


- 1Mucosal Biology Laboratory, School of Human Life Sciences, University of Tasmania, Launceston, TAS, Australia
- 2Laboratory of Experimental Immunopathology, IRCCS “de Bellis”, Castellana Grotte (BA), Italy
One of the most significant challenges of cell biology is to understand how each type of cell copes with its specific workload without suffering damage. Among the most intriguing questions concerns intestinal epithelial cells in mammals; these cells act as a barrier between the internally protected region and the external environment that is exposed constantly to food and microbes. A major process involved in the processing of microbes is autophagy. In the intestine, through multiple, complex signaling pathways, autophagy including macroautophagy and xenophagy is pivotal in mounting appropriate intestinal immune responses and anti-microbial protection. Dysfunctional autophagy mechanism leads to chronic intestinal inflammation, such as inflammatory bowel disease (IBD). Studies involving a number of in vitro and in vivo mouse models in addition to human clinical studies have revealed a detailed role for autophagy in the generation of chronic intestinal inflammation. A number of genome-wide association studies identified roles for numerous autophagy genes in IBD, especially in Crohn’s disease. In this review, we will explore in detail the latest research linking autophagy to intestinal homeostasis and how alterations in autophagy pathways lead to intestinal inflammation.
Introduction
Autophagy literally means “self-eating.” Autophagy describes a process by which our cells are able to cope with damaged organelles and malformed proteins by directing them to lysosomal degradation. Autophagy may be divided into three classes; macroautophagy, macroautophagy, and chaperone-mediated autophagy (1, 2). Of these three classes, macroautophagy, or simply autophagy, as it will be referred to hereafter, is the most studied and is the form implicated in the regulation of chronic inflammation and is therefore the focus of this review. Induction of autophagy promotes cell survival during nutrient deprivation, oxidative stress, and endoplasmic reticulum (ER) stress, and is therefore critically important for the maintenance of cellular homeostasis (3–5). Since autophagy involves the degradation of bulk cytoplasm it is minimally active under normal conditions but may be readily induced by a diverse range of intracellular stressors. It is now evident that the mechanisms which target specific structures for degradation are in fact quite selective. Selective degradation of mitochondria (mitophagy), ribosomes (ribophagy), peroxisomes (pexophagy), and ER has been reported (6–9). An additional form of selective autophagy is xenophagy. Xenophagy serves to degrade invasive pathogens in a specialized form of autophagy termed xenophagy (10). Xenophagy involves the recruitment of the autophagic machinery to degrade invasive viral or bacterial pathogens. The myriad roles of autophagy in regulating homeostasis make it of particular interest in study of the intestinal mucosa, where all the aforementioned stresses are likely to converge.
The intestinal mucosa is constantly exposed to dietary and microbial antigens. The intestinal epithelium and its associated mucus layer serves as a physical barrier against the external environment. However, in order to fulfill its function in the absorption of nutrients a degree of permeability to the luminal contents is required. Naturally, not all antigens to which the intestinal mucosa is exposed are pathogenic; the correct balance between inflammation and tolerance is a complex mechanism that involves hematopoietic and non-hematopoietic cells. A form of intestinal homeostasis allowing the protection against pathogens whilst tolerating food antigen and the beneficial, non-pathogenic commensal microbes is required. The need to maintain intestinal homeostasis has seen a number of complex signaling pathways evolve between a diverse range of cell types. Deficiencies in this signaling network may result in a loss of homeostasis and in the development of chronic inflammatory diseases such as the inflammatory bowel diseases (IBD). Two chronic relapsing and remitting disorders, namely Crohn’s disease (CD) and ulcerative colitis (UC), constitute the majority of IBD cases. Both of these conditions cause severe abdominal discomfort and lead to significant morbidity and loss of quality of life. Currently no cure for IBD exists; the treatment of IBD involves the alleviation of symptoms using anti-inflammatory agents such as steroids and non-steroid anti-inflammatory drugs (NSAID). Despite intensive research, the exact pathogenesis of IBD remains elusive.
Recently, a number of studies, genome-wide association studies (GWAS) in particular, have identified a link between genes involved in autophagy regulation and IBDs. Given its importance of autophagy as a regulator of cellular homeostasis and of innate immunity, understanding the contribution of defective autophagy to the pathogenesis of chronic inflammation may be of considerable value to human health. In this review, we aim to initially discuss the general mechanisms of autophagy, and then describe in detail the link between autophagy machinery and the intestinal epithelium in gut homeostasis and inflammation.
Autophagy Mechanisms
Although autophagy has been reported since the 1960s, detailed description of the molecular mechanisms regulating autophagy has only occurred relatively recently (11). We know now that the autophagy pathway is reliant upon the proteins encoded by autophagy-related genes (Atg), the first of which, Atg1, was identified 20 years ago in the yeast Saccharomyces cerevisiae (12). Since then, over 30 Atg proteins and their functions have been identified (13, 14). The autophagy-related genes essential for the assembly of the autophagosome are highly conserved between yeasts, worms, flies, and mammals. Such high degree of conservation is presumably due to the importance autophagy in cell survival, therefore much of our knowledge of autophagy mechanisms obtained from yeast may be translated to mammalian cells. Several comprehensive reports detailing the current understanding molecular mechanisms and regulation of autophagy in physiology and disease in both yeasts and mammals already exist in the literature (15, 16). For the purpose of this review however, we will give a brief overview of the proposed general mechanisms of mammalian autophagy prior to describing the role of autophagy-regulating genes in the pathogenesis of CD.
The defining feature of macroautophagy, as opposed to the other classes of autophagy, is the formation of the double-membrane vesicle known as the “autophagosome.” The process of autophagy may be divided into several stages: induction, nucleation, elongation, endosomal/lysosomal docking and fusion with the autophagosome, and finally, degradation (Figure 1). The first of these stages, the initiation of autophagy, may occur through a range of signaling pathways, dependent upon the stimulus. The mammalian target of rapamycin complex 1 (mTORC1) appears to be the central regulator of autophagy induction. In nutrient-rich conditions mTORC1 is active, and represses autophagosome formation (Figure 2). Inactivation of mTORC1, e.g., by starvation, results in the de-repression of signaling pathways downstream of mTORC1 and results in initiation of autophagy. The importance of mTOR in autophagy stimulated by other stressors such as certain invasive pathogens however, may be limited (17). Under the control of mTORC1 is a complex composed of uncoordinated 51-like kinase 1 (ULK1; the mammalian ortholog of Atg1), Atg13, Atg101, and focal adhesion kinase family interacting protein of 200 kDa (FIP200; Atg17 ortholog) (18–20). The ULK1-Atg13-FIP200 complex is thought to be the earliest factor recruited to the autophagosome precursor. Repression of mTORC1 results in phosphorylation of Atg13 and FIP200 by ULK1 and the entire complex is relocated to the phagophore (21, 22). Activation of Atg13 and FIP200 is required for the formation of the phagophore under starvation conditions whereas ULK1 appears to be dispensable (23). It remains to be seen whether the role of ULK1 in autophagy extends beyond its kinase function. The ULK1 ortholog in yeast, Atg1, interacts with the lipid membranes of vesicles via its C-terminal domain, suggesting that it may recruit the first vesicles to the phagophore assembly site (PAS) following autophagy induction (24).
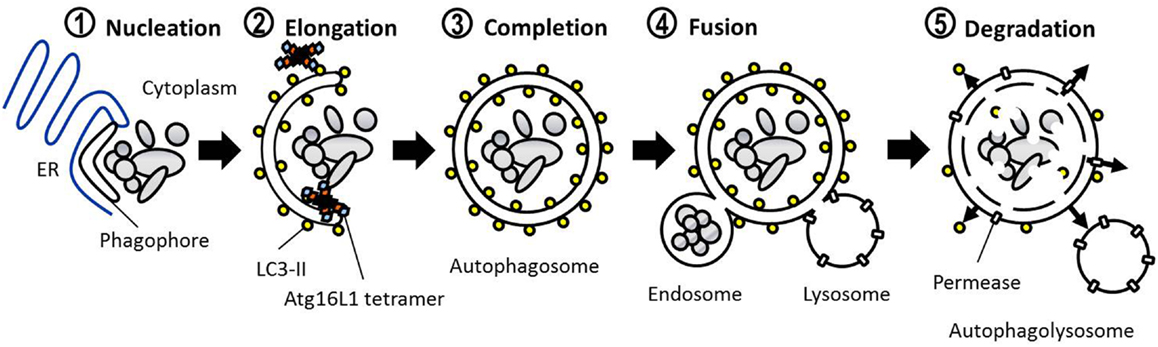
Figure 1. Basic steps involved in mammalian macroautophagy.
