
- Laboratory of Pathology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
T lymphocytes first carried foreign genes safely into humans over two decades ago. Since these pioneering studies, scientific techniques to better understand the genomic landscape of cells has directly led to a more sophisticated appreciation of the diversity, functional complexity, and therapeutic potential of T cells. Through the use of mouse models, we now know the function of the many genes that are critical for T cells to recognize foreign, mutated, or self-antigens and the factors responsible for the lineage diversification of T cells that lead to inhibitory or stimulatory immune responses. This knowledge combined with well-established modalities to introduce genes into T cells allows for the design of effector and memory CD8 and CD4 T lymphocytes specific for viral, fungal, bacterial, parasitic, and tumor-antigens and to design regulatory lymphocytes specific for the self-antigens responsible for autoimmune and inflammatory diseases. Here, I review strategies for designing the ideal T cell by introducing genes controlling (1) the secretion of cytokines/chemokines and their receptors, (2) T-cell receptor specificity, (3) chimeric-antigen receptors that enable for the recognition of surface antigens in an MHC-independent fashion, (4) co-stimulatory/inhibitory surface molecules, and (5) disease defining single-gene factors.
Introduction
The Deoxyribonucleic acid (DNA) molecule, perhaps one of biology’s greatest discoveries, helped unlock the secrets to the flow of genetic information that we now know forms the basis for the complexity of all life on earth. In the 1960s, the scientific community demonstrated for the first time that exogenous DNA could be taken up and ectopically expressed in mammalian cell lines (1). Shortly after, a growing understanding of viral reverse transcription processes and advances in recombinant DNA technologies paved the way for engineering viruses to carry therapeutic genes into cells (2).
Fast-forward 40 years and there now exists numerous viral and non-viral modalities to introduce therapeutic genes into cells. The most common viral vectors include retroviruses, adenovirus, and herpes simplex viral backbones with non-viral modalities centered on physical (DNA transfection/electroporation) or chemical (synthetic oligonucleotides, lipoplexes, nanoparticles) methods of delivery and transposon systems (3–6). Of these various modalities, gene therapy using retroviral based vectors is perhaps the most established methodology both in experimental models and in human clinical trials due to the ability to stably integrate genes into dividing cells (7–9).
In addition to the technologic advancements in gene therapy, a growing understanding of the genetic causes of human disease and the downstream function and network-like interactions between specific genes are enabling scientists to devise strategies to treat ailments once thought incurable (10, 11). While the in vivo delivery of genes targeting specific cell types remains a grand hope for the future, current methodologies readily enable for the stable introduction of foreign genes into cells ex vivo, allowing for the transfer of these cells back into patients (6).
T lymphocytes represent the ideal vehicle for carrying therapeutic genes into humans. T cells are easily obtained through peripheral blood draws or apheresis procedures and can be induced to divide robustly ex vivo, a characteristic that allows them to be highly permissible to retroviral introduction of ectopic genes (12). The first clinical trial to safely infuse a foreign gene into humans consisted of transducing tumor-infiltrating lymphocytes with a neomycin resistant gene that enabled for the detection of the transgene within a tumor biopsy several days following transfer (13). Today, the adoptive transfer of tumor-infiltrating lymphocytes combined with total-body irradiation, lymphodepleting chemotherapy, and high-dose IL-2 achieve response rates as high as 70% in patients with metastatic melanoma (14). The rapid development of gene therapy in this field promises to vastly improve current cellular therapies and opens the door to treat cancers of various histologies and wider arrays of human disease. Here, I discuss potential therapeutic genes that may improve current gene therapies, although rigorous pre-clinical testing and careful phase 1 clinical trials will be required for many of the suggestions in this review.
Cytokines, Chemokines, and Their Receptors
The theory of immune surveillance in cancer is controversial but there exists reproducible scientific data pointing to the importance of interferon-gamma as a critical mediator for the elimination of malignantly transformed cells (15). Furthermore, there is a clear increase in the incidence of cancer in patients with HIV, Immunodeficiency syndromes triggered by mutations in genes such as GATA-2 (MonoMAC) and post-transplant patients receiving immune-suppressive drugs (16, 17).
