accelerates autoimmunity to the melanoma antigen tyrosinase-related protein 1. J Immunol (2010) 185:2828–35. doi:10.4049/jimmunol.1000945
20. Sealy R, Chaka W, Surman S, Brown SA, Cresswell P, Hurwitz JL. Target peptide sequence within infectious human immunodeficiency virus type 1 does not ensure envelope-specific T-helper cell reactivation: influences of cysteine protease and gamma interferon-induced thiol reductase activities. Clin Vaccine Immunol (2008) 15:713–9. doi:10.1128/CVI.00412-07
21. West LC, Grotzke JE, Cresswell P. MHC class II-restricted presentation of the major house dust mite allergen Der p 1 Is GILT-dependent: implications for allergic asthma. PLoS ONE (2013) 8:e51343. doi:10.1371/journal.pone.0051343
22. Haque MA, Li P, Jackson SJ, Zarour HM, Hawes JW, Phan UT, et al. Absence of gamma-interferon-inducible lysosomal thiol reductase in melanomas disrupts T cell recognition of select immunodominat epitopes. J Exp Med (2002) 195:1267–77. doi:10.1084/jem.20011853
23. Phipps-Yonas H, Semik V, Hastings KT. GILT expression in B cells diminishes cathepsin S steady-state protein expression and activity. Eur J Immunol (2013) 43:65–74. doi:10.1002/eji.201242379
24. Sepulveda FE, Maschalidi S, Colisson R, Heslop L, Ghirelli C, Sakka E, et al. Critical role for asparagine endopeptidase in endocytic toll-like receptor signaling in dendritic cells. Immunity (2009) 31:737–48. doi:10.1016/j.immuni.2009.09.013
25. Shirahama-Noda K, Yamamoto A, Sugihara K, Hashimoto N, Asano M, Nishimura M, et al. Biosynthetic processing of cathepsins and lysosomal degradation are abolished in asparaginyl endopeptidase-deficient mice. J Biol Chem (2003) 278:33194–9. doi:10.1074/jbc.M302742200
26. Yi AK, Tuetken R, Redford T, Waldschmidt M, Kirsch J, Krieg AM. CpG motifs in bacterial DNA activate leukocytes through the pH-dependent generation of reactive oxygen species. J Immunol (1998) 160:4755–61.
27. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi:10.1146/annurev.immunol.20.083001.084359
28. Tian T, Avalos AM, Mao SY, Chen B, Senthil H, Wu H, et al. toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and rage. Nat Immunol (2007) 8:487–96. doi:10.1038/ni1457
29. Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, et al. The ectodomain of toll-like receptor 9 is cleaved to generate a functional receptor. Nature (2008) 456:658–62. doi:10.1038/nature07405
30. Matsumoto F, Saitoh S, Fukui R, Kobayashi T, Tanimura N, Konno K, et al. Cathepsins are required for toll-like receptor 9 responses. Biochem Biophys Res Commun (2008) 367:693–9. doi:10.1016/j.bbrc.2007.12.130
31. Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of toll-like receptor 9. Nat Immunol (2008) 9:1407–14. doi:10.1038/ni.1669
32. Asagiri M, Hirai T, Kunigami T, Kamano S, Gober HJ, Okamoto K, et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science (2008) 319:624–7. doi:10.1126/science.1150110
33. Ewald SE, Engel A, Lee J, Wang M, Bogyo M, Barton GM. Nucleic acid recognition by toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J Exp Med (2011) 208:643–51. doi:10.1084/jem.20100682
34. Maschalidi S, Hassler S, Blanc F, Sepulveda FE, Tohme M, Chignard M, et al. Asparagine endopeptidase controls anti-influenza virus immune responses through TLR7 activation. PLoS Pathog (2012) 8:e1002841. doi:10.1371/journal.ppat.1002841
35. Kanno A, Yamamoto C, Onji M, Fukui R, Saitoh S, Motoi Y, et al. Essential role for toll-like receptor 7 (TLR7)-unique cysteines in an intramolecular disulfide bond, proteolytic cleavage and RNA sensing. Int Immunol (2013) 25:413–22. doi:10.1093/intimm/dxt007
36. Qi R, Singh D, Kao CC. Proteolytic processing regulates toll-like receptor 3 stability and endosomal localization. J Biol Chem (2012) 287:32617–29. doi:10.1074/jbc.M112.387803
37. Toscano F, Estornes Y, Virard F, Garcia-Cattaneo A, Pierrot A, Vanbervliet B, et al. Cleaved/associated TLR3 represents the primary form of the signaling receptor. J Immunol (2013) 190:764–73. doi:10.4049/jimmunol.1202173
38. Garcia-Cattaneo A, Gobert FX, Muller M, Toscano F, Flores M, Lescure A, et al. Cleavage of toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc Natl Acad Sci U S A (2012) 109:9053–8. doi:10.1073/pnas.1115091109
39. Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol (2007) 177:265–75. doi:10.1083/jcb.200612056
40. Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature (2008) 452:234–8. doi:10.1038/nature06726
41. Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science (2006) 314:308–12. doi:10.1126/science.1128346
42. Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via toll-like receptors 3, 7 and 9. Nat Immunol (2006) 7:156–64. doi:10.1038/ni1297
Keywords: antigen processing, MHC class II, endosomal proteases, intracellular toll-like receptors
Citation: Manoury B (2013) Proteases: essential actors in processing antigens and intracellular toll-like receptors. Front. Immunol. 4:299. doi: 10.3389/fimmu.2013.00299
Received: 01 August 2013; Accepted: 09 September 2013;
Published online: 24 September 2013.
Edited by:
Laura Santambrogio, Albert Einstein College of Medicine, USAReviewed by:
Natalio Garbi, University of Bonn, GermanyJanice Blum, Indiana University School of Medicine, USA
Copyright: © 2013 Manoury. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bénédicte Manoury, Institut National de la Santé et de la Recherche Médicale, U1013, Hôpital Necker, 149 rue de Sèvres, 75015 Paris, France e-mail:YmVuZWRpY3RlLm1hbm91cnlAaW5zZXJtLmZy