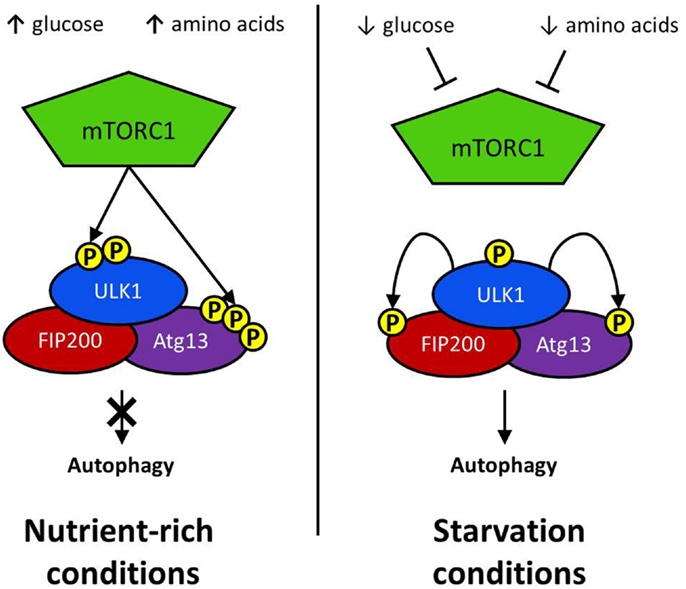
Figure 2. Initiation via ULK1 complex: the regulatory complex mTORC1 represses autophagy activation in nutrient rich conditions. mTORC1 phosphorylates a serine residue on ULK1 to prevent it interacting with positive regulators of autophagy induction. Atg13 activation is similarly repressed by mTORC1-mediated phosphorylation. Glucose or amino acid starvation results in the repression of mTOR activation. Consequently, ULK1 phosphorylates both FIP200 and Atg13, resulting in the activation of downstream autophagy effector proteins.
The second step in the autophagic process involves the formation of a phospholipid bilayer membrane known as the isolation membrane or phagophore. This early membrane structure is the precursor to the mature autophagosome membrane. The origin of the autophagosome precursor, known as the “phagophore,” is an aspect of autophagy about which little is currently known. Considerable divergence in the formation of the phagophore between mammalian and yeast cells exists. In S. cerevisiae autophagosome formation begins at a defined location known as the PAS (25). The PAS is associated with the yeast vacuole and the resultant autophagosome eventually fuses with the vacuole and the autophagosomal contents are degraded. In contrast, mammalian autophagosomes may instead form at multiple locations throughout the cell (26).
Autophagosome formation has been observed associated with various membranous structures such as the ER, plasma membrane, Golgi apparatus, and mitochondria (27–30). A growing body of evidence supports the ER as a starting point for phagophore formation in mammalian cells. Axe et al. identified a unique compartment of the ER involved in autophagosome formation marked by the presence of phosphatidylinositol-(3)-phosphate (PI (3)P)-binding double FYVE-containing protein (DFCP1), since termed the omegasome (27). Visualization of the DFCP1+ omegasome shows the formation of the phagophore surrounded by the cradle-like omegasome. Furthermore, the two membranes are directly connected suggesting that perhaps the phagophore arises from the ER (27, 31, 32). Further experiments are required to determine the precise origins of the initial isolation membrane and the functional significance of the DFCP1+ omegasome in autophagy.
An early event in the formation of the phagophore membrane is likely to be the recruitment of PI(3)P to the membrane. Although its precise role of PI(3)P in autophagy is unknown, it is a key requirement for the recruitment of many of the downstream effector proteins critical for extension and completion of the autophagosomal membrane (33). The autophagosome membrane is enriched with PI(3)P, particularly at the elongating tips of the phagophore (34, 35). Localization to these points most likely serves to recruit FYVE, PX, and WD-40 domain-containing autophagy effectors for efficient assembly of the phagophore membrane. Generation of PI(3)P requires the kinase activity of class III PI(3)K complexes to the phagophore (Figure 3). The most prominent of this class of PI(3)K complex in mammalian autophagy is the complex consisting of beclin 1 (BCN1), the PI(3)K Vps34, its regulatory subunit Vps15, and Atg14 (36). Following the localization of the ULK1 complex to the phagophore the next major step involves the recruitment of the beclin 1 complex to the phagophore. Atg14 is of critical importance in directing the beclin 1-Vps34 kinase to the phagophore due to its ability to interact with the ER membrane (35, 37, 38). Since the phagophore appears to form in close proximity to the ER membrane it is possible that the omegasome promotes autophagosome formation by recruiting PI(3)K activity to the adjacent membrane.
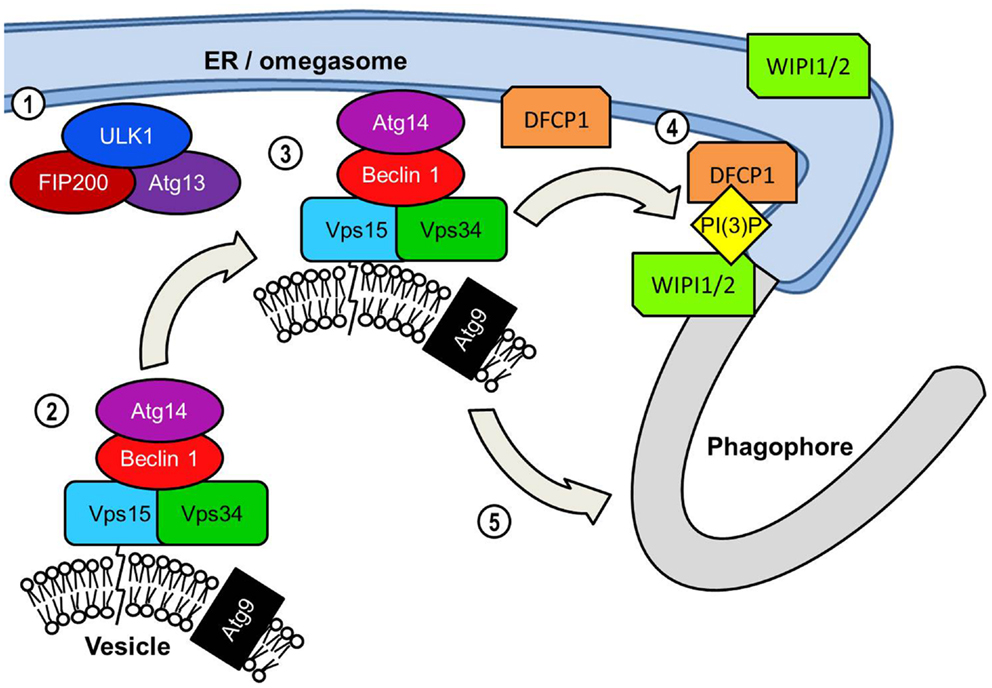
Figure 3. PI(3)K-mediated vesicle nucleation: The activated ULK1 complex translocates to the phagophore formation site. In starvation-induced autophagy this is likely to be adjacent to the ER initially but ULK1 also associates with the phagophore. ULK1 complex activation results in the translocation of the Beclin 1 lipid kinase complex to the phagophore assembly site. Beclin 1 acts as a scaffold protein forming a complex with Vps34 PI(3)K, its Vps15 subunit and Atg14. Similarly to orthologs yeast proteins the mammalian Vps15 subunit is predicted to interact with lipid membranes thereby tethering Vps34 to vesicles. Vps34 is the subunit of the Beclin1 lipid kinase complex that catalyzes the phosphorylation of the 3 region of the inositol head group of phosphatidylinositol to generate PI(3)P. The transmembrane Atg9 is found localized to post-Golgi vesicles but is depicted on the same vesicle as the Beclin 1 complex for convenience. The extent to which vesicular Beclin 1 and Atg9 overlap in their distribution is unclear. The inclusion of the Atg14 protein recruits the Beclin 1 complex specifically to the ER. This may serve two functions in assembly of the autophagosome. The first and perhaps most crucial function is the generation of PI(3)P at the omegasome and/or phagophore, which is essential for the recruitment of many effector proteins required for autophagosome assembly. PI(3)P production results in an accumulation of PI(3)P-binding protein DFCP1 at the omegasome membrane, the functional significance of which is unknown. ER-associated WIPI proteins, WIPI1 and WIPI2 also bind PI(3)P and are essential in the transition from omegasome to the double membrane phagophore.
The kinase activity of Vps34 is also required for the activation of another key step in the process of autophagosome formation. WD-40 repeat domain-containing phosophoinositide interacting proteins (WIPI) WIPI1 and WIPI2 (mammalian orthologs of the yeast Atg18) may be found localized to the ER, plasma membrane, and Golgi (39). WIPI1 and WIPI2 have also been observed in close proximity to DFCP1 suggesting that it is recruited to the omegasome or phagophore via its PI(3)P-binding domain (40–42). Cells deficient in WIPI2 displayed defective DFCP1+ omegasome formation in response to starvation (42). WIPI1 and WIPI2 are both later found localized to the LC3+ autophagosomal membrane supporting the theory that WIPI1 and WIPI2 regulate the transition of the omegasome to phagophore (39). In yeasts the WIPI ortholog, Atg18, interacts with both Atg2 and Atg9, and is involved in retrograde movement of Atg9 from the autophagosome (43). Orthologs of the yeast Atg2, mammalian Atg2A and Atg2B are known to be essential for the completion of the autophagosome in mammalian autophagy but their role is still poorly understood (44). However, Atg9 has been the subject of considerable investigation due to its unique transmembrane localization and its role in the early stages of autophagosome formation. Interaction between Atg9 and the phagophore membrane appears to be transient yet essential for autophagy (45, 46). Atg9 is likely carried on Golgi-derived secretory vesicles or on endosomes, which are trafficked between the phagophore and the trans-Golgi network (TGN) and endosomes (29, 47). The trafficking of Atg9+ vesicles from other membranous organelles and the essential role of Atg9 in autophagosome formation has led to speculation that these vesicles may transport lipids vital for extending the phagophore (48). An additional role for Atg9 has been suggested by study into autophagy-induced by Salmonella typhimurium. Kageyama et al. observed that Atg9 is required for recruitment of the PI(3)K complex to the Salmonella-containing vacuole and formation of the autophagosome (49). The observed absence of PI(3)P-binding WIPI1 from membrane-bound bacteria suggested that WIPI1 may instead function downstream of Atg9.