Additional support for the importance of an immune response for cancer elimination can be garnered from clinical data with robust long-term follow up showing the ability of systemic IL-2, anti-CTLA-4 antibodies, and anti-PD-1/anti-PD-L1 antibodies, and the adoptive transfer of T lymphocytes to induce tumor regression in patients with metastatic melanoma and metastatic renal cell carcinoma (18, 19). Three major factors are important for an effective immune response against cancer: (1) overcoming suppressive factors induced by mutated cancer cells within the tumor microenvironment, (2) the quality of the T cells transferred, and (3) polymorphic factors of an individual’s host immune response. Some of these factors can be readily modified by over-expressing cytokines, chemokines, and their receptors in transferred T cells, enabling lymphocytes to secrete supra-physiologic amounts of therapeutic immune-stimulatory molecules.
The IL-12/IFN-γ/TH-1 Axis
IL-12 is a hallmark inflammatory cytokine and is critical for driving an effective immunologic response against cancer and foreign pathogens (20). It is mainly produced by inflammatory cells such as dendritic cells, macrophages, and neutrophils and directly augments the functionality of multiple end effectors such as CD4+ T cells, CD8+ T cells, natural killer (NK) cells, and NKT cells (20). The anti-tumor effects of IL-12 are well documented (21). IL-12 enhances the ability of CD8+ T cells to cause the regression of large established tumors by potently stimulating the production of high-levels of IFN-γ, resulting in an increase in the cross-presentation of tumor-antigens and the reversal of suppressive functions of myeloid-derived suppressor cells, alternatively activated macrophages, and dendritic cells (22). These changes subsequently lead to the collapse of the tumor stroma and the regression of large established masses (22, 23).
Unlike activated lymph nodes stimulated by pathogen-activated molecular patterns, sterile conditions within tumors lead to low levels of IL-12 secretion by innate immune cells. This lack of a danger signal within the tumor microenvironment results in a skewing away from a Th-1 type effector immune response. One attractive approach is to increase the levels of IL-12 directly at the point of T-cell/Tumor cell and T-cell/Antigen-presenting contact within tumors (24) (Figure 1). Several studies show that over-expressing a single-chain, functionally active IL-12 gene in tumor-antigen-specific lymphocytes significantly increases the levels of IL-12 to supra-physiological levels within tumors, leading to the regression of large established masses (25–28). This modification enables for therapeutic anti-tumor immunity with smaller numbers of T cells and does not require the use of systemic gamma-chain cytokines to support the transfer of cells in vivo. Currently, clinical trials are determining if the benefits of IL-12 gene therapy outweigh the many risks associated with a systemic increase in IL-12 and IFN-γ.
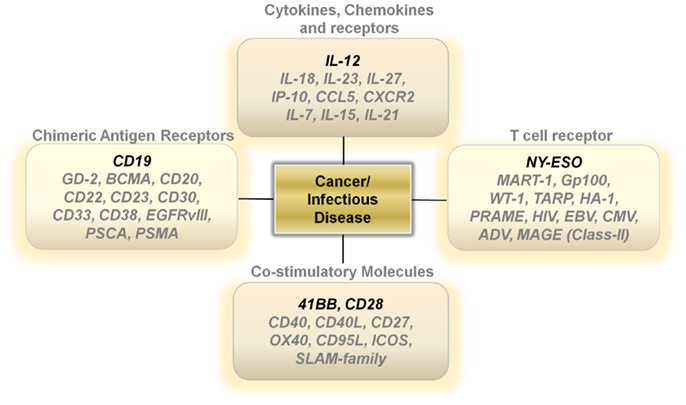
Figure 1. Potential therapeutic gene therapies in T cells for Cancer and Infectious Diseases.
Additional Cytokines, Chemokines, and Chemokine Receptors
The importance of gamma-chain cytokines in the proliferation and maintenance of memory T cells remains a critical and extremely important avenue of research for many investigators (29–32). However, clinical trials using TIL transduced with the IL-2 gene did not show a clinical benefit (33). Over-expressing the other gamma-chain cytokines such as IL-7, IL-15, or IL-21 in T cells may lead to better results. However, the constitutive expression of genes that drive T-cell proliferation carries the risk of causing an uncontrollable expansion of transferred T cells due to the stable integration of retrovirally transduced genes being expressed in every daughter cell (Figure 1). Designing viral vectors using a NFAT promoter for inducible gene expression upon T-cell receptor (TCR) ligation may provide an important degree of safety (26). Another strategy is to use adenoviral vectors or systems that only transiently express the genes that control T-cell memory formation.
Other potentially attractive cytokines include those within the IL-12 family, such as IL-23, and IL-27 (Figure 1). These cytokines may invoke beneficial downstream mechanisms for anti-tumor immunity without the heavy reliance on the induction of IFN-γ secretion. Furthermore, genetic polymorphisms within the human population may make certain individuals more likely to mount an anti-tumor response to one of the alternate members of the IL-12 family rather than IL-12 itself.
Another strategy that may turn out to be fruitful is the over-expression of chemokines and chemokine receptors in T cells (Figure 1). Melanomas can secrete chemokines such as CXCL1 and CXCL8 to aid in the recruitment of monocytes into local microenvironments and studies show that expressing the chemokine receptor CXCR2 on transferred T cells aids in the ability of T cells to infiltrate tumors and cause regression (34). This approach can be easily tailored to other tumor histologies depending on the chemokine secretion profile of the cancer. Additionally, over-expressing chemokines in T cells may also provide some benefit. Upon recognizing cognate antigens, T cells arrest their migration and accumulate at sites with productive antigen presentation. The over-expression of chemokines such as IP-10 or CCL5 in transferred T cells may enable antigen-specific T cells to secrete products that attract activated T cells to the local microenvironment they inhabit. This in turn may provide a positive feedback loop that enables for an increase in infiltrating antigen-specific T cells and an improved therapeutic outcome. Thus, the possibility to genetically alter the cytokine or chemokine profile of adoptively transferred T cells may prove to enhance and simplify current treatments requiring lymphodepletion and high-dose IL-2.
Chimeric-Antigen Receptors
The ability to generate a single fusion molecule that can bind surface antigens and trigger T-cell function holds great promise for the future of cell therapy. Chimeric-antigen receptors (CAR) are the latest form of gene therapy, where a single vector is constructed with a binding moiety recognizing a surface antigen [usually designed from a single-chain variable fragment (scFv) derived from a tumor-antigen-specific monoclonal antibody] (35–37). The beauty of CAR generated T cells is the ability to generate lymphocyte specificity in an MHC-independent fashion due to the ability to design receptors that recognize surface antigens. This is accomplished by cloning the sequences from the variable region of antibodies (many of which already exist) and adding T-cell signaling and co-stimulatory domains to the vector construct.
Early phase trials for CARs recognizing the antigen CD19, expressed on many B cell lymphomas and leukemias are showing promising results in adult and pediatric patients at multiple institutions (38–41). One of the major advantages of using CARs as the main platform for gene therapy is the ability to rapidly and clearly define the expression of the target protein. Often, the antibodies whose variable region is cloned into the CAR vector can also be used diagnostically to look for the expression of the desired target.
Other antigen targets that may be worthwhile exploring for CAR development includes GD-2 for neuroblastomas (42), CD20 (43), and CD22 for B cell lymphomas (44), BCMA (B cell maturation antigen) (45), and CD38 for multiple myeloma (46), CD23 for chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) (47), CD30 for Hodgkin’s lymphoma and anaplastic large cell lymphomas (48), CD33 for acute myeloid leukemias (49), EGFRvIII for glioblastomas (50) and PSCA (51) and PSMA (52) for prostate adenocarcinomas (Figure 1).
T-Cell Receptors
Although CAR-directed gene therapy remains a promising modality for the future, many cancers, especially carcinomas and sarcomas, do not possess known surface expression of unique non-shared antigens. Targeting surface proteins that may be expressed on normal tissue with CARs may cause serious end organ damage and toxicity. Gene therapy using high avidity TCR enables for the design of lymphocytes targeting epitopes from differentially expressed or mutated intra-nuclear and/or intra-cytoplasmic proteins such as transcription factors (22, 53). Emerging data now shows that tumor-infiltrating lymphocytes possess the ability to recognize mutated melanoma antigens (54, 55). This exciting finding opens up a large window of opportunity to develop effective TCR gene therapies. It is possible that in the future we may perform whole exome sequencing for every tumor for diagnostic purposes, enabling us to design TCR recognizing the most frequently mutated epitopes for different tumor histologies.
A great example of the success of TCR gene therapy was recently described with a clinical trial utilizing the NY-ESO TCR (56). This study led to significant tumor regression in four out of six patients with synovial sarcoma and five out of 11 patients with metastatic melanoma. Overall, the cancer-testis antigens represent an ideal set of target antigens due to their relatively low to negligible expression on normal tissue, except in the testis, where cells express low levels of MHC Class I. Identifying antigens with limited normal tissue distribution will be critical to extending TCR gene therapy to different types of cancer. Developing TCRs for breast, prostate, and thyroid cancer also seems reasonable since targeting of normal tissue in these organs might not be accompanied with serious life-threatening adverse events (Figure 1).