Elongation of the phagophore/early autophagosome membrane in yeasts requires the protein Atg8. In mammals there have been four Atg8 orthologs identified; microtubule-associated protein 1 light chain 3 (LC3), GABARAP, GATE-16, and Atg8L. Of these four proteins LC3 is the best characterized in autophagy. LC3 is unique in that, once converted from LC3-I to its lipidated form (LC3-II) it is incorporated into the elongating autophagosome and remains associated with the autophagosome even after the dissociation of other Atg proteins. Thus LC3-II serves as an accepted and widely used marker of autophagosomes (50). For the efficient processing of LC3 from pro-LC3 to LC3-II, eukaryotes have evolved a pair of ubiquitinylation-like reactions. The first ubiquitinylation-like reaction catalyzes the formation of an Atg16L1-Atg5-Atg12 complex (Figure 4) (51). The Atg7 homodimer forms a thioester bond between a cysteine residue within the Atg7 active site and the Atg12 C-terminal glycine, similar in its mechanism to an ubiquitin E1-like activating enzyme. The Atg12 C-terminal thioester bond is subsequently transferred from Atg7 to Atg10 (E2-like enzyme). Atg10 catalyzes the formation of an isopeptide bond which covalently links Atg5 to Atg12. Atg16L1 binds non-covalently to Atg5 and enables the formation of a Atg16L1-Atg5-Atg12 tetrameric complex through interaction between Atg16L1 coiled-coil domains (52). The second ubiquitinylation-like reaction involves the conversion of lipidated LC3-II from its precursor form (Figure 5). The protease Atg4 cleaves the C-terminal region of pro-LC3 immediately after the precursor is synthesized, releasing a soluble LC3-I into the cytosol (53). The proteolytic action of Atg4 exposes a C-terminal glycine residue which allows for interaction with the active site of Atg7 homodimers. Atg7 functions as a E1-like enzyme in this second ubiquitinylation-like reaction also, this time however, transferring the LC3-I C-terminal domain to Atg3 (54). Atg3 (E2-like enzyme) catalyzes the conjugation of LC3-I to the lipid phosphatidylethanolamine (PE) (55). Lipidation of LC3 is a critical step in the formation and completion of the autophagosomal membrane (56). The Atg16L1 complex may facilitate the lipidation and incorporation of LC3 into the expanding autophagsomal membrane. The Atg16L1-Atg5-Atg12 conjugate interacts with Atg3 and may also lipidate LC3 independent of Atg3 (57). The Atg16L1 complex may therefore assist in ensuring LC3 conversion occurs in close proximity to the membrane to accelerate autophagosome formation.
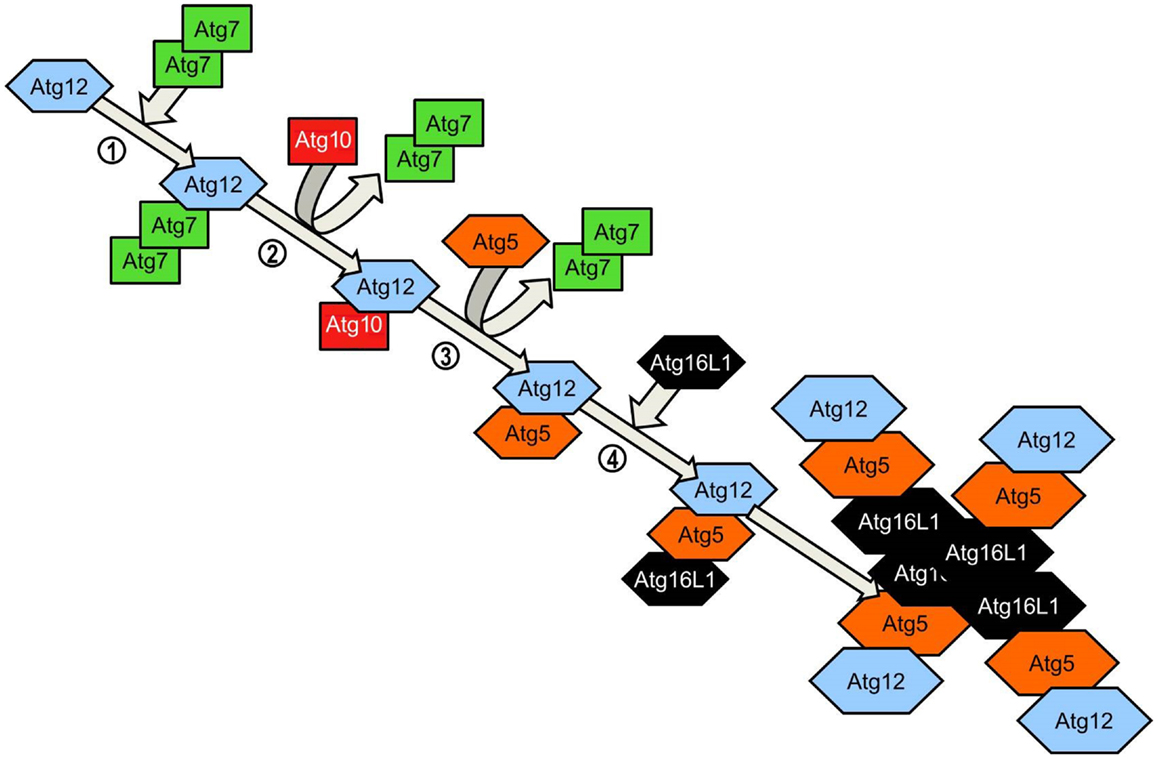
Figure 4. Atg5-Atg12 complex conjugation system. Agt12 is conjugated to Atg5 and Atg16L1 via a ubiquitinylation-like process. Homodimeric Atg7 functions as an ubiquitin-activating (E1)-like enzyme, activating Atg12 through the formation of a thioester bond between the Atg7 active site cysteine and the Atg12 C-terminal glycine. Atg7 transfers the activated Atg12 to the E2-like enzyme Atg10 via the C-terminal thioester linkage. Atg10 catalyzes the formation of an irreversible isopeptide bond between the Atg12 C-terminal glycine and Atg5. After the formation of the Atg5-Atg12 conjugate, Atg16L1 associates with the complex, binding non-covalently with Atg5. Interaction between Atg16 coiled-coil domains on adjacent Atg5-Atg12-Atg16L1 conjugates results in the formation of tetrameric complexes.
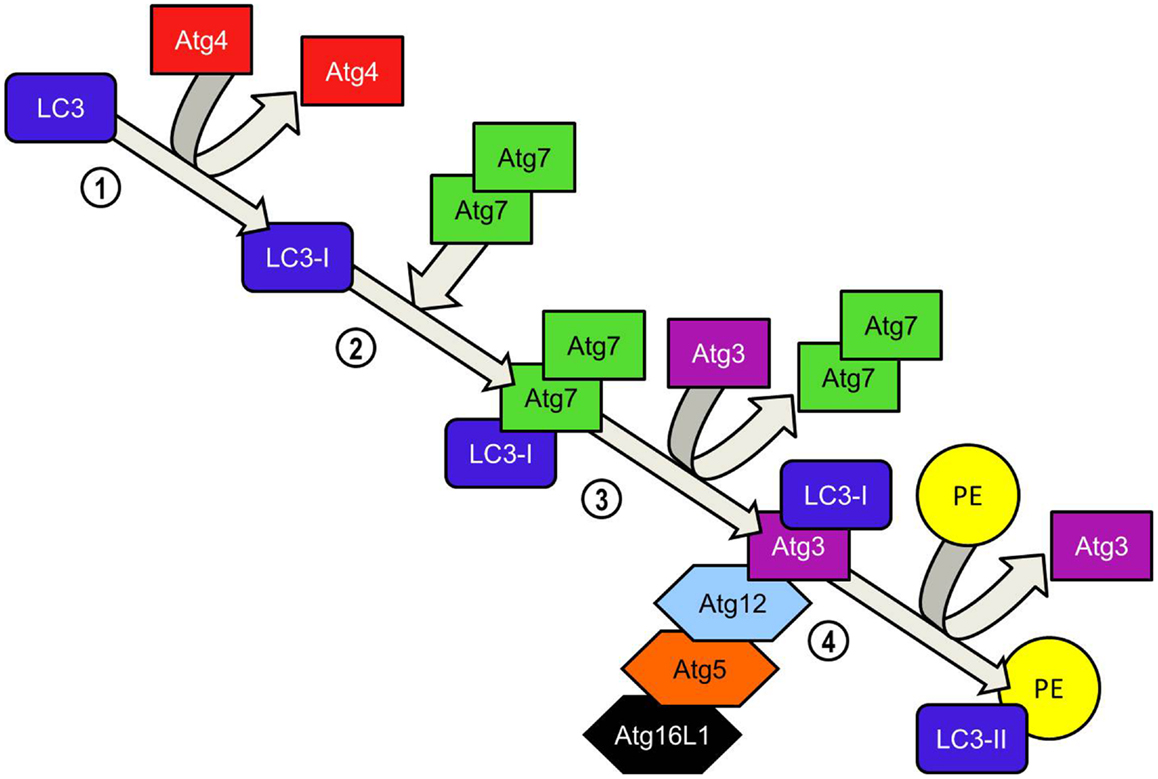
Figure 5. LC3-PE conjugation system. Atg4 cleaves the carboxyl terminal region of LC3 immediately after synthesis, generating a soluble LC3-I which now possesses a C-terminal glycine required for further modifications. Homodimeric Atg7 functions similarly to ubiquitin-activating (E1) enzymes and binds the LC3-I C-terminal exposed by Atg4 before recruiting Atg3 via its N-terminal domain. LC3-I is transferred to the Atg3 enzyme during the Atg3-Atg7 interaction. Atg7 dissociates from the LC3-I complex. LC3-II is formed when LC3-I is conjugated to the lipid phosphatidylethanolamine (PE) in a reaction catalyzed by Atg3. Binding of the Atg5-Atg12-Atg16L1 to Atg3 enhances the lipidation of LC3-I. The ability of the Atg5-Atg-12 complex to bind PE already inserted into the isolation membrane ensures that newly converted LC3-II is incorporated into the elongating isolation membrane. Unlike the formation of the Atg5-Atg12 complex, the conversion of LC3-I to LC3-II is reversible. Atg4 cleaves PE from LC3-II and LC3 is recycled.
The final stage in autophagy involves the fusion of endosomes and lysosomes with the mature autophagosome resulting in degradation and recycling of its contents. Dissociation of Atg proteins from the autophagosomal membrane may be required before fusion with endosomes or lysosomes can begin. Part of this process involves the depletion of PI(3)P associated with the autophagosome. Defective PI(3)P phosphatase activity prevents the dissociation of Atg proteins from the autophagosome and results in an accumulation of completed autophagosomes in the yeast cytoplasm (58). In mammals the fusion of endosomes with the autophagosome forms a multivesicular body (MVB), which appears to be an important process in the degradation of select protein accumulations (59). Fusion of endosomes is mediated by various proteins involved in endosomal trafficking including ESCRT, SNARE, and Rab7 proteins (59–61). The protein encoded by the ultraviolet radiation resistance-associated gene (UVRAG) regulates Rab7-mediated endosomal trafficking by forming a unique complex with beclin 1-Vps34-Vps15 (62). The association of UVRAG with beclin 1 recruits the Vps kinase complex to the membrane, thereby activating Rab7 promoting endosomal and lysosomal fusion with the autophagosome. Recent evidence suggests that LC3-II bound to the autophagosome membrane may also be a key element in Rab7-mediated endosomal/lysosomal fusion (63). Following lysosomal fusion, the inner autophagosomal membrane and its contents are degraded and amino acids transported into the cytoplasm. In contrast to yeasts which utilize Atg proteins such as Atg15 and Atg22 in the degradation and recycling process, to our knowledge no such equivalents have yet been discovered in mammalian autophagy. Instead common lysosomal proteases such as cathepsins B, D, and L are required for degradation of autophagosomal contents (64, 65). Recycling of nutrients from the degraded autophagosomal contents are presumably recycled via lysosomal permeases transferred to the autophagosomal membrane during lysosomal fusion. However, a recent study has identified a mammalian transmembrane lysosomal permease, Spinster, involved in transporting carbohydrate from the autophagosome into the cytoplasm (66). Of particular interest is the role of Spinster in the termination of autophagy, which may be marked by autophagic lysosome reformation (ALR), during which functional lysosomes are reconstituted from the autophagosome (67). Spinster is necessary for the reactivation of mTOR and the subsequent reformation of functional lysosomes from the autophagolysosome (66). The role of other lysosomal efflux transporters in signaling the termination of autophagy and its potential significance in human disease may be a promising area for future study.
Autophagy Genes Involved in Intestinal Homeostasis and Susceptibility to IBD
To date a number of polymorphisms in a wide range of genes have been identified as risk factors predisposing an individual to the development of IBD. Numerous genome-wide association scanning (GWAS) studies have attempted to identify the genes contributing to the development of CD. Two of the most prominent associations with CD have been autophagy-related gene 16-like (ATG16L1) and NOD2 (68–71). The identification of an ATG16L1 polymorphism as a genetic risk factor for CD by GWAS studies has drawn attention to the potential importance of autophagy as an etiologic factor in IBD. Since the identification of ATG16L1 as a risk factor for CD, more recent GWAS studies have implicated a number of other autophagy-related genes in susceptibility to CD (summarized in Table 1). Of particular interest in the etiology of CD are the genes which link innate immunity with autophagy such as IRGM and NOD2. It is noteworthy that autophagy appears to play a lesser role in UC. ER stress has been implicated in the development of UC and as such a strong association with XBP1 mutants and UC exists. However, one of the mechanisms which may be employed to relieve ER stress is autophagy, and as such defective XBP1 affect autophagy induction in UC. However, limited overlap in susceptibility genes between IBD does exist, with the autophagy-regulating gene, PTPN2 associated with both CD and UC (72).

Table 1. Genetics of aberrant autophagy in IBD.
ATG16L1
Several GWAS studies have linked polymorphisms in the gene encoding the mammalian ortholog of Atg16 (ATG16L1) to susceptibility to CD (69, 73). Functional ATG16L1 is crucial for the induction of autophagy. Atg5-Atg12 protein conjugates interact with Atg16L1 to form a high molecular weight protein complex essential for the elongation of the autophagosomal membrane (52, 74). Insight into the potential importance of ATG16L1 and autophagy in the pathogenesis of CD were revealed by the generation of the following two experimental models.
Atg16L1 deficiency
Due to the importance of autophagy in neonatal survival, very few mice bred with a deficiency in Atg16L1 survived 1 day post-partum (75). To examine the effect of an otherwise lethal Atg16L1 deficiency, two experimental approaches have been used. The first involves the creation of chimeric mice, in which fetal liver cells from Atg16L1-deficient mice were transferred to lethally irradiated wild-type mice. In contrast to wild-type embryonic cells, fibroblasts deficient in functional Atg16L1 failed to respond to starvation with autophagosome formation (75). Furthermore, Atg16L1-deficient fetal liver chimeric mice displayed normal bacterial colonization of the colon and no indication of spontaneous colitis (75). However, Atg16L1-deficient chimeric mice administered 5% DSS for 7 days exhibited a marked exacerbation of colitis with reduced survival.
ATG16L1 hypomorphic mice
The second model utilized gene trap mutations of the ATG16L1 gene resulting in hypomorphic gene expression. In addition to defective autophagosome assembly, ileal Paneth cells from ATG16L1 hypomorphic mice displayed abnormal exocytosis and secretion of anti-microbial peptides (76). The morphological abnormalities in Paneth cells generated by this mutation were similar to that obtained by targeted deletion of epithelial ATG5 (76). Apart from aberrant Paneth cell morphology, no histological differences could be observed between the ileum of ATG16L1 knockouts and wild-type mice. The results of these experiments strongly suggest that Paneth cells in particular are vulnerable to defects in autophagy.
The validity of these murine models is supported by observations in human patients with CD. Defective Paneth cell function in Atg16L1-deficient mice resembles the abnormalities in Paneth cell secretory granule exocytosis in patients with CD (76, 77). The function of autophagy mechanisms including Atg16L1 may therefore to extend to cytokine secretion. Pro-inflammatory stimuli such as lipopolysaccharide and muramyl dipeptide (MDP) are capable of enhancing IL-1β transcription, thereby increasing levels of intracellular pro-IL-1β (78). The major mechanism through which IL-1β regulation occurs is at the post-translational level, cleavage of pro-IL-1β to the biologically active IL-1β. Cleavage occurs through the activity of the enzyme caspase-1, which itself must first be activated by the inflammasome protein complex. The greatly enhanced IL-1β secretion in Atg16L1-deficient macrophages suggests that autophagy plays a role in modulating immune responses from these cells. Indeed autophagy has been demonstrated to regulate IL-1β secretion in vitro.
Recently, Lee et al. (79) showed that increased MDP-induced IL-1β secretion from Atg16L1-deficient PBMC was greatly diminished by type III PI3K autophagy inhibitors. Another more recent study has revealed an additional autophagy-dependent regulatory mechanism in IL-1β signaling. The protein p62 is thought to promote the secretion of IL-1β via the activation of TRAF6, an element of the IL-1β receptor signaling pathway, upstream of NF-κB (79). Degradation of p62 via Atg16L1-mediated autophagy would therefore be expected to act as a negative regulator of IL-1β signal transduction. A reduction in functional Atg16L1, such as may be expected to occur with the CD-associated ATG16L1 allele, may therefore lead to cellular hyper responsiveness to IL-1β secretion.
It seems plausible that autophagy mechanisms are required for secretion of Paneth cell granules via a non-conventional secretory pathway. Patients with CD carrying the ATG16L1 T300A risk alleles also displayed increased numbers of LC3+ autophagosomes within Paneth cells (77). Increased numbers of autophagosomes would suggest that either formation of autophagosomes is increased or alternatively, lysosomal fusion is inhibited and as a result autophagosomes accumulate in the cytoplasm. Defective ATG16L1 may therefore inhibit the completion of the autophagosome membrane by unknown mechanisms. However, Thachil and colleagues reported that neither the common ATG16L1 nor IRGM polymorphisms associated with CD, appeared to be responsible for the increased numbers of autophagosomes in Paneth cells (77). It is possible that other ATG16L1 variants exist, in the coiled-coil domain region for example, which would prevent efficient autophagosome degradation (80). Further characterization of the CD-associated ATG16L1 mutant proteins will likely explain the conflicting data. Overall, evidence still suggests that ATG16L1 mutation results in a consequent reduction in lysozyme secretion. Therefore defective autophagy could conceivably reduce the anti-microbial effectiveness of the intestinal barrier, permitting greater microbial invasion into the mucosa.