Co-Stimulatory Molecules
Generating both a specific and a productive T-cell response requires not only appropriate signaling through the TCR but an additional secondary co-stimulatory signal. The most well studied co-stimulatory molecule is CD28, a disulfide linked homodimer that is constitutively expressed on naive T cells (57). CD28 engagement with CD80 and CD86 on antigen-presenting cells enables T cells to differentiate and become functionally activated (57). However, after initial antigen encounter and under altered cytokine conditions, T cells can lose or decrease their expression of CD28, leading to replicative senescence and functional anergy. The lack of CD28 signaling can also result in an impaired memory response and activation induced cell death (AICD) (58). One strategy to circumvent these physiological restraints is to constitutively over-express CD28 in T cells. Currently, second and third generation CAR constructs use the intracellular domain of CD28 to improve the persistence, function, and activity of CAR transduced T cells (59). Other important co-stimulatory molecules include 41BB, CD27, OX40, CD40, CD27, ICOS, Fas ligand, and the Slam family of proteins (60, 61). These molecules all have been implicated in tipping the balance in favor of generating a functional T-cell response and helping avoid AICD during antigen re-stimulation. The intracellular domains of 41BB, OX40, and CD27 are currently being incorporated into various CAR constructs that are being developed. Thus, it is possible that the constitutive over-expression of these various co-stimulatory molecules may aid in designing long lived, functionally active T cells that are resistant to cellular senescence (Figure 1).
Severe Combined Immunodeficiency Syndromes
The first successful therapeutic gene therapy in humans in the early 1990s involved treating two children with severe combined immunodeficiency syndrome (SCID) caused by a genetic defect in the enzyme adenosine deaminase (SCID-ADA) (62). This syndrome resulted in defective T and B cells, leading to debilitating recurrent opportunistic infections. A normal/wild type ADA gene, enabling for the production of a functional enzyme, was introduced into T cells and infused back into the patient. The results were striking, and for the first time in these patients, there was evidence for IgM antibody production and the detection of tetanus antibody in the serum following immunization (62). In one patient, approximately 20% of the circulating lymphocytes still expressed the retrovirally inserted gene 10 years following transfer (63).
Although these initial results led to heightened optimism, attempts to develop gene therapies for SCID-X1, a disease characterized by a defective common gamma-c cytokine receptor subunit, by retroviral transfer of the corrected gene into CD34+ hematopoietic stem cells, led to the development of leukemias in some patients (64, 65). These setbacks sent shock waves through the scientific and medical communities. We now know that retroviral vectors can result in insertional mutagenesis, although this phenomenon still remains poorly understood (66). Five out of 20 patients treated in trials carried out in London and Paris developed leukemias secondary to the expansion of clones containing vector integration near proto-oncogenes such as CCND and LMO2 (65).
Despite the clear dangers of gamma-retroviral gene transfer into hematopoietic stem cells, transferring genes into T cells ex vivo appears to be much more resistant to oncogenic transformation. There now exists robust long-term follow up for over a 100 patients treated on various gene therapy trials utilizing ex vivo retroviral insertion of genes into T cells with no evidence of malignant transformation (9). The mechanisms for the differences in oncogenesis between transducing hematopoietic stem cells versus T cells is not well understood. Perhaps introducing genes into more differentiated cells that contain a vastly different genetic and epigenetic landscape from stem cells leads to retroviral integration away from oncogenes.
Currently, gene therapists are continuing to try to improve safety through vector design. One strategy gaining support includes creating self inactivating gamma (SIN) retroviral vectors and lentiviral vectors by deleting the U3 region in the 3′ LTR (67). This modification generates a pro-virus with defective transcriptional activity at both the 5′ and 3′ LTR end regions, preventing the possibility of transcriptionally activating cellular oncogenes near the site of viral integration. Importantly, an internal promoter will need to be designed to drive the expression of the desired transgene within the SIN vector construct. Additionally safety measures include the genetic modification of shorter lived cell populations or the use of suicide genes (68).