Exposure to the microbiota or an inappropriate response to the microbiota of the intestine is widely considered to be a causative factor in IBD. In DSS-induced colitis for example, inflammation may be improved by antibiotic therapy (81). Other experimental models of colitis such as T cell transfer and IL-10−/− colitis do not display overt inflammation when housed in sterile conditions. ATG16L1 hypomorphic mice, when rederived into a sterile barrier facility, displayed morphologically normal Paneth cells (82). The evidence for a microbial etiological factor was strengthened further by inoculating mice with a murine norovirus (MNV). The CR6 strain causes persistent gastrointestinal infections in mice. Inoculation of ATG16L1 hypomorphic mice with the CR6 strain reproduced the Paneth cell abnormalities observed in mice housed under less stringent conditions. Inoculation of a similar strain (CW3) that did not result in persistent infection, also failed to induce alteration to Paneth cell morphology. Interestingly when infected hypomorphic mice were challenged with a moderately high dose of DSS the resultant pathology was strikingly similar to CD. These mice exhibited transmural inflammation of the anorectal junction accompanied by mucosal lymphoid aggregates, thickening of the muscularis propria, and subserosal fibrosis (82). In addition to colonic inflammation CR6-infected Atg16L1-deficient mice also displayed ileal inflammation not observed with DSS challenge in wild-type mice. Inflammation of the ileum and colon could be attenuated by treatment with antibiotics, tumor necrosis factor-α (TNF-α), or interferon-γ (IFN-γ) blocking therapy, all of which can be used effectively in symptomatic treatment of CD. The experimental evidence suggests that chronic viral infection, when combined with epithelial injury and defective autophagy, may be the prelude to IBD.
The individual contribution of reduced functional Atg16L1 to inflammation in the MNV-DSS model remains to be elucidated. It is known that macroautophagic mechanisms are involved in immunity. It is possible that persistent enteric infections caused by some pathogens are poorly limited when macroautophagic mechanisms are deficient. However, alternative autophagy pathways may compensate in the absence of functional macroautophagy.
NOD2
The nucleotide-binding oligomerization domain-containing (NOD) pattern recognition receptors NOD1 and NOD2 are key elements in immunity against intracellular pathogens. Cytoplasmic NOD1 and NOD2 recognize cell wall components of invasive bacteria such as Shigella flexneri and Listeria monocytogenes. The association of NOD2 variants with susceptibility to CD has highlighted the role of autophagy in innate immunity. The 3020insC SNP (rs2066847) frameshift mutation in NOD2 is the most prevalent of the three identified NOD2 risk variants in patients with CD (83). This mutation produces a Nod2 protein with a truncated leucine-rich repeat (LRR) domain (84). The consequence of this mutation is a loss of Nod2 functionality. Expression of the 3020insC NOD2 variant in human cells resulted in attenuated activation of NF-κB in response to MDP or peptidoglycan (85). Not only is NF-κB activation abrogated but NOD2-mediated xenophagy is also affected.
Both NOD1 and NOD2 appear to be critical components in degradation of intracellular bacteria as part of a process referred to as xenophagy. Nod2 acts a sensor for invasive intracellular bacteria. As such Nod2 proteins have been show to accumulate adjacent to the plasma membrane after cells are co-incubated with S. flexneri (86). During S. flexneri infection, Nod2 appeared to recruit Atg16L1 to the site of bacterial entry, adjacent the plasma membrane. Mice expressing a frameshift mutation in Nod2 comparable to that of the 3020insC NOD2 SNP, exhibited marked reduction in bacterial autophagy. Lymphoblasts homozygous for the CD-associated ATG16L1 T300A allele obtained from patients with CD, displayed a marked impairment in autophagosome formation in response to the Nod2 agonist muramyl dipeptide (86). It is unclear however, whether NOD2 mutation was present in the same cells.
These findings suggest that the observed impairment in autophagy in the intestine of patients with CD may occur as a result of a combined defect in NOD2 and ATG16L1. It is noteworthy that the interaction between Nod2 and Atg16L1 appeared to be unaffected by the ATG16L1 T300A substitution associated with CD. However, this CD-associated Atg16L1 T300A variant was no different substitution. However, these cells were not genotyped for NOD2 mutations. These findings link NOD2 and ATG16L1, both genes identified as risk factors for CD, in a shared pathway with implications for the pathogenesis of CD.
The combined effect of these mutations may have profound implications for innate immunity. The role of xenophagy in the epithelium may be twofold. Firstly, degradation of intracellular bacteria such as S. flexneri, Salmonella spp., and L. monocytogenes may reduce spread of these pathogens to uninfected cells. However, the significance of autophagic clearance of intracellular bacteria in limiting infections is unclear. Secondly, autophagy is a key element in the processing of bacterial antigen for presentation to T lymphocytes (87). It therefore seems plausible that combined NOD2 and ATG16L1 mutations may result in impaired adaptive immune responses to potentially pathogenic bacteria in the gut. Conversely, the effect could conceivably result in inappropriate immune responses directed against bacterial commensals that are normally tolerated in most people. The role for a defect in innate immunity due to defective autophagy is strengthened by the association of immunity-related GTPase family M (IRGM) with CD.
IRGM
The small GTPase encoded by the human IRGM gene is thought to direct autophagy in the degradation of intracellular pathogens. So far, the only cellular proteins, which include Atg5 and Atg10, that have been shown to interact with Irgm, are all involved in the initial stages of autophagosome assembly (88). IRGM may limit viral infection directly, through autophagic clearance, and indeed a plethora of viral proteins target IRGM to disrupt autophagy and promote viral replication (88). Perhaps more important is the role of Irgm-mediated xenophagy in antigen processing and presentation to generate effective adaptive immune responses against bacteria invading the cell. Aside from antiviral immunity, IRGM is a significantly limiting intracellular bacterial infections has been well demonstrated. In mice, Irgm1 mediates IFN-γ-induced autophagy required for the degradation of intracellular mycobacteria in macrophages (89). Similarly, human THP-1 macrophages overexpressing IRGM displayed decreased numbers of intracellular adherent-invasive E. coli (AIEC) (90).
Early studies initially associated genetic variation in the IRGM region with a susceptibility to CD (68). Further analysis revealed three IRGM coding sequence variants, two of which could not be associated with CD (91). The third IRGM risk variant identified was a synonymous exonic SNP which does not result in alteration to amino acid sequence or splicing sites (92). It is a deletion polymorphism located upstream of the IRGM coding region. The effect of this deletion polymorphism was cell type-dependent, and in a colonic cell line actually increased IRGM expression compared to the reference sequence used (92). Conversely, the aforementioned synonymous IRGM variant does in fact contain an altered miRNA binding site (93). As a result, binding of regulatory miR-196 is significantly impaired. To link a novel mechanism by which IRGM expression is potentially dysregulated in CD, Brest et al. analyzed the expression and localization of IRGM and miR-196 in the colonic mucosa (93). Expression of IRGM within the colonic epithelium is high under normal conditions, but is reduced during inflammation while epithelial miR-196 expression increases. In those patients with CD possessing the IRGM risk allele, no similar reduction of IRGM occurred during active inflammation, presumably due to inability of miR-196 to bind IRGM transcript. While overexpression of IRGM results in increased autophagic activity during AIEC infection in human macrophages, epithelial cells appear to respond differently (90). Increased autophagy in IRGM overexpressing HEK293 cells appeared to encourage proliferation of AIEC within autophagic vacuoles (93).
The functional consequence of IRGM overexpression in CD remains unclear. Invasive species of E. coli, including AIEC, have been observed to colonize the ileal mucosa of patients with CD in greater numbers than normal controls (94, 95). Inefficiency in the clearance of invasive pathogens via autophagy in the intestinal mucosa of patients with CD may result in persistent infection. This is supported by in vitro evidence suggesting that impaired autophagy due to ATG16L1, NOD2 and IRGM mutation in macrophages results in increased TNF-α and IL-6 secretion (90). This could conceivably lead to exacerbation of the initial inflammation and tissue injury allowing greater bacterial penetrance into the mucosa in IBD.
PTPN2
Another gene recently identified as a risk factor in the development of IBD is T cell protein tyrosine phosphate non-receptor type 2 (PTPN2). An SNP (rs2542151) upstream of the coding region of PTPN2 has been associated with susceptibility to both CD (70, 96) and UC (72). A second SNP (rs1893217) located within intron 7 of the PTPN2 gene has also been associated with CD (97, 98). The protein encoded by PTPN2 appears to be an important negative regulator of signaling induced by pro-inflammatory cytokines such as IFN-γ and IL-6 through its ability to dephosphorylate STAT1 and STAT3 in vitro (99). Ptpn2 knock-out mice display severe defects in T cell development and from an early age develop a lethal systemic inflammation, with high serum IFN-γ and TNF-α (100–102). It is interesting to note that Ptpn2+ mice display greater susceptibility to DSS than wild-type Ptpn2 mice, suggesting that Ptpn2 negatively regulates inflammation (103). Expression of PTPN2 in human T84 colonocytes is enhanced by IFN-γ and TNF-α and accordingly PTPN2 mRNA levels are increased in the inflamed mucosa of patients with CD (104, 105). Since the link between defective autophagy and CD was identified, a potential role for PTPN2 in the regulation of autophagy has also been investigated. Autophagy in wild-type T84 colonic epithelial cells was induced by 24 h challenge with IFN-γ and TNF-α, indicated by increased LC3B-II protein and LC3B-labeled puncta (106). Knock-down of PTPN2 with specific siRNA in the T84 cell line increased both mTOR phosphorylation and beclin 1 expression but simultaneously abrogated IFN-γ/TNF-α-induced up-regulation of Atg5 and Atg7. Consequently, fewer larger LC3B-labeled autophagosomes were observed following PTPN2 siRNA knock-down, suggesting autophagosome formation was defective in PTPN2 deficient cells. To assess the contribution of the rs2542151 PTPN2 variant to the autophagy impairment observed in CD, Scharl et al. cultured lamina propria fibroblasts from the colon of patients with CD. Stimulation of fibroblasts with both IFN-γ and TNF-α increased autophagosome formation in wild-type PTPN2 cells but this effect was absent in fibroblasts carrying the PTPN2 CD-associated variant. The mechanisms by which PTPN2 influences autophagy remain unclear. It has been suggested that PTPN2 exerts its autophagy-promoting effect by repressing mTOR activation via the EGFR-PI(3)K-AKT pathway following its activation by IFN-γ and TNF-α (107).
The role of PTPN2 in coupling autophagy to pro-inflammatory signaling may mean that PTPN2 is crucial in maintaining the integrity of the intestinal barrier against pathogens. Scharl et al., showed that PTPN2 mutation or deficiency reduced the localization of L. monocytogenes to autophagosomes in colonic epithelial cells or fibroblasts (107). Whether the inclusion and/or clearance of L. monocytogenes, or other invasive bacteria such as AIEC, in autophagosomes in PTPN2 deficient cells is affected by IFN-γ or TNF-α is yet to be determined. It is plausible therefore, that PTPN2 risk allele associated with CD, in combination with ATG16L1 and NOD2 mutants, permits greater persistence and spread of intracellular pathogens, possibly contributing to a an increased susceptibility to chronic inflammation (76, 82). Aside from the role of PTPN2 in handling intracellular bacteria, PTPN2 may also regulate the permeability of the intestinal barrier. Deficiency of PTPN2 in T84 colonocytes increased the permeability of in vitro epithelial monolayers to ionic and macromolecular flux. Increased permeability was associated with a reduction in the expression of occludin and zona-occludens-1, proteins typically associated with epithelial tight junctions and key regulators of paracellular permeability. Recent evidence indicates a role for autophagy in the regulation of cell–cell adhesion in human intestinal epithelial cells stimulated with TNF-α. Pharmacological inhibition of TNF-α-induced autophagy resulted in a reduction in the protein levels of functional integrin β1 subunit (108). It is possible therefore that autophagy is responsible for the removal and recycling of cellular adhesion and tight junction proteins to facilitate regeneration of the intestinal epithelium after acute inflammation. Determining the full extent to which the CD-associated PTPN2 mutation and the resultant autophagic defect contributes to chronic inflammation promises to yield valuable insight into the cellular physiology of the intestine in both health and chronic disease.
XBP1
XBP1 is induced by ER stress and forms part of the unfolded protein response. One of the mechanisms used to regulate ER stress induced by misfolded protein in the ER is autophagy (9). The identification of two XBP1 SNPs associated with CD and UC suggests a link between IBD and the ER stress response (109). The two XBP1 variants were associated with hypomorphic XBP1 expression indicating that the XBP1-mediated ER stress response by is defective in patients with IBD.
Knock-out mice generated with an XBP1 deletion restricted to the epithelial cells of the large and small intestine spontaneously develop mild small intestinal inflammation. XBP1−/− Paneth cells and to a lesser extent, goblet cells, undergo ER stress and consequently undergo apoptosis. ER stress induced by XBP1 deletion resulted in the depletion of Paneth cells in the small intestine but not in the colon. However, XBP1−/− mice were vulnerable to the induction of DSS-colitis. XBP1−/− mice displayed wasting, rectal bleeding, mucosal erosion, and leukocyte inflammation to a greater extent than did wild-type mice following 5 days of 4.5% DSS-administration (109). The consequence of hypomorphic XBP1 mutation in human IBD patients may therefore result in impaired anti-microbial activity of Paneth cells. When combined with the aforementioned ATG16L1 mutation in CD, defective XBP1 may further promote deleterious interactions with the intestinal microbiota.
ULK1
Following the identification of autophagy-related genes such as ATG16L1, NOD2, and IRGM a GWAS study was conducted to identify additional risk factors among known autophagy genes. An SNP in one these candidate genes, ULK1, was significantly more frequent among patients with CD (110). Mammalian ULK1 is homologous to the yeast Atg1 and its expression is ubiquitous in most tissues. Together with Atg13, and FIP200, ULK1 is an essential member of a large protein complex whose activation initiates the formation of the autophagosome. Autophagy induction under conditions of high glucose abundance is repressed by interaction between the autophagy regulator mTORC1 and ULK1, preventing phosphorylation of the latter. During glucose starvation, ULK1 associates instead with AMPK, resulting in phosphorylation of ULK1 and subsequent activation of autophagy (19, 111).
To best assess the biological function of ULK1 in vivo, a knock-out mouse model was developed. Interestingly, ULK1−/− mice exhibited normal autophagy in response to nutrient deprivation suggesting that autophagy induction may still occur through Ulk2 signaling (112). The effect of ULK1 deletion in autophagy-induced by other mechanisms however is yet to be determined. Invasion of intestinal epithelial cells by bacterial pathogens such as AIEC may be facilitated by impairment ULK, thereby promoting persistent intestinal inflammation.
LRRK2
A polymorphism in the LRR kinase 2 gene (LRRK2) has been associated with susceptibility to CD (70, 71). However, knowledge of its contribution to the pathogenesis of IBD is limited. The expression of LRRK2 is predominantly limited to monocytes, dendritic cells, and lymphocytes (113). Furthermore LRRK2 expression can be up regulated in human PBMCs through IFN-γ stimulation. Since IFN-γ dominates the mucosal cytokine profile of patients with CD (114), it follows that LRRK2 expression is higher in the inflamed mucosa of patients with CD relative to non-inflamed regions (113). Although mutations in LRRK2 are known to confer risk of developing CD and also Parkinson’s disease, little evidence for its pathological function exists. However, LRRK2 has relatively recently been implicated in the regulation of autophagic pathways. LRRK2 co-localized with autophagolysosomes and multivesicular bodies, both structures formed by autophagy (115). In contrast to the other autophagy-related genes associated with CD, LRRK2 appears to function as a negative regulator of autophagy. Cells transfected with siRNA specific for LRRK2 exhibited enhanced basal autophagic activity, and enhanced starvation-induced autophagy (115). In neuronal cells, poorly controlled autophagy can result in autophagic stress. Neuronal cells transfected with Parkinson’s disease-associated LRRK2 variants exhibited increased autophagic vacuole formation and accumulation of protein aggregates (116). However, the mutations associated with both Parkinson’s and CD is quite different. Mutation in the LRRK2 kinase domain is associated with Parkinson’s disease whereas the CD-associated LRRK2 variant results in less functional protein (117). To assess the functional consequences of the CD-associated mutant an experimental animal model was utilized. C57BL/6 mice generated with Lrrk2 deficiency did not develop spontaneous intestinal pathology (117). However, following 8 days of DSS-administration, Lrrk2−/− mice exhibited more severe colitis than mice with functional Lrrk. Notably, the pathology induced by DSS in Lrrk2−/− mice was penetrated into the muscle layer in a similar fashion to CD lesions. Liu et al. identified a potential role for LRRK2 in regulating pro-inflammatory cytokine secretion that likely contributes to the pathology of CD. However, the effect of reduced LRRK2 on autophagy, and the mechanism through which it occurs, remains to be investigated in this model.
The bulk of our understanding of autophagy mechanisms is derived from experimental work performed using starvation-induced autophagy as a model. It is assumed that the general mechanisms of autophagy will be conserved for the most part among autophagic processes induced by other stimuli. Naturally, alternative autophagy pathways have been observed. For example, the induction of autophagy by invasive bacteria may occur through the mTORC1 pathway, or independently of this pathway depending on bacterial species (17). It is noteworthy that starvation-induced autophagy also contributes to host defense in the event of infection with Shigella or Salmonella species (118). The mechanisms for starvation-induced autophagy, which may be somewhat non-specific and those regulating degradation of specific proteins and intracellular structures may also differ. Given the involvement of autophagy in a broad spectrum of cellular processes, it is likely that a wide range of receptors specific for various proteins and organelles exists. Several receptors mediating the targeting of protein aggregates to the internal membrane of the autophagosome have already been identified, perhaps the most prominent being p62, and more receptors are likely to be identified in the future (119).
Conclusions and Future Directions
In this review, we have discussed in detail, the most important gene pathways that have been shown to be important in mediating autophagy in intestinal homeostasis. Research work in the area of autophagy suggests that autophagy is not only an important player in the pathogenesis of IBD but also a pathway that merges intestinal innate immune sensing, microbial immune response, and ER stress response. In all the above mentioned cellular responses the intestinal epithelium, including the goblet cells and Paneth cells, seems to orchestrate major signaling events. While current studies are not conclusive about the inter-mingled pathways connecting innate immunity, autophagy, and ER stress, future studies will unravel the common pathways. While the overall gene mutations/polymorphisms associated with autophagy and their importance in predisposition to sub types of IBD are understood, the exact triggers for defective autophagy are not understood yet. For example, mice hypomorphic for Atg16L1 need a viral infection trigger for inducing colitis-like pathology. Future research work will focus on identifying specific environmental triggers that elicit defective autophagy responses in disease.