The current progress in improving the safety of gene therapies is helping the field move forward. In regard to SCID, although mutations in the common gamma-c chain receptor is the most common cause of the disease, a broad range of single-gene mutations can result in a similar disease pattern of recurrent opportunistic infections (Figure 2). Theoretically, for all the various defects that may occur, introducing a correctly functioning gene ex vivo into T cells or hematopoietic stem cells/monocyte/dendritic cell populations may re-capitulate the early excitement seen in the SCID-ADA trials and build on the recent successes of gene therapy for cancer.
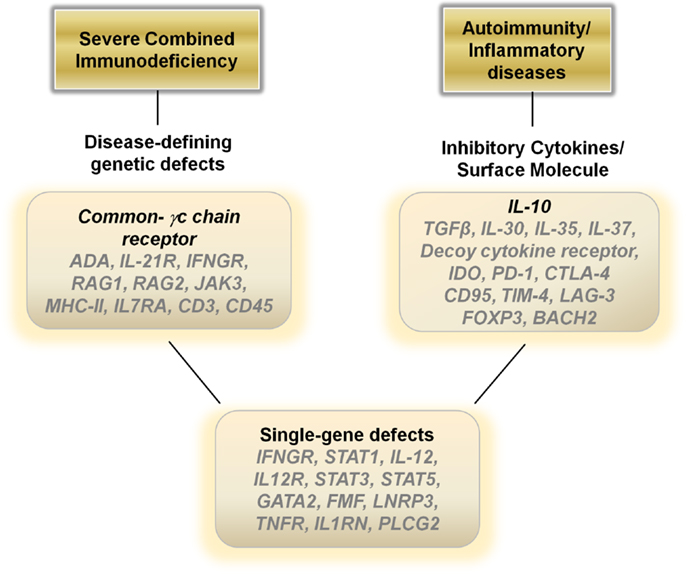
Figure 2. Potential therapeutic genes to over-express in hematopoietic stem cells, monocytes, dendritic cells or T cells for autoimmune, inflammatory, and single-gene disorders.
Autoimmune and Inflammatory Diseases
Although genetic modifications to stimulate the immune system is beneficial for battling infectious organisms and cancer, there also exists a set of devastating diseases that are caused by an over-zealous and unchecked immune response. The targeting of self-antigens under normal physiologic conditions can cause a range of serious ailments including type 1 diabetes, multiple sclerosis, rheumatoid arthritis, systemic lupus erythematosus, and autoimmune encephalomyelitis. Recently, a relatively new set of autoimmune diseases categorized as autoinflammatory diseases are beginning to be characterized such as familial Mediterranean fever (FMF), neonatal onset multisystem inflammatory disease (NOMID), tumor necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS), deficiency of the Interleukin-1 receptor antagonist (DIRA) and Behcet’s disease (69, 70). Additional inflammatory diseases that cause morbidity and mortality in a large number of patients include inflammatory bowel disease (Crohn’s disease and ulcerative colitis), chronic granulomatous disease (CGD), and the various forms of vasculitis (71, 72).
In general, dampening the immune response is the ideal treatment for autoimmune and inflammatory diseases and current therapies revolve around the use of steroids, cytokine antagonists, or directly down regulating the immune system utilizing various modalities. Gene therapies may provide a viable biological alterative to directly blunt an over-active immune response. Over-expressing anti-inflammatory cytokines such as IL-10, TGF-β, IL-30, IL-35, or IL-37 in T cells or monocyte/dendritic cell populations ex vivo with a re-infusion of the modified cells may aid in decreasing inflammatory driven symptoms (Figure 2). Another alternative may be to construct a decoy cytokine receptor that contains the correct receptor sequence to enable for binding of pro-inflammatory cytokines such as IL-12 combined with a non-functioning cytoplasmic signal transducing sequence. Over-expressing these “dominant-negative” receptors would enable re-infused immune cells to function as sinks for the inflammatory cytokines responsible for the pathophysiology of the disease. Other genes that may aid in dampening the immune response include over-expressing indoleamine 2,3-dioxygenase in monocytes/dendritic cells or CTLA-4, PD-1, CD95, LAG-3, FOXP3, and BACH2 in T cells (73) (Figure 2).
Single-Gene Defects
Although many human diseases are caused by complex genetic polymorphisms, perhaps the greatest potential for gene therapy is in the ability to treat diseases caused by single Mendelian gene defects. Mutations in genes such as IFNGR1, STAT1, IL-12, and IL-12R can lead to immune dysfunction and recurrent mycobacterial infections (74). Genetic disruptions also cause many of the autoinflammatory diseases, such as mutations in the FMF gene in FMF, the LNRP3 gene in NOMID, the TNFR gene in TRAPs, and IL1RN gene in DIRA. Inserting the corrected sequence for these genes into hematopoietic cells or more safely into differentiated immune cells may result in dramatic improvements in the health of these patients (Figure 2).
Conclusion
T lymphocytes represent one of the first vehicles to carry therapeutic genes into humans, and its current use, centered on the adoptive transfer of T cells, is proving to be a promising cancer therapeutic modality. However, logistic hurdles still exist for the wider use of this technology due to costs associated with GMP quality viral production and the requirement of significant technologic infrastructure and expertise. Increased collaboration between industry and academia for developing gene therapies may help overcome current financial limitations by developing viable business models.
There exist over 4000 known single-gene disease causing disorders in addition to the innumerable genetic polymorphisms that increase susceptibilities for diseases. Gene therapy in T cells is paving the way for a broader application of this therapeutic modality in human disease. The ability to stably introduce functional genes into hematopoietic stem cells or differentiated cells ex vivo provides hope for the thousands of patients diagnosed with a wide range of devastating genetic diseases, highlighted by recent successes in childhood cerebral adrenoleukodystrophy (75) and hemophilia B (76). Gene therapy represents the ultimate form of personalized medicine, and in the future, it is conceivable to imagine that diseases that were once considered untreatable will be readily controlled or eradicated with a single specialized treatment.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The author has no conflicting financial interests.
References
2. Varmus HE. Form and function of retroviral proviruses. Science (1982) 216(4548):812–20. doi:10.1126/science.6177038
3. Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med (2001) 7(1):33–40. doi:10.1038/83324
5. Gao X, Kim KS, Liu D. Nonviral gene delivery: what we know and what is next. AAPS J (2007) 9(1):E92–104. doi:10.1208/aapsj0901009
6. Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet (2011) 12(5):301–15. doi:10.1038/nrg2985
7. Hwu P, Rosenberg SA. The genetic modification of T cells for cancer therapy: an overview of laboratory and clinical trials. Cancer Detect Prev (1994) 18(1):43–50.
8. Brenner MK. Gene transfer and the treatment of haematological malignancy. J Intern Med (2001) 249(4):345–58. doi:10.1046/j.1365-2796.2001.00807.x
9. Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med (2012) 4(132):132ra153. doi:10.1126/scitranslmed.3003761
10. Rao MS, Collins FS. Steering a new course for stem cell research: NIH’s intramural center for regenerative medicine. Stem Cells Transl Med (2012) 1(1):15–7. doi:10.5966/sctm.2011-0032
11. Varmus H, Kumar HS. Addressing the growing international challenge of cancer: a multinational perspective. Sci Transl Med (2013) 5(175):175cm172. doi:10.1126/scitranslmed.3005899
12. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer (2013) 13(8):525–41. doi:10.1038/nrc3565
13. Rosenberg SA, Aebersold P, Cornetta K, Kasid A, Morgan RA, Moen R, et al. Gene transfer into humans – immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med (1990) 323(9):570–8. doi:10.1056/NEJM199008303230904
14. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol (2012) 12(4):269–81. doi:10.1038/nri3191
15. Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity (1994) 1(6):447–56. doi:10.1016/1074-7613(94)90087-6
16. Rosenzweig SD, Holland SM. Recent insights into the pathobiology of innate immune deficiencies. Curr Allergy Asthma Rep (2011) 11(5):369–77. doi:10.1007/s11882-011-0212-9
17. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med (2013) 368(2):161–70. doi:10.1056/NEJMra1202117
18. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature (2011) 480(7378):480–9. doi:10.1038/nature10673
19. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer (2011) 11(11):805–12. doi:10.1038/nrc3153
20. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol (2003) 3(2):133–46. doi:10.1038/nri1001
21. Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev (2002) 13(2):155–68. doi:10.1016/S1359-6101(01)00032-6
22. Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest (2011) 121(12):4746–57. doi:10.1172/JCI58814
23. Kerkar SP, Leonardi AJ, van Panhuys N, Zhang L, Yu Z, Crompton JG, et al. Collapse of the tumor stroma is triggered by IL-12 induction of Fas. Mol Ther (2013) 21(7):1369–77. doi:10.1038/mt.2013.58
24. Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res (2010) 70(17):6725–34. doi:10.1158/0008-5472.CAN-10-0735
25. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res (2011) 71(17):5697–706. doi:10.1158/0008-5472.CAN-11-0103
26. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther (2011) 19(4):751–9. doi:10.1038/mt.2010.313
27. Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res (2012) 18(6):1672–83. doi:10.1158/1078-0432.CCR-11-3050
28. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119(18):4133–41. doi:10.1182/blood-2011-12-400044
29. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17(10):1290–7. doi:10.1038/nm.2446
30. Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood (2011) 117(3):808–14. doi:10.1182/blood-2010-05-286286
31. Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res (2011) 17(16):5343–52. doi:10.1158/1078-0432.CCR-11-0503
32. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity (2011) 35(6):972–85. doi:10.1016/j.immuni.2011.09.019
33. Heemskerk B, Liu K, Dudley ME, Johnson LA, Kaiser A, Downey S, et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum Gene Ther (2008) 19(5):496–510. doi:10.1089/hum.2007.0171
34. Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res (2010) 16(22):5458–68. doi:10.1158/1078-0432.CCR-10-0712
35. Lee DW, Barrett DM, Mackall C, Orentas R, Grupp SA. The future is now: chimeric antigen receptors as new targeted therapies for childhood cancer. Clin Cancer Res (2012) 18(10):2780–90. doi:10.1158/1078-0432.CCR-11-1920
36. Riddell SR, Jensen MC, June CH. Chimeric antigen receptor – modified T cells: clinical translation in stem cell transplantation and beyond. Biol Blood Marrow Transplant (2013) 19(1 Suppl):S2–5. doi:10.1016/j.bbmt.2012.10.021
37. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov (2013) 3(4):388–98. doi:10.1158/2159-8290.CD-12-0548
38. Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood (2010) 116(19):3875–86. doi:10.1182/blood-2010-01-265041
39. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med (2011) 3(95):95ra73. doi:10.1126/scitranslmed.3002842
40. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365(8):725–33. doi:10.1056/NEJMoa1103849
41. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5(177):177ra138. doi:10.1126/scitranslmed.3005930
42. Mujoo K, Cheresh DA, Yang HM, Reisfeld RA. Disialoganglioside GD2 on human neuroblastoma cells: target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor growth. Cancer Res (1987) 47(4):1098–104.
43. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood (2012) 119(17):3940–50. doi:10.1182/blood-2011-10-387969
44. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood (2013) 121(7):1165–74. doi:10.1182/blood-2012-06-438002
45. Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res (2013) 19(8):2048–60. doi:10.1158/1078-0432.CCR-12-2422
46. Mihara K, Bhattacharyya J, Kitanaka A, Yanagihara K, Kubo T, Takei Y, et al. T-cell immunotherapy with a chimeric receptor against CD38 is effective in eliminating myeloma cells. Leukemia (2012) 26(2):365–7. doi:10.1038/leu.2011.205
47. Giordano Attianese GM, Marin V, Hoyos V, Savoldo B, Pizzitola I, Tettamanti S, et al. In vitro and in vivo model of a novel immunotherapy approach for chronic lymphocytic leukemia by anti-CD23 chimeric antigen receptor. Blood (2011) 117(18):4736–45. doi:10.1182/blood-2010-10-311845
48. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood (2009) 113(25):6392–402. doi:10.1182/blood-2009-03-209650
49. Dutour A, Marin V, Pizzitola I, Valsesia-Wittmann S, Lee D, Yvon E, et al. In vitro and in vivo antitumor effect of anti-CD33 chimeric receptor-expressing EBV-CTL against CD33 acute myeloid leukemia. Adv Hematol (2012) 2012:683065. doi:10.1155/2012/683065
50. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther (2012) 23(10):1043–53. doi:10.1089/hum.2012.041
51. Morgenroth A, Cartellieri M, Schmitz M, Günes S, Weigle B, Bachmann M, et al. Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate (2007) 67(10):1121–31. doi:10.1002/pros.20608
52. Ma Q, Safar M, Holmes E, Wang Y, Boynton AL, Junghans RP. Anti-prostate specific membrane antigen designer T cells for prostate cancer therapy. Prostate (2004) 61(1):12–25. doi:10.1002/pros.20073
53. Merhavi-Shoham E, Haga-Friedman A, Cohen CJ. Genetically modulating T-cell function to target cancer. Semin Cancer Biol (2012) 22(1):14–22. doi:10.1016/j.semcancer.2011.12.006
54. Lu YC, Yao X, Li YF, El-Gamil M, Dudley ME, Yang JC, et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol (2013) 190(12):6034–42. doi:10.4049/jimmunol.1202830
55. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19(6):747–52. doi:10.1038/nm.3161
56. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29(7):917–24. doi:10.1200/JCO.2010.32.2537
57. Shahinian A, Pfeffer K, Lee KP, Kündig TM, Kishihara K, Wakeham A, et al. Differential T cell costimulatory requirements in CD28-deficient mice. Science (1993) 261(5121):609–12. doi:10.1126/science.7688139
58. Radvanyi LG, Shi Y, Vaziri H, Sharma A, Dhala R, Mills GB, et al. CD28 costimulation inhibits TCR-induced apoptosis during a primary T cell response. J Immunol (1996) 156(5):1788–98.
59. Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res (2006) 66(22):10995–1004. doi:10.1158/0008-5472.CAN-06-0160
60. Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, et al. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Sci Transl Med (2010) 2(55):55ra78. doi:10.1126/scitranslmed.3000448
61. Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood (2012) 119(3):696–706. doi:10.1182/blood-2011-03-344275
62. Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science (1995) 270(5235):475–80. doi:10.1126/science.270.5235.475
63. Muul LM, Tuschong LM, Soenen SL, Jagadeesh GJ, Ramsey WJ, Long Z, et al. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood (2003) 101(7):2563–9. doi:10.1182/blood-2002-09-2800
64. Cavazzana-Calvo M, Hacein-Bey S, de SaintBasile G, Gross F, Yvon E, Nusbaum P, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science (2000) 288(5466):669–72. doi:10.1126/science.288.5466.669
65. Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M. Gene therapy of primary T cell immunodeficiencies. Gene (2013) 525(2):170–73. doi:10.1016/j.gene.2013.03.092
66. Knight S, Collins M, Takeuchi Y. Insertional mutagenesis by retroviral vectors: current concepts and methods of analysis. Curr Gene Ther (2013) 13(3):211–27. doi:10.2174/1566523211313030006
67. Romano G. Development of safer gene delivery systems to minimize the risk of insertional mutagenesis-related malignancies: a critical issue for the field of gene therapy. ISRN Oncol (2012) 2012:616310. doi:10.5402/2012/616310
68. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365(18):1673–83. doi:10.1056/NEJMoa1106152
69. Aksentijevich I, Kastner DL. Genetics of monogenic autoinflammatory diseases: past successes, future challenges. Nat Rev Rheumatol (2011) 7(8):469–78. doi:10.1038/nrrheum.2011.94
70. Park H, Bourla AB, Kastner DL, Colbert RA, Siegel RM. Lighting the fires within: the cell biology of autoinflammatory diseases. Nat Rev Immunol (2012) 12(8):570–80. doi:10.1038/nri3261
71. Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest (2007) 117(3):514–21. doi:10.1172/JCI30587
72. Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis (2010) 45(3):246–65. doi:10.1016/j.bcmd.2010.07.012
73. Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature (2013) 498(7455):506–10. doi:10.1038/nature12199
74. Milner JD, Holland SM. The cup runneth over: lessons from the ever-expanding pool of primary immunodeficiency diseases. Nat Rev Immunol (2013) 13(9):635–48. doi:10.1038/nri3493
75. Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science (2009) 326(5954):818–23. doi:10.1126/science.1171242
Keywords: gene therapy, cancer, immunotherapy, T cells, inflammation, chimeric-antigen receptors, cytokines, severe combined immunodeficiency
Citation: Kerkar SP (2013) “Model T” cells: a time-tested vehicle for gene therapy. Front. Immunol. 4:304. doi: 10.3389/fimmu.2013.00304
Received: 09 August 2013; Accepted: 12 September 2013;
Published online: 27 September 2013.
Edited by:
Bruno Laugel, Cardiff University School of Medicine, UKReviewed by:
David L. Wiest, Fox Chase Cancer Center, USABruno Laugel, Cardiff University School of Medicine, UK
Copyright: © 2013 Kerkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sid P. Kerkar, Laboratory of Pathology, Center for Cancer Research, National Cancer Institute, NIH Clinical Center, Room 2B38, Building 10, 10 Center Drive, Bethesda, MD 20892, USA e-mail:a2Vya2Fyc0BtYWlsLm5paC5nb3Ysc3BrZXJrYXJAeWFob28uY29t