While much of our understanding of autophagy mechanisms comes from in vitro systems using either yeast or mammalian cell lines, the significance of autophagy in pathology has mostly been derived from experimental animal models. Using animals to model defective autophagy mechanisms may however, introduce an additional layer of complexity in our attempts to understand cell type-specific defects in autophagy. Autophagy appears to be ubiquitous through virtually all cell types and tissues and is employed in a wide range of functions. The mammalian intestinal mucosa includes several types of structural cells in addition to a large concentration of resident immune cells. Functions such as maintaining cytoplasmic stores of amino acids and removal of defective mitochondria are likely to be employed by most cell types. However autophagy is also involved in more specialized processes which are restricted to certain types of other cells. For example, autophagic mechanisms contribute to the secretion of anti-microbial peptides and cytokines. Mutation of a single autophagy gene may therefore be predicted to result in a broad spectrum of pathological phenotypes across the various cells of a single tissue. Considering the ubiquity of autophagy throughout human tissues future attempts to modulate pathologic immune responses caused in part by dysfunctional autophagy, may be difficult. Investigating the contribution of dysfunctional autophagy in each cell type of the intestinal mucosa may therefore be necessary. Nevertheless, autophagy and its key role in regulating homeostasis in the intestinal mucosa make it an attractive target for exploring future avenues for therapy. Given the rapidly growing interest in the role autophagy in human disease it seems likely that the next few years will bring about a wealth of exciting new research findings in IBD and autoimmune disease in general.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kaushik S, Cuervo AM. Chaperones in autophagy. Pharmacol Res (2012) 66(6):484–93. doi: 10.1016/j.phrs.2012.10.002
2. Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy (2011) 7(7):673–82. doi:10.4161/auto.7.7.14733
3. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature (2004) 432(7020):1032–6. doi:10.1038/nature03029
4. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol (2005) 169(3):425–34. doi:10.1083/jcb.200412022
5. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol (2006) 26(24):9220–31. doi:10.1128/MCB.01453-06
6. Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys (2007) 462(2):245–53. doi:10.1016/j.abb.2007.03.034
7. Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol (2008) 10(5):602–10. doi:10.1038/ncb1723
8. Hutchins MU, Veenhuis M, Klionsky DJ. Peroxisome degradation in Saccharomyces cerevisiae is dependent on machinery of macroautophagy and the Cvt pathway. J Cell Sci (1999) 112(Pt 22):4079–87.
9. Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol (2006) 4(12):e423. doi:10.1371/journal.pbio.0040423
10. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469(7330):323–35. doi:10.1038/nature09782
11. Deter RL, Baudhuin P, De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol (1967) 35(2):C11–6. doi:10.1083/jcb.35.2.C11
12. Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett (1993) 333(1–2):169–74. doi:10.1016/0014-5793(93)80398-E
13. Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol (2008) 3:427–55. doi:10.1146/annurev.pathmechdis.2.010506.091842
14. Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem (2011) 80:125–56. doi:10.1146/annurev-biochem-052709-094552
15. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev (2010) 90(4):1383–435. doi:10.1152/physrev.00030.2009
16. Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol (2009) 335:1–32. doi:10.1007/978-3-642-00302-8_1
17. Zullo AJ, Lee S. Mycobacterial induction of autophagy varies by species and occurs independently of mammalian target of rapamycin inhibition. J Biol Chem (2012) 287(16):12668–78. doi:10.1074/jbc.M111.320135
18. Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol (2008) 181(3):497–510. doi:10.1083/jcb.200712064
19. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13(2):132–41. doi:10.1038/ncb2152
20. Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy (2009) 5(5):649–62. doi:10.4161/auto.5.5.8249
21. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell (2009) 20(7):1992–2003. doi:10.1091/mbc.E08-12-1249
22. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell (2009) 20(7):1981–91. doi:10.1091/mbc.E08-12-1248
23. Alers S, Loffler AS, Paasch F, Dieterle AM, Keppeler H, Lauber K, et al. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy (2011) 7(12):1423–33. doi:10.4161/auto.7.12.18027
24. Ragusa MJ, Stanley RE, Hurley JH. Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell (2012) 151(7):1501–12. doi:10.1016/j.cell.2012.11.028
25. Suzuki K, Ohsumi Y. Current knowledge of the pre-autophagosomal structure (PAS). FEBS Lett (2010) 584(7):1280–6. doi:10.1016/j.febslet.2010.02.001
26. Chen Y, Klionsky DJ. The regulation of autophagy – unanswered questions. J Cell Sci (2011) 124(Pt 2):161–70. doi:10.1242/jcs.064576
27. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol (2008) 182(4):685–701. doi:10.1083/jcb.200803137
28. Moreau K, Rubinsztein DC. The plasma membrane as a control center for autophagy. Autophagy (2012) 8(5):861–3. doi:10.4161/auto.20060
29. Yen WL, Shintani T, Nair U, Cao Y, Richardson BC, Li Z, et al. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J Cell Biol (2010) 188(1):101–14. doi:10.1083/jcb.200904075
30. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell (2010) 141(4):656–67. doi:10.1016/j.cell.2010.04.009
31. Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy (2009) 5(8):1180–5. doi:10.4161/auto.5.8.10274
32. Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol (2009) 11(12):1433–7. doi:10.1038/ncb1991
33. Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem (2000) 275(2):992–8. doi:10.1074/jbc.275.2.992
34. Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, et al. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J (2000) 19(17):4577–88. doi:10.1093/emboj/19.17.4577
35. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol (2009) 11(4):468–76. doi:10.1038/ncb1854
36. Volinia S, Dhand R, Vanhaesebroeck B, MacDougall LK, Stein R, Zvelebil MJ, et al. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J (1995) 14(14):3339–48.
37. Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol (2010) 190(4):511–21. doi:10.1083/jcb.200911141
38. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol (2009) 11(4):385–96. doi:10.1038/ncb1846
39. Proikas-Cezanne T, Robenek H. Freeze-fracture replica immunolabelling reveals human WIPI-1 and WIPI-2 as membrane proteins of autophagosomes. J Cell Mol Med (2011) 15(9):2007–10. doi:10.1111/j.1582-4934.2011.01339.x
40. Baskaran S, Ragusa MJ, Hurley JH. How Atg18 and the WIPIs sense phosphatidylinositol 3-phosphate. Autophagy (2012) 8(12):1851–2. doi:10.4161/auto.22077
41. Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy (2010) 6(6):764–76. doi:10.4161/auto.6.6.12709
42. Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy (2010) 6(4):506–22. doi:10.4161/auto.6.4.11863
43. Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell (2004) 6(1):79–90. doi:10.1016/S1534-5807(03)00402-7
44. Velikkakath AK, Nishimura T, Oita E, Ishihara N, Mizushima N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell (2012) 23(5):896–909. doi:10.1091/mbc.E11-09-0785
45. Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol (2010) 190(6):1005–22. doi:10.1083/jcb.200912089
46. Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell (2012) 23(10):1860–73. doi:10.1091/mbc.E11-09-0746
47. Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci (2006) 119(Pt 18):3888–900. doi:10.1242/jcs.03172
48. Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol (2012) 198(2):219–33. doi:10.1083/jcb.201202061
49. Kageyama S, Omori H, Saitoh T, Sone T, Guan JL, Akira S, et al. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol Biol Cell (2011) 22(13):2290–300. doi:10.1091/mbc.E10-11-0893
50. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy (2012) 8(4):445–544. doi:10.4161/auto.19496
51. Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem (1998) 273(51):33889–92. doi:10.1074/jbc.273.51.33889
52. Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci (2003) 116(Pt 9):1679–88. doi:10.1242/jcs.00381
53. Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL. A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J Biol Chem (2003) 278(51):51841–50. doi:10.1074/jbc.M308762200
54. Taherbhoy AM, Tait SW, Kaiser SE, Williams AH, Deng A, Nourse A, et al. Atg8 transfer from Atg7 to Atg3: a distinctive E1-E2 architecture and mechanism in the autophagy pathway. Mol Cell (2011) 44(3):451–61. doi:10.1016/j.molcel.2011.08.034
55. Nemoto T, Tanida I, Tanida-Miyake E, Minematsu-Ikeguchi N, Yokota M, Ohsumi M, et al. The mouse APG10 homologue, an E2-like enzyme for Apg12p conjugation, facilitates MAP-LC3 modification. J Biol Chem (2003) 278(41):39517–26. doi:10.1074/jbc.M300550200
56. Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell (2008) 19(11):4762–75. doi:10.1091/mbc.E08-03-0309
57. Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem (2007) 282(52):37298–302. doi:10.1074/jbc.C700195200
58. Cebollero E, van der Vaart A, Zhao M, Rieter E, Klionsky DJ, Helms JB, et al. Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr Biol (2012) 22(17):1545–53. doi:10.1016/j.cub.2012.06.029
59. Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol (2007) 179(3):485–500. doi:10.1083/jcb.200702115
60. Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell (2012) 151(6):1256–69. doi:10.1016/j.cell.2012.11.001
61. Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci (2004) 117(Pt 13):2687–97. doi:10.1242/jcs.01114
62. Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol (2008) 10(7):776–87. doi:10.1038/ncb1740
63. Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol (2010) 188(2):253–69. doi:10.1083/jcb.200907015
64. Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am J Pathol (2005) 167(6):1713–28. doi:10.1016/S0002-9440(10)61253-9
65. Tatti M, Motta M, Di Bartolomeo S, Scarpa S, Cianfanelli V, Cecconi F, et al. Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts. Hum Mol Genet (2012) 21(23):5159–73. doi:10.1093/hmg/dds367
66. Rong Y, McPhee CK, Deng S, Huang L, Chen L, Liu M, et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci U S A (2011) 108(19):7826–31. doi:10.1073/pnas.1013800108
67. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature (2010) 465(7300):942–6. doi:10.1038/nature09076
68. Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature (2007) 447(7145):661–78. doi:10.1038/nature05911
69. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2007) 39(2):207–11. doi:10.1038/ng1954
70. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet (2008) 40(8):955–62. doi:10.1038/ng.175
71. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet (2010) 42(12):1118–25. doi:10.1038/ng.717
72. Glas J, Wagner J, Seiderer J, Olszak T, Wetzke M, Beigel F, et al. PTPN2 gene variants are associated with susceptibility to both Crohn’s disease and ulcerative colitis supporting a common genetic disease background. PLoS ONE (2012) 7(3):e33682. doi:10.1371/journal.pone.0033682
73. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39(5):596–604. doi:10.1038/ng2032
74. Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J (1999) 18(14):3888–96. doi:10.1093/emboj/18.14.3888
75. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456(7219):264–8. doi:10.1038/nature07383
76. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456(7219):259–63. doi:10.1038/nature07416
77. Thachil E, Hugot JP, Arbeille B, Paris R, Grodet A, Peuchmaur M, et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn’s disease. Gastroenterology (2012) 142(5):1097e–9e. doi:10.1053/j.gastro.2012.01.031
78. Plantinga TS, Crisan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, et al. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut (2011) 60(9):1229–35. doi:10.1136/gut.2010.228908
79. Lee J, Kim HR, Quinley C, Kim J, Gonzalez-Navajas J, Xavier R, et al. Autophagy suppresses interleukin-1beta (IL-1beta) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J Biol Chem (2012) 287(6):4033–40. doi:10.1074/jbc.M111.280065
80. Jiang T, Qin B, He J, Lin S, Ding S. Three isoforms of the Atg16L1 protein contribute different autophagic properties. Mol Cell Biochem (2013) 378(1-2):257–66. doi:10.1007/s11010-013-1616-8
81. Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol (2002) 20:495–549. doi:10.1146/annurev.immunol.20.100301.064816
82. Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell (2010) 141(7):1135–45. doi:10.1016/j.cell.2010.05.009
83. Waterman M, Xu W, Stempak JM, Milgrom R, Bernstein CN, Griffiths AM, et al. Distinct and overlapping genetic loci in Crohn’s disease and ulcerative colitis: correlations with pathogenesis. Inflamm Bowel Dis (2011) 17(9):1936–42. doi:10.1002/ibd.21579
84. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411(6837):603–6. doi:10.1038/35079114
85. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278(11):8869–72. doi:10.1074/jbc.C200651200
86. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11(1):55–62. doi:10.1038/ni.1823
87. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med (2010) 16(1):90–7. doi:10.1038/nm.2069
88. Petkova DS, Viret C, Faure M. IRGM in autophagy and viral infections. Front Immunol (2012) 3:426. doi:10.3389/fimmu.2012.00426
89. Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science (2006) 313(5792):1438–41. doi:10.1126/science.1129577
90. Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol (2012) 14(6):791–807. doi:10.1111/j.1462-5822.2012.01768.x
91. Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet (2007) 39(7):830–2. doi:10.1038/ng2061
92. McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet (2008) 40(9):1107–12. doi:10.1038/ng.215
93. Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet (2011) 43(3):242–5. doi:10.1038/ng.762
94. Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology (2004) 127(2):412–21. doi:10.1053/j.gastro.2004.04.061
95. Martinez-Medina M, Aldeguer X, Lopez-Siles M, Gonzalez-Huix F, Lopez-Oliu C, Dahbi G, et al. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis (2009) 15(6):872–82. doi:10.1002/ibd.20860
96. Fisher SA, Tremelling M, Anderson CA, Gwilliam R, Bumpstead S, Prescott NJ, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat Genet (2008) 40(6):710–2. doi:10.1038/ng.145
97. Amre DK, Mack DR, Morgan K, Israel D, Deslandres C, Seidman EG, et al. Susceptibility loci reported in genome-wide association studies are associated with Crohn’s disease in Canadian children. Aliment Pharmacol Ther (2010) 31(11):1186–91. doi:10.1111/j.1365-2036.2010.04294.x
98. Scharl M, Mwinyi J, Fischbeck A, Leucht K, Eloranta JJ, Arikkat J, et al. Crohn’s disease-associated polymorphism within the PTPN2 gene affects muramyl-dipeptide-induced cytokine secretion and autophagy. Inflamm Bowel Dis (2012) 18(5):900–12. doi:10.1002/ibd.21913
99. McCole DF. Regulation of epithelial barrier function by the inflammatory bowel disease candidate gene, PTPN2. Ann N Y Acad Sci (2012) 1257:108–14. doi:10.1111/j.1749-6632.2012.06522.x
100. Heinonen KM, Nestel FP, Newell EW, Charette G, Seemayer TA, Tremblay ML, et al. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood (2004) 103(9):3457–64. doi:10.1182/blood-2003-09-3153
101. You-Ten KE, Muise ES, Itie A, Michaliszyn E, Wagner J, Jothy S, et al. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J Exp Med (1997) 186(5):683–93. doi:10.1084/jem.186.5.683
102. Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Invest (2011) 121(12):4758–74. doi:10.1172/JCI59492
103. Hassan SW, Doody KM, Hardy S, Uetani N, Cournoyer D, Tremblay ML. Increased susceptibility to dextran sulfate sodium induced colitis in the T cell protein tyrosine phosphatase heterozygous mouse. PLoS ONE (2010) 5(1):e8868. doi:10.1371/journal.pone.0008868
104. Scharl M, McCole DF, Weber A, Vavricka SR, Frei P, Kellermeier S, et al. Protein tyrosine phosphatase N2 regulates TNFalpha-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut (2011) 60(2):189–97. doi:10.1136/gut.2010.216606
105. Scharl M, Paul G, Weber A, Jung BC, Docherty MJ, Hausmann M, et al. Protection of epithelial barrier function by the Crohn’s disease associated gene protein tyrosine phosphatase n2. Gastroenterology (2009) 137(6):2030e–40e. doi:10.1053/j.gastro.2009.07.078
106. Scharl M, Wojtal KA, Becker HM, Fischbeck A, Frei P, Arikkat J, et al. Protein tyrosine phosphatase nonreceptor type 2 regulates autophagosome formation in human intestinal cells. Inflamm Bowel Dis (2012) 18(7):1287–302. doi:10.1002/ibd.21891
107. Scharl M, Rogler G. The role for protein tyrosine phosphatase nonreceptor type 2 in regulating autophagosome formation. Ann N Y Acad Sci (2012) 1257:93–102. doi:10.1111/j.1749-6632.2012.06578.x
108. Saito M, Katsuno T, Nakagawa T, Sato T, Noguchi Y, Sazuka S, et al. Intestinal epithelial cells with impaired autophagy lose their adhesive capacity in the presence of TNF-alpha. Dig Dis Sci (2012) 57(8):2022–30. doi:10.1007/s10620-012-2133-4
109. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134(5):743–56. doi:10.1016/j.cell.2008.07.021
110. Henckaerts L, Cleynen I, Brinar M, John JM, Van Steen K, Rutgeerts P, et al. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm Bowel Dis (2011) 17(6):1392–7. doi:10.1002/ibd.21486
111. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol (2010) 22(2):124–31. doi:10.1016/j.ceb.2009.11.014
112. Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood (2008) 112(4):1493–502. doi:10.1182/blood-2008-02-137398
113. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol (2010) 185(9):5577–85. doi:10.4049/jimmunol.1000548
114. Zorzi F, Monteleone I, Sarra M, Calabrese E, Marafini I, Cretella M, et al. Distinct profiles of effector cytokines mark the different phases of Crohn’s disease. PLoS ONE (2013) 8(1):e54562. doi:10.1371/journal.pone.0054562
115. Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O, et al. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet (2009) 18(21):4022–34. doi:10.1093/hmg/ddp346
116. Plowey ED, Cherra SJ III, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem (2008) 105(3):1048–56. doi:10.1111/j.1471-4159.2008.05217.x
117. Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol (2011) 12(11):1063–70. doi:10.1038/ni.2113
118. Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe (2012) 11(6):563–75. doi:10.1016/j.chom.2012.04.012
Keywords: IBD, autophagy, intestinal epithelium, ATG16L1, IRGM
Citation: Randall-Demllo S, Chieppa M and Eri R (2013) Intestinal epithelium and autophagy: partners in gut homeostasis. Front. Immunol. 4:301. doi: 10.3389/fimmu.2013.00301
Received: 08 May 2013; Accepted: 10 September 2013;
Published online: 30 September 2013.
Edited by:
Yasmin Thanavala, Roswell Park Cancer Institute, USAReviewed by:
Wim Van Den Broeck, Ghent University, BelgiumRita Carsetti, Ospedale Pediatrico Bambino Gesù, Italy
Copyright: © 2013 Randall-Demllo, Chieppa and Eri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajaraman Eri, School of Human Life Sciences, University of Tasmania, Locked Bag 1320, Launceston, TAS 7250, Australia e-mail:cmRlcmlAdXRhcy5lZHUuYXU=